

Abstract
Low intensity maintenance therapy with 6-mercaptopurine (6-MP) limits the occurrence of acute lymphoblastic leukemia (ALL) relapse and is central to the success of multi-agent chemotherapy protocols. Activating mutations in the cytosolic 5’ nucleotidase II (NT5C2) gene drive resistance to 6-MP in over 35% of early relapse ALL cases. Here we identify CRCD2 as a first-in-class small molecule NT5C2 nucleotidase inhibitor broadly active against leukemias bearing highly prevalent relapse-associated mutant forms of NT5C2 in vitro and in vivo. Importantly, CRCD2 treatment also enhanced the cytotoxic activity of 6-MP in NT5C2 wild type leukemias leading to the identification of NT5C2 S502 phosphorylation as a novel NT5C2-mediated mechanism of 6-MP resistance in this disease. These results uncover an unanticipated role of non-genetic NT5C2 activation as a driver of 6-MP resistance in ALL and demonstrate the potential of NT5C2 inhibitor therapy for enhancing the efficacy of thiopurine maintenance therapy and overcoming resistance at relapse.
Keywords: Acute lymphoblastic leukemia, ALL, relapse, chemotherapy resistance, NT5C2, targeted therapy, nucleotidase inhibitor
Introduction
High intensity multi-agent chemotherapy protocols combining glucocorticoids, DNA-damaging agents, mitotic poisons and L-asparaginase, followed by a prolonged lower intensity maintenance therapy with oral 6-mercaptopurine (6-MP) have become the gold-standard treatment for acute lymphoblastic leukemia (ALL) (1,2). These regimens achieve over 80% cure rates in children and almost a 50% long-term remission in adults with this disease (3,4). However, despite these overall favorable results, patients with incomplete responses and the even more sizeable number who experience relapse after a transient remission face a very poor prognosis. The unfavorable outcomes of relapsed ALL are linked to the development of secondary chemotherapy resistance, which hampers the efficacy of salvage therapies (5,6). Genomic analyses have identified specific genetic drivers of chemotherapy resistance and relapse in support of a Darwinian selection of resistance-mutation bearing clones at relapse (7–13). Prominent among these relapse-associated genetic alterations, activating mutations in the 5’-Nucleotidase Cytosolic II (NT5C2) gene are present in ~10% of relapsed B-precursor ALL cases and ~20% of T-ALLs (7,8) and are selectively more frequent in early relapses occurring during or early after maintenance therapy (7,8). Detailed genetic, enzymatic, structural and functional analyses of relapsed leukemia-associated NT5C2 mutant alleles identified three distinct NT5C2 gain-of-function mechanisms implicating direct stabilization of the catalytically active enzymatic configuration, the disruption of an intramolecular switch-off mechanism responsible for returning the enzyme to its basal inactive state following allosteric activation, and finally enhanced allosteric activation normally limited by the stabilization of the basal inactive form of the enzyme by the insertion of the C-terminal acidic tail in the basic interface separating the two NT5C2 dimer subunits (14–16). Despite this molecular heterogeneity, structural analyses of wild type and relapse-associated NT5C2 mutant proteins reveals a markedly convergent configuration for NT5C2 mutant alleles overlapping with that of the allosterically activated wild type enzyme (14–16). Mechanistically, relapse-associated gain-of-function NT5C2 mutant proteins induce resistance to treatment with 6-MP by facilitating the dephosphorylation and subsequent degradation of thio-purine nucleotides generated via incorporation of 6-MP in the salvage pathway of purine biosynthesis (17,18). Based on these results, we hypothesized that small molecule NT5C2 inhibitors could overcome thiopurine resistance at relapse and enhance the efficacy of 6-MP maintenance therapy in the clinic.
Results
CRCD2 inhibits NT5C2 activity in vitro
Besides its role in the inactivation of cytotoxic metabolites of 6-MP and 6-thioguanine, NT5C2 regulates the purine nucleotide intracellular pool by dephosphorylating 6-hydroxypurine nucleotide monophosphates such as inosine monophosphate (IMP), guanosine monophosphate (GMP) and xanthosine monophosphate (XMP) (19,20). To identify small molecule compounds with NT5C2 inhibitory activity, we screened a chemical library against recombinant NT5C2 R367Q-mutant protein, the most prevalent relapsed ALL-associated NT5C2 allele present in over 90% of NT5C2-mutant relapsed leukemia samples (14) (Fig. 1A–C). Analysis of 60,640 small molecules enriched in compounds with drug-like features in a high-throughput malachite-green-based nucleotidase assay using IMP as substrate identified 225 potential active small molecules with 33.21% inhibitory activity or greater. Following secondary screening of compounds with the highest Z’ scores, we validated the activity of top hits in dose-response curves (Fig. 1D). Independent analysis of top candidates with newly sourced compounds further confirmed the NT5C2 inhibitory activity of these compounds (Fig. 1E) and identified N-(3-carbamoyl-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-1H-benzo[d]imidazole-5-carboxamide (hereafter termed CRCD2) as the most active NT5C2 inhibitor in this screen. To further evaluate the specific inhibitory activity of CRCD2 and to verify its chemical structure, we tested an additional independently commercially sourced lot of this molecule and two batches of in-house synthesized compound generated using two different synthesis routes. All four sources of CRCD2 showed uniform dose-dependent inhibitory activity against NT5C2 R367Q (Fig. 1F).
Figure 1. High-throughput compound screen for the identification of NT5C2 inhibitors.
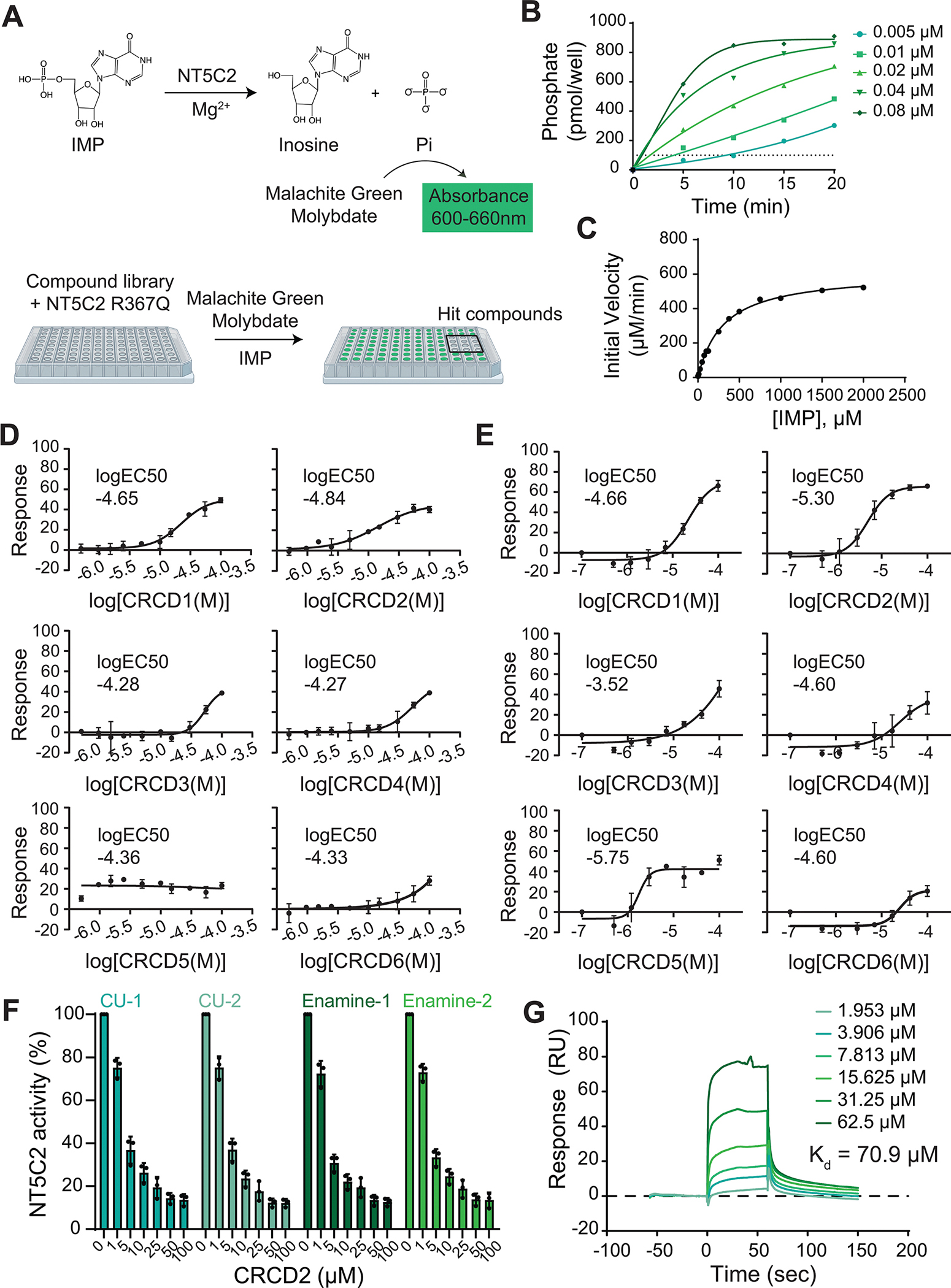
(A) Malachite green NT5C2 enzyme assay reaction and colorimetric measurement. (B) Reaction progress curve of NT5C2 R367Q recombinant protein at five protein concentrations. (C) Initial velocity versus substrate concentration of 0.02 μM NT5C2 R367Q recombinant protein with IMP as substrate. Vmax= 610 μM IMP/min, Km = 308 μM IMP. (D) Malachite green assays showing dose-response curves of top six compounds from the screening library. Graphs show technical replicates from a single experiment performed in triplicate. logEC50 for each compound is shown. (E) Malachite green assays showing dose-response curves for top hits sourced from an independent synthesis batch sourced from Enamine as in (E). (F) Malachite green assay analysis of CRCD2 synthesized at the Columbia Probe Synthesis facility by two different synthesis routes (CU-1, CU-2) and two different lots provided by Enamine (Enamine-1, Enamine-2) incubated with recombinant NT5C2 R367Q protein. Graph shows mean ± SD of three independent experiments performed in triplicate. (G) Sensorgram for CRCD2 binding to NT5C2 D52N R367Q recombinant protein generated with Biacore surface plasmon resonance technology. Equilibrium dissociation constant (Kd) is shown.
Expression of the Nt5c2 R367Q relapse-associated mutation in ALL cells results in a NT5C2-mediated increase in degradation of purine monophosphate nucleotides with consequent depletion of IMP and accumulation of purine degradation products compared with isogenic wild type controls (21). In this model, CRCD2 treatment of Nt5c2 R367Q expressing lymphoblasts resulted in increased levels of IMP and decreased accumulation of deoxyxanthosine congruent with the NT5C2 inhibitory activity of this compound (Fig. S1A–C). Furthermore, and in concert of a direct inhibitory effect, surface plasmon resonance analysis demonstrated direct binding of CRCD2 to NT5C2 R367Q recombinant protein with a KD of 70.9 μM (Fig. 1G).
Increased nucleotidase activity in the NT5C2 R367Q mutant results from loss of an intramolecular switch-off mechanism responsible for returning the enzyme to its basal inactive configuration following allosteric activation (14). This is in contrast with rare but highly active class I NT5C2 mutants, which reconfigure the environment of the catalytic center locking this region in its active open configuration, resulting in high levels of constitutive NT5C2 activity in the absence of allosteric effectors (14). To test the span of NT5C2 inhibition by CRCD2 (Fig. 2A), we evaluated the activity of this compound against NT5C2 R367Q in comparison with wild type NT5C2 and the class I NT5C2 K359Q mutant protein, the most active relapse-associated form of NT5C2. In these assays, CRCD2 displayed dose-dependent low micromolar inhibitory capacity against both NT5C2 wild-type and the R367Q mutant and significantly reduced NT5C2 K359Q nucleotidase activity, albeit with lower potency (Fig. 2B).
Figure 2. Binding and inhibition of wild type, R367Q and K359Q recombinant protein by CRCD2.
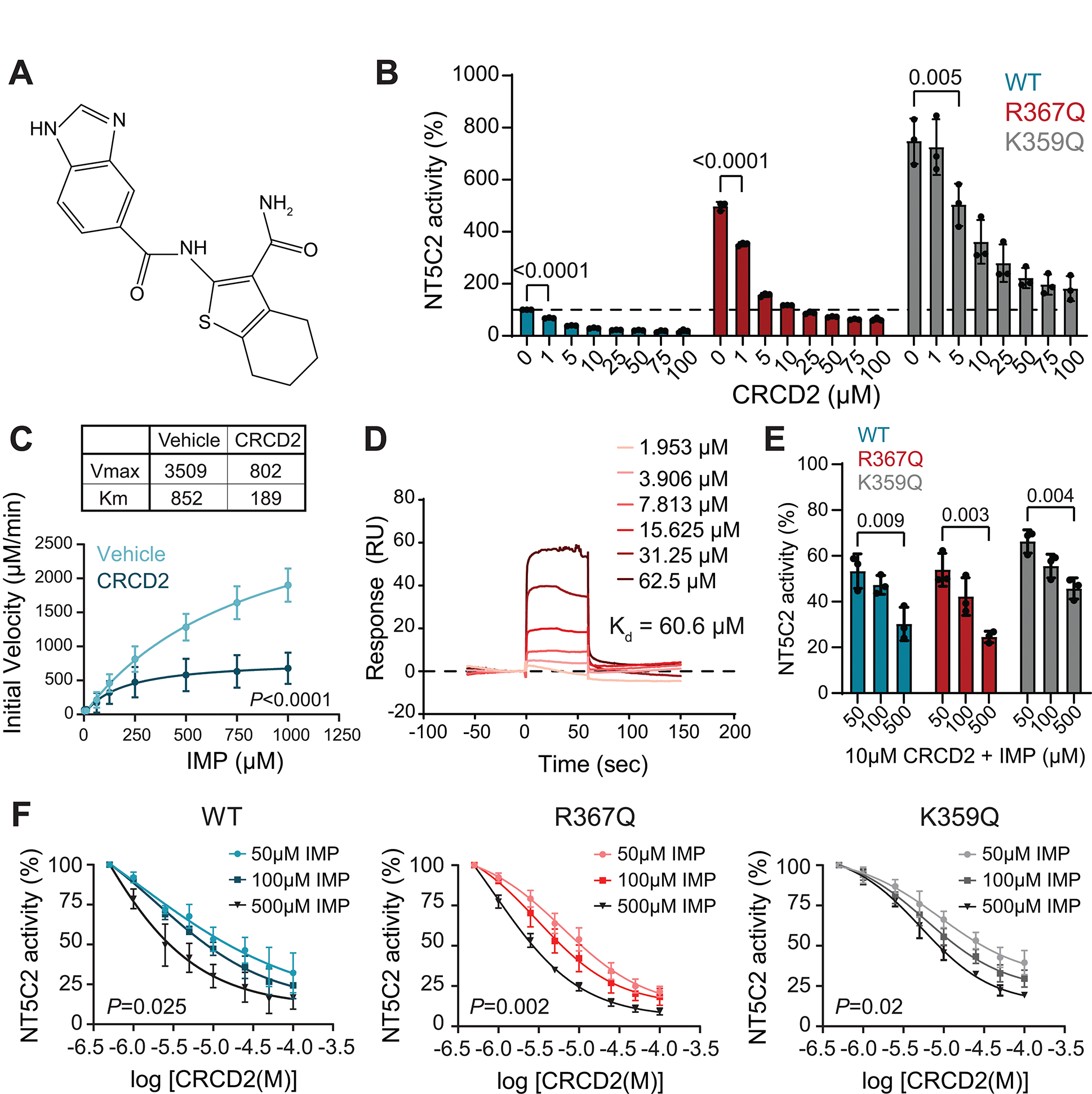
(A) Chemical structure of CRCD2 compound. (B) Malachite green assay of wild type, R367Q and K359Q NT5C2 recombinant protein incubated with increasing concentrations of CRCD2. Graph shows average ± SD of three independent experiments performed in triplicate. P values were calculated using ordinary one-way ANOVA and Dunnett’s multiple comparisons test. (C) Michaelis-Menten curve showing NT5C2 enzyme kinetics in the absence and in the presence of 10μM CRCD2. Graph shows average ± SD of three independent experiments performed in triplicate. We calculated the P value using a Michaelis-Menten non-linear regression curve fit test. (D) Sensorgram for CRCD2 binding to NT5C2 D52N R367Q recombinant protein in the presence of 500μM IMP, generated with Biacore surface plasmon resonance technology. Equilibrium dissociation constant (Kd) is shown. (E) Malachite green assay of wild type, R367Q and K359Q recombinant protein incubated with 10 μM CRCD2 and increasing concentrations of IMP. Graph shows average ± SD of three independent experiments performed in triplicate. P values were calculated using ordinary one-way ANOVA and Dunnett’s multiple comparisons test. (F) Malachite green assay of wild type, R367Q and K359Q recombinant protein incubated with increasing concentrations of both CRCD2 and IMP. Graphs show average ± SD of three independent experiments performed in triplicate. We calculated P values using two-tailed Student’s t-test of the AUC of 500 mM vs 50 mM IMP.
To evaluate the mechanism of NT5C2 inhibition by CRCD2 we tested the effect of this compound against NT5C2 R367Q in the presence of increasing concentrations of substrate (IMP). Michaelis-Menten curve analyses of these results showed a reduction in the maximum rate of reaction (Vmax) and in the Michaelis constant (Km) in support of an uncompetitive mode of action (Fig. 2C), in which inhibitory effects are enhanced in the presence of substrate. Consistent with this model, surface plasmon resonance analysis of CRCD2 binding to NT5C2 R367Q in the presence of IMP revealed a 14.5% decrease in KD compared with substrate free conditions (Fig 2D). In agreement, in vitro enzymatic assays testing CRCD2 against NT5C2 wild type, R367Q and K359Q recombinant proteins in the presence of increasing concentrations of IMP showed a significantly higher dose-dependent inhibitory capacity in the presence of substrate (Fig. 2E,F). To get a deeper insight into this mechanism of action, we performed a hydrogen-deuterium exchange mass spectrometry (HDX-MS) analysis of NT5C2 R367Q in the presence of IMP as substrate and in the presence or absence of CRCD2 (Fig. S2A,B). Consistent with their high dynamic activity, we observed high deuterium exchange rates in regulatory regions of the protein implicated in allosteric activation and return to the basal inactive state including the Arm domain and the N-terminal segment (Fig. S2C). Moreover, we detected several peptides showing a different Deuterium exchange kinetics in the presence of CRCD2 (Fig. S2D). Most of these locate in the Arm domain of NT5C2, a result consistent with decreased dynamics of this region consequent to decreased enzymatic activity in the presence of inhibitor. In addition, and most interestingly, we also observed changes in Deuterium exchange kinetics in 4 peptides in the HAD III catalytic domain of NT5C2 (Fig. S2E), a region critical for NT5C2 activity located near the substrate binding area (Fig. S2F). These results suggest that CRCD2 binds to or induces a reconfiguration of the catalytic domain environment in the presence of substrate, a mechanism congruent with the uncompetititive nature of this inhibitor in enzymatic assays. In all, these results identify CRCD2 as a first in class small molecule uncompetitive inhibitor of wild type and relapsed leukemia-associated gain of function mutant forms NT5C2.
Exploration of structure-activity relationships of CRCD2 led to the identification of several active analogs and revealed relevant moieties needed for the NT5C2 inhibitory activity. Methylation of the benzimidazole ring on C-2 (CRCD47) or nitrogen (CRCD48) gave active analogs. However, replacement of one of the benzimidazole ring nitrogens with sulfur (CRCD18) or CH (CRCD74) was not tolerated (Fig. S3A). Replacement of the benzimidazole ring with a diamino phenyl group (CRCD54) or a phenylpyrrolidine-2,5-dione (CRCD9) led to an active analogs, however, replacement with benzotriazole (CRCD57) or 2-aminobenzimidazole (CRCD58) led to loss of potency (Fig. S3B). Methylation of the amide nitrogen in CRCD2 led to an analog (CRCD40) with a loss of potency, as did replacement of the amide with a methyl ester (CRCD43) or carboxylic acid moiety (CRCD76). In contrast, replacement of the amide group in CRCD2 with a nitrile (CRCD22) gave an active analog. Modification of the saturated six-membered ring fused to the thiofuran with a five-membered ring gave an active analog (CRCD41), however, the seven-membered ring analog (CRCD49) was not potent. Other modifications such as conversion of the six-membered ring to a pyran ring (CRCD42) or methylation of the ring (CRCD39) were also not tolerated (Fig. S3C). Removal of the saturated six-membered ring (CRCD50) or replacement with one (CRCD51, CRCD53) or two methyl groups (CRCD52) also led to loss of potency (Fig. S3D). Exploration of replacement of the 4,5,6,7-tetrahydrobenzo[b]thiophene moiety with other moieties (for example alkyl, aromatic, and heteroaromatic systems) did not lead to potent analogs (CRCD59-CRCD73) (Fig. S3E).
These structure activity relationship analyses reveal several regions of the CRCD2 scaffold that can be further modified to enhance potency and physicochemical properties (Fig. S3F). For example, the benzimidazole ring can be substituted at carbon or nitrogen (CRD47 and CRCD48, respectively) and functionalized substituents could make additional binding interactions such as electrostatic or hydrogen bonding with the enzyme to increase potency and/or contain water solubilizing groups to enhance solubility. The amide on the thiophene ring of CRCD2 can be replaced with a nitrile group (CRCD22), suggesting that exploration of other substituents at this position could lead to increased activity. Similarly, the amines in active analog CRCD54 could be alkylated or acylated to explore additional chemical space with the potential to increase potency. In addition, other modifications like fused aromatics, use of other heterocycles (furan, pyrrole) or linking longer groups or with other functionalities (ether, alkyl) are worth being explored.
CRCD2 reverses 6-MP resistance in vitro and in vivo
Following structure activity relationship analyses and given that the original hit compound CRCD2 showed the highest level of NT5C2 inhibitory activity we evaluated the capacity of this small molecule to reverse 6-MP resistance driven by relapsed-leukemia associated NT5C2 mutations in cellular assays. Towards this goal, we analyzed the response to 6-MP in NT5C2 wild-type Jurkat and CUTLL1 T-ALL cells and in PEER and BE13 lines, which harbor the relapse-associated activating NT5C2 R29Q mutation. These analyses revealed increased sensitivity to 6-MP following CRCD2 treatment of wild type cells and effective reversal of 6-MP resistance driven by the NT5C2 R29Q allele (Fig. 3A,B). Similar, results were obtained in REH (NT5C2 wild-type) and 697 (NT5C2 R368W mutant) B-precursor ALL cell lines (Fig. 3C). In addition, CRCD2 treatment effectively reversed 6-MP resistance induced by lentiviral expression of a K359Q, L375F, R367Q and D407A mutant forms of NT5C2 in Jurkat cells (Fig. 3D–G). Moreover, CRCD2 treatment of NT5C2 R367Q mutant patient-derived xenograft cells from a pediatric T-ALL patient at relapse (11) (Supplementary Table 1) induced increased sensitivity to 6-MP treatment (Fig. 3H).
Figure 3. CRCD2 sensitizes to 6-MP chemotherapy in human ALL cell lines harboring wild-type and mutant NT5C2 and in ALL patient-derived xenografts.
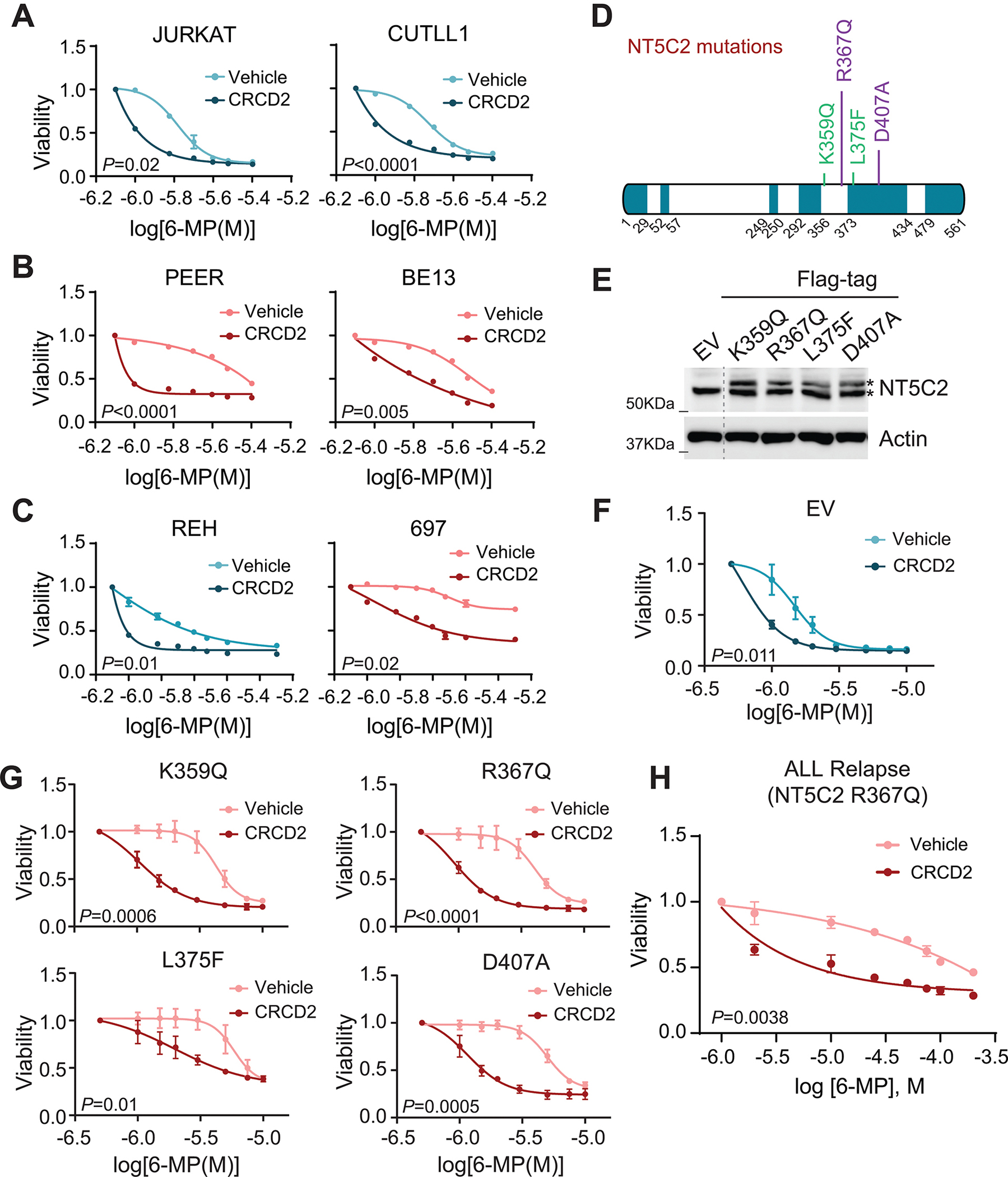
(A) Jurkat and CUTLL1 NT5C2 WT ALL cells treated with vehicle or 10 μM CRCD2 and increasing doses of 6-MP. Graph shows mean ± SD of three independent experiments performed in triplicate. (B) PEER and BE13 NT5C2 mutant ALL cells treated with vehicle or 10 μM CRCD2 and increasing doses of 6-MP. Graph shows mean ± SD of three independent experiments performed in triplicate. (C) REH (NT5C2 wild type) and 697 (NT5C2 R278W mutant) B-precursor ALL cells treated with vehicle or 10 μM CRCD2 and increasing doses of 6-MP. Graph shows mean ± SD of three independent experiments performed in triplicate. (D) Schematic representation of the NT5C2 mutants selected for the following assays. Mutants resulting in constitutive allosteric independent activation are shown in green Mutants with impaired intramolecular enzymatic switch off are shown in purple. (E) Immunoblot analysis of Jurkat cells expressing infected with empty vector or lentiviruses driving the expression of Flag-tagged mutants of NT5C2. Expression levels were verified in three independent experiments. (F) Viability assay of Jurkat cells infected with empty vector lentiviruses treated with vehicle or 10 μM CRCD2 and increasing doses of 6-MP. Graphs show mean ± SD of three independent experiments performed in triplicate. (G) Viability assay of Jurkat cells infected with mutant NT5C2 expressing lentiviruses treated with vehicle or 10 μM CRCD2 and increasing doses of 6-MP as in (F). (H) Patient-derived xenograft relapsed-T-ALL NT5C2-mutant lymphoblasts treated with vehicle or 10μM CRCD2 and increasing doses of 6-MP. Graph shows mean ± SD of two independent experiments performed in triplicate. All P values were calculated using IC50 values and two-tailed Student’s t-test over wild-type.
Increased sensitivity to 6-MP in wild type and NT5C2 mutant-expressing cells is consistent with a broad inhibitory activity of CRCD2 against wild type and different relapse-associated resistance-driving forms of NT5C2 and supports that basal levels of NT5C2 in wild type cells negatively impact the cytotoxic capacity of 6-MP. To further test this possibility and to evaluate the specificity of the effects of CRCD2 in 6-MP resistance, we tested the thiopurine sensitivity of isogenic NT5C2 wild type and knockout CUTLL1 cells (Fig. S4A,B) in presence and absence of inhibitor. In these experiments, treatment with CRCD2 and genetic inactivation of NT5C2 induced similar increased sensitivity to 6-MP further supporting a role for the wild type enzyme in modulating the response to thiopurine therapy (Fig. S4C–E). Moreover, NT5C2 knockout cells showed similar response to 6-MP in the presence and absence of CRCD2 (Fig. S4C–E). The epistatic interaction of CRCD2 with NT5C2 knockout in the response to 6-MP corroborates the NT5C2-specific mechanism of action of this inhibitor.
Next, we aimed to directly test the differential interaction of CRCD2 with 6-MP in the context of isogenic Nt5c2 wild type and Nt5c2 R367Q mutant primary leukemia lymphoblasts. Towards this goal, we isolated mouse leukemia cells generated via bone marrow transplantation of hematopoietic progenitors from conditional inducible Nt5c2 knockin mice (Rosa26+/CreERT2Nt5c2+/co-R367Q) infected with retrovirus particles expressing a constitutively active form of NOTCH1 (ΔE-NOTCH1). In this validated model of resistance (21), tamoxifen treatment activates the expression of the Nt5c2 R67Q mutant knockin allele in leukemia lymphoblasts resulting in overt resistance to 6-MP (Fig. 4A). In this experiment, treatment of Nt5c2 wild type ALL cells with CRCD2 induced increased sensitivity to 6-MP, and treatment of their isogenic Nt5c2 R367Q counterparts resulted in effective reversal of 6-MP resistance (Fig. 4B–D). Further analyses of drug response curves across multiple dose levels revealed mild synergism (combination index = 0.8) between CRCD2 and 6-MP in drug-sensitive Nt5c2 wild type cells, and a strong synergistic interaction in 6-MP resistant tamoxifen-treated Nt5c2 R367Q lymphoblasts (combination index = 0.32) (Fig. 4E–G).
Figure 4. NT5C2 inhibitor CRCD2 sensitizes both NT5C2 WT (Nt5c2+/co-R367Q) and NT5C2 mutant (Nt5c2+/R367Q) mouse lymphoblasts to 6-MP.
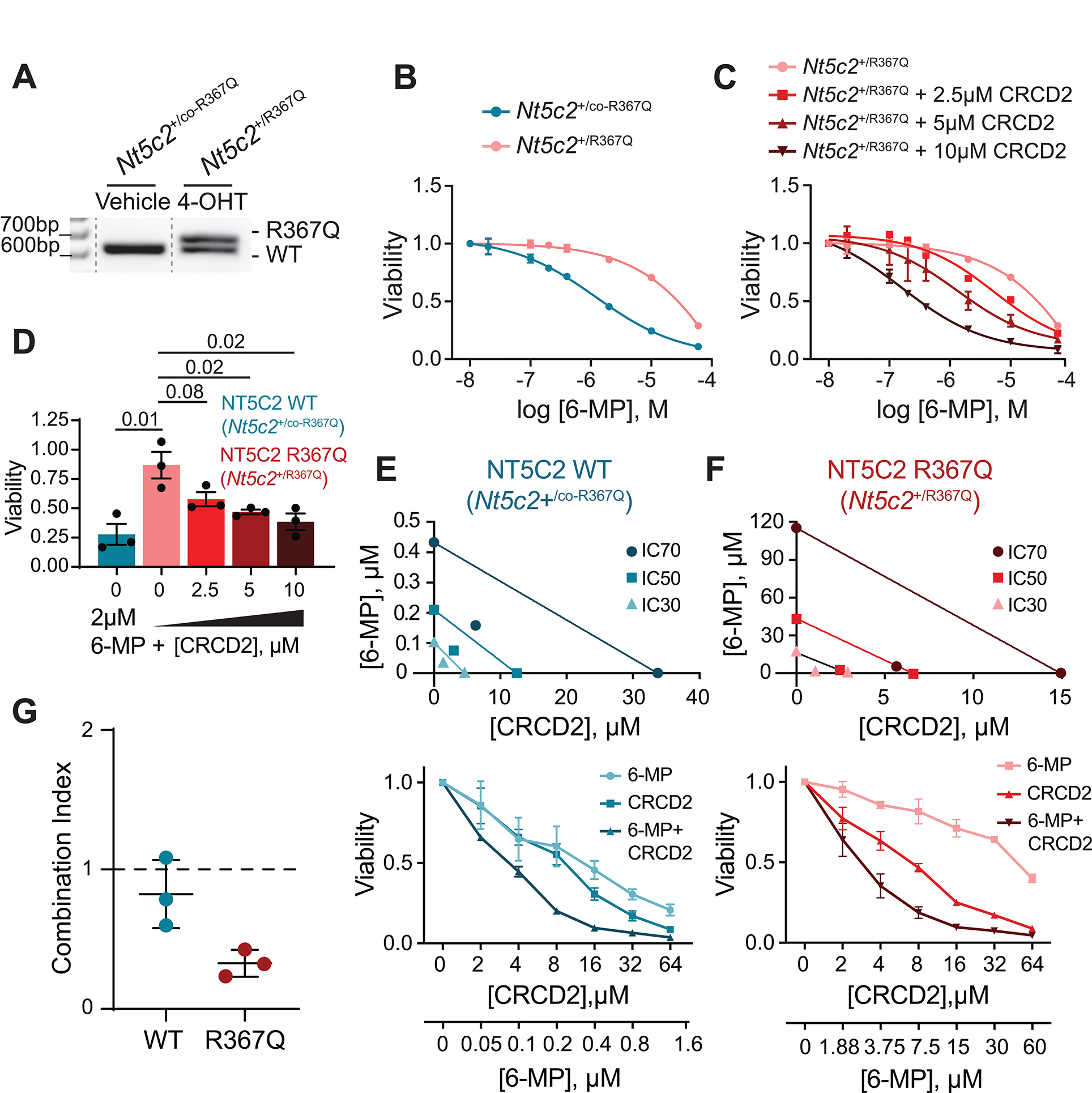
(A) PCR amplification of Nt5c2 wild-type and R367Q mutant alleles after 4-OHT treatment. Isobologram analysis of NT5C2 WT (Nt5c2+/co-R367Q). (B) Cell viability of NT5C2 wild type (Nt5c2+/co-R367Q) and NT5C2 mutant (Nt5c2+/R367Q) mouse lymphoblasts treated with increasing doses of 6-MP and CRCD2. Graph shows one representative experiment with technical replicates. Two additional experiments showed similar results. (C) Cell viability of NT5C2 mutant (Nt5c2+/R367Q) mouse lymphoblasts treated with increasing doses of 6-MP and CRCD2. Graph shows one representative experiment with technical replicates. Two additional experiments showed similar results. (D) Cell viability of NT5C2 wild type (Nt5c2+/co-R367Q) and NT5C2 mutant (Nt5c2+/R367Q) mouse lymphoblasts treated with 2 μM 6-MP and increasing doses of CRCD2. Graph shows mean ± SD of three independent experiments performed in triplicate. P values were calculated using two-tailed Student’s t-test. (E) Isobologram analyses and cell viability of mouse wild type (Nt5c2+/co-R367Q) lymphoblasts treated with 6-MP, CRCD2, or a combination of 6-MP and CRCD2. Graphs show mean of three technical replicates. We repeated the experiment two additional times with similar results. (F) Isobologram analyses and cell viability of NT5C2 mutant (Nt5c2+/R367Q) mouse lymphoblasts treated with 6-MP, CRCD2, or a combination of 6-MP and CRCD2. Graphs show mean of three technical replicates. We repeated the experiment two additional times with similar results. (G) Combination Index of isogenic wild type and Nt5c2 R367Q mutant mouse ALL lymphoblasts treated with 6-MP and CRCD2. Dots represent mean values from three independent experiments performed in triplicate.
Following on these in vitro results, we evaluated the therapeutic activity of CRCD2 for reversal of 6-MP resistance in vivo. Analysis of plasma clearance kinetics following CRCD2 administration revealed favorable pharmacokinetics with a half-life of 3.2 hours in female C57/BL6 mice (Fig. S5A). Consistently, we documented a significant reduction in plasma 5’ nucleotidase activity in vivo 1 hour after CRCD2 injection (Fig. S5B). In addition, intraperitoneal injection of CRCD2 in a 5 days on-2 days off dose escalation scheme revealed no weight loss or apparent toxicities at the limit of solubility of this drug (Fig. S5C,D). Moreover, histopathologic analysis of CRCD2 treated animals revealed no specific alterations in intestine, kidney, liver and brain compared with controls in support of a favorable safety profile (Fig. S5E). Evaluation of the toxicity profile of CRCD2 with 6-MP in combination over 5 days of treatment (Fig. S5F) compared with 6-MP alone revealed that adding CRCD2 to thiopurine treatment results in a modest effect in body weight and no differences in (Fig. S5G) in bone marrow cellularity (Fig. S5H) and peripheral blood cell counts (Fig. S5I–J). We noted no specific histological changes in kidney and intestine. Finally, similar alterations including a decrease in bone marrow cellularity and focal, mild sinusoidal dilatation and congestion in the liver (Fig. S5K), consistent with the toxicity profile of 6-MP were noted in both groups.
To test the therapeutic activity of CRCD2 we allografted luciferized NOTCH1-induced conditional inducible Nt5c2 R367Q (Rosa26+/CreERT2 Nt5c2+/co-R367Q) T-ALL tumor cells into isogenic mice and treated these with vehicle only or tamoxifen to generate isogenic wild type and Nt5c2 R367Q leukemias, respectively (21). Consistent with the role of Nt5c2 R367Q in driving 6-MP resistance, treatment of Nt5c2 wild-type leukemia-bearing mice with 6-MP at 50 mg/kg and 100 mg/kg induced overt and complete therapeutic responses, respectively; while treatment of Nt5c2 R367Q expressing tumors resulted in significantly decreased therapeutic effects (Fig. S6A,B) (21). In this setting, treatment of Nt5c2 wild type leukemias with vehicle only, 6-MP, CRCD2, and 6-MP plus CRCD2 in combination (Fig 5A) revealed significantly improved antitumor activity in the CRCD2 plus 6-MP cotreatment arm after 5 days of treatment as evidenced by quantification of tumor burden by luciferase bioimaging, spleen size and spleen and bone marrow tumor cell content (Fig 5B–E). In addition, treatment of mice harboring Nt5c2 R367Q tumors documented effective reversal of 6-MP resistance in animals cotreated with 6-MP and CRCD2 in combination (Fig 5F–I). Moreover, in vivo treatment of our relapsed-leukemia-derived T-ALL xenograft harboring the R367Q NT5C2 mutation (Fig S7A) verified enhanced antitumor response for the 6-MP plus CRCD2 combination (Fig. S7B and S7C) when compared to 6-MP treatment alone.
Figure 5. Response of NT5C2 wild type and R367Q tumors to CRCD2 and 6-MP combinatorial chemotherapy in vivo.
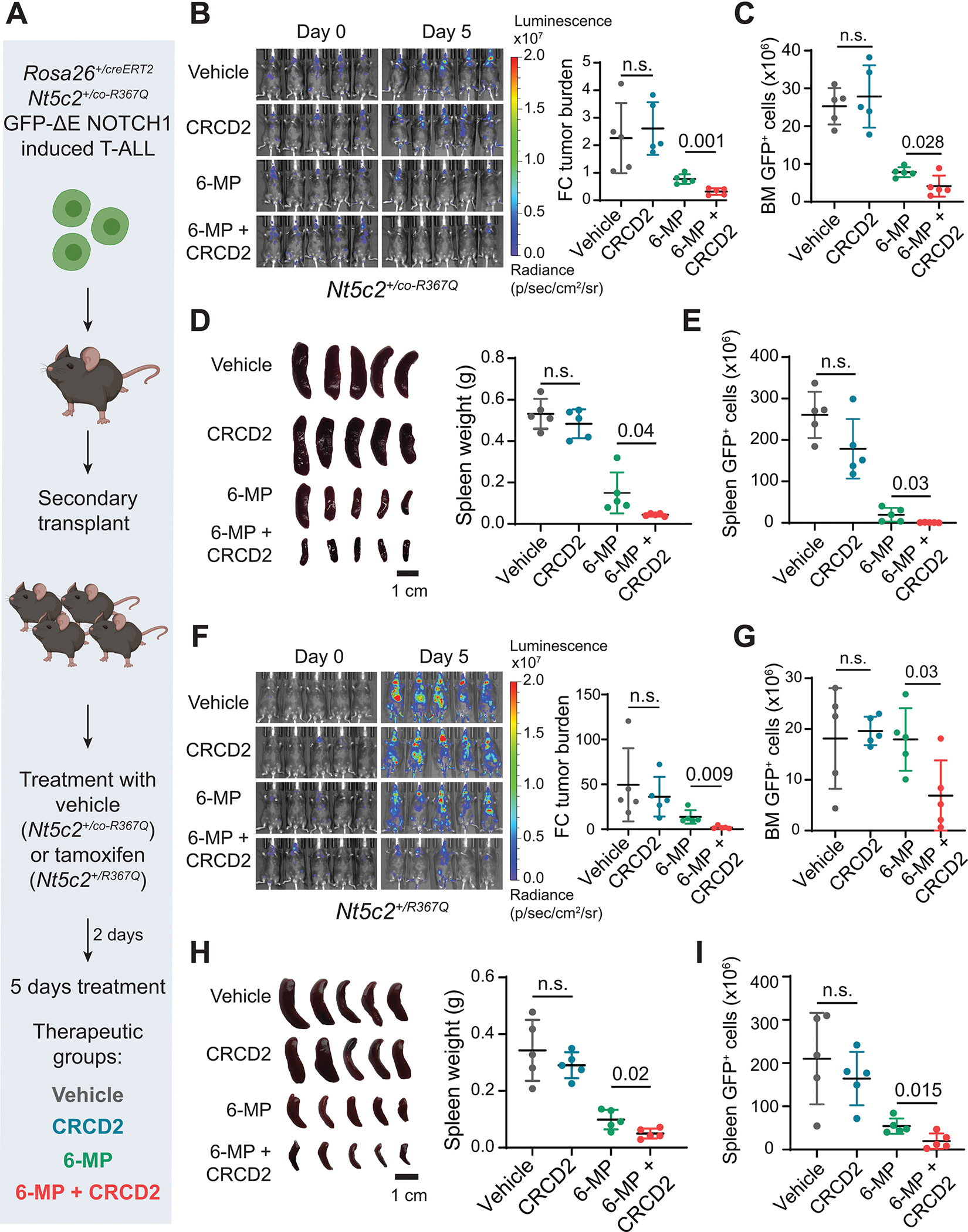
(A) Schematic illustration of 6-MP and CRCD2 combination experimental therapeutic treatment in Nt5c2+/co-R367Q (wild type) and Nt5c2+/R367Q (mutant) NOTCH1- induced ALL mouse model. (B) Luciferase in vivo bioimaging indicative of tumor burden and quantitative analysis of tumor response (fold change in bioluminescence relative to the basal signal before treatment) in Nt5c2 wild-type ALL tumors treated with vehicle, single-drug or 6-MP and CRCD2 combination. (C) Femoral bone marrow leukemia burden (GFP+ cells) analyzed by flow cytometry following treatment of wild type leukemias as in (A). (D) Images of spleens and quantitation of spleen weight following treatment of wild type leukemias as in (A). (E) Spleen leukemia burden (GFP+ cells) analyzed by flow cytometry following treatment of wild type leukemias as in (A). (F) Luciferase in vivo bioimaging indicative of tumor burden and quantitative analysis of tumor response (fold change in bioluminescence relative to the basal signal before treatment) in Nt5c2 R367Q ALL tumors treated with vehicle, single-drug or 6-MP and CRCD2 combination as in (A). (G) Femoral bone marrow leukemia burden (GFP+ cells) analyzed by flow cytometry following treatment of Nt5c2 R367Q ALL bearing mice as in (A). (H) Images of spleens and quantitation of spleen weight following treatment of in Nt5c2 R367Q ALL tumors as in (A). (I) Spleen leukemia burden (GFP+ cells) analyzed by flow cytometry following treatment of wild type leukemias as in (A). N = 5 independent mice per treatment condition. Data are presented as mean values ± SD. P values were calculated applying two-sided Student’s t-test.
Altogether, these results demonstrate effective CRCD2-mediated inhibition of Nt5c2 in wild type and R367Q mutant expressing leukemia cells in vivo, which results in increased therapeutic response to 6-MP and effective reversal of 6-MP resistance, respectively.
NT5C2 Ser502 phosphorylation induces NT5C2 activation and drives resistance to 6-MP at relapse
Early relapsed ALLs, those with disease progression under 6-MP chemotherapy, have a particularly high prevalence of NT5C2 mutations pointing to increased NT5C2 activity as a prominent mechanism of resistance. However, about 65% early relapsed ALL cases are wild type for NT5C2 and are devoid of other known 6-MP resistance-driver mutations. Across our experimental therapeutic assays we observed that, even though NT5C2 wild type protein shows limited enzymatic activity in cell free systems, treatment with CRCD2 resulted in increased sensitivity to 6-MP in NT5C2 wild type leukemia cells in vitro and in vivo. Given the dynamic role of NT5C2 configuration changes in the regulation of NT5C2 nucleotidase activity, we hypothesized that post-translational modifications involving NT5C2 regulatory regions could induce increased nucleotidase activity, phenocopy the effects of NT5C2 mutations, and drive 6-MP resistance by convergent therapeutically relevant non-genetic mechanisms in ALL that could also be pharmacologically targeted for enhanced efficacy during maintenance.
To explore this possibility, we performed mass spectrometry analysis of NT5C2 wild type protein immunoprecipitated from Jurkat human ALL cells. These analyses identified Ser418 and Ser502 as NT5C2 phosphorylation sites and residues Lys217 and Lys344 as modified by lysine acetylation (Fig. S8A,B). Each of these NT5C2 phosphorylation and acetylation sites are highly evolutionarily conserved across vertebrate species (Fig. S8C). To evaluate the potential regulatory role and functional relevance of these modifications as drivers of 6-MP resistance we expressed acetylation mimic (K217Q, K344Q), acetylation disruptive (K217R, K344R), phosphorylation mimic (S418D, S502D) and phosphorylation disruptive (S418A, S502A) mutant forms of NT5C2 in Jurkat ALL cells and assessed their impact in response to 6-MP (Fig. S8D). These analyses revealed not significant changes in the response to 6-MP in cells expressing K217, K344 and S418 modification mutant forms of NT5C2 (Fig. S8E). In contrast, expression of Ser502 disruptive (S502A) and phosphomimic (S502D) NT5C2 mutants in this system resulted in 6-MP resistance in support of a regulatory role for Ser502 in NT5C2 activity (Fig. 6A–C). Consistently, enzymatic analysis of recombinant NT5C2 S502D protein revealed increased nucleotidase activity in response to allosteric activation compared with wild type NT5C2 control, further supporting a role for S502 phosphorylation in the regulation of NT5C2 function (Fig. 6D). NT5C2 is a tetrameric protein organized as a dimer of dimers with well-defined regulatory elements implicated in allosteric activation and the resolution of enzymatic activity and the return of the protein to its basal inactive closed configuration (14). Close examination of S502 in the crystal structure of wild-type NT5C2, revealed a hydrogen bond between this residue and Asp229 in the neighbor NT5C2 subunit selectively present in the absence of allosteric activators (Fig. 6E). Moreover, analysis of the crystal structure of the NT5C2 Ser502 phosphomimic mutant revealed a conformational change in which residues 494–561 in the C-terminal region of NT5C2 adopt a disordered conformation (Fig. 6F). These results suggest that the Ser502-Asp229 hydrogen bond contributes to stabilize this protein in its inactive closed configuration. To test the potential regulatory role of this Ser502-Asp229 hydrogen bond in NT5C2 regulation, we generated NT5C2 mutants disrupting this interaction and tested their capacity to induce resistance to 6-MP when expressed in ALL cells. In these experiments, expression of hydrogen bond disruptive mutants (S502D, D229A) induced resistance to 6-MP compared to the wild-type protein (Fig. 6G–I). In contrast, a dual mutant (S502D/D229S) in which the Ser and Asp residues are swapped to preserve the formation of the inter-monomeric hydrogen bond failed to induce 6-MP resistance over the wild-type (Fig. 6J). In all, these results identify the Ser502-Asp229 inter molecular hydrogen bond as a novel regulatory element in the control of NT5C2 nucleotidase activity and supports a role of NT5C2 Ser502 phosphorylation in the regulation of 6-MP metabolism and clearance.
Figure 6. Molecular and structural characterization of NT5C2 phosphorylation at Ser502.
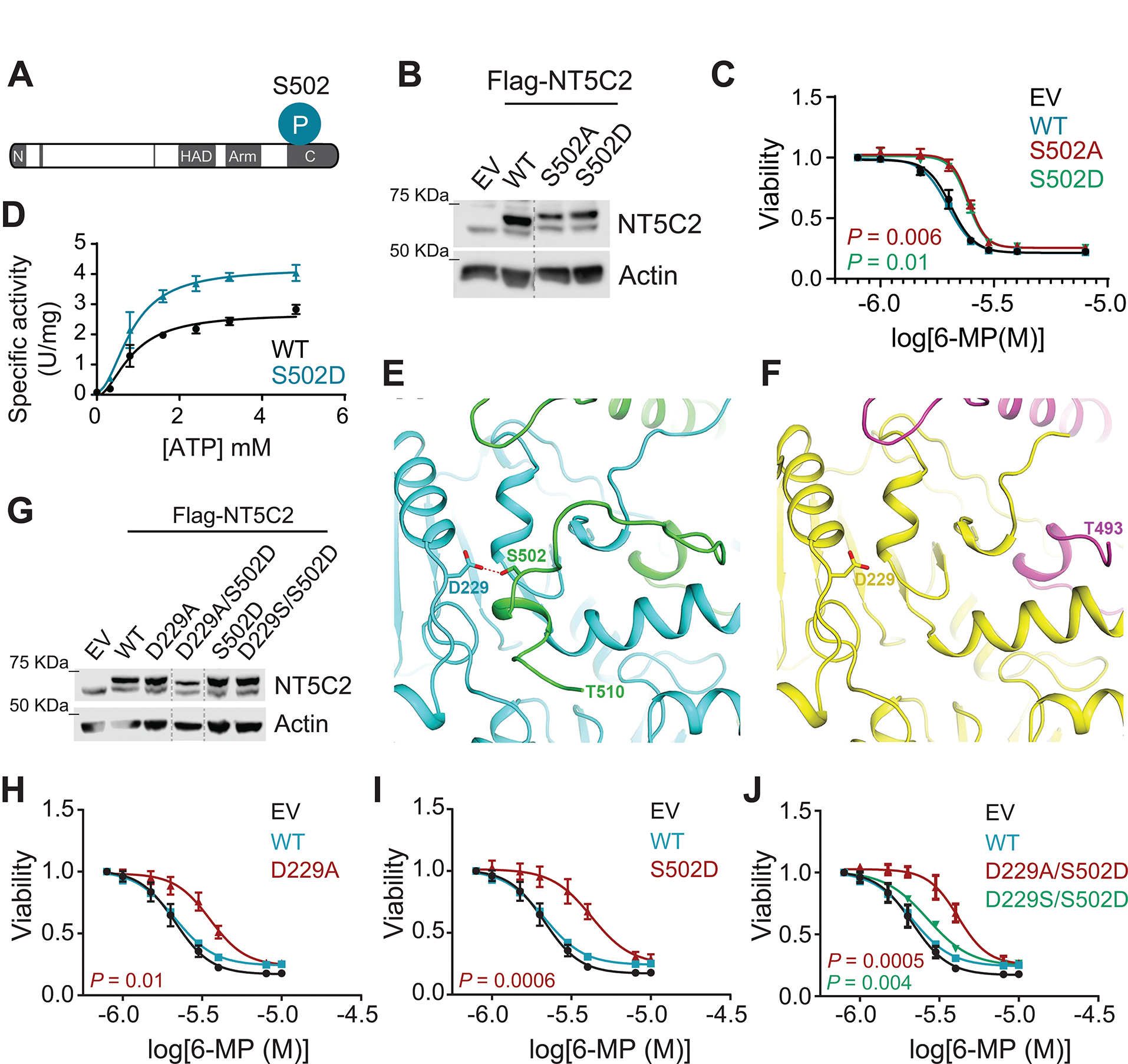
(A) Schematic illustration of Ser502 phosphorylation at the C-terminal domain of NT5C2. (B) Immunoblot analysis of Jurkat cells infected with empty vector or lentiviruses driving the expression of Flag-tagged Ser502 mutants of NT5C2. Expression levels were verified in three independent experiments. (C) Viability assay of Jurkat cells infected with mutant Ser502 NT5C2 expressing lentiviruses treated with increasing doses of 6-MP. (D) In vitro nucleotidase assays assessing the enzymatic activity of wild-type and S502D NT5C2 using increasing concentrations of ATP represented as specific activity. (E) Close-up view of the crystal structure of full-length wild-type NT5C2 (PDB id: 6DDO) showing residue S502 (green) from protomer A forms a hydrogen bond (red dash line) with D229 from protomer B (cyan). (F) Close-up view of the crystal structure of the full-length NT5C2 mutant S502D showing that mutation of S502 to D results in destabilization of C-terminal region (494–561). Protomers A and B are shown in magenta and yellow, respectively. (G) Immunoblot analysis of Jurkat cells infected with empty vector or lentiviruses driving the expression of Flag-tagged Asp-229 and Ser502 mutants of NT5C2. Expression levels were verified in three independent experiments. (H) Viability assay of Jurkat cells infected with mutant D229A NT5C2 expressing lentiviruses treated with increasing doses of 6-MP. (I) Viability assay of Jurkat cells infected with mutant S502D NT5C2 expressing lentiviruses treated with increasing doses of 6-MP. (J) Viability assay of Jurkat cells infected with double Asp-229 and Ser502 mutants expressing lentiviruses treated with increasing doses of 6-MP. All graphs show mean ± SD of three independent experiments performed in triplicate. P values were calculated using IC50 values and two-tailed Student’s t-test over wild-type.
To further explore the potential role of NT5C2 Ser502 phosphorylation as non-genetic mechanism of NT5C2 activation and the relevance of this posttranslational modification as a driver of 6-MP resistance in ALL we generated and characterized a NT5C2 Ser502 phospho-specific antibody. Western blot analysis of Jurkat cell lysates with anti-pSer502 NT5C2 revealed a single band corresponding to the predicted size of NT5C2 (65 kDa), which was ablated by preincubation with alkaline phosphatase in support of a specific phosphorylation-dependent signal (Fig. 7A). In addition, we observed no reactivity against NT5C2 S502A or S502D mutant proteins immunoprecipitated from Jurkat cells expressing Flag-NT5C2 constructs, further supporting the specificity of this antibody against NT5C2 Ser502 phosphorylation (Fig. 7B). Quantitative Western blot analysis of matched diagnosis and relapse patient derived xenografts revealed increased NT5C2 S502 phosphorylation levels in 50% (6/12) relapsed ALL xenografts analyzed compared to the respective diagnosis xenograft counterpart samples (Fig. 7C and D). We observed no effects of CRCD2 or 6-MP treatment on NT5C2 Ser502 phosphorylation in short treatment (Fig. 7E). In this context, the increased sensitivity to 6-MP of ALL cells expressing NT5C2 wild type following treatment with CRCD2 could result from inhibition of NT5C2 activity induced at least in part by Ser502 phosphorylation (Fig. 7F and G). To formally test this possibility, we evaluated the ability of CRCD2 to enhance the therapeutic response to 6-MP in resistant cells expressing the gain-of-function NT5C2 S502D phosphomimic mutant. In these experiments NT5C2 inhibition with CRCD2 effectively reversed 6-MP resistance induced by NT5C2 S502D to the same extent as that induced by expression of the relapse-associated NT5C2 R367Q mutant allele (Fig. 7F and G). These results support a role for non-genetic activation of NT5C2 nucleotidase activity as contributing factor to impaired response to 6-MP in ALL and argue for a therapeutic role of NT5C2 inhibition with CRCD2 in the reversal of genetic and non-genetic mechanisms of 6-MP resistance in this disease.
Figure 7. Prevalence and treatment of NT5C2 Ser502 phosphorylation in relapse ALL patient-derived xenografts.
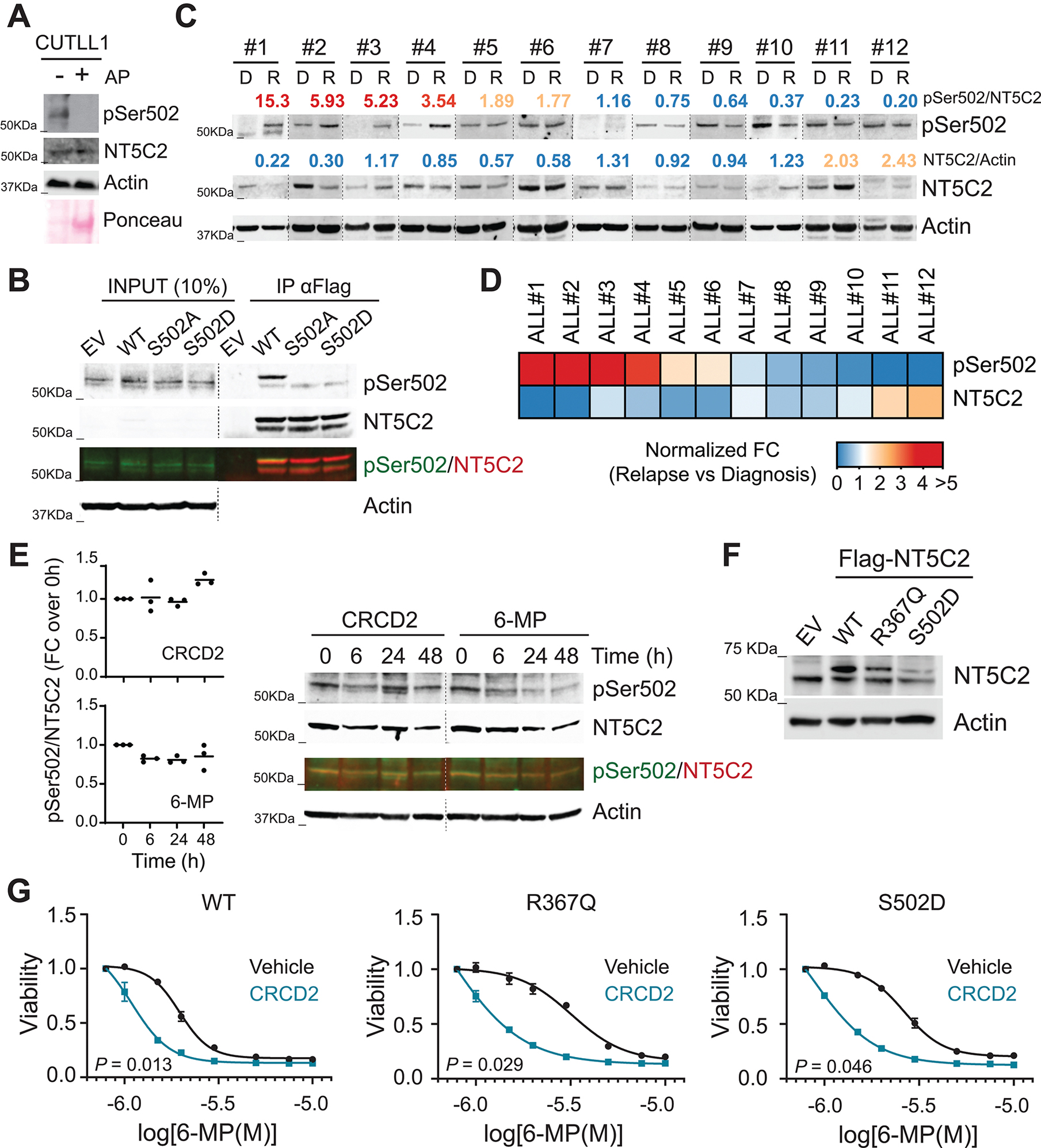
(A) Western-blot detection of NT5C2 pSer502 in CUTLL1 T-ALL cells. AP: alkaline phosphatase. (B) Western-blot detection of NT5C2 Ser502 phosphorylation after anti-Flag immunoprecipitation in Jurkat cells infected with empty vector or lentiviruses driving the expression of Flag-tagged wild-type, S502A and S502D mutants of NT5C2. Two additional experiments showed similar results. (C) NT5C2 Ser502 phosphorylation analysis in diagnostic and relapsed ALL patient derived xenografts. Numbers show normalized fold change of pSer502-NT5C2 and total levels of NT5C2 in relapse compared to the matched diagnosis xenograft. (D) Heatmap representation of normalized fold change of pSer502-NT5C2 and total levels of NT5C2 as in (C). (E) Western-blot detection of NT5C2 pSer502 levels in CUTLL1 cells treated with 10 μM CRCD2 or 1.5 μM 6-MP for 0, 6, 24 or 48 hours. A representative immuno-blot is shown. (F) Immunoblot analysis of Jurkat cells infected with empty vector or lentiviruses driving the expression of Flag-tagged wild-type, R367Q and S502D mutants of NT5C2. Expression levels were verified in three independent experiments. (G) Viability assay of Jurkat cells infected with wild-type or R367Q or S502D mutant NT5C2 expressing lentiviruses treated with vehicle or CRCD2 and increasing doses of 6-MP. Graphs show mean ± SD of three independent experiments performed in triplicate. P values were calculated using IC50 values and two-tailed Student’s t-test over wild-type.
Discussion
Low dose maintenance therapy with 6-MP effectively curtails the risk of relapse in ALL following high dose combination chemotherapy and represents a core component of the treatment of this disease (22–25). The critical role of 6-MP in the clinic is highlighted by the importance of duration, dose intensity and compliance during maintenance for the successful control of relapse driving leukemia clones (26,27). Indeed, therapy compliance monitoring has established decreased adherence to 6-MP therapy during maintenance as a prominent risk factor of relapse (2,28). In concert with this central role of 6-MP in the control of disease progression, relapsed ALL frequently show positive selection of resistance driving mutations antagonizing the cellular effects of this agent (7,8,29). These include, most prominently, gain-of-function point mutations in the NT5C2 gene (7,8,29–31), and in rare cases activating mutations in PRPS1 (29) and haploinsuficiency of the MSH6 mismatch DNA repair gene (30). In this context, NT5C2 stands out as a clear therapeutic target to improve the efficacy of ALL maintenance therapy by curtailing the emergence of 6-MP resistance clones responsible for relapse. However, and despite much effort (32–36), no specific NT5C2 inhibitor with validated activity in reversal of 6-MP resistance in vivo has been identified to date. Here, we leveraged a high throughput inhibitor screen against relapse-associated NT5C2 R367Q mutant protein and an array of preclinical cellular and animal models of NT5C2-driven 6-MP resistance towards the identification of CRCD2, as a first-in-class NT5C2 specific inhibitor with validated activity for reversal of 6-MP resistance in vitro and in vivo.
Interestingly, CRCD2 behaved as an uncompetitive inhibitor with increased NT5C2 binding and inhibitory activity in the presence of substrate suggesting that this small molecule engages the NT5C2 protein in its active (substrate accessible) configuration and may facilitate the transition from the active state to the inactive basal conformation. Across cell free system analyses, we observed broad inhibitory activity of CRCD2 against wild type NT5C2 protein and against relapse-associated mutations, which induce increased NT5C2 activity as a result of the reconfiguration of the catalytic center environment (K359Q) or disruption of intramolecular self-inactivation mechanisms (R367Q) (14). This observation is consistent with the largely overlapping structures of these relapse-associated NT5C2 mutant proteins with that of the wild type enzyme following allosteric activation (14). Even though a mutant specific inhibitor could offer in principle an improved therapeutic window, an important consideration here is that the NT5C2 protein adopts a tetrameric configuration and NT5C2 mutations are characteristically heterozygous, which results in the expression of an array of wild type and mutant NT5C2 heteromers with variable composition (7,8,37). Moreover, analysis of hetero-oligomeric complexes combining wild type and R367Q mutant NT5C2 subunits supports that the enzymatic activation induced by this amino acid substitution can be transmitted from the mutated to the wild type subunit (37). Thus, the capacity of CRCD2 to inhibit wild type NT5C2 activity may be of relevance to overcome overactivation of the wild type subunit in tumors harboring the R367Q NT5C2 mutation.
Treatment with CRCD2 resulted in increased sensitivity to 6-MP not only in ALL cells expressing mutant forms of NT5C2, but also in NT5C2 wild type leukemia, in agreement with the proposed role for the basal activity of the wild type enzyme in metabolizing the thiopurine mononucleotide metabolites of 6-MP (17,18). Indeed, genome-wide CRISPR screen-based drug-gene interaction mapping of modulators of 6-MP response in ALL demonstrates that genetic inactivation of NT5C2 can increase the sensitivity of NT5C2 wild type cells to 6-mercaptopurine, in support of therapeutically relevant non-genetic mechanisms of NT5C2 activation in ALL (11), that could also be pharmacologically targeted for enhanced efficacy during maintenance. In this setting, it is worth noting that NT5C2 is sensitive to allosteric activation by phosphate-containing cellular metabolites such as diadenosine polyphosphates, 2,3-bisphosphoglycerate and ATP, which connect its function with the metabolic state of the cell (38). Moreover, increased levels of NT5C2 expression and 6-MP metabolism have been reported in association with single nucleotide polymorphism germline variants resulting in increased enhancer activity at the NT5C2 locus (39,40). In addition, post-translational modifications could play an important role in the control of NT5C2 function, regulating intramolecular interactions between effector and regulatory domains, multiprotein complex assembly, subcellular localization and protein turn over. In this regard, our identification of NT5C2 Ser502 phosphorylation as a prevalent modification resulting in increased nucleotidase activity and resistance to 6-MP argues for a relevant role of non-genetic mechanisms of NT5C2 activation as drivers of reduced therapeutic response to this drug. The ability of CRCD2 to inhibit both wild type and mutant forms of NT5C2 and to counteract the effects of NT5C2 Ser502 phosphorylation offers opportunity to target both genetic and non-genetic mechanisms of NT5C2 activation in the clinic.
Finally, the identification of NT5C2 Ser502 phosphorylation as a relevant regulatory mechanism that influences the response to 6-MP argues that leukemia cell persistence during maintenance therapy may be initially mediated by non-genetic mechanisms resulting in increased NT5C2 activity creating opportunity for the occurrence of secondary activating mutations in NT5C2, which would then constitutively enhance enzymatic activity enabling the emergence of resistant disease, progression and relapse (10,21). In this setting, we propose a therapeutic role for NT5C2 inhibitors in combination with 6-MP for the treatment of NT5C2-mutant leukemia, but also to prevent the occurrence of relapsed disease by enhancing the antileukemic effects of 6-MP against NT5C2 wild type tumors before NT5C2 mutant clones emerge. A relevant open question for further research is the identification of the kinases responsible for NT5C2 S502 phosphorylation, as these could potentially serve as therapeutic targets complementing NT5C2 inhibitor therapies. The proposed physiologic role of NT5C2 is to balance intracellular nucleotide pools via degradation of excess purine nucleotide monophosphate nucleotides. It is plausible that NT5C2 S502 phosphorylation functions downstream of signaling networks implicated in the regulation of purine metabolism, cell cycle and DNA synthesis and repair.
We propose that in the context of a heterogeneous leukemia cell populations with variable levels of NT5C2 S502 phosphorylation, 6-MP treatment will favor the positive selection of cells with increased levels of this posttranslational modification. However, it did not escape our attention that while a significant fraction of relapsed ALL xenografts show increased NT5C2 S502 phosphorylation other samples displayed reduced levels of this modification. NT5C2 activity is tightly regulated, as excess purine degradation in the context of deregulated NT5C2 enzymatic activity impairs leukemia cell growth and leukemia initiating cell activity (21). As a result, expression of the Nt5c2 R367Q resistance-driving allele is negatively selected in the absence of 6-MP (21). Thus, it is possible that downregulation of NT5C2 S502 phosphorylation may happen during xenograft expansion when cells are not exposed to 6-MP. However, it is possible that that decreased NT5C2 S502 in these cases may reflect an adaptive mechanism to maintain cell homeostasis in relapsed samples in which other factors such as increased levels of allosteric regulators drive NT5C2 activation. Further biochemical characterization of nucleotide pools, NT5C2 post-translational modifications and NT5C2 activity in serial primary samples from patients treated with 6-MP could help clarify the mechanisms at play.
Altogether, our results provide a framework for the development of new combination therapies aimed at curtailing the emergence of thiopurine-resistant relapse-driving clones in ALL and support the development of CRCD2 as the first resistance-directed targeted therapy for the treatment of ALL.
Materials and Methods
Drugs and small-molecule compounds.
We treated human cell lines with inducible expression of NT5C2 for 48 hours with doxycycline (1 mg ml−1). We purchase 4-hydroxytamoxifen from Sigma-Santa Cruz Biotechnology (#SC-3542) and we dissolve it in 100% ethanol for in vitro assays. We purchased 6-mercaptopurine monohydrate (6-MP, #AC226520050) from Thermo Fisher, inosine 5’-monophosphate disodium salt hydrate (IMP, #57510), adenosine 5′-triphosphate disodium salt hydrate (ATP, #A3377) and tamoxifen (# T5648–1G) from Sigma-Aldrich. For in vitro assays, we dissolved 6-MP in DMSO, 4-hydroxytamoxifen in 100% ethanol and ATP and IMP in Tris gel filtration buffer (50 mM Tris-HCl, 100 mM NaCl, 10% Glycerol, 5 mM β-mercaptoethanol). For intraperitoneal injections of tamoxifen, we resuspended 100 mg tamoxifen in 100 μl of ethanol and added corn oil to reach a final concentration of 30 mg ml-1. We then rotated the tamoxifen suspension for 1 h at 55 °C and froze it in aliquots at - 20 °C. We administered tamoxifen as a single 100 μl intraperitoneal injection per mouse. For in vivo studies with 6-MP, we prepared frozen aliquots of 5 mg ml−1 6-MP in 0.1 M NaOH and immediately before each round of treatment we prepared fresh final solutions of 6-MP by buffering the stock solution down to pH 8 with 0.2 M NaH2PO4. This resulted in a 6-MP concentration of 3.53 mg ml−1, which we diluted to various final concentrations using a solution made from 0.05 M NaOH and 0.2 M NaH2PO4 adjusted to pH 8. We administered 6-MP as 50 mg kg−1 or 100 mg kg−1 twice a day. We prepared vehicle by dissolving 0.254 g NaCl in 50 ml 0.05 M NaOH and adjusting the pH to 8 with 0.2 M NaH2PO4. We purchased CRCD2 from Enamine (Z27358589) and dissolved in DMSO for in vitro studies. For in vivo experiments, we dissolved CRCD2 to 50 mM in DMSO and then we further dissolved to 5 mM in a 30% PEG PBS solution. We adjust injection volume to correct for any differences in weight between individual mice.
We purchased CRCD2 from Enamine (Z27358589) or we synthesized it using two different routes. CRCD2 was synthesized by treatment of 1H-benzo[d]imidazole-5-carbonyl chloride with 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide and diisopropylethylamine in DMF followed by purification by reverse phase HPLC (Fig S9A). Alternatively, we obtained CRCD2 by treating the diamine CRCD54 with formic acid (Fig S9B). The diamine CRCD54 was synthesized by reaction of 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide with 3,4-dinitrobenzoyl chloride and triethylamine in dichloromethane followed by reduction of the dinitro compound with hydrogen and palladium on carbon in methanol (Fig S9C). Treatment of the diamine CRCD54 with NaNO2 in HOAc gave the benzotriazole CRCD57, and treatment with cyanogen bromide in aqueous methanol gave the 2-aminobenzoimidazole CRCD58 (Fig S9D). The remaining CRCD2 analogs were purchased from commercial suppliers (Enamine, ChemBridge) or synthesized by treatment of acid chlorides with 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide or analogous amines in DMF in the presence of Hunig’s base followed by purification by reverse phase HPLC. The acid chlorides were commercially available or the commercially available carboxylic acid was converted to the corresponding acid chloride by treatment with oxalyl chloride and catalytic DMF in dichloromethane.
Human primary leukemia xenograft cells.
We generated and expanded ALL xenograft cells by intravenous injection of relapsed ALL lymphoblasts in NRG (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ) immunodeficient mice (Jackson Labs) from a relapsed ALL sample provided by the Children’s Oncology Group and Princess Maxima Center (Netherlands) leukemia tissue banks. Written informed consent was obtained at study entry and samples were collected under the supervision of local Institutional Review Boards for participating institutions and analyzed under the supervision of the Columbia University Medical Center Institutional Review Board (Protocol Number: IRB-AAAB3250) and in compliance with ethical regulations.
Cell culture.
We performed cell culture in a humidified atmosphere at 37°C under 5% CO2 and we regularly tested for mycoplasma contamination. We purchased HEK293T cells for viral production from American Type Culture Collection (ATCC) and grew them in DMEM media supplemented with 10% fetal bovine serum (FBS), 100 U ml−1 penicillin G and 100 μg ml−1 streptomycin for up to two weeks. The CUTLL1 cell line, which was generated by continuous culture of a T-cell lymphoblastic pleural effusion cells from a patient at relapse, has been characterized and reported before(41). We obtained Jurkat and REH cells from American Type Culture Collection (ATCC) and PEER, BE13 and 697 cells from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). We cultured CUTLL1, Jurkat and REH cells in RPMI-1640 media supplemented with 10% FBS, 100 U ml−1 penicillin G and 100 μg ml−1 streptomycin and PEER, BE13 and 697 cells in RPMI-1640 media supplemented with 20% FBS, 100 U ml−1 penicillin G and 100 μg ml−1 streptomycin. Mouse Rosa26+/CreERT2 Nt5c2+/co-R367Q T-ALL tumor cells were previously described(14), and were cultured in OptiMEM media supplemented with 10% fetal bovine serum (FBS), 100 U ml−1 penicillin G, 100 μg ml−1 streptomycin, 55 μM β-mercaptoethanol, and 10 ng ml−1 mouse IL7. Primary human xenograft ALL cells were passaged and collected from the spleens of NRG mice (NOD.Cg-Rag1tm1Mom/I2rgtm1Wjl/SzJ, Jackson Laboratory) and cultured in RPMI medium supplemented with 20% FBS, 100 U ml−1 penicillin G, 100 μg ml−1 streptomycin and 10 ng ml−1 human IL-7.
Plasmids and vectors.
We obtained the pET28aLIC (Plasmid #26094) and pL-CRISPR.efs.gfp (Plasmid #57818) plasmids from Addgene and pLVXTRE3GZsGreen1 vector from Clontech. We amplified the coding sequence of the NT5C2 cDNA from pLOC-NT5C2(7) and cloned it into the pET28aLIC and pLVXTRE3GZsGreen1 vectors using In-fusion cloning using the In-Fusion HD Cloning Kit (Clontech) following manufacturer guidelines. We generated lentiviral vectors expressing CAS9 and gRNAs targeting exon 3 or 8 of Nt5c2 by cloning the corresponding gRNA oligonucleotides (Sigma-Aldrich) into pL-CRISPR.efs.gfp as reported(42). We cloned NT5C2 R238W, K359Q, R367Q, L375F, D407A, K217R, K217Q, K344R, K344Q, S418A, S418D, S502A, S502D, D229A and D229S mutations into the pLOC-NT5C2(7) or pET28aLIC-NT5C2 by site directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies) according to manufacturer’s guidelines.
Lentiviral production and infection.
We transfected lentiviral plasmids together with gag-pol (pCMV ΔR8.91) and V-SVG (pMD.G VSVG) expressing vectors into HEK293T cells using JetPEI transfection reagent (Polyplus). We collected viral supernatants after 48h and used them to infect the CUTLL1 human cell line by spinoculation with 4 μg ml−1 Polybrene Infection/Transfection Reagent (Fisher Scientific). We selected infected human cell lines with 1 mg/ml blasticidin (InvivoGen, #ant-bl-1) for 14 days or with 1 mg/ml puromycin (Sigma-Aldrich, #P8833) for 7 days.
NT5C2 recombinant protein purification.
For 5’-nucleotidase assays in the absence and presence of allosteric activators we cloned, expressed and purified recombinant wild-type and mutant NT5C2 proteins as previously described(14). Briefly, we cloned full-length NT5C2 cDNA constructs with an N-terminal hexahistidine (His6) tag in the pET28a-LIC expression vector. We expressed recombinant proteins from Rosetta 2 (DE3) Escherichia coli cells by induction with 0.5 mM isopropyl-β-D-thiogalactopyranoside overnight at 16 °C. We resuspended harvested cells in lysis buffer (50 mM Tris-HCl pH 7.4, 500 mM sodium chloride, 10% glycerol, 0.5 mM TCEP, 20 mM imidazole) supplemented with Complete EDTA-free protease inhibitor (Roche) and lysed cells by sonication. We purified recombinant proteins using an ӒKTA fast protein liquid chromatography system (GE Healthcare) using a 2-step protocol adapted from one previously described(43). We first performed affinity chromatography using a 1 ml Ni2+-charged His-Trap HP column (GE Healthcare) equilibrated in lysis buffer. We eluted NT5C2 proteins from the His-Trap column in a step-wise method with elution buffer (lysis buffer with 500mM imidazole) by first setting the buffer ratio to 25% elution buffer for 8 column volumes and then switching to a linear gradient to 100% elution buffer over 10 column volumes. We pooled NT5C2-containing fractions and purified further by size exclusion chromatography using a HiLoad 16/60 Superdex 200 gel filtration column (GE Healthcare) equilibrated in 50mM Tris-HCl, pH 7.4, 100 mM NaCl, 10% glycerol and 0.5 mM TCEP (for in vitro nucleotidase assays using Malachite Green kit) or in 50 mM sodium phosphate, pH 7.4, 100 mM NaCl, 10% glycerol and 0.5 mM TCEP (for protein crystallization or nucleotidase assays using Diazyme kit). We assessed protein expression and purity by SDS-PAGE and Coomassie staining. For crystallography studies, we concentrated protein samples to 4–9 mg/ml.
5’-Nucleotidase assays.
For high-throughput screenings, the malachite green enzymatic assay was adapted from a previously published protocol (32) and optimized for NT5C2 R367Q enzyme kinetics. To optimize the assay, initial velocity and linear reaction conditions were performed following NIH enzyme assay guidelines (44). Briefly, we incubated 0.02 μM or 2.6 ng ml−1 purified recombinant NT5C2 R367Q protein with 100 μM inosine monophosphate (IMP) as substrate for 15 minutes at 37°C. We terminated the enzymatic reaction (IMP→ Pi +inosine) by adding the malachite green reagent following manufacturer’s guidelines (Sigma Aldrich, #MAK307). Colorimetric analysis of malachite green reaction with free phosphate (Pi) was measured after 15 minutes at 600 nM. We performed these assays in Tris gel filtration buffer (50 mM Tris-HCl, 100 mM NaCl, 10% Glycerol, 5 mM β-mercaptoethanol) with a final concentration of 10 mM MgCl2. For in vitro nucleotidase assays with inhibitors or small molecule compounds, we added compounds dissolved in DMSO to the protein and incubated for 10 minutes at room temperature prior to adding IMP substrate. We next incubated the plate at 37°C for 10 minutes and measure malachite green reaction after 30 minutes. For Michaelis-Menten curve analyses, we used a phosphate standard curve following manufacturer’s guidelines. We assessed 5’-NT activity of purified recombinant wild-type and mutant NT5C2 proteins using the 5’-NT Enzymatic Test Kit (Diazyme) according to the manufacturer’s instructions as described previously (7). We calculated 5’-NT activity levels using a calibrator of known 5’-NT activity as standard. We performed assays in triplicate in a Glomax Multidetection System plate reader (Promega). For assays with allosteric activators ATP, was dissolved directly in Reagent 2 of the test kit (containing the substrate IMP) and made serial dilutions to achieve a range of concentrations. We evaluated 5’-nucleotidase activity in plasma from mice treated with 17 mg kg−1 CRCD2 using the 5’-NT Enzymatic Test Kit (Diazyme) according to the manufacturer’s instructions.
Enamine Library.
The Enamine Library consists of 60,640 compounds filtered for traditional physicochemical descriptors such as the rule of five, rotatable bond count, topological polar surface, and suitable aqueous solubility. The Enamine library was curated by assigning a score for each compound relative to the calculated value in relationship to a standard value, a method modified from previous works (45). For example, the number of rotatable bonds was calculated for each compound, then using a standard value of 6, deviations from that value are penalized based on distance from 6. After all criteria are calculated, the distribution of scores is then used to stratify the population with the highest scoring compounds being eliminated. We clustered the resulting compounds and we selected two compounds from each cluster for purchase.
High-throughput inhibitor screen.
We performed the inhibitor screen on a Cell:Explorer robotic station (Perkin Elmer) using the following modules: Janus liquid handling platform with NanoHead, FlexDrop liquid dispenser, Liconic microplate hotel, Liconic 500 microplate incubator (37°C), Envision plate reader. Briefly, we plated 20 μL of 0.4 μM NT5C2 R367Q recombinant protein in Tris Gel Filtration Buffer or buffer alone controls in clear bottom low binding black 384 well microplates (Greiner, #781906). We delivered compounds from the Enamine library to the plates by the NanoHead from 10mM stock library microplates to a final concentration of 100 nM and we incubated the plates at room temperature in the Liconic incubator for 10 minutes. Then, we added 20 μL of 200 mM Inosine monophosphate (IMP), substrate, to the wells with the Flexdrop and plates were incubated at 37 °C in the Liconic incubator for 15 minutes. Following incubation, we added 5 μL of the malachite green reagent (Sigma, #MAK-307) to all of the wells. We incubated the plates for 15 minutes and then we read on an Envision plate reader at 650 nM. We added MgCl2 to all buffers for a final concentration of 10 mM in the assay. In screening the Enamine library, each plate had two columns for a non-treatment control containing all elements of the assay without the addition of library compounds and two columns serving as negative controls that had neither substrate nor drugs added. We normalized the raw data for all compound containing wells to the average of all the wells screened. From the primary screen, 3 standard deviations away from the mean was used as a cutoff to select 225 compounds that had 33.21% inhibitory activity or higher. Next, those 225 compounds were subject to confirmatory assay by cherry-picking them from the original library and testing again in the same assay. We confirmed 28 compounds out of 225. Next, we chose 6 compounds, which showed best inhibition and tested them in dose-response curves.
Plasmon Surface Resonance (SPR).
SPR analysis were performed by Creative Biolabs using a Biacore T200 instrument (Cytiva). NT5C2 D52N R367Q recombinant protein was directly immobilized on the CM5 chip using an amine coupling kit (Cytiva). Before immobilization, the CM5 sensor surface was activated using a mixture of 400 mM 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC) and 100 mM N-hydroxysuccinimide (NHS). Then, 50 μg ml−1 of NT5C2 protein in immobilization buffer (10 mM NaAc (pH 4.0)) was then injected into Fc2 sample channel at a flow rate of 10 μL min-1. The amount of ligand immobilized was about 15,000 RU. The chip was deactivated by 1 M Ethanolamine hydrochloride-NaOH (GE Healthcare Life Sciences) at a flow rate of 10 μL/min for 420 s. The reference Fc1 channel underwent similar procedures but without injecting the ligand. The analyte (CRCD2) was serially diluted with the running buffer, in the absence or the presence of 500 μM IMP, to give a concentration of 62.5, 31.25, 15.625, 7.813, 3.906, 1.953, and 0 μM, respectively. Different concentrations of analytes were then injected into the Fc2-Fc1 of channels at a flow rate of 30 μL min−1, with a contact time of 60 s, followed by a dissociation time of 90 s. Data analysis was performed on the Biacore T200 computer and with the Biacore T200 evaluation software, using the steady state affinity model.
Hydrogen-Deuterium exchange mass spectrometry analysis (HDX-MS).
HDX-MS studies were performed at the City University of New York Mass Spectrometry Facility. All subsequent sample handling was performed in an ice bath. The quenched sample was digested online using Enzymate BEH Pepsin column (Waters). The digestion was performed at a flow rate of 0.15 ml min−1 using 0.15% formic acid/3% acetonitrile as the mobile phase. The resulting peptides were collected and desalted with an inline 4 μl C8-Opti-lynx II trap cartridge (Optimize Technologies) and then eluted through a C-18 column (Thermo Fisher Scientific, 50 × 1 mm Hypersil Gold C-18) using a rapid gradient from 2 to 90% acetonitrile containing 0.15% formic acid and a flow rate of 0.04 ml min−1, leading directly into a maXis-II ETD ESI-QqTOF mass spectrometer. The total time for the digest and desalting was 3 min, and all peptides had eluted from the C-18 column by 15 min. To avoid cross-contamination from carry-over peptides, comprehensive pepsin and C-18 column wash steps were included after each run. The peptide fragments were identified using Bruker Compass and Biotools software packages. The level of deuterium incorporation was assessed using the commercial software HDExaminer (Trajan Scientific).
Crystallization and Structure Determination.
The protein solution of the full-length mutant (S502D) NT5C2 at concentration 2.5 mg/ml in a protein buffer comprising 50 mM ammonium phosphate (pH 7.5), 100 mM sodium chloride, 10% (v/v) glycerol, and 1 mM TCP was initially subjected to extensive robotic screening at the High-Throughput Crystallization Screening Center (46) of the Hauptman-Woodward Medical Research Institute (HWI) (https://hwi.buffalo.edu/high-throughput-crystallization-center/). The only crystal hit was reproduced using under oil micro batch method at 4°C.
Small block-shaped crystals of NT5C2 appeared after three weeks in a crystallization condition comprising 100 mM sodium acetate trihydrate (pH 4.6), 30% (v/v) MPD, and 20 mM calcium chloride dihydrate with protein to crystallization reagent at a ratio 2:1 μl. The crystals were subsequently transferred into a similar crystallization reagent that was supplemented by 20% (v/v) glycerol and flash-frozen in liquid nitrogen. A similar methodology was used for growing crystals of truncated (537X) mutant (S502D) NT5C2. A native dataset was collected on each crystal of the full-length NT5C2 at the NE-CAT24-ID-C beam line of Advanced Photon Source (APS) in Lemont, IL, and NYX beam line of NSLSII was used for data collection on crystals of the truncated (537X) mutant (S502D) NT5C2. The best crystal for the full-length and truncated NT5C2 diffracted the X-ray beam to resolution 2.77 Å and 2.12 Å, respectively. The images were processed and scaled in space group C2 using XDS (47). Both structures were determined by molecular replacement method using program MOLREP (48) and the crystal structure of wild-type full-length (PDB id: 6DDO) and truncated NT5C2 (PDB id: 6DDC) was used as a search model for each. The geometry of each crystal structure was subsequently fixed and modeled by programs XtalView (49) and Coot (50) and refined using Phenix (51). There are two protomers of the full-length and truncated NT5C2 in the asymmetric unit of each crystal. The crystallographic statistics is shown in Supplementary Table 2.
Targeted metabolomic analysis.
For conditional tamoxifen-inducible expression of R367Q NT5C2 in vitro we treated cells with 1 μM 4-hydroxytamoxifen for 48h and tested induction of R367Q allele by PCR as previously described (21). We next add 100μl MeOH to pellets containing at least 1×106 live cells and flash-freeze the samples. Analysis of purine and pyrimidine metabolites was performed at UVic-Genome BC Proteomics Centre (Victoria, Canada). Serially diluted standard solutions containing standard substances of the targeted compounds were prepared. Each sample was lysed on a MM 400 mill mixer with the aid of two beads at a shaking frequency of 30 Hz for 1 min, twice. The mixtures were homogenized and then placed at −20 °C for 1 hour, followed by centrifugation at 21,000 g and 0 °C for 10 min. Protein pellets were used for protein quantification and normalization. Then, supernatant was dried under a nitrogen gas flow and reconstituted in 100 μL of the internal standard solution. 10-μL aliquots were injected into a C18 LC column (2.1×150 mm, 1.8 μm) to run UPLC-MRM/MS on a Waters Acquity UPLC system coupled to a Sciex QTRAP 6500 Plus mass spectrometer operated in the negative-ion mode for detection of nucleotides. The mobile phase was a tributylamine buffer (A) and acetonitrile/methanol (B) for binary gradient elution (5% to 40% B in 25 min), at 0.25 mL min−1 and 45 °C. For quantitation of nucleosides and nucleobases, 10 μL aliquots of the sample solutions and standard solutions were injected onto a polar reversed-phase C18 column (2.1×100 mm, 2.0 μm) to run UPLC-MRM/MS on a Waters Acquity UPLC system coupled to a Sciex QTRAP 6500 Plus mass spectrometer operated in the positive-ion mode. The mobile phase was 0.1% formic acid (A) and methanol (B) for binary gradient elution (0% to 60% B in 15 min), at 0.30 mL min−1 and 40 °C. Concentrations of detected analytes were calculated with internal-standard calibration by interpolating the constructed linear-regression curves of individual compounds, with the analyte-to-internal standard peak area ratios measured from the sample solutions.
In vitro cell viability and chemotherapy response assays.
We analyzed chemotherapy responses of human leukemia cell lines or murine mouse lymphoblasts following 72-hour incubation with increasing concentrations of 6-mercaptopurine or CRCD2 by measurement of the metabolic reduction of the tetrazolium salt MTT using the Cell Proliferation Kit I (Roche) following the manufacturer’s instructions. We performed isobologram synergy assays as above with 6-MP, CRCD2 or the combination of 6-MP and CRCD2 at a consistent ratio. We perfomed isobologram analysis using Calcusyn software.
CRISPR/CAS9 NT5C2 Knockout in ALL cell lines.
CUTLL1 cells were infected with pL-CRISPR.efs.gfp lentiviral particles containing a Nt5c2 exon 3 targeted sgRNA (GCAAAGCTGAGCAACTCCTG), Nt5c2 exon 8 targeted sgRNA (GTCCTACCGGAGTATGTTCC) or empty vector controls. We sorted infected cells based on GFP expression using a SONY SH800S cell sorter (SONY) and subsequently grew single cell clones. We confirmed NT5C2 knockout via western blot analysis.
pSer502-NT5C2 antibody generation.
We generated rabbit polyclonal antisera directed against Keyhole Limpet Hemocyanin-conjugated NT5C2 peptides corresponding to the Ser502 region (CDINEMESPLATR). Phospho-specific immunoglobulins were purified from rabbit sera by positive affinity purification using the corresponding immobilized peptide columns (Covance).
Immunoprecipitation and western blot analysis.
We lysed cells in RIPA buffer and cleared of cell debris. We performed BCA protein quantification according to manufacturer guidelines (BCA Protein Assay Kit, Fisher Scientific). For immunoprecipitation of Flag-tag, we rotated 1mg lysate with Flag affinity beads (EZview™ Red ANTI-FLAG M2 Affinity Gel clone M2, Sigma-Aldrich) overnight, and then wash the unbound material with RIPA buffer. For Western-Blot analysis we loaded whole IP or equal amounts of lysate onto a 4–12% Bis-Tris gel (Life technologies), separated by SDS PAGE, and transferred to a nitrocellulose membrane for western blot analysis. We detected NT5C2 with mouse anti-NT5C2 (Sigma Aldrich, #WH0022978M2) and rabbit anti-pSer502-NT5C2 (dilution 1:1000) (Covance) antibodies and β-actin with a mouse monoclonal anti-β-actin antibody (Sigma Aldrich, #A5441).
Post-translational modification mass spectrometry analysis.
We collected 500 million Jurkat cells expressing HA-Flag-NT5C2 in lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, 1% Triton, 5% glycerol), lysed for 30 min 4C in rotating shaker and spun down in ultracentrifuge at 2,500g 1h 4C. Then, we incubated lysate with anti-FLAG beads (EZview™ Red ANTI-FLAG® M2 Affinity Gel clone M2; Sigma Aldrich # F2426–1ML; 1 mg ml−1) for more than 8 h at 4C and eluted twice with FLAG peptide (1 mg/mL). We next incubated FLAG eluate anti-HA beads (EZview™ Red Anti-HA Affinity Gel; Sigma Aldrich # E6779–1ML; 1 mg ml−1) for more than 8 h at 4C and eluted twice with HA peptide (1 mg ml−1).
For the identification of post-translational modifications in NT5C2, HA eluates were analyzed by mass spectrometry at Taplin Mass Spectrometry Facility a Harvard Medical School. Briefly, we electrophoresed on a 4–12% Bis-Tris gel, stained with Simply Blue Stain (Invitrogen), excised, reduced with dithiothreitol, alkylated with iodoacetamide, digested with trypsin. Peptides were later extracted by removing the ammonium bicarbonate solution and then dried in a speed-vac (~1 hr). On the day of analysis, the samples were reconstituted in 5 – 10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid) and eluted using a nano-scale reverse-phase HPLC capillary column and increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid). As each peptide was eluted they were subjected to electrospray ionization and then they entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Eluting peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein or translated nucleotide databases with the acquired fragmentation pattern by the software program, Sequest (ThermoFinnigan, San Jose, CA). Modification assignments were determined by the Ascore algorithm {Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. Nat Biotechnol. 2006 Oct;24(10):1285–92. Epub 2006 Sep 10}. All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate.
Mouse organs histopathology.
We fixed mouse organs in 10% buffered formalin. The Molecular Pathology shared resource facility at the Herbert Irving Cancer Comprehensive Center proceed to embedding of fixed mouse organs in paraffin blocks, sectioning and hematoxylin and eosin- stain by following standard procedures. Slides were scanned using a Leica SCN 400 scanner, and photomicrographs were examined with Aperio ImageScope software (Leica Biosystems).
Mice and animal procedures.
All animals were maintained in specific-pathogen-free facilities at the Irving Cancer Research Center at Columbia University Medical Center. The Columbia University Institutional Animal Care and Use Committee approved all animal procedures. Animal experiments were conducted in compliance with all relevant ethical regulations. Animals were euthanized upon showing symptoms of clinically overt disease (not feeding, lack of activity, abnormal grooming behavior, hunched back posture) or excessive weight loss (15% body-weight loss over a week).
For systemic toxicity evaluation in vivo, we treated C57BL/6 mice with increasing concentrations of CRCD2 (8.5, 17, 34mg/kg/day) in 5-days ON 2-days OFF schedule until we reached maximum deliverable dose. We monitored the mice for low activity and weight loss. To evaluate toxicity of the combination of 6-MP and CRCD2 in vivo, we treated C57BL/6 mice with therapeutic doses of 6-MP (50 mg kg−1) and CRCD2 (34 mg kg−1) for 5 consecutive days and evaluated: blood cell counts, bone marrow cellularity and tissue histopathology.
We performed pharmacokinetic analysis of CRCD2 in plasma following as previously described (52). We injected 17 mg kg−1 into C57BL/6 mice and we collected samples at 0, 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 24 h and 48 h from three mice per time point. We collected blood from the mouse via cardiac puncture and placed into tubes containing EDTA anti-coagulant on ice. We next centrifuged EDTA tubes at 2100 g for 10 minutes at 4 °C, flash froze samples and stored them at −80°C. Then, we added 900 μL of acetonitrile to 100 μL of plasma, we mixed the sample and sonicated it. After spinning for 10 minutes at 4,000 g at 4°C, the supernatant is collected and analyzed by liquid chromatography mass spectrometry (LC-MS) on a platform comprising a Thermo Scientific Dionex Ultimate 3000RS controlled by Chromeleon (Dionex) and a Bruker amazon SL ESI ion-trap mass spectrometer. Chromatographic separation was performed at 20°C on an Agilent Eclipse Plus C18 column (2.1 × 50 mm, 3.5 mm) at 20°C over a 12 minute gradient elution. Mobile phase A consisted of water with 0.1% acetic acid v/v and mobile phase B was methanol with 0.1% acetic acid v/v. Mass spectrometry analysis was performed on a Bruker Amazon SL (Billerca, MA) in positive ESI mode. Trap Control was used to control the ESI settings with the inlet capillary held at −4500 V and the end plate offset at −500 V. Nitrogen was used as the desolvation gas. Hystar v3.2 was used to integrate the UHPLC and MS applications, and data analysis was performed with the Compass DataAnalysis software. The base peak chromatogram at m/z 655.2 with a width of ± 0.1 was integrated and peak area quantified by standard curve. Pk of CRCD2 was assessed using Prism fitted with lognormal of one phase exponential decay.
For experimental therapeutics treatment studies, we allografted luciferized NOTCH1-induced conditional inducible Nt5c2 R367Q (Nt5c2+/co-R367Q Rosa26+/CreERT2) T-ALL tumor cells into isogenic mice. We harvested fresh luciferased tumor cells and transplanted them into sublethally irradiated (500 Rads) C57BL/6 recipients by retro-orbital injection. We monitored tumor development by in vivo luminescence bioimaging with the In vivo Imaging System (IVIS, Xenogen). Once mice had a detectable baseline tumor burden by bioluminescence, we randomly assigned animals into different treatment groups and we treated them with tamoxifen or corn oil vehicle by intraperitoneal injection as described above. Two days later, we initiated treatment with vehicle only, 34 mg kg−1 of CRCD2, 50 or 100 mg kg−1 of 6-MP, or the combination treatment, via intraperitoneal injection for five consecutive days (n = 5 mice per group). We monitored disease progression and response to chemotherapy by bioluminescence imaging on days 0 and 5 after the start of treatment. We euthanized mice on day 5 and analyzed bioluminescence, GFP+ tumor infiltration in the spleen and bone marrow by flow cytometry and spleen weight.
For therapeutic studies in primary human leukemia xenografts, luciferized NT5C2 R367Q bearing T-ALL PDX was transplanted into secondary NRG recipients (NOD.Cg-Rag1tm1Mom/I2rgtm1Wjl/SzJ) by retro-orbital injection. We monitored tumor development by in vivo luminescence bioimaging with the In vivo Imaging System (IVIS, Xenogen). Once mice had a detectable baseline tumor burden by bioluminescence, we randomly assigned animals into different treatment groups and we introduced treatment with vehicle only, 34 mg kg−1 of CRCD2, 100 mg kg−1 of 6-MP, or the combination via intraperitoneal injection for five days. After this first cycle mice were allowed to recover for 9 days after which we reintroduced therapy at the same level for two consecutive days evaluating disease progression and therapeutic response by in vivo bioimaging at day 16.
Statistics and reproducibility.
We conducted statistical analyses using Prism software v8.0 (GraphPad software) and considered statistical significance at P < 0.05. We reported results as mean ± s.d. with significance annotated by P value calculated as indicated in the figure legends using Student’s t-tests assuming equal variance and normal distribution or using ANOVA and Dunnett’s multiple-comparison tests. The investigators were not blinded to allocation during the experiments and outcome assessment. The experiments were not randomized. No data were excluded from the analyses.
Data availability.
No datasets were generated or analyzed during the current study.
Supplementary Material
Statement of Significance.
Relapse-associated NT5C2 mutations directly contribute to relapse in acute lymphoblastic leukemia by driving resistance to chemotherapy with 6-mercaptopurine (6-MP). Pharmacologic inhibition of NT5C2 with CRCD2, a first-in-class nucleotidase inhibitor, enhances the cytotoxic effects of 6-MP and effectively reverses thiopurine resistance mediated by genetic and non-genetic mechanisms of NT5C2 activation in ALL.
Acknowledgements
This work was supported by the Chemotherapy Foundation (AAF); NIH grants P30 CA013696 (Genomics and High Throughput Screen Shared Resource, Oncology Precision Therapeutics Shared Resource), R35 CA210065 (A.A.F.), R01 CA206501 (AAF and BRS), U10 CA98543 (JMG, MLL), Human Specimen Banking Grant U24 CA114766 (JMG), a Translational Research Grant (AAF, LLS 6455–15) and a Screen to Lead grant (8011–18, AAF) by the Leukemia & Lymphoma Society, an Innovative Research Award from the Alex Lemonade Stand Foundation (AAF) and a Accelerating Cancer Therapeutics (ACT) Pilot Award by the Irving Institute for Clinical and Translational Research of Columbia University. C.R. is supported by a Leukemia and Lymphoma Society Special Fellow award. R. A. is supported by the Leukemia and Lymphoma Society Postdoctoral Fellowship award.
Footnotes
Declaration of Interests
The authors declare no competing financial interests relevant for the work reported here. Financial disclosures for Adolfo Ferrando: Employment Regeneron Genetics Center, Consulting for VantAI, and Brystol Myers Squibb. B.R.S. is an inventor on patents and patent applications involving small molecule therapeutics, co-founded and serves as a consultant to Inzen Therapeutics, Nevrox Limited, Exarta Therapeutics, and ProJenX, Inc. and serves as a consultant to Weatherwax Biotechnologies Corporation and Akin Gump Strauss Hauer & Feld LLP. Arie Zask is a co-founder of and consultant to ProJenX, Inc. A provisional patent protecting composition of matter on lead compounds has been filed by Columbia University.
References
- 1.Koren G, Ferrazini G, Sulh H, Langevin AM, Kapelushnik J, Klein J, et al. Systemic exposure to mercaptopurine as a prognostic factor in acute lymphocytic leukemia in children. N Engl J Med 1990;323(1):17–21 doi 10.1056/NEJM199007053230104. [DOI] [PubMed] [Google Scholar]
- 2.Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999;93(9):2817–23. [PubMed] [Google Scholar]
- 3.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med 2015;373(16):1541–52 doi 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 4.Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet (London, England) 2020;395(10230):1146–62 doi 10.1016/s0140-6736(19)33018-1. [DOI] [PubMed] [Google Scholar]
- 5.Moricke A, Zimmermann M, Reiter A, Henze G, Schrauder A, Gadner H, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 2010;24(2):265–84 doi 10.1038/leu.2009.257. [DOI] [PubMed] [Google Scholar]
- 6.Salzer WL, Devidas M, Carroll WL, Winick N, Pullen J, Hunger SP, et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984–2001: a report from the children’s oncology group. Leukemia 2010;24(2):355–70 doi 10.1038/leu.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med 2013;19(3):368–71 doi 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 2013;45(3):290–4 doi 10.1038/ng.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. P Natl Acad Sci USA 2016;113(40):11306–11 doi 10.1073/pnas.1608420113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020;135(1):41–55 doi 10.1182/blood.2019002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oshima K, Zhao J, Perez-Duran P, Brown JA, Patino-Galindo JA, Chu T, et al. Mutational and functional genetics mapping of chemotherapy resistance mechanisms in relapsed acute lymphoblastic leukemia. Nat Cancer 2020;1(11):1113–27 doi 10.1038/s43018-020-00124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schroeder MP, Bastian L, Eckert C, Gökbuget N, James AR, Tanchez JO, et al. Integrated analysis of relapsed B-cell precursor Acute Lymphoblastic Leukemia identifies subtype-specific cytokine and metabolic signatures. Scientific reports 2019;9(1):4188 doi 10.1038/s41598-019-40786-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richter-Pechańska P, Kunz JB, Hof J, Zimmermann M, Rausch T, Bandapalli OR, et al. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood cancer journal 2017;7(2):e523 doi 10.1038/bcj.2017.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dieck CL, Tzoneva G, Forouhar F, Carpenter Z, Ambesi-Impiombato A, Sanchez-Martin M, et al. Structure and Mechanisms of NT5C2 Mutations Driving Thiopurine Resistance in Relapsed Lymphoblastic Leukemia. Cancer Cell 2018;34(1):136–47 e6 doi 10.1016/j.ccell.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dieck CL, Ferrando A. Genetics and mechanisms of NT5C2-driven chemotherapy resistance in relapsed ALL. Blood 2019;133(21):2263–8 doi 10.1182/blood-2019-01-852392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hnízda A, Fábry M, Moriyama T, Pachl P, Kugler M, Brinsa V, et al. Relapsed acute lymphoblastic leukemia-specific mutations in NT5C2 cluster into hotspots driving intersubunit stimulation. Leukemia 2018;32(6):1393–403 doi 10.1038/s41375-018-0073-5. [DOI] [PubMed] [Google Scholar]
- 17.Brouwer C, Vogels-Mentink TM, Keizer-Garritsen JJ, Trijbels FJ, Bökkerink JP, Hoogerbrugge PM, et al. Role of 5’-nucleotidase in thiopurine metabolism: enzyme kinetic profile and association with thio-GMP levels in patients with acute lymphoblastic leukemia during 6-mercaptopurine treatment. Clin Chim Acta 2005;361(1–2):95–103 doi 10.1016/j.cccn.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Moriyama T, Liu S, Li J, Meyer J, Zhao X, Yang W, et al. Mechanisms of NT5C2-Mediated Thiopurine Resistance in Acute Lymphoblastic Leukemia. Molecular cancer therapeutics 2019;18(10):1887–95 doi 10.1158/1535-7163.mct-18-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spychala J, Madrid-Marina V, Fox IH. High Km soluble 5’-nucleotidase from human placenta. Properties and allosteric regulation by IMP and ATP. J Biol Chem 1988;263(35):18759–65. [PubMed] [Google Scholar]
- 20.Oka J, Matsumoto A, Hosokawa Y, Inoue S. Molecular cloning of human cytosolic purine 5’-nucleotidase. Biochem Biophys Res Commun 1994;205(1):917–22 doi 10.1006/bbrc.1994.2752. [DOI] [PubMed] [Google Scholar]
- 21.Tzoneva G, Dieck CL, Oshima K, Ambesi-Impiombato A, Sanchez-Martin M, Madubata CJ, et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018;553(7689):511–4 doi 10.1038/nature25186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S, et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010;24(2):345–54 doi 10.1038/leu.2009.251. [DOI] [PubMed] [Google Scholar]
- 23.Conter V, Arico M, Basso G, Biondi A, Barisone E, Messina C, et al. Long-term results of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88, 91 and 95 for childhood acute lymphoblastic leukemia. Leukemia 2010;24(2):255–64 doi 10.1038/leu.2009.250. [DOI] [PubMed] [Google Scholar]
- 24.Gaynon PS, Angiolillo AL, Carroll WL, Nachman JB, Trigg ME, Sather HN, et al. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: a Children’s Oncology Group Report. Leukemia 2010;24(2):285–97 doi 10.1038/leu.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006;354(2):166–78 doi 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 26.Childhood ALLCG. Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomised children. Lancet (London, England) 1996;347(9018):1783–8 doi 10.1016/s0140-6736(96)91615-3. [DOI] [PubMed] [Google Scholar]
- 27.Pinkel D, Hernandez K, Borella L, Holton C, Aur R, Samoy G, et al. Drug dosage and remission duration in childhood lymphocytic leukemia. Cancer 1971;27(2):247–56 doi . [DOI] [PubMed] [Google Scholar]
- 28.Bhatia S, Landier W, Shangguan M, Hageman L, Schaible AN, Carter AR, et al. Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children’s oncology group. J Clin Oncol 2012;30(17):2094–101 doi 10.1200/JCO.2011.38.9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B, Li H, Bai Y, Kirschner-Schwabe R, Yang JJ, Chen Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med 2015;21(6):563–71 doi 10.1038/nm.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evensen NA, Madhusoodhan PP, Meyer J, Saliba J, Chowdhury A, Araten DJ, et al. MSH6 haploinsufficiency at relapse contributes to the development of thiopurine resistance in pediatric B-lymphoblastic leukemia. Haematologica 2018;103(5):830–9 doi 10.3324/haematol.2017.176362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood 2008;112(10):4178–83 doi 10.1182/blood-2008-06-165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallier F, Lallemand P, Meurillon M, Jordheim LP, Dumontet C, Perigaud C, et al. Structural insights into the inhibition of cytosolic 5’-nucleotidase II (cN-II) by ribonucleoside 5’-monophosphate analogues. PLoS Comput Biol 2011;7(12):e1002295 doi 10.1371/journal.pcbi.1002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordheim LP, Marton Z, Rhimi M, Cros-Perrial E, Lionne C, Peyrottes S, et al. Identification and characterization of inhibitors of cytoplasmic 5’-nucleotidase cN-II issued from virtual screening. Biochem Pharmacol 2013;85(4):497–506 doi 10.1016/j.bcp.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 34.Meurillon M, Marton Z, Hospital A, Jordheim LP, Bejaud J, Lionne C, et al. Structure-activity relationships of beta-hydroxyphosphonate nucleoside analogues as cytosolic 5’-nucleotidase II potential inhibitors: synthesis, in vitro evaluation and molecular modeling studies. Eur J Med Chem 2014;77:18–37 doi 10.1016/j.ejmech.2014.02.055. [DOI] [PubMed] [Google Scholar]
- 35.Marton Z, Guillon R, Krimm I, Preeti, Rahimova R, Egron D, et al. Identification of Noncompetitive Inhibitors of Cytosolic 5’-Nucleotidase II Using a Fragment-Based Approach. J Med Chem 2015;58(24):9680–96 doi 10.1021/acs.jmedchem.5b01616. [DOI] [PubMed] [Google Scholar]
- 36.Guillon R, Rahimova R, Preeti, Egron D, Rouanet S, Dumontet C, et al. Lead optimization and biological evaluation of fragment-based cN-II inhibitors. Eur J Med Chem 2019;168:28–44 doi 10.1016/j.ejmech.2019.02.040. [DOI] [PubMed] [Google Scholar]
- 37.Hnizda A, Fabry M, Moriyama T, Pachl P, Kugler M, Brinsa V, et al. Relapsed acute lymphoblastic leukemia-specific mutations in NT5C2 cluster into hotspots driving intersubunit stimulation. Leukemia 2018;32(6):1393–403 doi 10.1038/s41375-018-0073-5. [DOI] [PubMed] [Google Scholar]
- 38.Wallden K, Nordlund P. Structural basis for the allosteric regulation and substrate recognition of human cytosolic 5’-nucleotidase II. J Mol Biol 2011;408(4):684–96 doi 10.1016/j.jmb.2011.02.059. [DOI] [PubMed] [Google Scholar]
- 39.Jiang C, Yang W, Moriyama T, Liu C, Smith C, Yang W, et al. Effects of NT5C2 Germline Variants on 6-Mecaptopurine Metabolism in Children With Acute Lymphoblastic Leukemia. Clinical pharmacology and therapeutics 2021;109(6):1538–45 doi 10.1002/cpt.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tulstrup M, Grosjean M, Nielsen SN, Grell K, Wolthers BO, Wegener PS, et al. NT5C2 germline variants alter thiopurine metabolism and are associated with acquired NT5C2 relapse mutations in childhood acute lymphoblastic leukaemia. Leukemia 2018;32(12):2527–35 doi 10.1038/s41375-018-0245-3. [DOI] [PubMed] [Google Scholar]
- 41.Palomero T, Barnes KC, Real PJ, Glade Bender JL, Sulis ML, Murty VV, et al. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to gamma-secretase inhibitors. Leukemia 2006;20(7):1279–87 doi 10.1038/sj.leu.2404258. [DOI] [PubMed] [Google Scholar]
- 42.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014;343(6166):84–7 doi 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallden K, Stenmark P, Nyman T, Flodin S, Graslund S, Loppnau P, et al. Crystal structure of human cytosolic 5’-nucleotidase II: insights into allosteric regulation and substrate recognition. J Biol Chem 2007;282(24):17828–36 doi 10.1074/jbc.M700917200. [DOI] [PubMed] [Google Scholar]
- 44.Brooks HB, Geeganage S, Kahl SD, Montrose C, Sittampalam S, Smith MC, et al. Basics of Enzymatic Assays for HTS. In: Markossian S, Sittampalam GS, Grossman A, Brimacombe K, Arkin M, Auld D, et al., editors. Assay Guidance Manual. Bethesda (MD)2004. [Google Scholar]
- 45.Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem 2012;4(2):90–8 doi 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. Journal of structural biology 2003;142(1):170–9 doi 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 47.Integration Kabsch W., scaling, space-group assignment and post-refinement. Acta crystallographica Section D, Biological crystallography 2010;66(Pt 2):133–44 doi 10.1107/s0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta crystallographica Section D, Biological crystallography 2010;66(Pt 1):22–5 doi 10.1107/s0907444909042589. [DOI] [PubMed] [Google Scholar]
- 49.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. Journal of structural biology 1999;125(2–3):156–65 doi 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography 2010;66(Pt 4):486–501 doi 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography 2010;66(Pt 2):213–21 doi 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell chemical biology 2019;26(5):623–33.e9 doi 10.1016/j.chembiol.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analyzed during the current study.