

Abstract
Purpose:
Mucoepidermoid carcinoma (MEC) is a poorly understood salivary gland malignancy with limited therapeutic options. Cancer stem cells (CSC) are considered drivers of cancer progression by mediating tumor recurrence and metastasis. We have shown that clinically relevant small molecule inhibitors of MDM2-p53 interaction activate p53 signaling and reduce the fraction of CSC in MEC. Here we examined the functional role of p53 in the plasticity and self-renewal of MEC CSC.
Experimental design:
Using gene silencing and therapeutic activation of p53 we analyzed the cell cycle profiles and apoptosis levels of CSCs in MEC cell lines (UM-HMC-1, −3A, −3B) via flow cytometry and looked at the effects on survival/self-renewal of the CSCs through sphere assays. We evaluated the effect of p53 on tumor development (N=51) and disease recurrence (N=17) using in vivo subcutaneous and orthotopic murine models of MEC. Recurrence was followed for 250 days after tumor resection.
Results:
Although p53 activation does not induce MEC CSC apoptosis, it reduces stemness properties such as self-renewal by regulating Bmi-1 expression and driving CSC towards differentiation. In contrast, downregulation of p53 causes expansion of the CSC population while promoting tumor growth. Remarkably, therapeutic activation of p53 prevented CSC-mediated tumor recurrence in preclinical trials.
Conclusion:
Collectively, these results demonstrate that p53 defines the stemness of MEC and suggest that therapeutic activation of p53 might have clinical utility in patients with salivary gland mucoepidermoid carcinoma.
Keywords: Cancer stem cells, Self-renewal, Differentiation, Mucoepidermoid carcinoma, Cell fate, MDM2, Bmi-1
Introduction
Salivary gland cancers are morphologically and clinically diverse neoplasms that account for 6% of all head and neck cancers (1). The World Health Organization has categorized these tumors into 20 distinct subtypes (1). The rarity and diversity of these malignancies presents diagnostic and treatment challenges for clinicians (2). Among these subtypes, mucoepidermoid carcinoma (MEC) is the most common malignant salivary gland tumor in adults and children (3). Little is known about the pathobiology of MEC, limiting the development of effective therapies. Chemotherapy is of no benefit to these patients (2). Consequently, no systemic or targeted therapy is currently approved for MEC. Treatments often involve radical head and neck surgery and can include radiation therapy if there is high risk of recurrence or in cases of unresectable tumors(2). These treatments typically result in high patient morbidity with major facial disfigurement and most patients with recurrent/metastatic cancers succumb to disease. As such, a safe and effective, mechanism-based therapy is urgently needed for treatment of patients with this orphan, albeit deadly, disease.
Few recurrent genetic alterations have been found in MEC, with CRCT1/MAML2 fusions being the most common (4, 5). In this fusion, the NOTCH-binding domain of MAML2 is replaced by the CREB-binding domain of CRCT1 resulting in disruption of NOTCH signaling and activation of c-AMP responsive target genes (6, 7). Although this fusion is thought to play a role in tumorigenesis, there has been conflicting evidence as to how its presence affects patient outcomes (8–11). Unlike other head and neck cancers, p53 is not frequently mutated in MEC suggesting that p53 activating therapeutics may have clinical utility for the treatment of this disease (12–14). One such therapeutic involves using MDM2 inhibitors, which activate p53 signaling by disrupting the binding between MDM2 (Mouse double minute 2) and p53. MDM2 is an E3 ubiquitin ligase that targets p53 for proteasomal degradation and is also a transcriptional target of p53, resulting in a negative feedback loop that keeps p53 levels in check (15). p53 is the most commonly mutated gene in cancer. Cancers that do not directly have mutated p53 inactivate p53 signaling by overexpressing MDM2 or deregulating downstream p53 effectors. Importantly, p53 is considered a master regulator of cell fate by controlling processes involved in cell survival, cell division, and stem cell and cancer stem cell self-renewal and differentiation (16).
MEC cancer stem cells (CSC) are a subset of cells marked by high ALDH enzymatic activity and CD44 expression that are highly tumorigenic, have self-renewal capacity, and can generate the different cell phenotypes that make up MEC tumors (17, 18). Studies have shown cancer stem cells are resistant to conventional therapeutics and are thought to be responsible for tumor recurrence and metastasis (19, 20). We have previously demonstrated that treatment of MEC cells with the MDM2 inhibitor MI-773 activates p53 signaling and decreases the MEC CSC population (21, 22). However, the mechanisms underlying this effect are not known. It has been suggested that p53 can play a role in regulating cell fate decisions (e.g. apoptosis/survival, asymmetric division) (16). Here, we evaluated the functional role of p53 in regulating MEC cancer stem cell fate and unveiled the therapeutic potential of targeting this pathway in a pre-clinical trial performed in murine models of MEC.
Materials and Methods
Cell Culture and Reagents
University of Michigan Human Mucoepidermoid Carcinoma cell lines (UM-HMC-1, UM-HMC-3A, and UM-HMC-3B) (RRID:CVCL_Y473, RRID:CVCL_Y471, RRID:CVCL_Y472, respectively) were generated from surgical specimens (23). These cells were cultured in high glucose Dulbecco’s Modified Eagle’s Medium (Invitrogen; Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Atlanta Biologicals; Flowery Branch, GA, USA), 1% L-Glutamine (MilliporeSigma; Burlington, MA, USA), 1% Antibiotic-Antimycotic (MilliporeSigma), 400 ng/mL hydrocortisone (StemCell Technologies; Vancouver, BC, Canada), 20 ng/mL recombinant human epidermal growth factor (R&D Systems; Minneapolis, MN), and 5 μg/mL recombinant human insulin (Sigma-Aldrich; Burlington, MA, USA) at 37°C and 5% CO2. Low passage primary human microvascular endothelial cells (HDMEC) (Lonza; Morristown, NJ, USA) were cultured in endothelial growth medium-2 for microvascular cells (Lonza). Small molecule inhibitors of MDM2-p53 interaction (i.e., MI-773, APG-115, and MI-1061) and MDM2 degrader (MD-224) were provided by Shaomeng Wang (University of Michigan) (22, 24, 25). MG132 (MilliporeSigma) was used to inhibit proteasomal degradation. PTC596 (MedChemExpress; Monmouth Junction, NJ), a small molecule inhibitor of Bmi-1, was used to evaluate the direct impact of Bmi-1 on MEC stemness.
Cell Line Authentication
Genomic DNA was extracted from reference tissues and from the UM-HMC-1, UM-HMC-3A, and UM-HMC-3B cell lines using Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). DNA genotyping by STR profiling and testing for mycoplasma contamination were performed by an independent company (Genetica Cell Line Testing; Burlington, NC, USA) and confirmed the authenticity and lack of contamination of the cell lines used here (Supplementary Fig. S1).
Animals
Naïve female CB17 SCID mice (Charles River, Wilmington, MA, USA) were used to generate Mucoepidermoid carcinoma xenografts. Mouse sex was matched to the original cell line donor. Mice were randomly assigned to their surgical and/or treatment groups. For the recurrence study, tumors were randomly distributed to obtain comparable tumor size means and distribution between both treatment groups. Surgeons were blinded during tumor resections. All studies were performed according to the experimental protocols approved by the University of Michigan Institutional Animal Care and Use Committee (IACUC), and all procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Salisphere Assays
UM-HMC-1,−3A,−3B (RRID:CVCL_Y473, RRID:CVCL_Y471, RRID:CVCL_Y472, respectively) cells (23) were cultured in ultra-low attachment plates or flasks (Corning; Corning, NY, USA) in DMEM/F12 (Invitrogen; Waltham, MA) supplemented with 1% N2 Supplement (Invitrogen), 1% GlutaMAX (Invitrogen), 1% antibiotic-antimycotic (Millipore Sigma, St. Louis, MO), 20 ng/mL rhEGF (R&D Systems; Minneapolis, MN), 20 ng/mL recombinant human basic FGF (R&D Systems), 10 ng/mL recombinant human insulin (Sigma-Aldrich, St. Louis, MO), and 1 μM Dexamethasone (Sigma-Aldrich). 4,000 cells/well were plated in 6-well ultra-low attachment plates and treated the following day with either MI-773 or APG-115, unless otherwise stated. Secondary salispheres were generated by dissociating primary spheres into single cell suspensions with Accutase (StemCell Technologies; Vancouver, Canada) and re-plating 4,000 cells/well in 6-well ultra-low attachment plates. Spheres were quantified 7–9 days after being plated, unless otherwise stated. Salispheres were defined as non-adherent spheres containing ≥30 cells, as observed through 100–200x magnification. Results are representative of a minimum of two independent experiments performed in triplicate experimental conditions.
Mucoepidermoid carcinoma xenografts (subcutaneous)
Mucoepidermoid carcinoma subcutaneous xenograft tumors were generated, as we described (17). In brief, poly-L-lactic acid scaffolds were seeded with 600,000 UM-HMC-3A or UM-HMC-3B cells with or without 400,000 primary human HDMEC cells (Lonza; Basel, Switzerland) in a cell growth media and Matrigel (Corning) mix. The scaffolds were implanted subcutaneously in the dorsal region of CB17 SCID mice (Charles River; Wilmington, MA, USA). Tumor measurements were taken along the x and y axis using a caliper and volumes were calculated using the equation V=(height*width2)/2. For tumor growth studies, mice were monitored twice a week until tumor volumes reached 2 cm3, at which point all groups were euthanized.
Mucoepidermoid carcinoma xenografts (orthotopic)
Orthotopic xenograft tumors were generated by injecting 500,000 UM-HMC-3A or UM-HMC-3B cells into the submandibular gland of CB17 SCID mice. Mice were euthanized 80 days post-injection or upon significant adverse events such as weight loss. For flow cytometry analysis of the cancer stem cell population, tumor tissues were digested using collagenase (StemCell Technologies) and single cell suspensions were stained for ALDH and CD44 as described above. All animal experiments were performed under an IACUC-approved protocol.
Lentiviral knockdown
Lentiviral particles were produced in HEK293T cells (RRID:CVCL_HA71) using the calcium phosphate method by co-transfecting pMD2.G (RRID:Addgene_12259) and psPAX2 (RRID:Addgene_12260) packaging vectors with either shRNA-control (pGIPZ scrambled or pLKO scrambled), shRNA-p53 constructs on a pGIPZ backbone (RRID:Addgene_121488; University of Michigan Vector Core; seq −1: TACACATGTAGTTGTAGTG, seq −2: TAACTGCAAGAACATTTCT, seq #3: TACACATGTAGTTGTAGTG), or shRNA-p21 constructs on a pLKO backbone (Sigma, seq 11: GACAGATTTCTACCACTCCAACTCGAGTTGGAGTGGTAGAAATCTGTC, seq 22: CGCTCTACATCTTCTGCCTTACTCGAGTAAGGCAGAAGATGTAGAGCG, seq 33: GACACCACTGGAGGGTGACTTCTCGAGAAGTCACCCTCCAGTGGTGTC). The UM-HMC-1,−3A,−3B cells were infected with supernatant containing the lentiviral particles and with 4 μg/mL polybrene (Sigma-Aldrich) overnight. Infected cells were selected with 1 μg/mL puromycin (InvivoGen; San Diego, CA, USA) for at least one week. pGIPZ constructs were subsequently sorted via FACS for GFP positive cells. Immunoblotting was used to verify p53 and p21(CDKN1A) knockdown of the selected cells.
Cytotoxicity Assay
Sulforhodamine B assays were used to measure the cytotoxicity of small molecule inhibitors of MDM2-p53 interaction in UM-HMC cells. Here, 800–1,000 cells/well were plated in 96-well plates and were exposed the following day to either vehicle, MI-773, or APG-115 for 24 to 72 hours. Cells were fixed in 10% trichloroacetic acid for 1 hour at 4°C. After drying, plates were stained with a 0.4% Sulforhodamine B solution (Sigma Aldrich) at room temperature for 30 minutes. Unbound dye was washed away with 1% acetic acid. The absorbed dye was resolubilized in 10 mM unbuffered Tris base and the plates were read in a microplate reader at 565 nm (GENios, TECAN). Results were normalized to vehicle control and IC50 values were calculated using nonlinear fit variable slope function in GraphPad PRISM. All conditions were evaluated in triplicate and results are representative of at least two independent experiments.
Single-cell sphere devices
To ensure clonal spheres are grown from single cancer cells, a microfluidic high-throughput sphere culture assay was introduced to improve single-cell capture rate. Each microfluidic device contains 3,200 individual sphere culture chambers that are isolated from each other. During cell loading process, cancer cells were plated at device inlet and flew into microfluidic chambers driven by gravity flow. Single cells were guided by hydrodynamic flow channel structures and captured at the trapping site in each sphere culture chamber. The sphere culture chambers were designed to be 150 μm × 150 μm × 150 μm to provide enough room for sphere growth. A 40 μm-high meander escape channel was designed at the end of main flow channels to release the residue flow and avoid multiple cell capture cases. Sphere culture media was exchanged every 24 hours. Microfluidic device images were taken right after cell loading, as well as on day 6 and day 12, to keep track of sphere growth. The cells were stained with Cell Tracker green (cat# C2925; ThermoFisher) for automated sphere size analysis with Matlab program (RRID:SCR_001622).
The microfluidic sphere culture chip was fabricated with a patterned PDMS (polydimethylsiloxane; Sylgard 184, Dow Corning) bonded to another piece of blank PDMS. Standard soft-lithography process was used to pattern PDMS piece. A silicon wafer patterned with SU8 photoresist (MicroChem) was used as mold for soft lithography. The mold was created by a 3-layer photolithography process with a 5 μm-thick-layer for cell capture site, a 40 μm-thick-layer for meander escape channel, and a 100 μm-thick-layer for sphere chambers and flow channels. 25 grams of PDMS reagent was poured on silicon mold. After curing at 100 °C for 1 day, the PDMS piece was peeled off. Then, an inlet and an outlet were created using a biopsy punch. Finally, the patterned PDMS pieces were activated by oxygen plasma treatment (80 Watts, 60 seconds) and bonded to another piece of blank PDMS. The microfluidic chips were sanitized using UV radiation and primed using a 5% (w/w) PEO-terminated triblock polymer (Pluronic® F108, BASF) 1 day before usage.
Western blot
Whole-cell lysates from UM-HMC cells were prepared using a 1% Nonidet P-40 (NP-40) lysis buffer. Lysates were loaded onto 9–15% SDS-PAGE gels for protein separation. Proteins were transferred to nitrocellulose membranes (GE Healthcare Life Sciences; Marlborough, MA) and probed with the following primary antibodies: mouse anti-p53 (cat# sc-126; RRID:AB_628082), mouse anti-MDM2 (cat# sc-965; RRID:AB_627920), HRP-conjugated mouse anti-beta-Actin (cat# sc-47778; RRID:AB_626632), mouse anti-NOXA (cat# sc-56169; RRID:AB_784877) (Santa Cruz Biotechnology; Santa Cruz, CA); rabbit anti-p21(cat#2947; RRID:AB_823586), rabbit anti-Bmi-1 (cat#6964; RRID:AB_10828713), rabbit anti-BIM (cat#2933; RRID:AB_1030947), rabbit anti-PUMA (cat#12450; RRID:AB_2797920) (Cell Signaling; Danvers, MA, USA); or mouse anti-GAPDH (cat# MAB374) (MilliporeSigma). Membranes were exposed to HRP-conjugated anti-mouse or anti-rabbit secondary antibodies (Jackson Laboratories; West Grove, PA) and proteins were visualized by SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL). Protein band densitometry was calculated with ImageJ version 2.0.0. (RRID:SCR_003070).
Cycloheximide treatment
Cells were pre-treated with DMSO vehicle control or 10 μM MI-773 for 3 hours before being treated with 25–50 μM Cycloheximide (Sigma-Aldrich) to stop protein synthesis. Whole cell lysates were generated at different timepoints using an NP-40 lysis buffer and subsequently analyzed through western blot. Protein degradation rates were calculated using one-phase decay function (Y0=1, Plateau=0) in GraphPad PRISM.
RT-PCR
RNA was isolated from cells with Quick-RNA Miniprep (Zymogen). 1 μg of RNA was used to generate cDNA using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories; Hercules, CA, USA) and PCR was performed with Platinum Taq DNA Polymerase (Invitrogen). The following primers were used to generate the PCR products: Bmi-1 (Sense 5’- CAGCGGTAACCACCAATCTT -3’, Antisense: 5’- AAAGTCTTGCCTGCTTTCCA -3’), p21 (Sense: 5’- AGTCAGTTCCTTGTGGAGCC -3’, Antisense: 5’- GAAGGTAGAGCTTGGGCAGG -3’), and GAPDH (Sense: 5′- GACCCCTTCATTGACCTCAACT -3′, Antisense: 5′- CACCACCTTCTTGATGTCATC -3′). RT-PCR products were verified through gel electrophoresis.
Flow Cytometry
All flow cytometry analysis was conducted in a BD LSRFortessa instrument and iCyt Synergy SY3200 cell sorter was used for fluorescence-activated cell sorting at the University of Michigan Flow Cytometry Core. Staining for ALDH enzymatic activity was carried out using Aldefluor Kit (StemCell Technologies) or AldeRed ALDH Detection Assay (MilliporeSigma). For analysis of the cancer stem cell fraction in vitro, 5×105 cells were incubated with 2.5 μL of activated Aldefluor or Aldered substrate in 250 μL of buffer at 37°C for 40 minutes. DEAB controls for all treatment conditions were included. After incubation, cells were washed with PBS and subsequently stained for CD44 using one of the following antibodies: CD44-PE (cat#566803; RRID:AB_2869876), CD44-APC (cat#559942; RRID:AB_398683), CD44-BV450 (cat#560451; RRID:AB_1645273) (BD Pharmingen; San Diego, CA, USA), or CD44-APC-Cy7 (cat#103028; RRID:AB_830785; BioLegend; San Diego, CA). DEAB controls were incubated with the corresponding isotype controls: PE Mouse IgG (cat#554680; RRID:AB_395506), APC Mouse IgG (cat#562025; RRID:AB_10892809), BV450 Rat IgG (cat#560457; RRID:AB_1645681; BD Pharmingen; Franklin Lanes, NJ), or APC-Cy7 Rat IgG (cat#400624; RRID:AB_326566) (BioLegend). CD44 staining was done at 4°C for 15–20 minutes. Cells were washed with PBS and DAPI (Molecular Probes; Eugene, OR) was added for live/dead discrimination. To look at the proportion of cancer stem cells undergoing apoptosis, cells stained for ALDH and CD44 were subsequently incubated with 10 μL of Annexin V-PE (cat#556421; RRID:AB_2869071; BD Pharmigen) in 100 μl of 1X binding buffer (BD Pharmingen) for 15 minutes at room temperature. Staining was immediately quenched with 200 μL of 1X binding buffer and DAPI was added. To analyze cell cycle of the ALDHhighCD44high cells, 1.5×106 cells/tube were co-incubated with 7.5 μL of activated Aldefluor substrate and 3 μL of Vibrant DyeCycle-Orange Stain (cat#V35005) (Invitrogen) in 1 mL of PBS for 35 minutes at 37°C. As previously described, DEAB was used as a negative control for Aldefluor. After incubation, cells were spun down at 800 rpm for 5 min, the supernatant was removed, and cells were resuspended in 2.5 μl anti-human CD44-APC-Cy7 or APC-Cy7 IgG control in 300 μL PBS for 15 minutes at 4°C. DAPI was added right after incubation and cells were taken immediately for flow cytometry analysis. All conditions were evaluated in triplicate and results are representative of at least two independent experiments. Flow cytometry analysis was done with FlowJo software (RRID:SCR_008520; LLC; Ashland, OR).
Immunohistochemistry
Paraffin embedded tissue section slides were incubated with Trypsin (Sigma) at 37°C for antigen retrieval followed by 0.1% Triton-x100 (Sigma) at room temperature. Endogenous peroxidase activity was inhibited by incubating with 3% hydrogen peroxide (Fisher) and nonspecific background antibody binding was blocked using Background Sniper (Biocare Medical) at room temperature. For ALDH staining, slides were incubated overnight at 4°C with mouse anti-human ALDH1 (cat #611195; RRID:AB_398729; BD Biosciences). Following incubation, sections were washed with 1X Immunohistochemistry (IHC) Wash Buffer (Dako) and afterwards incubated with MACHI 3 Probe (Biocare Medical). For Keratin (cytokeratin)-7 staining, antigen retrieval was performed using 1x Citrate Buffer, ph.6.0 (Fisher) for 20” at 95°C. Primary rabbit anti-human Keratin (Cytokeratin)-7 antibody (cat # 4465; RRID:AB_11178382; Cell Signaling) was diluted in a range from 1:100 in Antibody Diluent Solution (Dako) and slides were incubated at 4°C overnight. Sections were washed again with 1X IHC wash buffer followed by incubation with MACH 3 HRP (Biocare Medical) and washed again with IHC wash buffer. DAB chromogen kit (Biocare Medical, Concord, CA) was used for chromogenic development at room temperature. Finally, sections were incubated with hematoxylin, dehydrated, and permanent mounting solution (Vectamount, Vector) was used to fix the slide coverslip on the stained tissue section. Quantification of the fraction of Cytokeratin-positive cells was performed by a trained oral pathologist (FN) blinded for experimental conditions.
Immunocytochemistry
Cells were plated in 4-well chamber slides and incubated overnight before any treatment. Cells were fixed in formaldehyde/glutaraldehyde. After fixation, chambers were removed from the slides and slides were incubated first with 0.1% Triton-X 100 (Sigma), followed by 3% hydrogen peroxide, then background sniper (Biocare Medical; Pacheco, CA, USA), and finally overnight at 4°C with one of the following primary antibodies: mouse anti-human pan-cytokeratin (sc8018; RRID:AB_627396; Santa Cruz Biotechnology) and rabbit anti-human Bmi-1 (cat#6964; RRID:AB_10828713; Cell Signaling). The following day, slides were washed with 1X IHC wash buffer and incubated with the corresponding secondary antibodies: Alexa Fluor 488 anti-rabbit IgG (A32723, RRID:AB_2633275) or Alexa Fluor 594 anti-mouse IgG (A11037; RRID:AB_2534095) (Life Technologies; Carlsbad, CA, USA) for 1 hour at room temperature. Isotype-matched IgG was used as a negative control (Jackson Laboratories). Slides were washed again with 1X IHC wash buffer and coverslip was placed with DAPI-containing mounting solution (Vectashield, Vector). Imaging was achieved using an inverted Leica SP5 confocal microscope (Leica microsystems; Germany). To excite fluorescence in all channels, the 405 nm laser and the tunable white light laser were used. The detectors spectral range were set to blue spectral range from 415 to 478 nm; green spectral range from 498 to 550 nm and red spectral range was from 571 to 727 nm bandpass. Channels were acquired sequentially between lines. Scanning speed was set to 50 Hz in bidirectional mode with line average set to 2. All Images were recorded in 1,024 × 1,024 format and pinhole size was set to 60 μm.
Statistical Analysis
All statistical analysis was done using GraphPad PRISM (RRID:SCR_002798; GraphPad, San Diego, CA, USA). Sample sizes for both in vitro and in vivo experiments were determined from power calculations using data published in previous publications. All studies were done with a minimum of three biological replicates. Variance was similar between comparison groups. Pre-established outlier criteria for mouse tumors were defined as tumor volumes greater than two standard deviations away from the group mean. For cancer stem cell analysis of digested tumors, samples were excluded if less than 3,000 events were obtained per sample. Data is summarized as mean ± SD. Two-tailed student’s t-test was conducted for two group comparisons and analysis of variance (ANOVA) followed by appropriate post hoc tests (Tukey or Bonferroni) were conducted on comparisons between more than two groups. Kaplan-Meier graphs were analyzed using the Gehan-Breslow-Wilcoxon test. Significance level was set at p<0.05 for all experiments.
Data availability
All data that support the findings of this study are available from the corresponding author on reasonable request.
Results
Activation of p53 by MDM2 inhibitors decreases the fraction of cancer stem cells
We evaluated the baseline levels of several proteins involved in p53 signaling using a panel of human mucoepidermoid carcinoma cell lines i.e., UM-HMC-1, UM-HMC-3A, and UM-HMC-3B (Fig. 1A; Supplementary Fig. S1). The UM-HMC-1 cell line was generated from a primary tumor; meanwhile, the UM-HMC-3A cell line was generated from a recurrent tumor and the UM-HMC-3B cell line was generated from a lymph node metastasis in the same patient as UM-HMC-3A (23). The authenticity of these cell lines was confirmed by STR profiling against reference tumor tissues from the patients (Supplementary Fig. S1). Interestingly, the UM-HMC-3B cell line expressed higher p53 and lower MDM2 levels then UM-HMC-3A, suggesting p53 signaling might be regulated differently in the metastatic line (Fig. 1A). Whole-exome sequencing on these cells revealed no mutations in either MDM2 or TP53 that might explain this differential expression (21).
Figure 1: p53 regulates the fraction of mucoepidermoid carcinoma cancer stem cells in vitro.

A, Western blot showing baseline protein levels for MDM2, p53, p21 and Bmi-1 in human mucoepidermoid carcinoma (MEC) cell lines (UM-HMC-1, UM-HMC-3A, UM-HMC-3B). B, Western blots of MEC cells treated for 24 and 48 hours with MI-773, a 1st generation small molecule inhibitor of the MDM2-p53 interaction. C, Graph depicting the fraction of cancer stem cells (ALDHhighCD44high) measured by flow cytometry of MEC cell lines treated with MI-773 for 72 hours. D, Western blots verifying the impact on p53 protein expression in cells transduced with shRNA-p53 sequences. E, Graphs depicting cell density measurements of p53-silenced or vector control cells treated with MI-773 for 48 hours. Data was normalized against vehicle control. Graph inserts: half-maximal inhibitory concentrations (IC50) for MI-773. F, Western blots for MDM2, p53 and p21 of p53-silenced and control cells treated with MI-773 for 48 hours. G, Graph summarizing flow cytometry analysis of the ALDHhighCD44high cell fraction in vector control and p53-silenced cells treated with MI-773 for 72 hours. All results are representative of at least two independent experiments. Data was analyzed by two-tailed student’s t-test (α=0.05) in two group comparisons or one-way ANOVA followed by post-hoc Tukey (α=0.05) for multiple group comparisons. * P<0.05, ** P<0.001, *** P<0.0001, ns=not significant.
A small molecule inhibitor of the MDM2-p53 interaction (MI-773) was used as a tool to activate p53 signaling in MEC cells. Cytotoxicity assays revealed that UM-HMC-1 and UM-HMC-3A cells are more responsive than UM-HMC-3B cells to this inhibitor (Supplementary Fig. S2A). Western blotting showed that p53 signaling was activated in a dose-dependent manner in the UM-HMC-1 and UM-HMC-3A cells as evidenced by accumulation of transcriptional targets such as p21 and its negative regulator MDM2 (Fig. 1B, Supplementary Fig. S2B). Consistent with the cytotoxicity assays, UM-HMC-3B cells were less responsive to MI-773 than UM-HMC-1 or UM-HMC-3A. We hereon refer to the UM-HMC-1 and the UM-HMC-3A cell lines as sensitive while UM-HMC-3B as resistant to inhibition of the MDM2-p53 interaction. Of note, MI-773 treatment inhibited Bmi-1 expression in all three cell lines. This polycomb protein is an important regulator of stemness in other systems and is frequently used as a CSC marker (26). Confirming this observation, low cytotoxic doses of MI-773 caused a significant decrease in the CSC fraction (ALDHhighCD44high) in the sensitive MEC cells, while no significant difference was observed in the resistant UM-HMC-3B cells (Fig. 1C).
To confirm the results obtained with MI-773, a second-generation inhibitor of MDM2-p53 interaction called APG-115 was used to activate p53 signaling and verify the effects on MEC CSC (24). IC50 values calculated from the cytotoxicity assays confirmed that MEC cells are more sensitive to APG-115 than MI-773 (Supplementary Fig. S2A, S2C). As expected, UM-HMC-3B cells were more resistant than UM-HMC-1 and UM-HMC-3A. p53 pathway activation as well as decreased Bmi-1 expression was confirmed via Western blot (Supplementary Fig. S2D), and a decrease in the CSC fraction was observed in the sensitive UM-HMC-1 cells (Supplementary Fig. S2E).
p53 depletion leads to an expansion of the cancer stem cell population
To confirm that the decrease in MEC CSC is due to activation of p53 signaling and not due to off target effects of the MDM2-p53 inhibitors, we used short hairpin RNAs (shRNA) to silence p53 expression in MEC cells. Successful p53 silencing was obtained with sequences −1 and −2, while sequence −3 was used as a control (Fig. 1D). In p53 knockdown cells, MI-773 failed to activate p53 signaling and caused less cytotoxicity compared to scrambled vector-transduced cells, confirming the specificity of the inhibitor (Fig. 1E and 1F). Of note, there was no significant difference in the growth curves and doubling times (DT) between the vector-control and p53-silenced cells (Supplementary Fig. S2F). Importantly, in the absence of activated p53 we no longer observed a decrease in the CSC fraction upon inhibition of the MDM2-p53 interaction (Fig. 1G and Supplementary Fig. S2G), suggesting that p53 protein levels regulate the fate of MEC CSC.
To verify these in vitro observations, we evaluated the effects of p53 silencing in subcutaneous and orthotopic xenograft models of MEC. UM-HMC cells transplanted into the subcutaneous space or in the submandibular glands (SMG) of immunodeficient mice were monitored for tumor growth and all mice were euthanized when the first tumor reached endpoint (2 mm3). p53 knockdown dramatically enhanced tumor growth in xenograft tumors generated with the sensitive UM-HMC-3A cell line (Fig. 2A–F). Outlier tumor was excluded from analysis in Figures 2B, 2C, and 2H. Due to limited tumor growth with the vector control cells at study endpoint, there was insufficient tissue to perform flow cytometry analysis of the ALDHhighCD44high CSC population. As an alternative, immunohistochemistry was performed for ALDH1, a marker of CSC in MEC (17, 18). While all tumors in the shRNA-p53 group contained CSC as determined by ALDH1 expression, very few ALDH1-positive cells were found in the control group (Fig. 2G and 2H). These experiments were repeated with the resistant UM-HMC-3B cells and no detectable difference in the establishment and growth of the xenograft tumors was found between the control and the shRNA-p53 groups (Fig. 2I–L, 2M–P). Nevertheless, even in tumors generated with these less responsive cells we observed a trend towards a higher percentage of CSC in the shRNA-p53 tumors when compared to vector-control tumors (Fig. 2L, 2P).
Figure 2: p53 silencing increases tumor growth and expands the fraction of cancer stem cells in vivo.

A, Macroscopic image of subcutaneous xenograft tumors generated with UM-HMC-3A cells transduced with shRNA-p53 or vector controls. B, Kaplan-Meier curves depicting tumor-free survival from (A). Failure was defined as tumors that reached a volume of 200 mm3. C, Graph depicting tumor weights at study endpoint. D, Macroscopic image of orthotopic xenograft tumors generated with UM-HMC-3A cells transduced with shRNA-p53 or vector control and injected in the submandibular glands of mice. E, Kaplan-Meier curves depicting tumor-free survival from (D). Failure was defined when salivary gland tumors were palpable. F, Graph depicting the weight of the submandibular glands at study endpoint. G, Photomicrographs of H&E and immunohistochemical analysis of ALDH1 in subcutaneous tumors from (A), scale bar = 100 μM. H, Average ALDH1 scores of 5 randomly selected microscopic fields per tumor (G). Microscopic fields were scored as follows: 0 – no cells with ALDH1 staining; 1 – up to 10 cells stained for ALDH1; 2 – more than 10 cells with high ALDH1 expression. I, Macroscopic image of subcutaneous xenograft tumors generated with UM-HMC-3B cells. J, Kaplan-Meier curves depicting tumor-free survival. Failure was defined as tumors that reached a volume of 200 mm3. K, Graph depicting tumor weight at study endpoint. L, Fraction of cancer stem cells (ALDHhighCD44high) in tumors generated with UM-HMC-3B cells transduced with shRNA-p53 or vector control. M, Macroscopic image of orthotopic xenograft tumors generated with UM-HMC-3B cells. N, Kaplan-Meier curves depicting tumor-free survival. Failure was defined when salivary gland tumors were palpable. O, Weight of the submandibular glands at study endpoint. P, Orthotopic xenograft experiment with UM-HMC-3B cells was repeated (n=13) and the fraction of cancer stem cells was analyzed. Graph depicting the fraction of cancer stem cells (ALDHhighCD44high) in tumors generated with UM-HMC-3B cells transduced with p53 shRNA or vector control. Samples with less than 3,000 tumor cells obtained from digested tissues were excluded from flow cytometry analysis. Kaplan-Meier graphs were analyzed using Gehan-Breslow-Wilcoxon test. All other data was analyzed by two-tailed student’s t-test. *P<0.05.
p53 activation does not preferentially kill cancer stem cells
To understand how induction of p53 decreases the population of MEC CSC, we first assessed whether activating p53 signaling using inhibitors of the MDM2-p53 interaction depletes this population by preferentially inducing apoptosis. Annexin V staining is commonly used to detect early apoptotic events in viable cells and is compatible with ALDH enzymatic detection assays such as ALDEFLUOR™ and ALDERED™. This approach allowed the examination of apoptotic events specifically in the CSC. Cells were treated with low dose MI-773 (1 μM) to activate p53 signaling and apoptosis was measured at different time points. Under these conditions, modest increase in apoptosis was observed only in the bulk cell population of the UM-HMC-3A cells (Fig. 3A). In addition, we evaluated apoptosis and cell cycle with propidium iodide followed by flow cytometry, which demonstrated a modest increase (from ~1% to about 4%) in the fraction of bulk tumor cells in sub-G0/G1 (i.e. apoptotic cells) with increasing concentrations of MI-773 up to 1 μM (Supplementary Fig. S3A, S3C, S3E). Concomitantly, we observed an increase in the fraction of cells in G1 and a decrease in the fraction of cells in S-phase, which correlates with a slowdown in cell proliferation and a G1 cell cycle arrest of the bulk cells (Supplementary Fig. S3A, S3B, S3D). These changes in cell cycle and cell death correlated with the decrease in bulk cell density when cells were treated with MI-773 particularly in the longer time points, i.e. 48 and 72 hours (Supplementary Fig. S2A). In addition, evaluation of pro-apoptotic proteins by Western blots showed a modest increase in expression of PUMA but no changes in BIM or NOXA expression (Supplementary Fig. S3F) in bulk cells treated with MI-773. We observed modest changes in bulk cell death in response to 1 μM MI-773 (Fig. 3A, Supplementary Fig. S3C and S3E). Importantly, we showed that the fraction of cancer stem cells was significantly decreased in UM-HMC-1 and UM-HMC-3A cells treated with 1 μM MI-773 (Fig. 3B) despite the fact that MI-773 does not induce cancer stem cell apoptosis (Fig. 3C). Collectively, these data indicated that MI-773-mediated decrease in the fraction of cancer stem cells in mucoepidermoid carcinoma is not caused by preferential induction of apoptosis of these stem cells (Fig. 3C).
Figure 3: p53 activation does not preferentially kill cancer stem cells.

UM-HMC cell lines were treated with either vehicle or MI-773 (1μM) and subsequently analyzed for apoptotic cells using Annexin V staining. A, Graphs showing Annexin V staining in the bulk cell population 24–72 hours after MI-773 treatment. B, Graphs depicting the fraction of cancer stem cells (ALDHhighCD44high) in (A). C, Graphs showing the fraction of cancer stem cells (ALDHhighCD44high) undergoing apoptosis after treatment with MI-773 or vehicle control. All results are representative of at least two independent experiments. Data was analyzed by two-way ANOVA followed by post-hoc Bonferroni. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns=not significant.
p53 signaling through p21 induces accumulation of cancer stem cells in G1
A non-toxic cell permeable DNA dye (DyeCycle-Orange) compatible with the ALDH enzymatic assay was used to evaluate the effect of p53 activation on the cell cycle of MEC CSC. The MEC sensitive cells (UM-HMC-1 and UM-HMC-3A) were treated with sub-lethal doses of MI-773 (1 μM) to activate p53 signaling without appreciable cytotoxicity (Supplementary Fig. S2A) and the cell cycle profiles were analyzed 24 hours after treatment. MI-773 increased the proportion of bulk cells in G1 suggestive of a G1 cell cycle arrest (Fig. 4A, 4B), an effect that correlates with increased p21 expression. When we focused our cell cycle analysis on the CSC, we observed that a high percentage of CSC are in G2/M at baseline, which is the opposite of the bulk cell population in which most untreated cells are in G1 (Fig. 4A, 4B). Nevertheless, activation of p53 with MI-773 increased the proportion of CSC in G1 when compared to untreated cells, a response similar to that seen in the bulk cell population (Fig. 4A, 4B).
Figure 4: Activation of p53-p21 signaling shifts the cell cycle of mucoepidermoid carcinoma stem cells.

The cell cycle of UM-HMC cells was analyzed with DyeCycle-Orange after being treated with vehicle or MI-773 for 24 hours. A, Representative cell cycle plots for bulk (top) and cancer stem cells (bottom), ALDHhighCD44high, treated with vehicle or 1 μM MI-773 for 24 hours. Gates for the different cell cycle states were set based on the general cell population and kept the same for the cancer stem cells. B, Quantification of bulk and cancer stem cells in each phase of the cell cycle from (A). C, Western blot depicting the effect of shRNA-p21 or vector controls on p21 protein levels in UM-HMC cells. D, Western blot depicting the impact of increasing concentrations of MI-773 for 48 hours on p53 pathway activation in UM-HMC cells transduced with shRNA-p21 or vector control. E, Representative cell cycle plots of bulk cells or cancer stem cells (ALDHhighCD44high) in UM-HMC cells transduced with shRNA-p21 or vector control and treated with vehicle or MI-773 for 24 hours. F, Quantification of the cell cycle phases of bulk or cancer stem cells (ALDHhighCD44high) in UM-HMC cells transduced with shRNA-p21 or vector control. All results are representative of at least two independent experiments. Means not sharing any lower-case letters are significantly different by two-way ANOVA followed by post-hoc Tukey (α=0.001). ***P<0.001, ****P<0.0001, ns=not significant.
Since p53 affects the cell cycle by transcriptional activation of p21, we knocked down p21 using shRNA to determine its role in cell cycle regulation in the CSC. Knockdown was confirmed with two different shRNA constructs i.e., sequence −2 and −3 (Fig. 4C). Sequence −2 was used for the remaining experiments. As expected, p21 silencing did not affect p53 protein accumulation and activation of its downstream signaling by MI-773, as evidenced by the accumulation of MDM2 (Fig. 4D). Additionally, Bmi-1 protein levels decreased with MI-773 treatment, suggesting that the regulation of Bmi-1 is independent of the p21 signaling axis. Although p21 knockdown had modest effect on the cell cycle of the bulk cell population, it attenuated the MI-773-mediated G2/M to G1 shift in the CSC (Fig. 4E, 4F). Collectively, these results demonstrate that MI-773-induced activation of p53 causes a shift towards G1 in the cell cycle of the CSCs that is dependent on downstream p21 signaling.
p53 activation blocks self-renewal and induces differentiation of cancer stem cells
To understand how p53 activation affects the stemness phenotype of MEC, protein expression of the CSC and bulk cell fractions were analyzed after treatment with MI-773 for 72 hours (Fig. 5A). As expected, we observed higher baseline Bmi-1 expression in the CSC fraction when compared to non-CSC. Although a modest decrease in Bmi-1 was observed in the resistant UM-HMC-3B cell line, we observed a significant reduction in Bmi-1 protein levels in the sensitive UM-HMC-3A CSC treated with MI-773. These results indicate that p53 activation results in decreased CSC self-renewal and/or increased differentiation. To further explore this finding, pan-Cytokeratin was used as a marker of differentiation. We observed significantly higher expression of pan-Cytokeratin in CSC treated with MI-773, when compared to controls (Fig. 5B, 5C). CSC were co-stained for Bmi-1 and pan-Cytokeratin to identify a possible inverse relationship between these markers (Fig. 5D–F). As expected, activation of p53 with MI-773 caused a progressive loss in Bmi-1 expression and a gain in pan-Cytokeratin over time (Fig. 5E, 5F). Remarkably, cells expressing high levels of pan-Cytokeratin rarely co-expressed high levels of Bmi-1 (Fig. 5D). These results indicate activation of p53 signaling with MI-773 induces differentiation of MEC CSC.
Figure 5: p53 activation blocks self-renewal and induces differentiation of cancer stem cells.

A-C, UM-HMC cell lines were sorted for cancer stem cells (ALDHhighCD44high) and non-cancer stem cells (ALDHlowCD44low) after being treated with MI-773 for 72 hours. Immediately after, (A) the collected cells were used to make whole-cell lysates for western blot analysis or (B) cultured in a 4-well chamber slide for two days prior to fixing and staining for pan-Cytokeratin and Dapi (scale bar=100μm). C, Graph depicting quantification of pan-Cytokeratin expression in (B). D-F, Cells were sorted for cancer stem cells and immediately plated in a 4-well chamber slide and cultured for 24 hours prior to treatment with 1 μM MI-773. D, Slides were fixed at 48- and 72-hours post-treatment and subsequently stained for pan-Cytokeratin and Bmi-1(scale bar=100μm). E, Graph depicting quantification of nuclear Bmi-1 expression in cancer stem cells or non-cancer stem cells. F, Graph depicting quantification of pan-Cytokeratin expression in cells treated with 1 μM MI-773 or vehicle. G, Representative micrographs of spheres formed from unsorted cells plated in sphere conditions and treated the following day with increasing doses of MI-773 for 7–9 days. H, Quantification of (G). I, Representative micrographs of spheres formed from unsorted UM-HMC cells plated in sphere conditions and treated 5 days after being plated. J, Quantification of (I). K, Representative micrograph of primary salispheres formed by UM-HMC cells transduced with shRNA-p53 or vector control cells. L, Quantification of (K). M, Microscopic view of single cell capture microfluidic device showing sphere growth over time (scale bar=100μm). N, Graph depicting sphere diameter from single cell salispheres generated by UM-HMC cells transduced with shRNA-p53 or vector control cells. Dotted lines depict cut-off for minimum sphere size (100μm). O, Graph depicting the number of secondary salispheres generated from UM-HMC-3B primary salispheres previously treated with MI-773 or vehicle for 7–9 days. P, Western blot of primary salispheres generated by UM-HMC cells transduced with shRNA-p53 or vector control cells. All results are representative of at least two independent experiments. Immunofluorescence was measured as the mean gray value normalized to DAPI. At least 5 arbitrary areas were selected for quantification. All sphere micrographs and quantification was done 7–9 days after being plated, unless noted otherwise. Two-tailed student’s t-test (α=0.05) was used for two group comparisons, two-way ANOVA with post-hoc Tukey (α=0.05) was used for pan-Cytokeratin time course, and one-way ANOVA with post-hoc Tukey (α=0.05) was used for all other comparisons. *P<0.05, ****P<0.0001. Means not sharing any lower-case letters are significantly different to each other.
Salispheres can be used to measure the stemness and self-renewal of salivary gland cancer stem cells (17). UM-HMC-1 cells have limited sphere forming capacity. For this reason, only UM-HMC-3A and UM-HMC-3B cells were used in these assays. Treatment with MI-773 decreased primary salispheres for both the sensitive UM-HMC-3A and the resistant UM-HMC-3B cells (Fig. 5G, 5H). Interestingly, while UM-HMC-3B cells are less sensitive to MI-773 than UM-HMC-3A, we observed that p53 protein accumulation regulates stemness even in resistant MEC cells. Results were validated with APG-115, showing a 100-fold higher sensitivity than MI-773 (Supplementary Fig. S4A). To confirm that MI-773 affects primary sphere development and is not just toxic to cells grown under sphere conditions, UM-HMC-3A spheres were allowed to form for 5 days prior to being treated with MI-773 (1 μM) and were monitored for 5 to 14 days post-treatment. No significant difference in primary salisphere formation was observed (Fig. 5I, 5J).
To confirm the data obtained with MI-773-induced p53 activation, we performed the reverse experiment with p53-silenced MEC cells. We observed that p53-silenced cells formed more primary salispheres than cells transduced with scrambled vectors (Fig. 4K, 4L). To confirm these results, high-throughput sphere culture microfluidic devices were used to monitor single-cell derived spheres with higher precision (Fig. 5M). Each microfluidic device contained 3,200 individual sphere culture chambers allowing for rapid assessment of sphere formation. Single cells were guided by hydrodynamic flow and captured at the trapping site in each sphere culture chamber. p53-silenced cells presented a higher proportion of cells capable of sphere formation as well as increased sphere size (Fig. 5N). Furthermore, spheres formed from shRNA-p53 cells have higher Bmi-1 protein levels than those formed from control cells, indicating a role for p53 in regulating CSC self-renewal (Fig. 5O). Self-renewal was measured by evaluating secondary salisphere formation from primary spheres treated with MI-773. Treatment of primary salispheres caused a significant decrease in secondary sphere formation while p53 knock-down abrogated that effect (Fig. 5P). Meanwhile, no difference was found in the formation, size, or Bmi-1 protein levels of shRNA-p21-derived salispheres when compared to vector controls (Supplementary Fig. S4B, S4C). Furthermore, p21 knockdown did not impact the MI-773-mediated reduction of primary salisphere formation (Supplementary Fig. S4D). Collectively, these results demonstrate that p53 regulates CSC self-renewal and drives these cells towards a more differentiated state.
p53 regulates Bmi-1 expression independently of MDM2
Although Bmi-1 protein levels decrease with MI-773 treatment, no difference was found in Bmi-1 mRNA levels (Fig. 6A, 6B). Nevertheless, treatment with MI-773 resulted in increased p21 mRNA confirming activation of p53’s transcriptional activity (Fig. 6A). This prompted us to look at Bmi-1 protein stability. To do this, we pre-treated UM-HMC-3A cells with MI-773 for 3 hours to allow for p53 protein accumulation. Protein synthesis was halted with cycloheximide, and protein levels were measured at several timepoints. Similarly to what others have shown, an increase was observed in the molecular weight of Bmi-1 upon cycloheximide treatment possibly due to its phosphorylation (Fig. 6C; Supplementary Fig. S5A) (27, 28). In addition, protein accumulation was observed in vehicle-treated cells during the first 40 minutes of cycloheximide treatment, likely because of incomplete halt in protein synthesis (Fig. 6C; Supplementary Fig. S5A). Nevertheless, Bmi-1 protein turnover was slightly faster in the MI-773 treated cells (Fig. 6C). To find out whether MDM2 plays a role in Bmi-1 protein turnover, we treated UM-HMC cells with MD-224, an MDM2 degrader that activates p53 signaling while reducing MDM2 protein accumulation (25). We compared MD-224 treated cells to those treated with its control, MI-1061, and found no correlation between Bmi-1 and MDM2 protein levels (Fig. 6D). Furthermore, this p53-driven Bmi-1 decrease is proteosome-dependent as evidenced by treatment with MG-132 (Fig. 6E; Supplementary Fig. S5B). Notably, direct inhibition of Bmi-1 with the small molecule inhibitor PTC-596 is sufficient to decrease the fraction of MEC CSCs in UM-HMC-1 and UM-HMC-3A cells. These results imply that reduced Bmi-1 expression is a cause of stemness loss in MEC cells (Supplementary Fig. S6A, S6B). Collectively, these results suggest that p53 (not MDM2) regulates Bmi-1 expression, indicating a potential mechanism through which p53 controls MEC stemness.
Figure 6: Therapeutic induction of p53 prevents MEC tumor recurrence in mice.
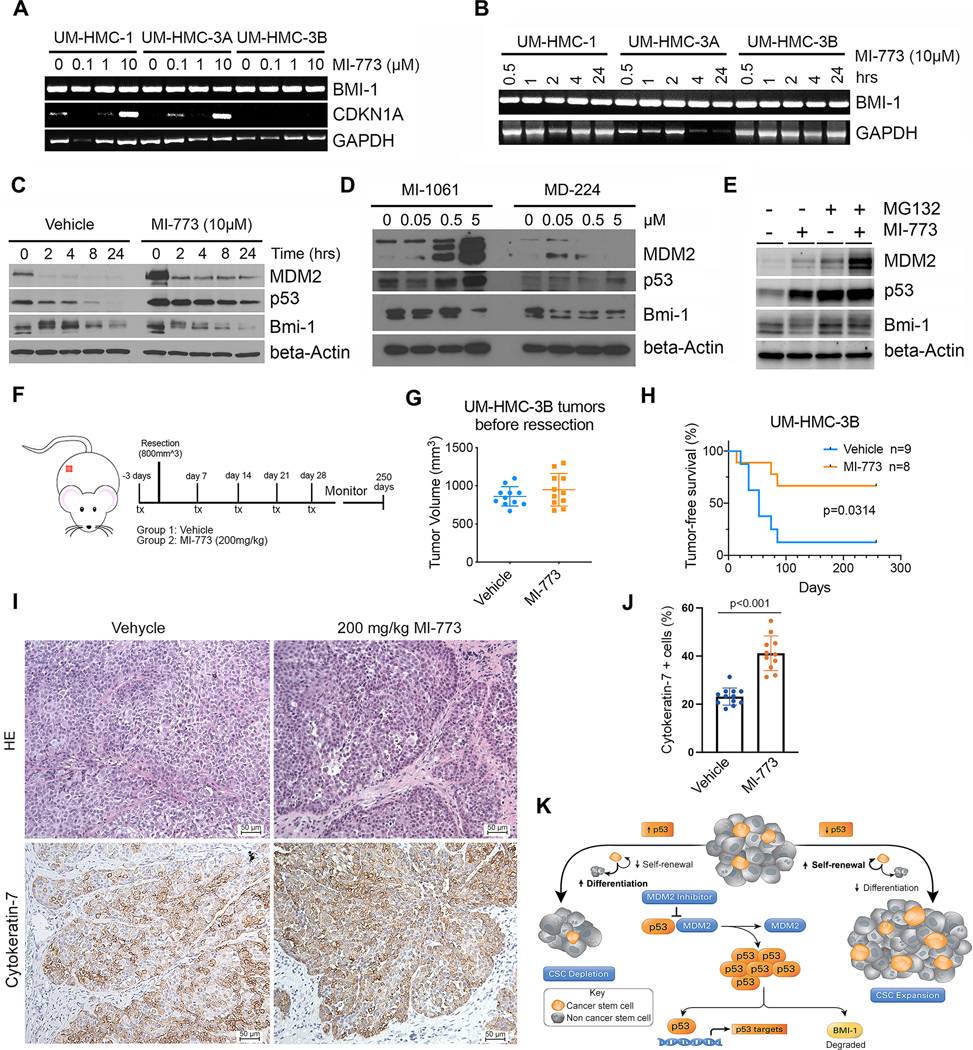
A, Reverse transcription polymerase chain reaction (RT-PCR) for Bmi-1, p21 and GAPDH of cells treated for 24 hours with increasing concentrations of MI-773. B, RT-PCR depicting time-course expression of Bmi-1 and GAPDH in cells treated with 10 μM MI-773. C, Western blot of cells treated with 10 μM MI-773 for 3 hours prior to addition of 25 μg/mL cycloheximide for up to 24 hours. D, Western blot of UM-HMC-3A cells treated for 24 hours with increasing concentrations of MD-224 (MDM2 degrader), or MI-1061 (MDM2 inhibitor used as a positive control for p53 activation). E, Western blot of UM-HMC-1 cells treated for 2 hours with 10 μM MG132 followed by 10 μM MI-773 for 4 hours. F, Schematic showing experimental design of tumor recurrence study. UM-HMC-3B subcutaneous xenograft tumors were allowed to grow to an average volume of 800 mm3 then randomly assigned to a treatment group. Mice were treated via oral gavage with either vehicle (N=9) or one dose of 200 mg/kg of MI-773 (N=8) three days prior to tumor resection. Weekly maintenance treatments of MI-773 (200 mg/kg) were given for four weeks after tumor resection. Mice were monitored weekly for tumor recurrence by palpability. G, Graph depicting tumor volumes in both experimental groups at start of treatment. H, Kaplan-Meier curves depicting tumor-free survival. Failure was defined as palpable subcutaneous tumors. Kaplan-Meier graphs were analyzed using the Gehan-Breslow-Wilcoxon test. I, Photomicrographs of hematoxylin/eosin staining (HE) and immunohistochemistry for Cytokeratin-7 of xenograft tumors three days after treatment with 200 mg/kg MI-773 (F). J, Graph depicting the quantification of Cytokeratin-7-positive cells in these tumors (F). K, Representation of the proposed effect of p53 on cancer stemness. p53 activation can be achieved by using small molecule inhibitors that block the interaction between p53 and its negative regulator MDM2. This leads to accumulation of p53 protein and activation of downstream signaling such as p53, the transcriptional target p21 and regulation of Bmi-1 protein expression. Downregulation of p53 results in increased cancer stem self-renewal leading to an expansion of the cancer stem cell population and increased tumor growth. Meanwhile, p53 activation results in decreased cancer stem cell self-renewal and increased differentiation resulting in depletion of the cancer stem pool and reduced tumor recurrence. This mechanism is partly mediated through the regulation of Bmi-1 protein expression by p53.
Therapeutic induction of p53 prevents MEC tumor recurrence in mice
To examine the therapeutic benefit of the small molecule inhibitor of MDM2-p53 interaction in a clinically relevant neoadjuvant setting simulating treatment of patients with advanced MEC, we transplanted MEC cells into mice and waited until tumors reached an average of 800 mm3 (Fig. 6F, 6G). At that stage, tumors were randomly assigned to receive either MI-773 or vehicle control (Fig. 6G). Treatment with 200 mg/kg MI-773 started 3 days prior to tumor resection and continued as weekly maintenance doses for 1 month (Fig. 6F). The treatment dose and weekly regimen were selected for its previously demonstrated negligible toxicity in mice (29). Tumor recurrence was monitored until palpable tumors were detected. At that stage, mice were euthanized, and the presence of recurrent tumors was verified macroscopically. While 7 out of 9 tumors (78%) recurred in the vehicle control group, only 3 out of 9 (33%) recurred in the MI-773-treated group (Fig. 6H). One event was censored from analysis due to unrelated death. All tumor-free mice were monitored for 250 days after which the experiment was terminated, and the absence of recurrent tumor lesions was verified upon autopsy. This pre-clinical trial demonstrated that neoadjuvant therapy with MI-773 prevents tumor recurrence in mice. Notably, post-surgical tumor progression involving local recurrence is the most common cause of treatment failure in human patients with salivary gland mucoepidermoid carcinoma (2). And finally, to understand the impact of MI-773 treatment on the differentiation status of these xenograft tumors, we used Cytokeratin-7 as a marker as it has been used extensively in MEC diagnosis and its expression is primarily found in more differentiated cells (e.g. mucous cells) (29). We observed a significant increase in the percentage of Cytokeratin-7 cells upon treatment with MI-773, when compared with vehicle-treated controls (Figure 6I and J).
Discussion
Much of the focus in understanding the pathobiology of mucoepidermoid carcinomas has been centered on the CRCT/MAML fusion. TP53 mutations have been described as rare events in these tumors, especially when compared to other head and neck cancers such as squamous cell carcinoma (12–14). Nevertheless, as more interest develops in understanding the genetic landscape of this rare malignancy and potential drivers of this disease, we find that p53 regulation might play an important role in MEC tumor biology. The first whole-exome sequencing study in MEC patients revealed that TP53 mutations are found in only about 30% of patients (5). Interestingly, TP53 mutations are present primarily in intermediate and high-grade disease and associated with higher number of overall mutations (30). Additionally, a group who studied serial tumor relapses from one MEC patient observed loss of expression of TP53 upon tumor evolution (18). Importantly they found that tumor recurrence in this patient correlated with a time-dependent increase in CSC fraction and higher tumorigenic potential. These findings suggest that a loss in p53 signaling might contribute to MEC progression. Consistent with this, we showed here that MEC xenograft tumors with silenced p53 expression grew faster than tumors with functional levels of p53. Importantly, these tumors also contained a higher proportion of CSC.
Throughout our study, we observed that UM-HMC-3B cells behaved differently than the UM-HMC-1 and UM-HMC-3A cells. Whole exome sequencing revealed the presence of a non-synonymous variant of TP53 (C215G; Pro72Arg) in both UM-HMC-3A and UM-HMC-3B (derived from the lymph node metastasis of the same patient that donated the tissue for the UM-HMC-3A cell line). This variant was not present in the UM-HMC-1. Sorting Intolerant from Tolerant (SIFT) analysis predicted minimal or no impact on p53 function. Considering that the same variant is present in both cell lines from the same patient (UM-HMC-3A and UM-HMC-3B), this variant cannot explain the difference in behavior and response to MI-773 observed in UM-HMC-3B (as compared to UM-HMC-3A). These findings inspired us to perform studies designed to understand mechanisms of resistance of UM-HMC-3B (metastatic) cells to therapy.
Some of the more commonly studied cell fate processes regulated by p53 are apoptosis and cell cycle progression. However, emergent evidence is providing support for a role for p53 in regulating self-renewal and differentiation. These cell fate processes are centered in a single mitotic event and a balance is needed to maintain the stem cell pool (16). For example, studies in mammary stem cells and breast cancer have shown p53 can regulate the asymmetric division of stem cells, where the absence of p53 can lead to an expansion of the stem cell pool through an increase in symmetric self-renewal while overexpression leads to stem cell depletion (31, 32). Our observations that MDM2 inhibitors cause a decrease in the cancer stem cell population while p53 knockdown causes an increase, led us to postulate that p53 signaling also regulates this balance in MEC. While MEC CSC were depleted upon activation of p53 via inhibition of its binding to MDM2, this decrease was drive through apoptosis. Surprisingly, most MEC CSC were in a G2/M state indicating these cells might have a longer G2 cell cycle phase or might be in a quiescent state in G2/M. Few studies have reported on G2-quiescent stem cells and on their biological role (33, 34). Stem cells in quiescence are considered to be in a “poised state” and serve as a reservoir that protects against stem cell depletion (35). Whether CSC in MEC are in a similar G2-quiescent state needs further exploration. Importantly, we showed that inhibitors of MDM2-p53 interaction shifted the cell cycle state of CSC towards G1. This G1 shift could be associated with an exit from G2-quiesence. An exit from G2-quiesence coupled with a loss in self-renewal could account for the decreased CSC fraction that we observed with therapeutic inhibition of MDM2-p53 interaction. Furthermore, this shift was mediated through the p53-p21 signaling axis. As p21 has been implicated in differentiation of stem cells (36), our data suggested that p53 activation might lead to decreased CSC fraction by promoting differentiation of the CSC.
Bmi-1 is an important regulator of stem cell self-renewal and is frequently used as a maker of stemness in different malignancies (37). We detected a progressive loss of Bmi-1 protein expression coupled with increased expression of pan-Cytokeratin (marker of epithelial differentiation) when MEC CSC were treated with inhibitors of the MDM2-p53 interaction. This suggested that p53 activation caused cells to lose their stem-like state whilst acquiring a more differentiated phenotype. We corroborated the loss of stemness in MEC CSC through sphere assays and found that p53 levels are an important regulator of the sphere forming ability of MEC cells. Importantly, while p53 did not affect Bmi-1 mRNA levels. It was previously reported that Bmi-1 and p53 can directly interact and that Bmi-1 can regulate p53 protein stability, but not the other way around (38). Here, we observed that p53 signaling regulates Bmi-1 expression independently of MDM2, although the mechanism by which this occurs still needs elucidation. Nevertheless, we propose here that p53 regulates CSC fate through parallel mechanisms that result from induced expression of p53 transcriptional targets such as p21 and decreased Bmi-1 expression.
Since CSC are thought to be responsible for recurrence and metastasis, having abrogated p53 signaling can give an important advantage to cancer cells and can partly explain why cancers with high p53 mutations have higher recurrence and metastatic rates (39, 40). Importantly, it also strengthens the rationale for using p53-activating therapies for the treatment of cancers that usually have functional p53 signaling, such as mucoepidermoid carcinomas. To date, small molecule inhibitors of MDM2-p53 such as MI-773 have been well tolerated in pre-clinical studies in mice (21, 22) and have passed phase I safety clinical trials for several solid malignancies (41). In these trials, although no objective response was observed, disease stabilization occurred in 58% of patients. Additionally, preclinical studies have shown therapeutic benefit of MI-773 treatment in salivary gland adenoid cystic carcinoma (42, 43). These results have led to a Phase I/II trial using APG-115 (second-generation small molecule inhibitor of MDM2-p53) for patients with salivary gland tumors (NCT03781986). In support of the rationale for using this class of drugs in salivary gland MEC, we observed here that tumor recurrence was prevented in mice treated with MI-773 in an adjuvant setting. Importantly, our studies monitored recurrence for a long period of time (250 days). This finding has major translational implications for the treatment of this orphan disease, as the most common reason of escape from therapy leading to MEC tumor progression and death is the high incidence of local and regional recurrences. However, considering that patients with MEC tend to present with delayed relapse many years after surgical resection of the primary tumor, the length of therapy with a small molecule inhibitor of MDM2-p53 after surgery becomes a very important factor to be considered. As such, more research will be needed before this neoadjuvant regimen can be translated into clinical use in patients with salivary gland MEC.
This work showed that activation of p53 signaling induces differentiation of MEC cancer stem cells and that a neoadjuvant regimen with a small molecule inhibitor of MDM2-p53 prevents MEC tumor relapse (Figure 6K). Our results give insight into the potential therapeutic benefits of small molecule inhibitors of MDM2-p53 for patients with salivary gland mucoepidermoid carcinomas. It also provides strong rationale for the exploration of combination therapies that target both the cancer stem cells (e.g., small molecule inhibitor of MDM2-p53) as well as bulk tumor cells (e.g., conventional chemotherapy, radiotherapy) to treat patients with unresectable or advanced salivary gland tumors.
Supplementary Material
Translational Relevance Statement.
There is no FDA-approved systemic therapy for salivary gland mucoepidermoid carcinoma (MEC). Here, we showed that clinically relevant small molecule inhibitors of MDM2 activate p53 signaling and reduce salivary gland cancer stemness by inducing differentiation of tumor cells. Remarkably, short-term neoadjuvant treatment with an MDM2 inhibitor prevented tumor recurrence in preclinical trials in mice. Collectively, our data demonstrate that p53 is a critical regulator of MEC stemness and suggest that these patients might benefit from therapeutic activation of p53.
Acknowledgements
We thank Drs. David Beer, Peter Polverini, and Mats Ljungman for their guidance and invaluable suggestions throughout this project. We thank Dr. Yu-Chih Chen for supporting the development of the microfluidic sphere formation assay used in this work. We would also like to thank Dr. Isabelle Lombaert and her research staff for their support in generating the orthotopic xenograft models of mucoepidermoid carcinoma, and Dr. Felipe Nor for photomicrographs and for the quantification of Cytokeratin-7 expression. We thank the University of Michigan Flow Cytometry, Imaging, Histology, and Vector Cores for their support. This work was funded by grants R01-DE021139S1 (C. Rodriguez-Ramirez) and R01-DE021139, R01-DE23220 (J.E. Nor) from the NIH/NIDCR.
Shaomeng Wang is the inventor of MI-773 and APG-115. MI-773 was licensed to Ascenta Therapeutics and Sanofi, while APG-115 has been licensed to Ascentage Pharma Group International, for which Shaomeng Wang receives royalties. Shaomeng Wang is a co-founder, owns stock, and has received research funding from Ascenta Therapeutics and Ascentage Pharma Group International. Max Wicha has financial holdings and is a scientific advisor for OncoMed Pharmaceuticals and MedImmune.
Competing Interests
Shaomeng Wang is the inventor of MI-773 and APG-115. MI-773 was licensed to Ascenta Therapeutics and Sanofi, while APG-115 has been licensed to Ascentage Pharma Group International, for which Shaomeng Wang receives royalties. Shaomeng Wang is a co-founder, owns stock, and has received research funding from Ascenta Therapeutics and Ascentage Pharma Group International. Max Wicha has financial holdings and is a scientific advisor for OncoMed Pharmaceuticals and MedImmune. The remaining authors declare no potential conflicts of interest.
Footnotes
Conflict of Interest: The remaining authors declare no potential conflicts of interest.
References
- 1.Seethala RR, Stenman G. Update from the 4th edition of the world health organization classification of head and neck tumours: tumors of the salivary gland. Head Neck Pathol 2017;11(1):55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chintakuntlawar AV, Okuno SH, Price KA. Systemic therapy for recurrent or metastatic salivary gland malignancies. Cancers Head Neck 2016;1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sultan I, Rodriguez-Galindo C, Al-Sharabati S, Guzzo M, Casanova M, Ferrari A. Salivary gland carcinomas in children and adolescents: a population-based study, with comparison to adult cases. Head Neck 2011;33(10):1476–81. [DOI] [PubMed] [Google Scholar]
- 4.Jee KJ, Persson M, Heikinheimo K, Passador-Santos F, Aro K, Knuutila S, et al. Genomic profiles and CRTC1-MAML2 fusion distinguish different subtypes of mucoepidermoid carcinoma. Mod Pathol 2013;26(2):213–22. [DOI] [PubMed] [Google Scholar]
- 5.Kang H, Tan M, Bishop JA, Jones S, Sausen M, Ha PK, et al. Whole-exome sequencing of salivary gland mucoepidermoid carcinoma. Clin Cancer Res 2017;23(1):283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet 2003;33(2):208–13. [DOI] [PubMed] [Google Scholar]
- 7.Coxon A, Rozenblum E, Park YS, Joshi N, Tsurutani J, Dennis PA, et al. Mect1-Maml2 fusion oncogene linked to the aberrant activation of cyclic AMP/CREB regulated genes. Cancer Res 2005;65(16):7137–44. [DOI] [PubMed] [Google Scholar]
- 8.Bell D, El-Naggar AK. Molecular heterogeneity in mucoepidermoid carcinoma: conceptual and practical implications. Head Neck Pathol 2013;7(1):23–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okumura Y, Miyabe S, Nakayama T, Fujiyoshi Y, Hattori H, Shimozato K, et al. Impact of CRTC1/3-MAML2 fusions on histological classification and prognosis of mucoepidermoid carcinoma. Histopathology 2011;59(1):90–7. [DOI] [PubMed] [Google Scholar]
- 10.Seethala RR, Dacic S, Cieply K, Kelly LM, Nikiforova MN. A reappraisal of the MECT1/MAML2 translocation in salivary mucoepidermoid carcinomas. Am J Surg Pathol 2010;34(8):1106–21. [DOI] [PubMed] [Google Scholar]
- 11.Birkeland AC, Foltin SK, Michmerhuizen NL, Hoesli RC, Rosko AJ, Byrd S, et al. Correlation of Crtc1/3-Maml2 fusion status, grade and survival in mucoepidermoid carcinoma. Oral Oncol 2017;68:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomes CC, Diniz MG, Orsine LA, Duarte AP, Fonseca-Silva T, Conn BI, et al. Assessment of TP53 mutations in benign and malignant salivary gland neoplasms. PLoS One. 2012;7(7):e41261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato S, Elkin SK, Schwaederle M, Tomson BN, Helsten T, Carter JL, et al. Genomic landscape of salivary gland tumors. Oncotarget 2015;6(28):25631–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross JS, Gay LM, Wang K, Vergilio JA, Suh J, Ramkissoon S, et al. Comprehensive genomic profiles of metastatic and relapsed salivary gland carcinomas are associated with tumor type and reveal new routes to targeted therapies. Ann Oncol 2017;28(10):2539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev 1993;7(7a):1126–32. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez-Ramirez C, Nor JE. p53 and cell fate: sensitizing head and neck cancer stem cells to chemotherapy. Crit Rev Oncog 2018;23(3–4):173–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams A, Warner K, Pearson AT, Zhang Z, Kim HS, Mochizuki D, et al. ALDH/CD44 identifies uniquely tumorigenic cancer stem cells in salivary gland mucoepidermoid carcinomas. Oncotarget 2015;6(29):26633–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keysar SB, Eagles JR, Miller B, Jackson BC, Chowdhury FN, Reisinger J, et al. Salivary gland cancer patient-derived xenografts enable characterization of cancer stem cells and new gene events associated with tumor progression. Clin Cancer Res 2018;24(12):2935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol Ther 2016;160:145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015;526(7571):131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrews A, Warner K, Rodriguez-Ramirez C, Pearson AT, Nor F, Zhang Z, et al. Ablation of cancer stem cells by therapeutic inhibition of the MDM2-p53 interaction in mucoepidermoid carcinoma. Clin Cancer Res 2019;25(5):1588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res 2014;74(20):5855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warner KA, Adams A, Bernardi L, Nor C, Finkel KA, Zhang Z, et al. Characterization of tumorigenic cell lines from the recurrence and lymph node metastasis of a human salivary mucoepidermoid carcinoma. Oral Oncol 2013;49(11):1059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguilar A, Lu J, Liu L, Du D, Bernard D, McEachern D, et al. Discovery of 4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2″-oxodispiro[ cyclohexane-1,2’-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane -1-carboxylic Acid (AA-115/APG-115): A potent and orally active Murine Double Minute 2 (MDM2) inhibitor in clinical development. J Med Chem 2017;60(7):2819–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J Med Chem 2019;62(2):448–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang L, Li J, Song L. Bmi-1, stem cells and cancer. Acta Biochim Biophys Sin 2009;41(7):527–34. [DOI] [PubMed] [Google Scholar]
- 27.Sahasrabuddhe AA, Dimri M, Bommi PV, Dimri GP. betaTrCP regulates BMI1 protein turnover via ubiquitination and degradation. Cell Cycle 2011;10(8):1322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yadav AK, Sahasrabuddhe AA, Dimri M, Bommi PV, Sainger R, Dimri GP. Deletion analysis of BMI1 oncoprotein identifies its negative regulatory domain. Mol Cancer 2010;9:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azevedo RS, Almeida OP, Kowalski LP, Pires FR. Comparative cytokeratin expression in the different cell types of salivary gland mucoepidermoid carcinoma. Head Neck Pathol 2008;2(4):257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, McDermott JD, Schrock AB, Elvin JA, Gay L, Karam SD, et al. Comprehensive genomic profiling of salivary mucoepidermoid carcinomas reveals frequent BAP1, PIK3CA, and other actionable genomic alterations. Ann Oncol 2017;28(4):748–53. [DOI] [PubMed] [Google Scholar]
- 31.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009;138(6):1083–95. [DOI] [PubMed] [Google Scholar]
- 32.Tao L, Roberts AL, Dunphy KA, Bigelow C, Yan H, Jerry DJ. Repression of mammary stem/progenitor cells by p53 is mediated by Notch and separable from apoptotic activity. Stem Cells 2011;29(1):119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otsuki L, Brand AH. Cell cycle heterogeneity directs the timing of neural stem cell activation from quiescence. Science 2018;360(6384):99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otsuki L, Brand AH. Dorsal-ventral differences in neural stem cell quiescence are induced by p57(KIP2)/Dacapo. Dev Cell 2019;49(2):293-300 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 2013;14(6):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreis NN, Louwen F, Yuan J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation, migration and cancer therapy. Cancers 2019;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao W, Li Y, Zhang X. Stemness-related markers in cancer. Cancer Transl Med 2017;3(3):87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calao M, Sekyere EO, Cui HJ, Cheung BB, Thomas WD, Keating J, et al. Direct effects of Bmi1 on p53 protein stability inactivates oncoprotein stress responses in embryonal cancer precursor cells at tumor initiation. Oncogene 2013;32(31):3616–26. [DOI] [PubMed] [Google Scholar]
- 39.Meric-Bernstam F, Zheng X, Shariati M, Damodaran S, Wathoo C, Brusco L, et al. Survival outcomes by TP53 mutation status in metastatic breast cancer. JCO Precis Oncol 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ecke TH, Schlechte HH, Schiemenz K, Sachs MD, Lenk SV, Rudolph BD, et al. TP53 gene mutations in prostate cancer progression. Anticancer Res 2010;30(5):1579–86. [PubMed] [Google Scholar]
- 41.de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ, et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer 2017;76:144–51. [DOI] [PubMed] [Google Scholar]
- 42.Nor F, Warner KA, Zhang Z, Acasigua GA, Pearson AT, Kerk SA, et al. Therapeutic inhibition of the MDM2-p53 interaction prevents recurrence of adenoid cystic carcinomas. Clin Cancer Res 2017;23(4):1036–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warner KA, Nor F, Acasigua GA, Martins MD, Zhang Z, McLean SA, et al. Targeting MDM2 for treatment of adenoid cystic carcinoma. Clin Cancer Res 2016;22(14):3550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study are available from the corresponding author on reasonable request.