

Abstract
Reduction of nitrite anions (NO2−) to nitric oxide (NO), nitrous oxide (N2O) and ultimately dinitrogen (N2) takes place in a variety of environments, including in the soil as part of the biogeochemical nitrogen cycle and in acidified nuclear waste. Nitrite reduction typically takes place within the coordination sphere of a redox-active transition metal. Here we show that Lewis acid coordination can substantially modify the reduction potential of this polyoxoanion to allow for its reduction under non-aqueous conditions (−0.74 V versus NHE). Detailed characterization confirms the formation of the borane-capped radical nitrite dianion (NO22−), which features a N(II) oxidation state. Protonation of the nitrite dianion results in the facile loss of nitric oxide (NO), whereas its reaction with NO results in disproportionation to nitrous oxide (N2O) and nitrite (NO2−). This system connects three redox levels in the global nitrogen cycle and provides fundamental insights into the conversion of NO2− to NO.
Oxyanions are a class of water pollutants of growing concern due to their high solubility in water, their potential toxicity and their negative environmental impacts1,2. Nature has evolved several biochemical pathways capable of facile, selective reduction of oxyanions. The constituent enzymes in these pathways offer inspiration for the design of catalysts purposed towards the remediation of oxyanion pollution3. For example, the nitrite anion (NO2−) serves as a terminal electron acceptor during denitrification, where it is progressively reduced to nitric oxide (NO), nitrous oxide (N2O) and ultimately to dinitrogen (N2). The initial, selective reduction of NO2− to NO is mediated by metalloenzymes including cytochrome c oxidase and nitrite reductases that leverage coordination of NO2− to Fe or Cu centres4–7. These redox-active metals transfer an electron to NO2−, facilitated both by the Lewis acidity of the metal centres and accompanied by binding of a proton to a nitrite O-atom in nitrite reductase8,9. Either of these binding events may lower the reduction potential of NO2−, rendering electron transfer more favourable (Fig. 1a,c).
Fig. 1 |. The role of protons and Lewis acids in nitrite reduction.
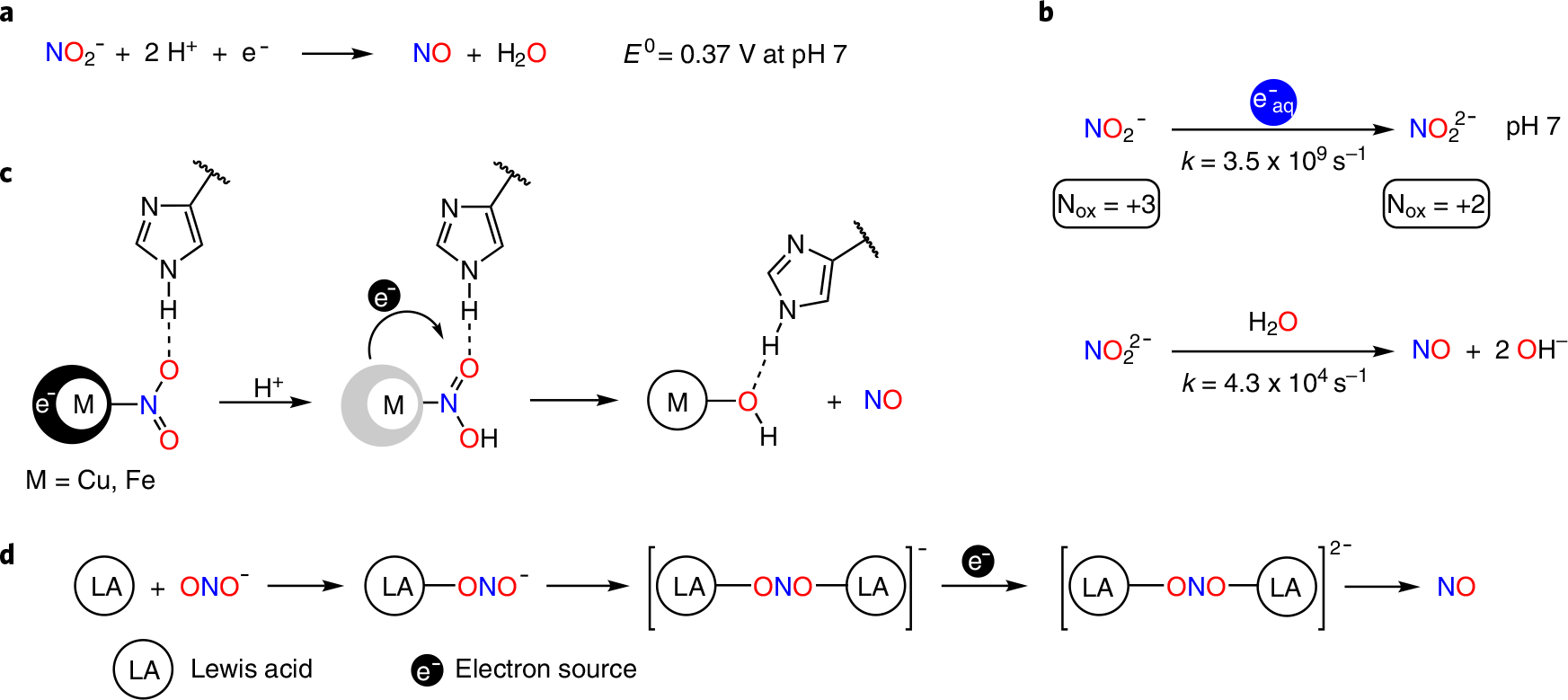
a, The standard reduction potential of nitrite to NO at pH 7 involves two protons. b, Rapid capture of β-particles (electrons) by nitrite from radioactive decay to form the nitrite dianion NO22− and subsequent proton-induced release of NO. c, Biological nitrite reduction to NO is facilitated by metal–nitrite coordination, hydrogen-bonding networks and proton transfer. d, Lewis acid coordination enables the isolation of nitrite dianion NO22− (this work).
Reduction of nitrogen oxyanions also occurs in high-level radioactive waste stored in nitrate- and nitrite-rich aqueous solutions10. Solvated electrons generated radiolytically undergo rapid capture by both nitrate and nitrite to form the dianions NO32− and NO22− at nearly diffusion-controlled rates of 9.7 × 109 s−1 and 3.5 × 109 s−1, respectively (Fig. 1b)10. Facile reaction of NO32− and NO22− with water to produce NO2 and NO at rates of 5.5 × 104 s−1 and 4.3 × 104 s−1, respectively, renders these dianions extremely short-lived species in aqueous solution, with lifetimes on the order of 20 μs (ref. 10).
To decouple the effects of Lewis acid coordination and proton transfer to an oxyanion during reduction11, we explored the effects of coordinating NO2− to a redox-innocent Lewis acid. We have previously shown that the coordination of the biological nitric oxide reservoir RSNOs to the Lewis acid B(C6F5)3 in RSNO–B(C6F5)3 adducts markedly raises the reduction potential for RSNOs (approximately −1.1 to −1.3 V versus normal hydrogen electrode (NHE)), which enables generation of the [RSNO–B(C6F5)3]•− radical anion at biologically relevant potentials (+0.1 versus NHE)12. Using a strong, yet chemically stable capping Lewis acid for each of the two O atoms of nitrite, this strategy enables the facile reduction of NO2− to its radical dianion NO22− without attendant proton transfer. This approach effectively decouples electron transfer and protonation11 to separately show the effect of Lewis acid activation on the reduction potential of the nitrite anion and on the proton-induced release of NO from the reduced nitrite dianion NO22−. Moreover, the use of capping Lewis acids enables the characterization and reactivity study of this nitrogen oxyanion that connects NO2−, NO and N2O representing three redox levels in the global nitrogen reduction cycle.
Results and discussion
Capture of the nitrite dianion, synthesis and characterization.
Addition of 1 equiv. B(C6F5)3 to [Cp*2Co][NO2] (1) in fluorobenzene leads to the formation of [Cp*2Co][(C6F5)3B–ONO] (2) in near quantitative yield (Fig. 2a). The X-ray structure of unsymmetrically capped 2 shows B(C6F5)3 bound to one O-atom of nitrite with markedly different N–O distances of 1.337(10) (capped O atom) and 1.200(10) Å (free O atom), respectively (Supplementary Fig. 30). Treating [Cp*2Co][(C6F5)3B–ONO] (2) with a second equivalent of B(C6F5)3 in fluorobenzene forms the doubly activated nitrite anion in [Cp*2Co][(C6F5)3B–ONO–B(C6F5)3] (3) (Fig. 2b). The X-ray structure of 3 shows more symmetric NO distances (1.261(2), 1.225(2) Å) that result upon coordination of a Lewis acid to each O atom in nitrite. Lewis acid coordination also results in a contraction of the nitrite O–N–O bond angle to 112.2(7)° and 109.3(17)° in mono- and di-capped species 2 and 3, respectively, compared to the free nitrite anion in 1 (120.6(6)°; Supplementary Figs. 29–31).
Fig. 2 |. Synthesis and structures of Lewis acid-capped nitrite mono- and dianions.
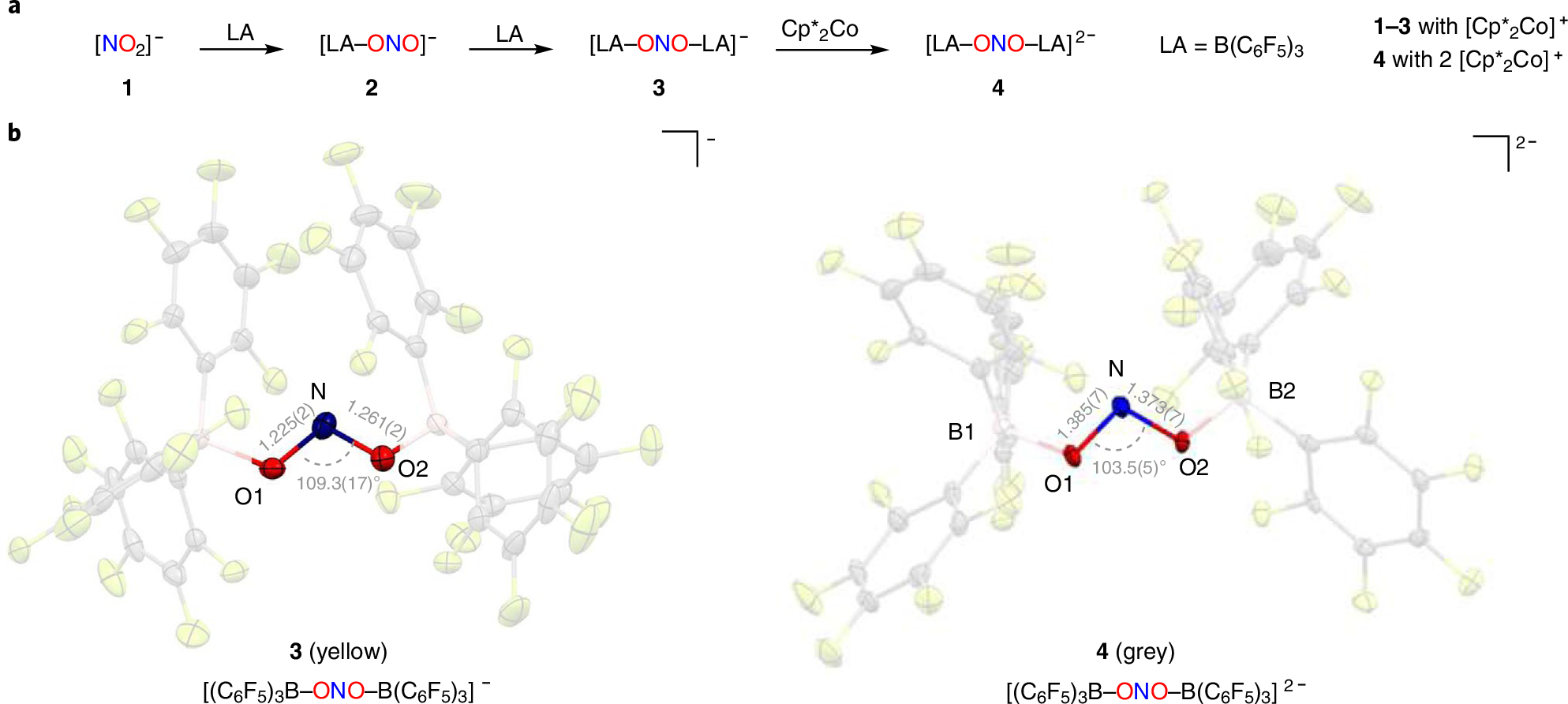
a, Synthesis of Lewis acid-capped nitrite anions [Cp*2Co][(C6F5)3B–ONO] (2), [Cp*2Co][(C6F5)3B–ONO–B(C6F5)3] (3) and [Cp*2Co]2[(C6F5)3B–ONO–B(C6F5)3] (4). b, X-ray structures of the nitrite monoanion [Cp*2Co] [(C6F5)3B–ONO–B(C6F5)3] (3) and dianion [Cp*2Co]2[(C6F5)3B–ONO–B(C6F5)3] (4) showing N–O distances (Å) and O–N–O angles (°) in the ONO moiety. Each anion is charge-balanced by the Cp*2Co+ cation. Atom colours: N, blue; O, red; B, pink; C, grey; F, yellow. LA, Lewis acid.
Cyclic voltammetry studies show the effect of the Lewis acid coordination on the reduction potential of free nitrite. Under aqueous conditions, the reduction potential for free nitrite is highly proton-dependent, ranging from +0.98 V and −0.48 versus NHE at pH 0.0 and 14.0, respectively13,14. In fluorobenzene with [PPN] [BArF4] (ArF = 3,5-(CF3)2C6H3) as electrolyte, however, neither [Cp*2Co][NO2] (1) nor [Cp*2Co][(C6F5)3B–ONO] (2) exhibits any reduction wave for nitrite or monocapped nitrite anion (Supplementary Fig. 26). Rather, only a wave for Cp*2Co+/Cp*2Co couple is evident which appears at −1.35 V in 2. In marked contrast, the cyclic voltammogram of [Cp*2Co][(C6F5)3B–ONO–B(C6F5)3] (3) shows a quasi-reversible wave centred at −0.74 V versus NHE that corresponds to the [(C6F5)3B–ONO–B(C6F5)3]−/[(C6F5)3B–ONO–B(C6F5)3]2− couple (Supplementary Fig. 27). The addition of a second Lewis acid to the nitrite anion induces a profound electronic effect that facilitates nitrite reduction.
Chemical reduction of [Cp*2Co][(C6F5)3B–ONO–B(C6F5)3] (3) with Cp*2Co in fluorobenzene effects a colour change from yellow to grey (Fig. 2a). Crystallization of the grey product confirms the formation of the borane-capped nitrite radical dianion [Cp*2Co]2[(C6F5)3B–ONO–B(C6F5)3] (4) (Fig. 2b). Although Lewis acid-capped mono- and dianions 3 and 4 possess similar overall structures, the bond lengths and angles in nitrite dianion 4 are greatly changed in comparison with nitrite 3. In nitrite dianion 4, the N–O bonds are markedly elongated (1.385(7), 1.373(7) Å) as compared to monoanion 3 (1.225(2), 1.261(2) Å) as a consequence of the extra electron present in the nitrite dianion 4. Furthermore, the nitrite O atoms in dianion 4 are more tightly bound to the capping boranes (B–O distances: 1.488(9), 1.490(9) Å), consistent with its greater negative charge in comparison to doubly capped nitrite anion 3 (B–O distances: 1.603(3), 1.628(3) Å). Moreover, the O–N–O bond angle in dianion 4 (103.5(5)°) is contracted relative to monoanion 3 (109.3(17)°). Infrared (IR) spectroscopy reports a markedly lower-energy N–O stretching frequency for dianion 4 (1,010 cm−1) in comparison with monoanion 3 (1,265 cm−1), consistent with a weakening of the N–O bonds of nitrite upon one-electron reduction.
Electronic structure of the nitrite dianion core.
Qualitative molecular orbital analysis for NO22−/1− predicts that the lowest unoccupied molecular orbital of 3/singly occupied molecular orbital (SOMO) of 4 should be an N-localized π* combination between N 2px and O 2px orbitals (Supplementary Fig. 38). This is confirmed by analysis of electron paramagnetic resonance (EPR) spectra. The continuous-wave (c.w.) X-band EPR spectrum obtained at room temperature for 4 (Fig. 3a) shows the expected isotropic three-line pattern arising from 14N hyperfine coupling, with the isotropic component of the hyperfine coupling (HFC) Aiso(14N) = 44.0 MHz. Accordingly, the 15N isotopologue exhibits a two-line pattern with Aiso(15N) = 62.0 MHz (Fig. 3b). The isotropic g-value (giso) value of 2.005 obtained from both measurements is consistent with a radical composed solely of light atoms. Frozen-solution spectra recorded at both the X- (Fig. 3c) and Q-band (Fig. 3d) (the latter obtained by field-swept echo detection) show substantial anisotropy of the 14N hyperfine values, with the best simulation of the X-band data giving Ax = 122 MHz, Ay = 4 MHz and Az = 6 MHz. Assignment of the larger HFC as Ax is supported by the corresponding Q-band data. The g values obtained from simulating the Q-band data of 4 are gx = 1.991, gy = 1.996 and gz = 1.996. For completeness, frozen-solution spectra and simulations for the 15N isotopologue are provided in the Supplementary Information.
Fig. 3 |. Probing the electronic structure of the nitrite dianion by ePR and X-ray absorption spectroscopy, as well as DFT calculations.
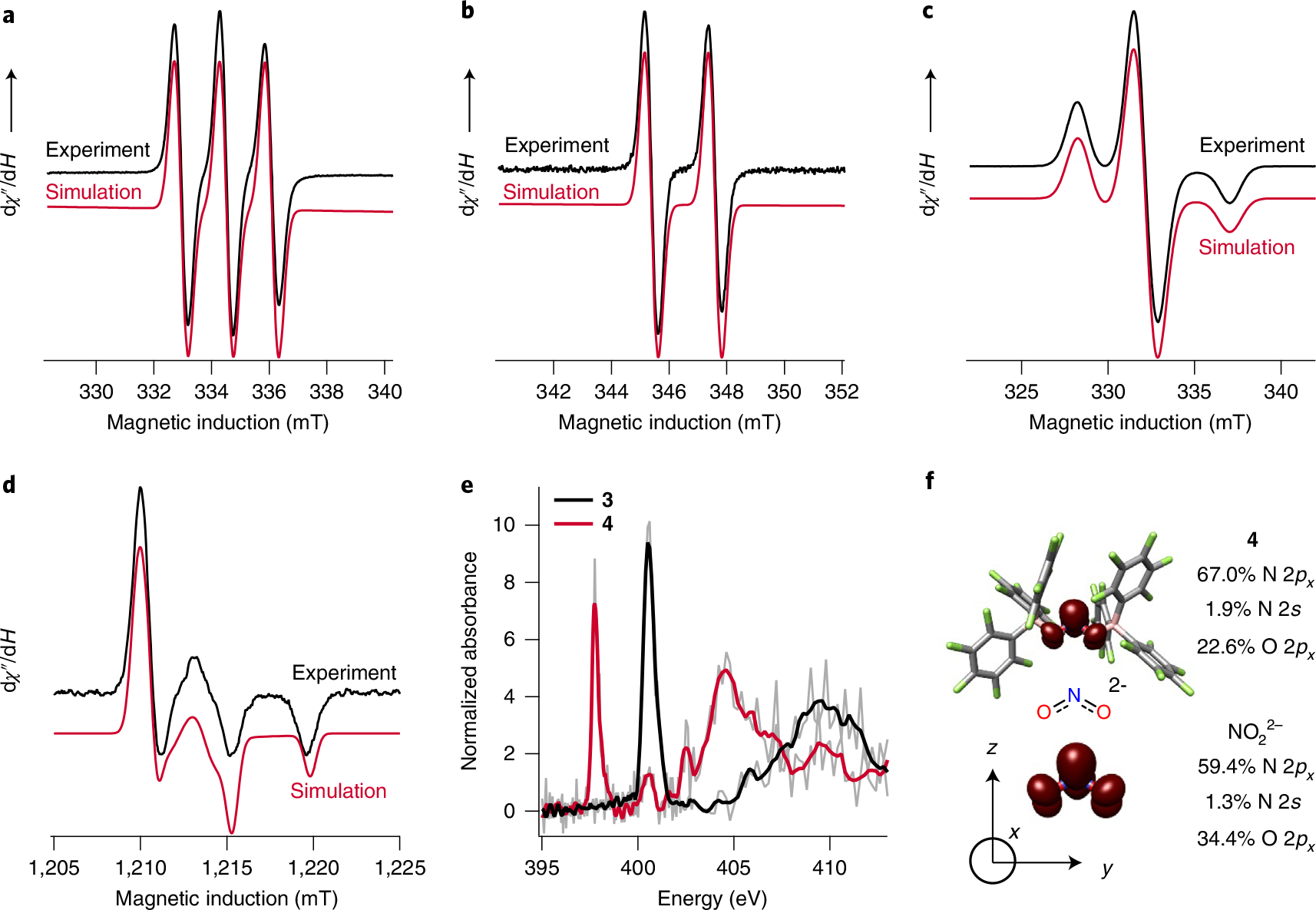
a, 298-K solution c.w. X-band (9.3886 GHz) EPR spectrum of 14N–4 (9.7249 GHz). b, Solution c.w. X-band EPR spectrum of 15N–4. c, 96-K frozen-solution c.w. X-band (9.2710 GHz) EPR of 14N–4. d, 10-K field-swept echo-detected Q-band (33.9544 GHz) EPR of 14N–4. dχ″/dH is the derivative of the imaginary component of the molecular magnetic susceptibility (χ″) with respect to the external static magnetic field H. Experimental data are shown in black and simulations in red. The simulated spectra are in very good agreement with the experimental results. e, Smoothed partial fluorescence yield-detected N K-edge XAS of 3 (black) and 4 (red). Raw data are in grey. f, B3LYP/def2−TZVP(-f) calculated spin density plots (isovalue 0.002 a.u., maroon) of the dianion in 4 and geometry-optimized NO22−. Atom colours: N, blue; O, red; B, pink; C, grey; F, yellow.
For an electron localized to a MO with substantial N 2px character, the A values [Ax, Ay, Az] are expected to be [Aiso + Tx, Aiso + Ty, Aiso + Tz], where T is the anisotropic value for 14N. Using values tabulated by Morton and Preston15, the value of aiso corresponding to an electron localized to N 2s is 1,811 MHz, whereas an electron localized to N 2px should have anisotropic [Tx, Ty, Tz] = [4/5, −2/5, −2/5] × 138.8 MHz = [111.04, −55.52, −55.52] MHz. Using the well-resolved Ax value to obtain Tx, the unpaired spin in 4 resides in a MO with 2.5% N 2s and 69% N 2px character.
N-localization of the reduction of 3 to 4 is further supported by N K-edge (1s → 2p) XAS (Fig. 3e). The spectrum obtained for 3 has an intense pre-edge feature at 400.6 eV this is shifted to 397.8 eV in 4. The intensity of this feature is diminished in 4, consistent with occupation of a formerly empty MO. The edge position in the spectrum obtained for 3 appears at ~5 eV higher energy relative to the edge in 4. Together, these are consistent with the reduction of N. The intensity of the pre-edge feature in 4 can be related to N 2p participation in the acceptor orbital via equation (1)16:
where D0 is the pre-edge peak intensity, h is the number of holes, n is the number of photoabsorbers (n and h = 1 in 4), Is is the radial dipole integral between N 1s and N 2p, and α2 is the percent N 2p character of the acceptor MO. Using the experimental D0 of 4.2 and the 69% N 2px character determined by EPR, a value of 18.3 is obtained for Is. This value is smaller than an estimate of Is obtained by studying a library of N-ligated coordination complexes17 and reflects decontraction of the 2p orbital of the highly electron-rich N-centre in 418.
Hybrid density functional theory (DFT) calculations using the B3LYP hybrid density functional and the scalar-relativistically recontracted ZORA-def2-TZVP(-f) basis set on all atoms further support the formulation of 4 as containing a NO22− core. The calculated spin density occupies a b1-symmetry O–N–O π* orbital composed 2% N 2s and 67% N 2px, in excellent agreement with values obtained from EPR (Fig. 3f). The residual spin density resides predominantly on O. Calculated spin Hamiltonian parameters exhibit high fidelity to values extracted from spectral simulations. Time-dependent DFT calculations of the N K-edge XAS confirm that the pre-edge feature in the spectrum obtained for 4 corresponds to a promotion from N 1s to the b1-symmetry SOMO. Additionally, these calculations reproduce the energy shifts observed between N K features of 3 and 4 (Supplementary Fig. 40).
Given the fidelity of the calculated electronic structure to that deduced from experiment, we sought to examine the role of the capping Lewis acids on the electronic structure of the NO22− core. Geometry optimization of the bare anion NO22− at this level of theory affords N–O distances of 1.372 Å and an angle of 111.5°, consistent with an electron in the O–N–O π* orbital. A single-point electronic structure calculation at the stationary point shows that the NO22− dianion is also a N-centred radical with a similar N orbital heritage (1.3% N 2s and 59% N 2px) as in Lewis acid-capped 4. The inductive effect of the Lewis acid bound to the nitrite dianion through the O atoms results in greater unpaired electron density on the less electronegative N atom in the O–N–O π* orbital (Fig. 3f).
Chemical reactivity of the nitrite dianion.
The nitrite radical dianion [Cp*2Co]2[(C6F5)3B–ONO–B(C6F5)3] (4) is stable in the solid state and at room temperature in fluorobenzene solution. Because the N atom of nitrite dianion 4 is at the same oxidation level as NO gas (Nox = +2), we explored avenues to produce NO gas from 4. Heating a fluorobenzene solution of 4 at 75 °C for 48 h leads to the formation of NO gas in 65% yield (Fig. 4a). 19F NMR analysis of this reaction indicates multiple B(C6F5)3 derived products, including [Cp*(η5-C5Me4CH2–B(C6F5)3)Co] (5) that has undergone deprotonation of the one Cp* methyl group followed by capture by the Lewis acid B(C6F5)3 (Supplementary Fig. 35). Mimicking proton transfer in nitrite reduction, however, addition of 2 equiv. trifluoroacetic acid to dianion 4 at room temperature triggers the instant release of NO gas in 72% yield with 2 equiv. [Cp*2Co][(C6F5)3B–OC(O)CF3] (6), isolated in 80% yield (Fig. 4a).
Fig. 4 |. Reactivity of the nitrite dianion.
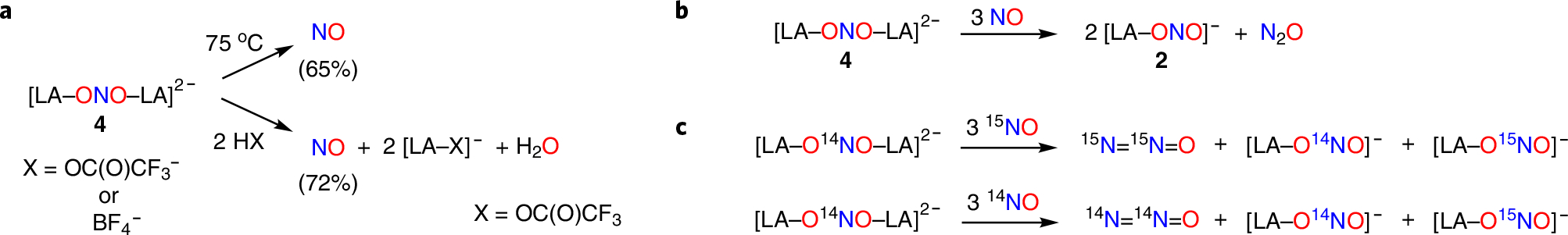
a, Release of NO from nitrite dianion 4 upon heating or addition of proton sources. b, Reduction of NO to N2O by nitrite dianion 4 generating 2 equiv. [(C6F5)3B–ONO] (2). c, Reaction with isotopically labelled NO shows a net outer-sphere reduction of NO to N2O.
Reaction of nitrite dianion 4 with 3 equiv. NO results in a disproportionation reaction that generates N2O and 2 equiv. [Cp*2Co] [(C6F5)3B–ONO] (2) quantified in 43% and 90% yields, respectively. Isotopic labelling experiments show that N2O forms solely from the added NOgas. Reaction of natural-abundance nitrite dianion 4–14N with 3 equiv. 15NOgas results in the exclusive formation of 15N15NO, and reaction of nitrite dianion 4–15N with 3 equiv. 14NOgas solely forms 14N14NO, as monitored by IR spectroscopy (Supplementary Fig. 25). In each case, a 1:1 mixture of monocapped nitrite anions [(C6F5)3B–O14NO] (2–14N) and [(C6F5)3B–O15NO] (2–15N) forms, according to 15N and 19F NMR analysis (Fig. 4c).
As an initial step, we propose net electron and Lewis acid transfer from nitrite dianion [(C6F5)3B–ONO–B(C6F5)3]2− to NO to form [(C6F5)3B–ONO]− and the nitroxide Lewis acid adduct [(C6F5)3B–NO]− (Supplementary Figs. 39 and 40). Hybrid DFT calculations that follow the model reaction of free nitrite dianion NO22− with NO via an inner-sphere pathway indicate favourable thermodynamics for conversion to the nitrite anion NO2− and NO− (ΔG = −45.9 kcal mol−1) with a minimal barrier (ΔG‡ = 6.9 kcal mol−1) via the [O2N–NO]2− intermediate (ΔG = −31.5 kcal mol−1) (Supplementary Fig. 39). Reaction of the borane-capped species [(C6F5)3B–ONO–B(C6F5)3]2− with NO to give [(C6F5)3B–ONO]− and [(C6F5)3B–NO]− is also exergonic (ΔG = −17.5 kcal mol−1) (Supplementary Fig. 40). Stepwise capture of 2 equiv. NO by [(C6F5)3B–NO]− provides the hyponitrite radical anion [(C6F5)3B–ONNO]− (ΔG = −7.2 kcal mol−1) followed by [(C6F5)3B–ONO]− and N2O (ΔG = −57.5 kcal mol−1). The overall reaction of [(C6F5)3B–ONO–B(C6F5)3]2− with 3 equiv. NO to give the experimentally observed 2 equiv. [(C6F5)3B–ONO]− and N2O (Fig. 4b) is highly exergonic (ΔG = −82.2 kcal mol−1 overall) (Supplementary Fig. 40). This latter sequence follows the previously studied reaction of NO− with NO to give [ONNO]− and [ONNONO]− intermediates en route to N2O and NO2− (refs. 19,20).
Conclusion
In summary, by using the Lewis acid B(C6F5)3, electron transfer and proton transfer steps in nitrite reduction become decoupled from N–O bond scission, permitting isolation of the nitrite dianion NO22− stabilized by a redox-innocent Lewis acid at each O atom. Although strong Lewis acids such as B(C6F5)3 enable the capture of reactive species such as O22− (ref. 21) or may combine with bulky, strong Lewis bases to form frustrated Lewis pair (FLP) adducts of N2O (ref. 22) and NO (ref. 23), this Lewis acid enables the isolation of the nitrite dianion 4. This new polyoxoanion with Nox = +2 provides fundamental insights into the conversion of NO2− to NO. Moreover, the nitrite dianion 4 is capable of further reducing NO to N2O with formation of the nitrite monoanion 2, connecting three nitrogen oxidation states in the global nitrogen cycle (Nox = +3, +2, +1). Use of potent, redox-innocent Lewis acids to enable the reduction of small molecules and ions without breaking bonds affords an opportunity to gain insight into the reduction of other environmentally relevant species, perhaps opening new approaches for catalysis. This is especially relevant for anions such as nitrate (NO3−) and perchlorate (ClO4−), for which transition metals mediate electron- and oxygen-atom transfer steps, yet bind to metal sites weakly, requiring assistance via non-covalent binding to facilitate oxyanion reduction3.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-022-01025-9.
Supplementary Material
Acknowledgements
This work was supported by the NIH (P41GM103521 to J.H.F., R35GM124908 to K.M.L. and R01GM126205 to T.H.W.). XAS data were obtained at SSRL, which is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy’s Office of Biological and Environmental Research and by NIH/NIGMS (including P41GM103393). The work at SSRL was also supported by the US Department of Energy Office of Basic Energy Sciences (proposal no. 100487).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41557-022-01025-9.
Peer review information Nature Chemistry and the authors thank Rebecca Melen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2074366 (1), 2074367 (2), 207468 (3), 2074369 (4), 2074370 (5), 2074371 (6) and 2074372 (7). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article and its Supplementary Information.
References
- 1.Camargo JA & Alonso Á Ecological and toxicological effects of inorganic nitrogen pollution in aquatic ecosystems: a global assessment. Environ. Int 32, 831–849 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Sebilo M, Mayer B, Nicolardot B, Pinay G & Mariotti A Long-term fate of nitrate fertilizer in agricultural soils. Proc. Natl Acad. Sci. USA 45, 18185–18189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ford CL, Park YJ, Matson EM, Gordon Z & Fout AR A bioinspired iron catalyst for nitrate and perchlorate reduction. Science 354, 741–743 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Maia LB & Moura JJG How biology handles nitrite. Chem. Rev. 14, 5273–5357 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Cosby K et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 9, 1498–1505 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Basu S et al. Nitrite reductase activity of cytochrome c. J. Biol. Chem. 283, 2590–32597 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Samouilov A, Liu X & Zweier JL Characterization of the effects of oxygen on xanthine oxidase-mediated nitric oxide formation. J. Biol. Chem. 276, 24482–24489 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Fukuda Y et al. Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography. Proc. Natl Acad. Sci. USA 113, 2928–2933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders BC, Hassan SM & Harrop TC NO2− activation and reduction to NO by a nonheme Fe(NO2)2 complex. J. Am. Chem. Soc. 136, 10230–10233 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Yakabuskie PA, Joseph JM, Stuart CR & Wren JC Long-term γ-radiolysis kinetics of NO3− and NO2− solutions. J. Phys. Chem. A 115, 4270–4278 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Nocera DG Proton-coupled electron transfer: the engine of energy conversion and storage. J. Am. Chem. Soc. 144, 1069–1081 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Hosseininasab V et al. Lewis acid coordination redirects S-nitrosothiol signaling output. Angew. Chem. Int. Ed. 59, 10854–10858 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bratsch SG Standard electrode potentials and temperature coefficients in water at 298.15 K. J. Phys. Chem. Ref. Data 18, 1–21 (1989). [Google Scholar]
- 14.Fukuto JM et al. Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide and their derived species. Chem. Res. Toxicol. 25, 769–793 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morton JR & Preston KF Atomic parameters for paramagnetic resonance data. J. Magn. Reson. 30, 577–582 (1978). [Google Scholar]
- 16.Neese F, Hedman B, Hodgson KO & Solomon EI Relationship between the dipole strength of ligand pre-edge transitions and metal–ligand covalency. Inorg. Chem. 21, 4854–4860 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Lukens JT, DiMucci IM, Kurogi T, Mindiola DJ & Lancaster KM Scrutinizing metal–ligand covalency and redox non-innocence via nitrogen K-edge X-ray absorption spectroscopy. Chem. Sci. 10, 5044–5055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarangi R et al. Sulfur K-edge X-ray absorption spectroscopy as a probe of ligand–metal bond covalency: metal vs ligand oxidation in copper and nickel dithiolene complexes. J. Am. Chem. Soc. 129, 2316–2326 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poskrebyshev GA, Sahirovich V & Lymar SV Hyponitrite radical, a stable adduct of nitric oxide and nitroxyl. J. Am. Chem. Soc. 126, 891–899 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Valiev M & Lymar SV Structural and mechanistic analysis through electronic spectra: aqueous hyponitrite radical (N2O2−) and nitrosyl hyponitrite anion (N3O3−). J. Phys. Chem. A 115, 12004–12010 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Henthorn JT & Agapie T Dioxygen reactivity with a ferrocene–Lewis acid pairing: reduction to a boron peroxide in the presence of tris(pentafluorophenyl)borane. Angew. Chem. Int. Ed. 53, 12893–12896 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Otten E, Neu RC & Stephan DW Complexation of nitrous oxide by frustrated Lewis pairs. J. Am. Chem. Soc. 131, 9918–9919 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Cardenas AJ et al. Capture of NO by a frustrated Lewis pair: a new type of persistent N-oxyl radical. Angew. Chem. Int. Ed. 50, 7567–7571 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2074366 (1), 2074367 (2), 207468 (3), 2074369 (4), 2074370 (5), 2074371 (6) and 2074372 (7). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article and its Supplementary Information.