

SUMMARY
It is currently accepted that cancer-associated fibroblasts (CAF) participate in T cell exclusion from tumor nests. To unbiasedly test this, we used single-cell RNA sequencing coupled with multiplex imaging on a large cohort of lung tumors. We identified four main CAF populations, of which two are associated with T cell exclusion: (i) MYH11+αSMA+ CAF, which are present in early-stage tumors and form a single-cell layer lining cancer aggregates, and (ii) FAP+αSMA+ CAF, which appear in more advanced tumors and organize in patches within the stroma or in multiple layers around tumor nests. Both populations orchestrate a particular structural tissue organization through dense and aligned fiber deposition compared to T cell permissive CAF. Yet they produce distinct matrix molecules, including collagen IV (MYH11+αSMA+ CAF) and collagen XI/XII (FAP+αSMA+ CAF). Hereby, we uncovered unique molecular programs of CAF driving T cell marginalization, whose targeting should increase immunotherapy efficacy in patients bearing T cell-excluded tumors.
INTRODUCTION
Lung cancer is the leading cause of cancer-related deaths worldwide, accounting for roughly 1.6 million deaths per year, with non-small cell lung carcinoma (NSCLC) being the most prevalent form (1). The partial success of immune checkpoint blockade in only a subset of NSCLC patients underscores the need for a better understanding of the determinants controlling anti-tumor immunity (2). In addition to high tumor mutational burden and PD-L1 expression levels in the tumor, CD8+ T cell density has been shown as a predictor of immunotherapy response (3,4). By analyzing the T cell localization within the tumor, recent studies have revealed the importance of T cell infiltration into the tumor nests relative to the surrounding stroma (3,5,6). Understanding the mechanisms regulating T cell exclusion are therefore crucial to improve T cell-based therapies and patient outcomes.
Using real-time imaging of T cell dynamics in human NSCLC, we previously found that dense fibers oriented parallel to the tumor-stroma interface form a barrier around the tumor mass and limit T cell contact with tumor cells (7). However, the cellular sources and their extracellular matrix (ECM) programs remain unknown. Fibroblasts are known to shape lymphocyte compartmentalization in secondary lymphoid organs, where they produce distinct sets of chemokines and a complex ECM conduit system that serves as a scaffold along which dendritic cells and lymphocytes migrate and engage (8–10). While the role of fibroblasts in restricting immune cell localization is well established in spleen and lymph nodes, only recently has the tumor stroma emerged as a player in regulating local immune responses (11–14).
Given the growing evidence indicating that cancer-associated fibroblasts (CAF) can regulate tumor immunity and progression(11–14), CAF are becoming an important target for cancer treatment. TGFβ blockade and NOX4 inhibition were shown to act on CAF and facilitate T cell infiltration, leading to better responses to anti-PD-1/PD-L1 treatment in murine cancer models (6,15,16). Yet modulating and depleting CAF have led to opposite results in other tumor systems (16,17) and has not yet managed to achieve clinical benefit in human cancer (18,19). How to manipulate fibroblast properties for therapeutic purpose remains challenging, largely due to our limited understanding of the tumor CAF compartment and the mechanisms by which distinct CAF populations modulate anti-tumor immunity, including immune cell spatial organization.
The initial characterization of functional heterogeneity of CAF included description of inflammatory CAF (iCAF) and myofibroblastic CAF (myCAF) in mouse models of pancreatic cancer(20). Transcriptional signatures of these distinct CAF phenotypes have subsequently been found in human pancreatic and breast cancer(21,22), as well as an additional subset, antigen-presenting CAF (apCAF)(22). iCAF are described as being found distal from the tumor site with a secretory phenotype whereas myCAF are characterized by activation and contractility genes and their close proximity to tumor cells(20). Prior studies have used single-cell RNA sequencing (scRNAseq) to profile CAF in various human cancers, including NSCLC(23–26), bladder(27), pancreas(22,28), breast(21), head and neck(29), and liver(30). While the diversity of CAF is increasingly appreciated, the molecular programs of human fibroblast subsets and their discrete functional contributions to the tumor organization and T cell compartmentalization have not been resolved.
We reasoned that pairing scRNAseq profiling with high resolution spatial mapping would enable unbiased identification of CAF transcriptional subsets and uncovering their spatial organization in the context of the tumor microenvironment (TME). Our scRNAseq analysis on 15 surgically resected NSCLC samples along with 12 paired adjacent tissue samples identified novel CAF subpopulations which we validated by profiling 35 tumors by multiplexed immunohistochemistry (IHC) (31). We analyzed the spatial organization of the stromal and immune cell populations, and revealed two CAF subsets with distinct ECM programs that were associated with CD3+ and CD8+ T cell exclusion from the tumor nests. Importantly, by applying high-resolution histological profiling on a large NSCLC cohort, our study characterizes both the intra-tumor and inter-tumor CAF and T cell heterogeneity.
RESULTS
Paired scRNAseq and IHC analysis identifies four CAF populations with distinct transcriptional profiles and structural organization in human NSCLC
To characterize the stromal cell compartment in NSCLC in an unbiased way, we profiled non-immune, non-tumor/epithelial cells isolated from 15 NSCLC samples and 12 paired adjacent tissue samples using the 10x Genomics scRNAseq platform (Figure 1A, Table S1). Using flow cytometry and mass cytometry by Time of Flight (CyTOF), we optimized the digestion and sorting protocols to maximize the stromal cell recovery while preserving cell integrity (Methods, Figures S1A-B). 33,742 cells were sequenced in total which contained 31,402 stromal cells after excluding contaminating immune cells, epithelial cells, and cells not passing quality control (Table S2). Using an unsupervised clustering, which integrates samples over different conditions and patients while modeling background noise (32,33), we identified 28 clusters, including; 24 stromal cell clusters of variable abundance shared among samples (Figure S1C, table S2) as well as 4 clusters, either containing contaminating immune cells or cells with high mitochondrial content, that were excluded from future analysis. mRNA counts (unique molecular identifiers, UMI) per cluster and mitochondrial content per sample were similar (Figures S1C-D).
Fig. 1 |. Paired scRNAseq and IHC analysis identifies four CAF populations with distinct transcriptional profiles and structural organization in human NSCLC.
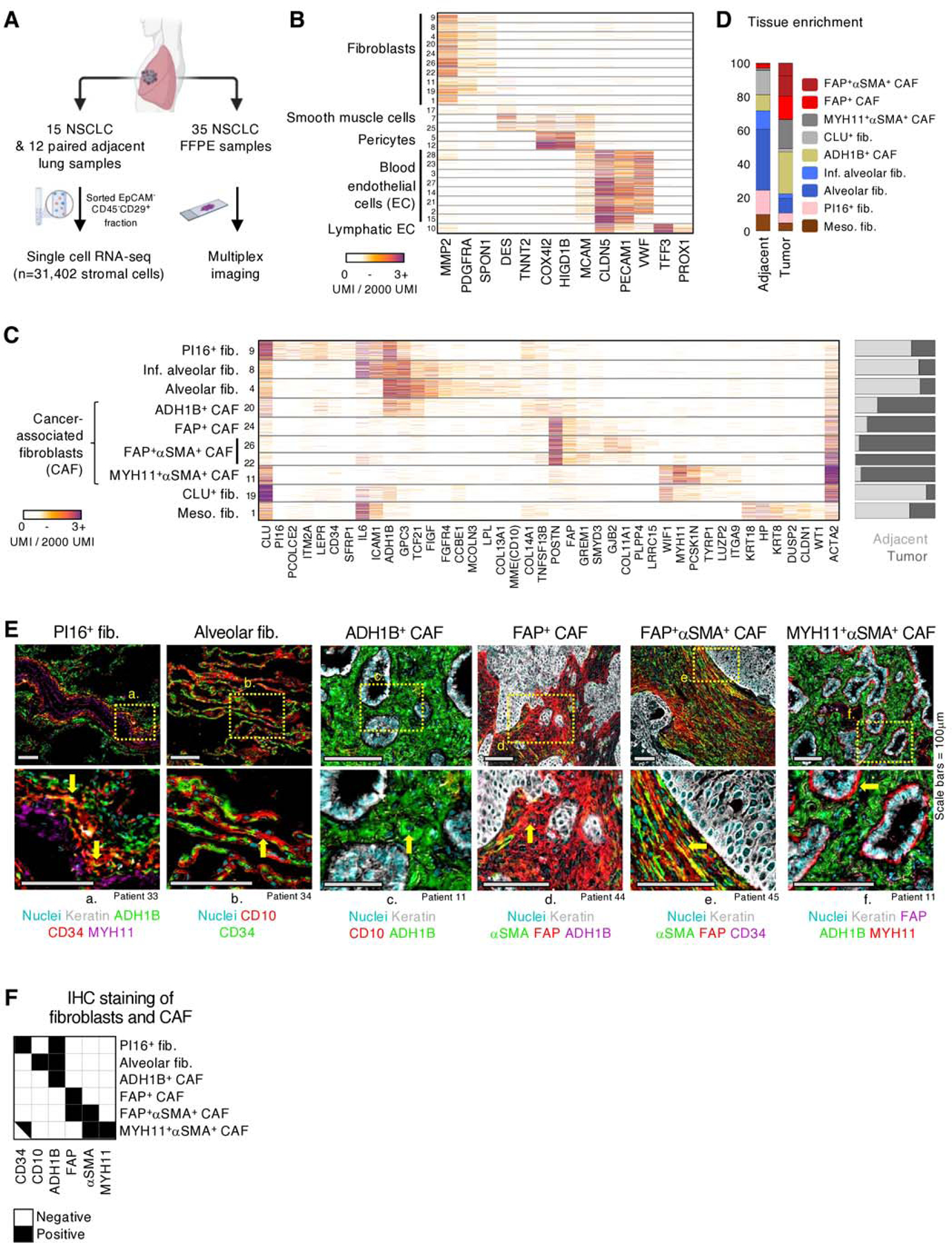
A, Tissue processing workflow for scRNAseq and IHC imaging of FFPE samples. B, scRNAseq mRNA counts (unique molecular identifiers, UMI) per cell (rows) of select stromal lineage marker genes (columns). Fibroblast, smooth muscle, pericyte, blood and lymphatic endothelial cell clusters are identified based on expression of marker genes such as, PDGFRA, DES, COX4I2, PECAM1, and TFF3, respectively. All cells displayed in this figure, and all subsequent similar scRNAseq figures, were downsampled to 2000 UMI. C, Extended gene lists highlighting gene expression profiles between the fibroblast subsets along with differing propensities for enrichment (right bar plot) in tumor (dark gray) or adjacent tissue (light gray). D, Averaged fibroblast composition in adjacent and tumor samples across all patients. The bar graph depicts the percentage of cells from each fibroblast subset among all fibroblasts. E, FFPE NSCLC sections were stained for fibroblast markers identified in scRNAseq results. All the scRNAseq-based fibroblast clusters (D) were detected utilizing IHC except meso. fib. and CLU+ fib., which were not in the scope of this study. Arrows highlight cells of interest (PI16+ fib.: CD34+ADH1B+MYH11neg, Alv. fib.: CD10+CD34neg, ADH1B+ CAF: ADH1B+CD10neg, FAP+ CAF: FAP+ADH1BnegαSMAneg; FAP+αSMA+ CAF: FAP+αSMA+CD34neg; MYH11+αSMA+ CAF: MYH11+FAPnegΑDH1Βneg). See Figure S3 for other stainings. All scale bars are 100μm. F, IHC staining presentation for the main identified fibroblast and CAF clusters.
To unbiasedly dissect cell identities, we analyzed the mRNA counts of variably expressed genes across the 24 stromal cell clusters (Figure S1E). The cell clusters represented 3 major stromal cell compartments and expressed well reported lineage markers: fibroblasts (PDGFRA+, MMP2+), endothelial cells [EC, including both blood (CLDN5+, PECAM1+) and lymphatic (TFF3+, PROX1+) EC], and perivascular cells [PvC, including pericytes (MCAM+, COX4I2+), and smooth muscle (SM) cells (MCAM+, DES+)] (Figures 1B, S1F, table S3). Blood EC clusters included arteries, venules, tip cells, as well as two lung capillary subsets recently described as aerocytes and general capillaries(34) (Figure S2A). The PvC clusters enriched in tumor lesions included tumor pericytes, which expressed high amounts of RGS5 and multiple collagens (COL1A1, COL3A1, COL6A3), and a cluster expressing multiple immunomodulatory genes including CCL19 and CCL21 (Figure S2B). To be noted, IHC showed that the MCAM+ cells were restricted to vascular areas and were not found in the rest of the stroma (Figure S2C).
Further dissection of fibroblast populations identified multiple subsets with distinct transcriptional profiles and uneven abundances in the tumor lesion or the adjacent tissue (Figures 1C-D, Table S4). Based on this scRNAseq analysis, we identified genes associated with each cluster and defined antibody panels (Table S5) that enabled further characterization by multiplexed IHC (Figures 1E, 1F, S3A-B). Two clusters enriched in the adjacent lung tissue were characterized by co-expression of MME (CD10), FIGF (VEGFD), FGFR4 (Figure 1C) and were annotated as alveolar fibroblasts (alv. fib.) based on their specific localization to the lung alveoli by IHC (Figures 1E, S3A). Interestingly, one of these clusters expressed high levels of inflammatory genes, including IL6 and ICAM1, and was thus referred to as inflamed alveolar fibroblasts (inf. alv. fib.) (Figure 1C). Another cluster enriched in the adjacent lung was annotated as PI16+ fibroblasts based on its co-expression of PI16, CD34 and LEPR (leptin receptor), localization to the blood vessel adventitia, and similarity to the universal PI16+ fibroblasts described in Buechler et al., 2021 (Figures 1C, 1E, S3A) (35–37). The last adjacent tissue cluster, CLU+ fib., was characterized by high expression of CLU (clusterin) (Figure 1C).
Fibroblast clusters enriched in tumor samples were annotated as CAF. One CAF cluster displayed an expression profile similar to that of alv. fib., including expression of the broad adjacent tissue fibroblast marker, ADH1B (alcohol dehydrogenase 1B), and lower expression of the canonical CAF marker, FAP, and was referred to as ADH1B+ CAF (Figures 1C, 1E, S3A). ADH1B+ CAF could be distinguished from alv. fib. in IHC by their lack of CD10 expression and localization in the tumor lesion (Figures 1C, 1E, S3A-C). Three clusters showed strong expression of canonical activated CAF markers, FAP, POSTN, LRRC15 and GREM1 (23,28) and were denoted as FAP+ CAF (Figures 1C, 1E, S3B). Another common CAF marker, ACTA2 (αSMA) (38), was differentially expressed among the FAP+ CAF (Figures 1C, 1E, S3B) and clusters with high ACTA2 expression were designated as FAP+αSMA+ CAF. Notably, a cluster that was highly enriched in a single patient (Table S2) shared both fibroblast genes (PDGFRA, MMP2, COL1A1, BGN) and mesothelial cell genes, such as keratins and WT1, and was therefore designated as mesothelial-like fibroblasts (meso. fib.) (39) (Figure 1C). An additional CAF cluster, MYH11+αSMA+ CAF, clearly distinct from the other CAF subsets, was characterized by the expression of MYH11 (myosin heavy chain 11), ACTA2 and intermediate levels of CD34, while lacking ADH1B and FAP expression (Figure 1C). Histological analysis of matched formalin-fixed paraffin-embedded (FFPE) tumor samples revealed a MYH11+αSMA+CD34+ADH1BnegFAPneg cell population observed as a single layer of elongated CAF encapsulating tumor nests, in contrast to ADH1B+ CAF and FAP+ CAF that are spread throughout the stroma (Figures 1E, 1F, S3A). CyTOF confirmed the presence of the main fibroblast subsets identified through scRNAseq, including alv. fib., PI16+ fib., MYH11+αSMA+ CAF and FAP+αSMA+ CAF (Figure 2A).
Fig. 2 |. Further characterization of CAF subsets in human NSCLC.
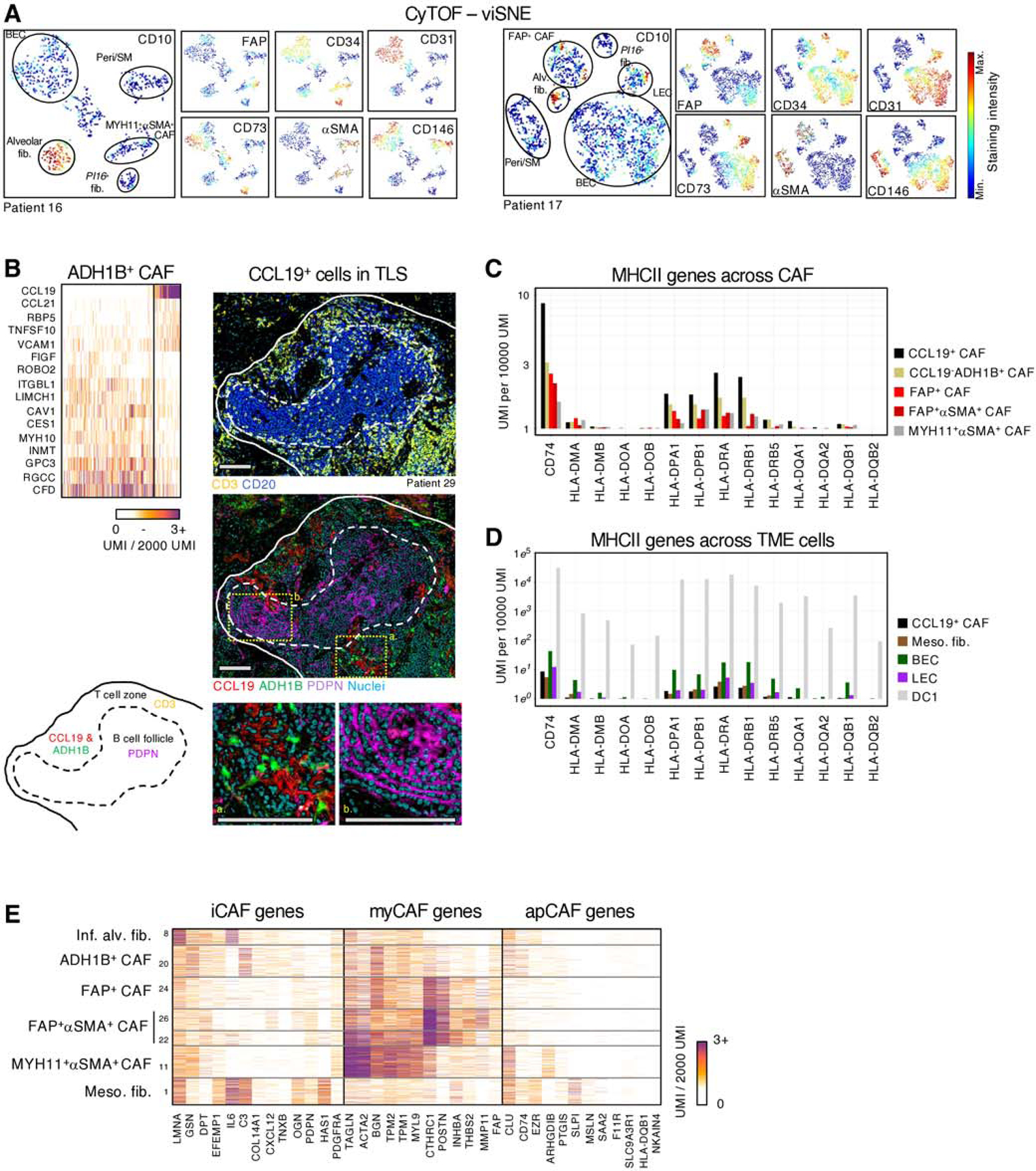
A, Stromal cell populations visualized with viSNE in CyTOF. EC, PvC and multiple fibroblast subsets can be distinguished with relatively few markers (CD10, CD31, CD34, CD73, FAP, CD146 and αSMA) B, (upper left panel) Highlighting CCL19 expressing cells within ADH1B+ CAF. These cells expressed high amounts of CCL19, CCL21, and VCAM1 and low levels of certain ADH1B+ CAF genes such as MYH10 and GPC3 (bottom and right panels). Multiplex IHC of a representative tertiary lymphoid structure. Podoplanin (PDPN) and CD20 marks follicular dendritic cells and B cells, respectively, in the B cell follicle, while the T cell zone is identified with CD3 staining. CCL19 and ADH1B staining show ADH1B+ fibroblasts surrounded by the secreted chemokine CCL19, specifically in the T cell zone. All scale bars are 100μm. C, Average expression of MHCII genes in each CAF subset. D, Average MHCII gene expression in classical antigen presenting cells, DC1, endothelial cells, and meso-like fibs. E, myCAF, iCAF and apCAF gene signatures (20,22) projected onto NSCLC CAF clusters.
Further analysis of ADH1B+ CAF revealed a subset of cells that expressed high levels of T cell-attracting and T cell retention genes CCL19, CCL21, and VCAM1, reminiscent of fibroblastic reticular cells present in secondary lymphoid organs (40) (Figure 2B). IHC staining of CCL19 confirmed the specific localization of these fibroblasts to tertiary lymphoid structures (TLS), with clear preferential enrichment for the T cell zone (Figure 2B). In some cases, the B cell zone was delineated by podoplanin expression, which marks follicular dendritic cells that were not captured by scRNAseq, likely due to their low abundance (Figure 2B). Given the report of MHCII-expressing CAF in human NSCLC tumors (41), we considered whether these CCL19+ TLS CAF may be involved in antigen presentation to T cells. The scRNAseq data showed MHCII gene expression among CAF, with CCL19+ADH1B+ CAF expressing the highest levels, suggesting that CCL19+ TLS CAF are involved in antigen presentation to T cells (Figure 2C). Notably, we compared CCL19+ CAF to other cells of the TME, including endothelial cells and mesothelial-like fibs captured in our study and immune cells from our NSCLC immune cell dataset (32). This analysis showed that MHCII expression in CCL19+ CAF is orders of magnitude lower than in endothelial cells, as well as the classical MHII-expressing cells, type 1 dendritic cells (DC1). Nevertheless, the high density of CCL19+ CAF in TLS may contribute to an effective antigen presentation in these areas (Figure 2D).
Taken together, our combined IHC and single-cell analysis has defined diverse fibroblast populations with distinct molecular and spatial patterns in human NSCLC. By enriching for stromal cells from a large NSCLC cohort, we achieved highly granular scRNAseq characterization and uncovered CAF populations undescribed to date, including a single layer of MYH11+αSMA+ CAF bordering tumor cells in a fraction of NSCLC lesions. The four CAF subsets described here expand upon the iCAF, myCAF, and apCAF profiles described in pancreatic tumors(20,22). Our analysis suggests that in human lung tumors, myCAF include both FAP+ CAF, FAP+αSMA+ CAF and MYH11+αSMA+ CAF highlighting the transcriptomic and spatial complexity of this population (Figure 2E). The full expression profiles of the different fibroblast populations and histology data are available at https://scdissector.org/grout that allows for multidimensional exploration.
ADH1B+ CAF and FAP+ CAF stratify NSCLC into two main stromal patterns associated with tumor stage and histology
Analysis of the fibroblast composition as determined by the scRNAseq indicated that low stage tumors were dominated by ADH1B+ CAF with or without MYH11+αSMA+ CAF, while higher stage tumors were enriched for FAP+ CAF and FAP+αSMA+ CAF (Figures 3A-B, S4A). To test the dichotomy between ADH1B+ and FAP+ CAF enrichment, we leveraged a larger cohort of 35-patient FFPE samples and quantified the tumor area covered by ADH1B and FAP using IHC. This unbiased analysis showed that the stroma of NSCLC is significantly dominated for either ADH1B+ or FAP+ CAF (hypergeometric test, p = 0.008) (Figure 3C, table S6). MYH11+αSMA+ CAF were observed in half of ADH1B+ CAF rich samples (9/18) (Figure 3C) but they were not observed in FAP+ CAF rich samples, corroborating the scRNAseq analysis that showed a correlation between ADH1B+ CAF and MYH11+αSMA+ CAF (Figure 3A). FAP+ CAF rich samples showed highly variable stroma coverage by FAP+αSMA+ CAF (Figures 3B, S4B), in line with the variable ACTA2 expression seen across the scRNAseq FAP+ CAF clusters (Figure 1C). While FAP+αSMA+ CAF could be found throughout the stroma, in a fraction of tumors they were organized as cell layers lining tumor nests (Figure S4C).
Fig. 3 |. ADH1B+ CAF and FAP+ CAF stratify NSCLC into two main stromal patterns associated with tumor stage and histology.
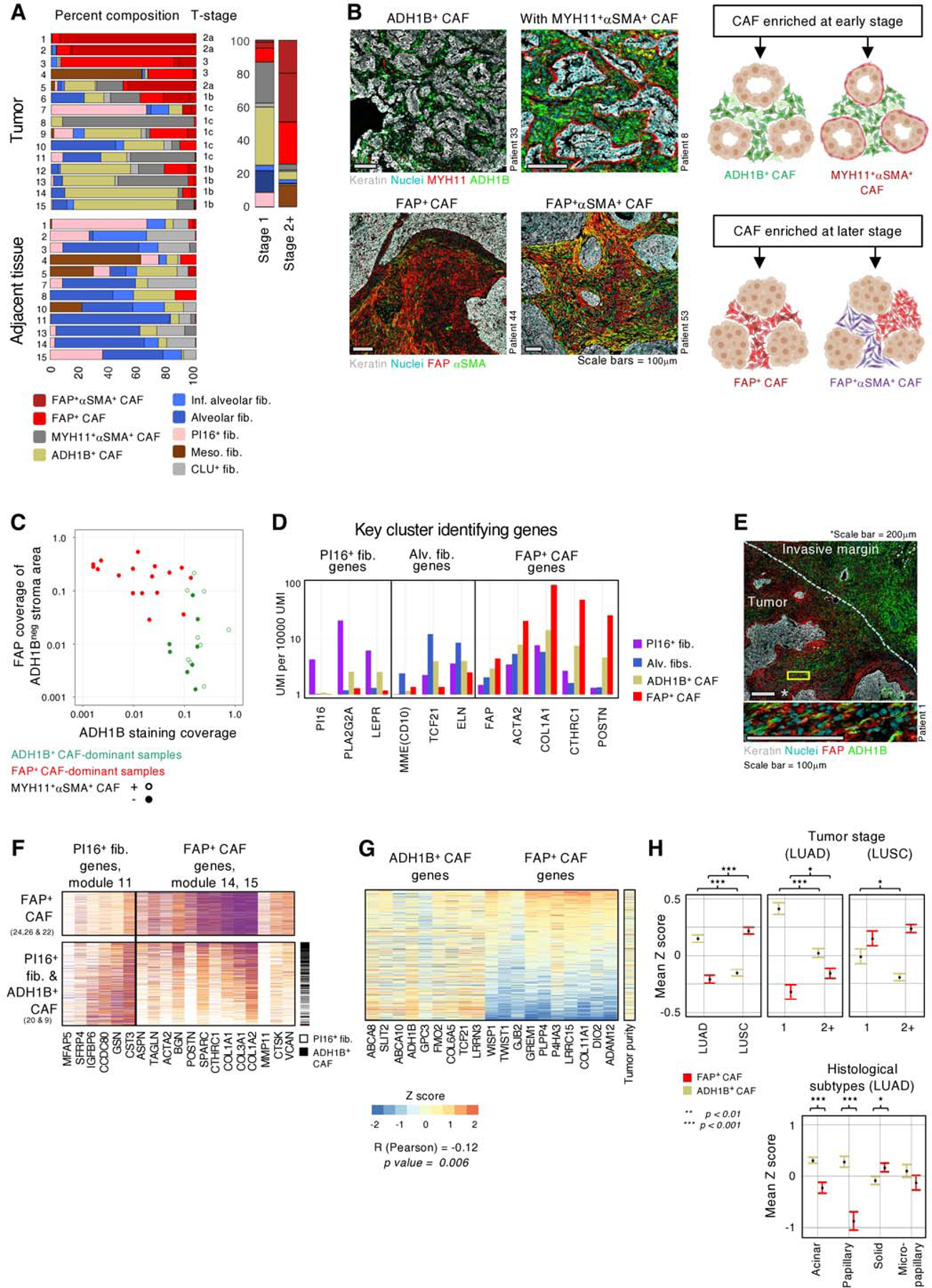
A, (left panel) Fibroblast subset composition, displayed by percentages, in individual tumor and adjacent tissue samples from the 15 scRNAseq patients. (right panel) Fibroblast distribution in stage 1 and stage 2+ tumors. The bar graph depicts the percentage of cells from each fibroblast subset among all fibroblasts. B, (Top left panels) ADH1B+ CAF rich patients showing ADH1B presence throughout the stroma. ADH1B+ CAF rich patients may present with (bottom left panel) or without (top left panel) a distinct single cell layer of MYH11+αSMA+ CAF at the tumor border. (Bottom left panels) FAP+ CAF rich patients with FAP staining throughout the stroma. The patients shown demonstrate the variable αSMA presentation in FAP+ cells. All scale bars are 100μm. (Right panels) Cartoon illustrating the observed presentation of multiple CAF subsets in NSCLC. C, ADH1B and FAP staining in the IHC cohort. ADH1B staining coverage in the stroma is shown on the X axis. FAP staining coverage in the stroma on regions that did not stain for ADH1B are shown on the Y axis. Tumors show significant preference for either ADH1B or FAP, with less than 5% coverage of the opposing stain. The 5% cutoff was selected after performing hypergeometric tests for 10 thresholds, at 5% increments, between 5% and 50%. The Bonferroni correction adjusted p value is 0.008. D, Mean expression of selected genes highlighting ADH1B+ CAF intermediate expression of PI16+ fib., alv. fib., and FAP+ CAF-associated genes. E, Tumor sample with an extensive invasive margin that displays a spectrum of ADH1B to FAP staining. (Zoom, bottom panel) Cells appearing to transition from ADH1B to FAP expression. Top panel scale bar is 200μm, bottom panel scale bar is 100μm. F, Expression of PI16+ fib. and FAP+ CAF module genes in PI16+ fib., ADH1B+ CAF, and FAP+ CAF. Based on gene expression patterns, ADH1B+ CAF appear to occupy an intermediate state of activation between PI16+ fib. and FAP+ CAF. G, Relative expression, displayed by Z score, of ADH1B+ CAF and FAP+ CAF-associated genes in TCGA LUAD bulk-RNAseq samples. ADH1B+ CAF and FAP+ CAF genes are significantly anticorrelated (Pearson) R = −0.12 and p = 0.006. The sample tumor nuclei count is used as a proxy of tumor purity and shows a relatively even distribution. H, TCGA LUAD mean Z score and standard error of mean (SEM) of ADH1B+ CAF and FAP+ CAF gene signatures stratified by tumor subtype (left and middle panels) or stage (right panel). Z score calculation is listed in methods and significance is calculated by independent t test (right panel).
To study the transcriptional programs behind ADH1B+ CAF and FAP+ CAF and to better understand their relationship to adjacent tissue fibroblasts, we analyzed gene expression covariance patterns across ADH1B+ CAF, FAP+ CAF, alv. fib. and PI16+ fib. We identified groups of co-expressed genes (gene modules) with distinct expression patterns across these fibroblast populations (Figure S4D). FAP+ CAF upregulated activation genes (modules 14, 15) including multiple collagen genes (COL1A1, COL3A1) that contribute to tissue stiffness(42) and other ECM genes such as biglycan (BGN) that can promote tissue mineralization (43) (Figure S4D). FAP+ CAF expressed low levels of the alv. fib. genes, including the fibroblast transcription factor TCF21 (44), the marker MME(CD10) as well as the ECM gene elastin (ELN) which is critical for normal lung physiology(45) (Figure 3D). ADH1B+ CAF expressed intermediate levels of FAP+ CAF activation genes (Figure 3D), and a subset of samples showed a spatial gradient of ADH1B+ CAF to FAP+ CAF from the invasive margin to the tumor center (Figure 3E) with some cells co-expressing both markers, suggesting that ADH1B+ CAF represent a range of lowly activated fibroblasts. ADH1B+ CAF also shared genes with both PI16+ fib. and alv. fib., which may point towards both lung fibroblast types as their potential cellular sources (Figures 3D, S5A). Interestingly, the scRNAseq data showed that ADH1B+ CAF cells express a gradient of FAP+ CAF and PI16+ fib. genes from cells with high expression of PI16 genes and low FAP genes to cells with low PI16 genes and high FAP genes (Figure 3F). This further supports the hypothesis that ADH1B+ CAF are a lowly activated form of fibroblast and may derive from PI16+ fib.
Leveraging our scRNAseq datasets, we created gene signatures for ADH1B+ CAF and FAP+ CAF by selecting for genes with highly specific expression in their corresponding CAF populations in contrast with all other cell types. With these signatures we scored the Cancer Genome Atlas (TCGA) lung adenocarcinoma (LUAD) samples by their expression of ADH1B+ CAF and FAP+ CAF genes and revealed an anticorrelation between the two scores (p = 0.006) (Figures 3G, S5B, table S7), supporting the two distinct CAF profiles observed across NSCLC patients in scRNAseq and histology (Figures 3A-C) (46). Analysis of tumor purity, estimated by tumor nuclei abundance, did not reveal clear association with ADH1B+ CAF or FAP+ CAF genes (Figure 3G), confirming that contaminating adjacent tissue was not a major contributor to the ADH1B+ CAF signal. Further analysis of TCGA data showed that ADH1B+ CAF genes were significantly increased in stage 1 tumors, LUAD and the papillary LUAD subtype, whereas FAP+ CAF were enriched in later stage tumors, LUSC, and the LUAD solid subtype (Figure 3H). LUAD across tumor stages confirmed our observation that ADH1B+ CAF and FAP+ CAF were correlated with lower and higher stage tumors, respectively. Similar associations were observed in our in-house FFPE cohort (Table S1). Altogether, we showed that ADH1B+ and FAP+ CAF phenotypes were correlated with tumor stage and clinically relevant histological subtypes (46,47), suggesting that molecular characterization of fibroblasts could refine clinical categorization of NSCLC tumors.
To validate the diverse transcriptional programs of CAF observed in our dataset, we studied their expression in four additional public scRNAseq datasets in NSCLC (23–26). This analysis identified two fibroblast subsets expressing ADH1B+ and FAP+ CAF genes, confirming their enrichment in early and late stage NSCLC tumors, respectively (Figures S6A-C, table S8). FAP+ cells display variable expression levels of the FAP+αSMA+ CAF program (Figure S6D), reflecting the observed heterogeneity within FAP+ cells (Figure S4D). Within Kim et al., 2020 (24) we identified a population resembling MYH11+αSMA+ CAF expressing multiple MYH11+αSMA+ CAF genes including MYH11, COL4A1 and COL4A2 (Figure S6E). As expected, these cells were not found in the other studies, which were predominantly composed of stage 2+ tumors and squamous cell carcinomas. Altogether, the external datasets examined validated the dominant CAF populations in NSCLC.
Next, we sought to determine if ADH1B+ and FAP+ CAF were present across different tumor types. To search for ADH1B+ and FAP+ CAF in other cancers we returned to TCGA to analyze the data available for breast, colon, pancreatic, prostate and ovarian cancer. Unsupervised hierarchical clustering grouped ADH1B+ and FAP+ CAF genes by their co-expression in each dataset separately (Figure S7). In pancreatic, breast and colon cancer we observed a significant separation of the two gene groups, suggesting that other cancer types also harbor lowly and highly activated CAF with similar transcriptional profiles as ADH1B+ and FAP+ CAF found in NSCLC.
ADH1B+ CAF and FAP+ CAF correlate with immune cell composition and not with T cell localization
Given the data showing that CAF contribute to regulating tumor immunity (11,12,48) we investigated the different ligands expressed by CAF populations found in human NSCLC (Figure 4A). Increased expression of the cytokines IL34 and CSF1 suggested macrophage regulation (49) by ADH1B+ CAF, whereas FAP+ CAF might attract eosinophils/basophils via CCL11 (50), as well as CCR5+ T cells and monocytes through CCL3 and CCL5 chemokines (51–53). Notably, the high levels of CCL21 and TNFSF13B (BAFF) in ADH1B+ CAF mainly come from the CCL19-expressing ADH1B+ cells specifically found in TLS (Figures 4A, 2B), likely contributing to naïve T cell attraction and B cell survival in these structures (40). MYH11+αSMA+ CAF expressed increased levels of TSLP (thymic stromal lymphopoietin) which can stimulate the maturation of immune cells that express both IL7R and CRLF2 genes forming the heterodimeric TSLP receptor, such as certain dendritic cells (54). MYH11+αSMA+ CAF also showed strong expression of TGFΒ1, which has been implicated in reducing cytotoxic T cell function (55). IHC confirmed expression of TGFβ1 by MYH11+αSMA+ CAF at the protein level and its lack of expression on ADH1B+ CAF or FAP+ CAF (Figures S8A-B).
Fig. 4 |. ADH1B+ CAF and FAP+ CAF correlate with immune cell composition and not with T cell localization.
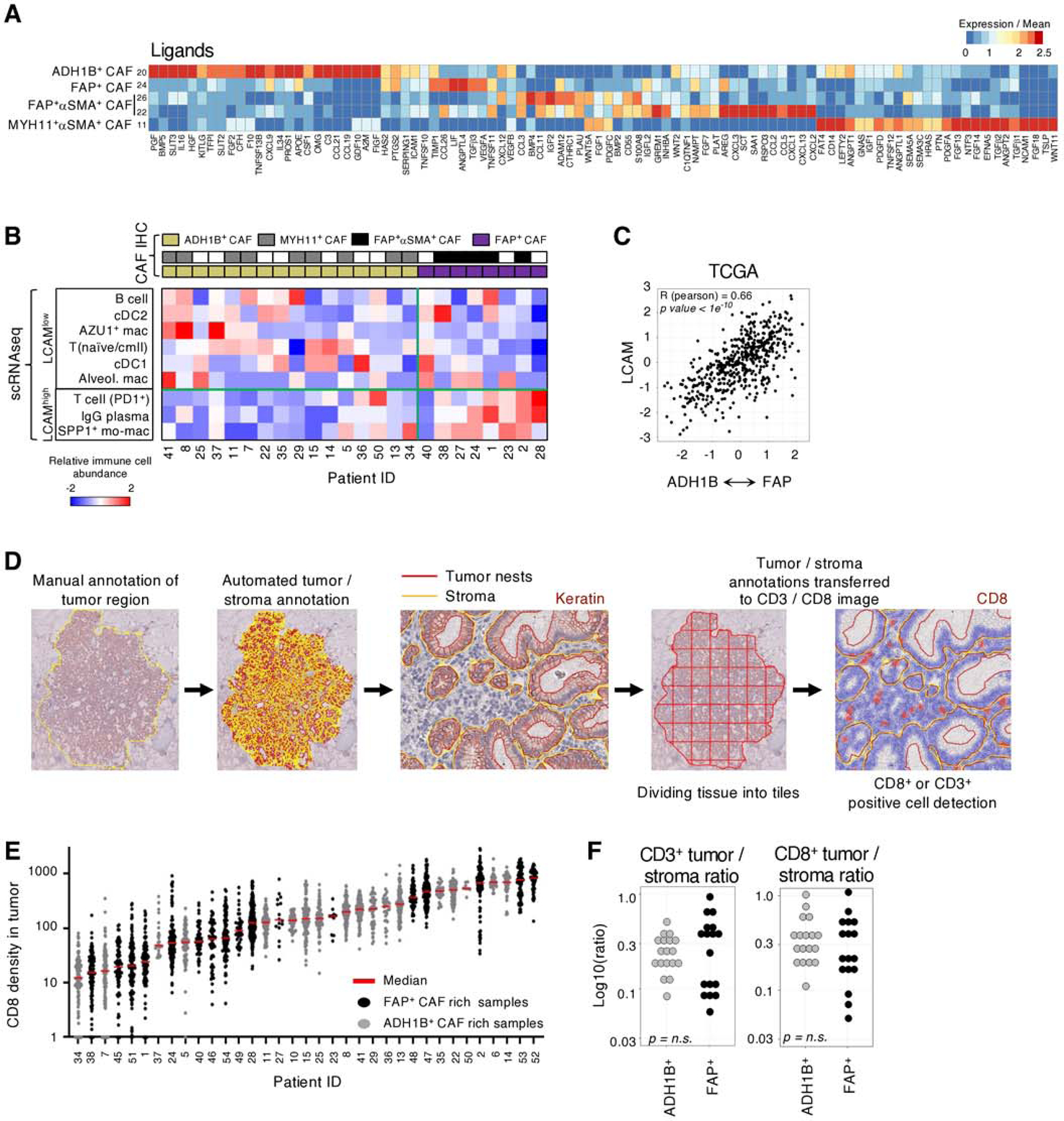
A, Gene expression over mean of highly variable immunomodulatory ligands in CAF clusters. B, Immune composition of scRNAseq tumor samples from (32). CAF phenotype is identified by IHC on the matched FFPE samples and then used to stratify samples. The relative abundance of each cell population within its respective compartment, i.e.; PD1+ T cells amongst all T cells, is calculated and then scaled across all tumors for the respective Z score value. LCAM score is significantly correlated with CAF phenotype (Pearson) R = 0.62, p = 0.01. C, Estimating the correlation between CAF phenotype and LCAM in TCGA LUAD samples. Each patients’ mean ADH1B+ CAF gene signature is subtracted from their mean FAP+ CAF gene signature and the resulting values are correlated with estimate LCAM score. The corresponding Pearson correlation values are shown. D, Schematic of QuPath methodology for tiling and T cell quantification. E, CD8+ cell infiltration into tumor nests in each patient (columns). Each point represents an individual 1000μm x 1000μm tile (all other tiling is 500μm x 500μm). F, IHC quantification of the tumor / stroma CD3+ or CD8+ cells per mm2 ratio. Tumor samples are stratified by their stroma profile (ADH1B+ CAF rich or FAP+ CAF rich) and no significant difference (t test) was observed.
To further investigate the contribution of different CAF populations to shaping the immune microenvironment, we used multiplex imaging on FFPE tissue to histologically profile the CAF subset composition of a large cohort of NSCLC samples that we had previously studied using scRNAseq of purified immune cells(32). We demonstrated a significant association (Pearson, R=0.62, p = 0.01) between the presence of FAP+ CAF and the enrichment of inflammatory SPP1+ monocyte-derived macrophages, IgG+ plasma cells, and PD1+ T cells (Figure 4B). These immune cell types were recently described as part of a cellular module termed Lung Cancer Activation Module (LCAM)(32). We then validated this CAF-immune association in the TCGA LUAD cohort. There was a significant correlation between CAF phenotype and the LCAM score (R = 0.66, p < 1e−10), supporting that FAP+ CAF rich samples are linked to more inflammatory and activated immune cells, LCAMhigh, in LUAD (Figure 4C).
Given that the spatial distribution of T cells is a predictor of clinical response to immune checkpoint blockade (56), we used an unbiased cell quantification method to measure T cell infiltration in the tumor nests and identified a wide range of infiltration levels across the cohort (Figures 4D-F). Importantly, there was no observed association between any of ADH1B+ and FAP+ CAF-rich profiles and CD3+, CD8+ T or FOXP3+ T cell localization (Figures 4E-F, S8C, table S9). Taken together, ADH1B+ CAF and FAP+ CAF stratify tumor lesions by two levels of fibroblast activation and correlate with the immune phenotype, but not with T cell spatial distribution.
MYH11+αSMA+ CAF are correlated with decreased T cell infiltration in tumor nests
The lack of correlation between ADH1B+ CAF or FAP+ CAF with T cell infiltration contrasts with the general idea that activated fibroblasts orchestrate T cell exclusion, raising the hypothesis that fibroblast subsets other than ADH1B+ or FAP+ CAF could be involved. To investigate if MYH11+αSMA+ CAF, which form a single cell layer around tumor nests in a fraction of early-stage tumors, could also impact T cell tumor infiltration, we subdivided stage 1 patients based on the presence of MYH11+αSMA+ CAF at the tumor border (Figure 5A). In tumor lesions containing MYH11+αSMA+ CAF, the tumor-to-stroma ratio of infiltrating CD3+ or CD8+ cells was significantly lower (Figure 5B, right graphs), consistent with a decreased infiltrating CD3+ or CD8+ T cell density in the tumor (Figure 5B, left graphs) (Table S9). FOXP3+ T cells showed a similar trend, although not significant (Figure S9A). In addition, high expression of TGFB1/TGFB2, WNT5A and WNT11 by MYH11+αSMA+ CAF (Figure 4A) is in line with previous findings linking TGFβ and WNT/βcatenin pathways with immune cell exclusion in tumors (6,57–59). These results suggested that MYH11+αSMA+ CAF, with their peri-tumoral location, may decrease T cell infiltration into tumor nests.
Fig. 5 |. MYH11+αSMA+ CAF are correlated with decreased T cell infiltration in tumor nests.
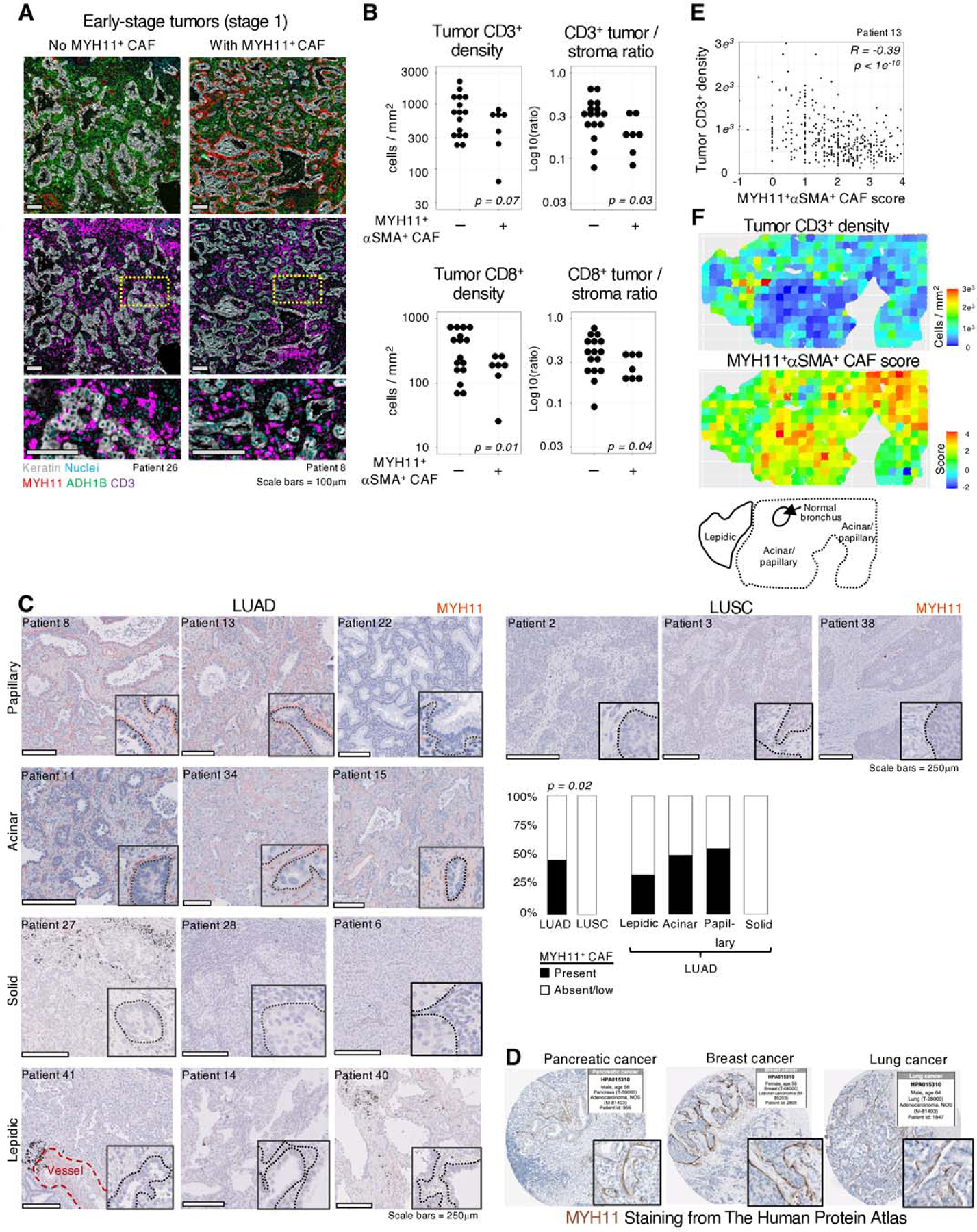
A, Representative examples of IHC stains from NSCLC tumors with and without MYH11+αSMA+ CAF present, showing CD3+ cell exclusion from tumor nests when MYH11+αSMA+ CAF are present. B, The presence or absence of MYH11+αSMA+ CAF demonstrates significant differences in tumor infiltrating CD3+ or CD8+ cells per mm2 (left) and the ratio of CD3+ or CD8+ cells per mm2 in the tumor versus stroma (right). Only early stage (tumor stage 1) patients were included to eliminate bias due to MYH11+αSMA+ CAF rarely being found at later stage. C, Representative images of MYH11 staining in multiple pathologies and histological subtypes. All scale bars are 250μm. (Barplot) MYH11+αSMA+ CAF distribution in different pathologies and histological subtypes in NSCLC. Significance determined by t test. D, MYH11 staining from The Human Protein Atlas. E, Quantification of 500×500μm tiles of both MYH11+αSMA+ CAF score, estimating tumor proximity of MYH11+αSMA+ cells by quantifying their enrichment within 10μm from tumor cells versus regions 20μm-30μm from tumor cells, and tumor infiltrating CD3+ cells per mm2. A high MYH11+αSMA+ CAF score is significantly anti-correlated (Pearson) with the number of tumor infiltrating CD3+ cells relative to the stroma. F, Visualization of the tiling described in E. (Bottom panel) Histological scoring of a tumor lesion highlighting that a high MYH11+αSMA+ CAF score is associated more with acinar/papillary phenotype, rather than lepidic.
Within our cohort, MYH11+αSMA+ CAF were found enriched in LUAD samples, especially in the acinar/papillary subtypes, while neither the solid subtype of LUAD, nor LUSC samples contained MYH11+αSMA+ CAF lining tumor nests (Figure 5C). This differential enrichment was also observed within tumor LUAD lesions displaying acinar and solid tumor regions, as annotated by a pathologist (Figures S9B-C). Interestingly, the IHC image bank of the Human Protein Atlas showed that a similar peritumoral MYH11 staining pattern as one layer was observed in a fraction of samples of pancreatic and breast cancer (Figure 5D), suggesting that these CAF may be present in additional cancer types.
While most tumor lesions were characterized by either high or low MYH11+αSMA+ CAF presence, a fraction of tumors showed local heterogeneity. We assessed the intensity of CAF barrier at the tumor boundary in 500μm-by-500μm tiles using the abundance of MYH11+αSMA+ cells in the stroma close to (<10μm) versus distant from (20μm-30μm) tumor cells, which is referred to as the MYH11+αSMA+ CAF score (Figures 5E-F, S10A-C, tables S10-11). This automated analysis found that locations where MYH11+αSMA+ CAF were present had significantly lower tumor T cell density in two independent samples, highlighting that local spatial organization may be driving inter-tumor differences. Additionally, histological analysis of the tumor lesion by a pathologist found that regions with high MYH11+αSMA+ CAF score were predominantly acinar/papillary, and lepidic regions at the tumor edge had a lower score (Figure 5F, bottom panel). Altogether, these results show that MYH11+αSMA+ CAF are a single layer of elongated cells associated with T cell marginalization both across NSCLC tumor samples and within tumor lesions.
FAP+αSMA+ CAF define regions of poor T cell infiltration within tumor lesions and are coupled with dense ECM deposition
Spatial analysis of FAP+ CAF rich samples revealed that FAP+αSMA+ CAF, a subset of FAP+ CAF in scRNAseq, could also explain CD3+ and CD8+ T cell infiltration within tumors. We measured αSMA coverage and T cell density in the stroma in 500μm-by-500μm sections across the tumor lesion (Figure 6A, table S12) and revealed that regions dense in αSMA are poorly infiltrated by T cells (R = −0.48, p = 1e−10) (Figure 6B). This anticorrelation was replicated across different tumors (Figures S11A-C, table S12-13) and suggested that FAP+αSMA+ CAF directly restrict T cell motility. Notably, a high fraction of FAP+ CAF rich samples presented with several layers of FAP+αSMA+ CAF lining tumor nests that delineated regions devoid of T cells (Figure 6C). In addition, inter-tumor αSMA heterogeneity showed a trend towards an anti-correlation with T cell infiltration in tumor nests (Figures S11D-E).
Fig. 6 |. FAP+αSMA+ CAF define patterns of poor T cell infiltration within tumor lesions.
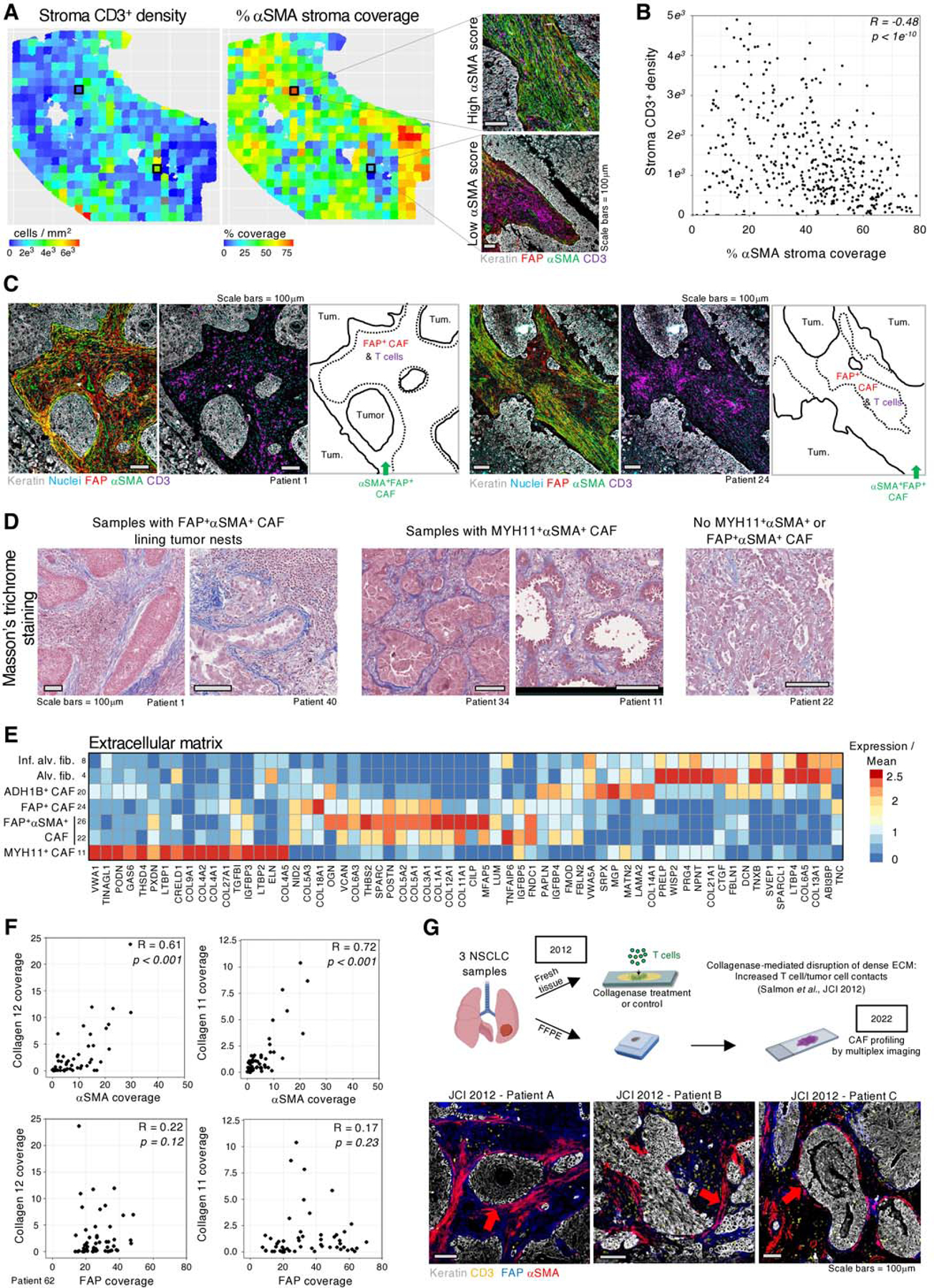
A, (Left panel) Intra-tumoral heterogeneity of αSMA coverage (middle panel) and CD3+ cell density in the stroma in 500×500μm tiles. (Right panel) Representative examples of tiles showing regions with high or low levels of αSMA. B, Quantification of αSMA coverage and CD3+ density in each tile (points) as defined in A, showing a significant anticorrelation (Pearson) of αSMA coverage and CD3+ cell density. C, Dense αSMA staining at tumor border associates with decreased CD3+ cell abundance. The green arrow highlights border regions with high αSMA and low CD3+ cells. D, Masson’s trichrome stains highlighting increased ECM at the tumor boundary in samples containing MYH11+αSMA+ or FAP+αSMA+ CAF. E, Averaged gene expression of highly variable ECM genes in CAF clusters. F, αSMA coverage of the stroma is significantly correlated (Spearman) with collagen XI and XII deposition, while FAP+ CAF show no correlation. G, (top panel) Our prior work in (7) showed that collagenase treatment of viable slices of NSCLC tumor tissue increased T cell access to tumor cells. FFPE sections from tumor samples of the same three patients were stained by multiplex IHC for markers of CAF identified in the present study. (bottom panel) FAP is found throughout the stroma and αSMA shows increased expression at the tumor border.
Based on prior studies showing that the ECM plays a role in T cell exclusion and immunosuppression(7,60,61), we postulated that FAP+αSMA+ CAF may also express a specific ECM profile involved in regulating T cell localization. Masson’s trichrome staining revealed a high density of fiber deposition at the tumor border in both MYH11+αSMA+ and FAP+αSMA+ CAF-containing tumor lesions, suggesting that these CAF are depositing a fibrillar barrier limiting T cell access to tumor cells (Figure 6D). Analysis of the scRNAseq showed that MYH11+αSMA+ CAF expressed COL9A1, COL27A1, and a distinct type of sheet-forming, basement membrane collagens, COL4A1 and COL4A2, which are found lining vessels and various epithelial layers(62,63) (Figure 6E). A thick layer of collagen IV fibers lining tumor nests was frequently found co-localized with MYH11+ CAF in tumor lesions (Figure S12A), whereas samples or tumor regions lacking MYH11+ CAF showed no to low collagen IV deposition (Figures S12B-C). There were rare exceptions to this observation (Figure S12D), indicating that additional ECM factors may contribute to the fiber density observed in MYH11+αSMA+ CAF rich samples by Masson’s trichrome (Figure 6D).
The ECM program of FAP+αSMA+ CAF was distinct from MYH11+αSMA+ CAF. FAP+ αSMA+ CAF expressed high levels of the fibrillar collagen COL11A1 (64), and COL12A1 (Figure 6E) compared to other CAF, including FAP+ CAF. We performed collagen fiber staining on FAP+ and FAP+αSMA+ CAF-rich samples and quantified the coverage of collagen XI, XII, and IV, as well as αSMA in 500μm x 500μm tiles. This analysis revealed strong correlation between the stroma coverage of αSMA and collagen XI/XII (Figures 6F & S13A-D, tables S14-16). Importantly, no positive correlation was observed between collagen XI/XII and FAP (αSMAneg) stroma coverage, nor between collagen IV with αSMA (Figures 6F & S13A-D). We observed strong alignment of collagen fibers in areas rich in FAP+αSMA+ CAF (Figures S13A, D, E), indicating that these CAF not only shape the local matrix composition, but also its structural organization.
In our prior study using live imaging of T cells in viable human NSCLC tumor slices (7), we showed that a dense matrix around tumor nests was anti-correlated with T cell motility. Collagenase treatment of the tumor tissue from three patients in that study led to increased T cell contact with tumor cells, demonstrating the functional role of dense matrix fibers in restricting T cell/tumor cell interactions (Figure 6G, upper panel). We hypothesized that the resolution of our current analysis could enable identification of the CAF subsets that generated this barrier. We therefore retrieved FFPE slides from the same three tumors and stained for CAF markers identified in the current study. Our staining shows multi-layer FAP+αSMA+ CAF localized around tumor nests, revealing the cellular source of the causal factor behind T cell exclusion in those samples (Figure 6G, bottom panel). Thus, the specific spatial distribution of FAP+αSMA+ CAF and their unique ECM profiles may drive T cell exclusion in NSCLC and represent potential therapeutic targets. Combined with MYH11+αSMA+ CAF, this suggests a refined model for CAF phenotypes in NSCLC and a potential mechanism for T cell exclusion (Figures 7A-B).
Fig. 7 |. Working model.
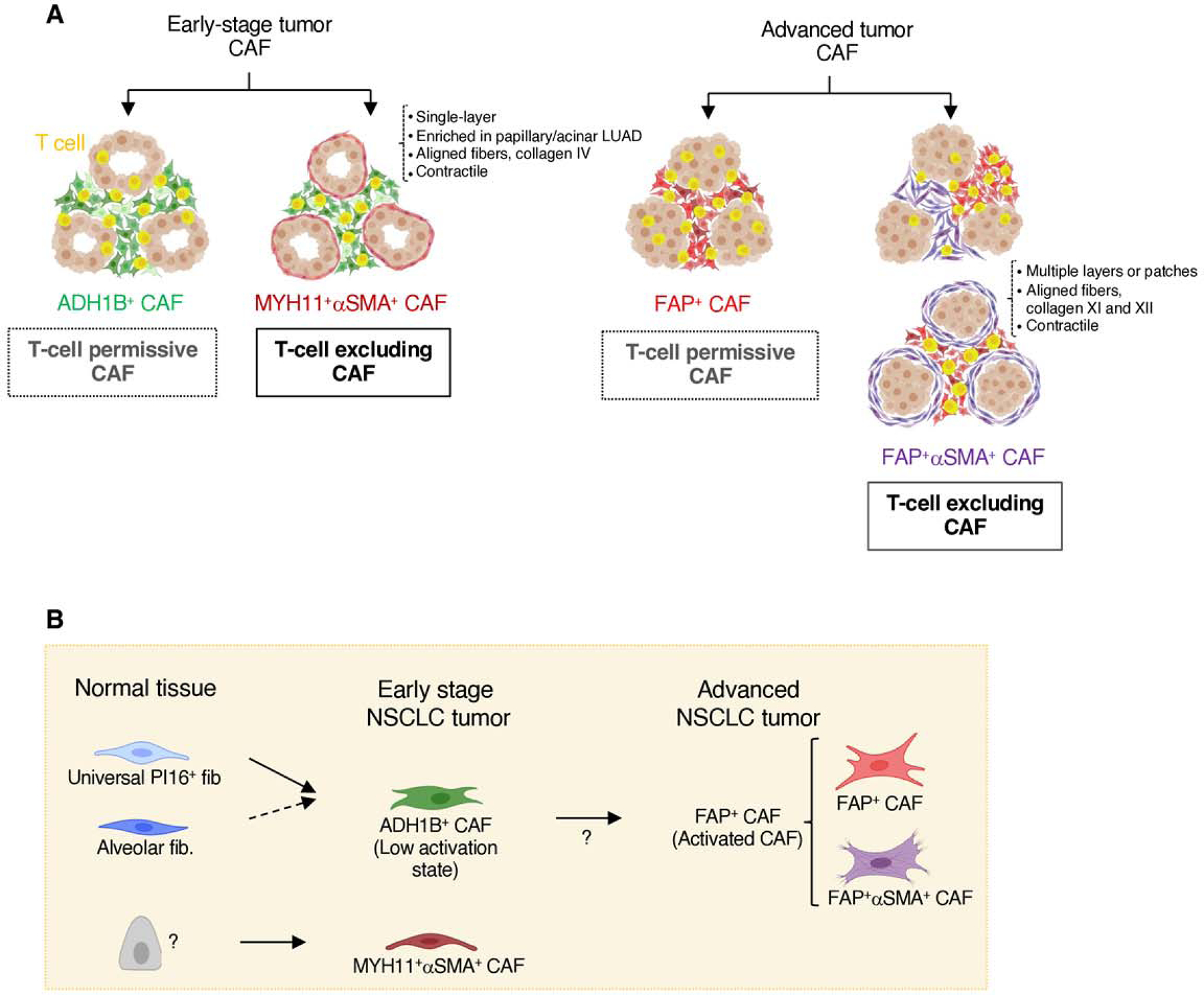
A, Graphical illustration of all stroma presentations found in this study. NSCLC samples enriched in ADH1B+ CAF throughout the stroma can be found with or without a single-cell layer of MYH11+αSMA+ CAF lining tumor cell aggregates. Those with MYH11+αSMA+ CAF show increased T cell exclusion from the tumor nests. NSCLC samples enriched in FAP+ CAF are found with variable abundance of FAP+αSMA+ CAF. Stromal regions with high αSMA have reduced T cell accumulation, and tumor nests surrounded by several layers of FAP+αSMA+ CAF show a lower T cell infiltration. B, Cartoon depicting the general distribution of fibroblast and CAF populations in adjacent lung tissue, early-stage NSCLC and advanced NSCLC, as well as the potential differentiation trajectories.
DISCUSSION
The majority of patients fail to achieve clinical benefit using standard immune checkpoint blockade, and as such, novel combination approaches are required to improve response (65). Patients with T-cell excluded tumors have poor response to immune checkpoint blockade compared to those with T-cell infiltrated tumors(3,5,6) which raises the possibility that targeting the mechanism of T cell exclusion would improve clinical responses. To this end, our study provides a comprehensive map of the fibroblast compartment in human lung tumors at the single cell level and with spatial resolution. We define the molecular and functional diversity of the fibroblast compartment of lung tumors and determine how distinct CAF subsets may influence the immune cell composition as well as T cell spatial organization.
Our analysis shows that the stroma in NSCLC lesions is dominated by either lowly activated ADH1B+ CAF, with or without MYH11+αSMA+ CAF, or highly activated FAP+ CAF, with variable αSMA levels (Figure 7). ADH1B+ CAF have higher activation levels than fibroblasts found in the normal lung tissue, as seen by their expression profile and enrichment in the tumor lesion, as well as by the clear spatial distinction between CD10+ alv. fib. in adjacent tissue and ADH1B+CD10neg CAF in the tumor as seen in multiplex IHC. FAP+ CAF, by contrast, show dramatic transcriptional differences from adjacent tissue fibroblasts, including high expression of many previously established CAF markers like FAP, POSTN, and COL1A1 (23,28). FAP+ CAF and FAP+αSMA+ CAF represent higher activation states compared to ADH1B+ CAF and occasionally form spatial gradients of ADH1B+-to-FAP+ CAF, suggesting ADH1B+ CAF may contribute to the FAP+ CAF pool. FAP+αSMA+ CAF are a subpopulation of FAP+ CAF with higher expression of contractility and ECM genes. Our spatial data frequently shows increased αSMA staining at the tumor nest boundary, suggesting that the αSMA program is upregulated in FAP+ CAF upon physical or molecular signals from tumor cells. In conjunction with our observation that ADH1B+ CAF transition to FAP+ CAF, this suggests that the tumor cells and TME play a critical role in CAF differentiation. ADH1B+ CAF express transcriptional programs of both alv. fib. and PI16+ fib., suggesting that these two lung tissue cell types could give rise to ADH1B+ CAF. In vivo fate-mapping experiments will be needed to further investigate this possibility.
We have also shown that ADH1B+ CAF and FAP+ CAF are associated with pathological and histological subtypes, and tumor stage, ADH1B+ CAF being associated with the adenocarcinoma papillary subtype and stage 1 whereas FAP+ CAF being enriched in the adenocarcinoma solid subtype and squamous cell carcinomas, and in later stage. This association between CAF populations and histological subtypes, which correlate with prognosis(47), may shed light on the molecular programs behind the different NSCLC subtypes, and inform clinical trial inclusion criteria when therapeutically targeting CAF subsets. Furthermore, our staining protocol for these CAF subsets may help refine the categorization of histological subtypes. Beyond subtype and stage, we found a significant association between FAP+ CAF and the LCAM inflammatory/activated immune phenotype (including SPP1+ Mo-Macs, IgG plasma cells, and PD1+ T cells) which we previously described (32). This observation, in conjunction with the distinct immunomodulatory profiles of ADH1B+ CAF and FAP+ CAF, suggests that CAF participate in shaping the immune response to the tumor.
Prior studies have suggested that activated CAF may play a role in T cell exclusion (6,15,16). Notably, FAP+ CAF do not correlate with the T cell distribution pattern in NSCLC, an important factor to keep in mind when developing therapeutics and may explain why strategies targeting FAP+ CAF have failed in human clinical trials so far (18,66). In contrast, we have found two distinct CAF populations with specific molecular programs and spatial organizations that contribute to T cell exclusion. First, FAP+αSMA+ CAF are significantly correlated with regions of T cell exclusion in the tumor stroma and can form multiple layers at the tumor boundary and restrict T cell contact with tumor cells. On the other hand, in a fraction of adenocarcinomas MYH11+αSMA+ CAF form a single cell layer lining tumor nests, and are significantly correlated with immune cell exclusion from tumor regions, both within cancer lesions and across tumor samples. The enrichment of MYH11+αSMA+ CAF in early-stage tumors may suggest that they respond to early tumor cell signals that may be lost upon tumor progression.
FAP+αSMA+ CAF and MYH11+αSMA+ CAF correspond to two clearly distinct fibroblast subsets, as observed through the scRNAseq data and in line with their presence in distinct tumors. Notably, they display similarities, including high expression levels of ECM and contractility genes, which implies that they influence T cell spatial distribution through similar mechanisms, with the production of fibers limiting T cell access to cancer cells (Salmon et al., 2012, (7)). The differences in their matrix deposition (including type IV collagen for MYH11+αSMA+ CAF and type XI and XII collagens for FAP+αSMA+ CAF) also indicate that they drive T cell marginalization by forming different types of barrier to lymphocytes. Our analysis of NSCLC tumor samples from Salmon et al. (7) demonstrates the causal role of FAP+αSMA+ CAF in excluding T cells, identifying them in areas of dense fiber deposition that were implicated in restricting T cell interactions with tumor cells (7). While ECM degradation can improve T cell infiltration (61), targeting ECM molecules is challenging in patients given the low specificity and the risk of on-target off-tumor toxicity. Here, our study paves the way to develop novel strategies to differently target the two distinct cellular sources of these ECM molecules.
In summary, our study has identified several CAF populations that show greater heterogeneity than previously established CAF classification, and provides novel therapeutic targets to pursue in order to augment response to cancer immunotherapies. We demonstrate that pairing molecular and spatial analysis is crucial to understanding the true organization of the human TME and to development of novel CAF targeting strategies for efficient anti-tumor combinations.
METHODS
Human subjects
In collaboration with the Biorepository and Department of Pathology tumor and adjacent non-involved lung samples were obtained from surgical specimens of patients undergoing resection at the Mount Sinai Medical Center (New York, NY). Written informed consent was obtained in accordance with U.S. common rule and the following protocol reviewed and approved by the Institutional Review Board at the Icahn School of Medicine at Mount Sinai, IRB Human Subjects Electronic Research Applications 10–00472 and 10–00135. Additional FFPE NSCLC samples were obtained from Institut Mutualiste Montsouris, Paris, in collaboration with Institut Curie, Paris. Collection of clinical NSCLC specimens at Institut Mutualiste Montsouris was conducted under the umbrella protocol of the pathological department and biospecimen core facility, established under the reference EUdract 2017-A03081–52 and approved by the Ethics Committee CPP SUD-EST I. FFPE blocks from NSCLC tumors used in (7) were obtained from Assistance Publique – Hôpitaux de Paris (Hôtel Dieu hospital), with the approval of the IRB CPP Ile de France II, 2008–133 and 2012 06–12, No. 2018 MS1.
Tissue processing
The non-involved lung and tumor tissue were weighed and cut into sections of 0.1–0.2 grams then placed into 5 mL microtube (Argos Technologies). Sections were minced with scissors and enzymatically digested in CO2-independent media (Fisher Scientific, 18045088) with Collagenase IV 0.25mg/ml (Sigma-Aldrich, C5138–1G), Collagenase D 200 U/ml (Sigma-Aldrich, 11088882001), and DNAse 0.1 mg/ml (Sigma-Aldrich, DN25–1G) for 40 minutes at 37°C under 80 rpm agitation. Cell suspensions were passed through a syringe with an 18-gauge needle 8–10 times, filtered through a 70µm cell strainer, then lysed in red blood cell (RBC) buffer (Fisher Scientific, NC9067514). The cells were resuspended in buffer comprising of DPBS (Corning, D8537–6X500ML) with 5% BSA (Equitech-Bio, BAH62–0500) and 1mM EDTA (Sigma-Aldrich, 46–034-CI) then counted using hemocytometer and Trypan blue (Fisher Scientific, MT25900CI).
Flow cytometry sorting
Cells were stained for EpCAM (Biolegend, clone 9C4), CD45 (Biolegend, clone HI30), CD29 (Biolegend, clone TS2/16), PDPN (Biolegend, clone NC-08), and LiveDead blue fluorescent dye (Thermo Scientific, L34963) for 30 minutes at 4°C. Among live cells, EpCAM and CD45 were used to remove epithelial and immune cells respectively, while CD29, present on all stromal cells, was used to enrich for cells with intact surface markers (see fig. S1B). The 1.5ml collection tubes (Fisher Scientific, 05–408-129) were coated with 10% BSA to improve cell survival post sorting.
Single cell RNA sequencing
For each sample, up to an estimated 5,000 cells were loaded directly from the flow cytometry sort onto 10X Chromium chemistry kits. Kit versions for each sample are indicated in Table S1. Processing downstream of cell loading was performed by the Human Immune Monitoring Core at Icahn School of Medicine at Mount Sinai. Libraries were prepared according to manufacturer instructions and QC of the cDNA and final libraries was performed using CyberGreen qPCR library quantification assay. Sequencing was performed on Illumina sequencers to a depth of at least 80 million reads per library.
Sequencing data analysis and unsupervised batch-aware clustering
Transcriptomic library reads were aligned to the GRCh38 reference genome and quantified using Cell Ranger (v3.1.0).
Stromal cells isolated from tumor and adjacent lung samples were analyzed using an unsupervised batch-aware clustering method we have recently described (33). First, stromal cells were filtered for cell barcodes recording > 800 UMI, with < 25% mitochondrial gene expression, and with less than defined thresholds of expression for genes associated with red blood cells, epithelial cells, macrophages, T cells and plasma cells (Table S17). 13 tumor and 11 adjacent samples were clustered jointly. This EM-like algorithm iteratively updates both cluster assignments and sample-wise noise estimates until it converges, using a multinomial mixture model capturing the transcriptional profiles of the different cell-states and sample specific fractions of background noise. We ran the algorithm described in Martin et al., 2019 (33) with minor modifications: Training and test set sizes per sample were 7500 and 2500 respectively. The best clustering initiation was selected from 1000 instead of 10000 k-means+ runs. For this clustering we included barcodes with more than 800 UMIs and used Kreg_ds = 0.2; (P1,P2) = (0th,50th) percentiles; Kreg = 5∙10−6; k=28. Genes with high variability between patients across were not used in the clustering. Those genes consisted of mitochondrial, stress, metallothionein genes, immunoglobulin variable chain genes, HLA class I and II genes and 3 specific genes with variable/noisy expression: MALAT1, JCHAIN and XIST (Table S17). Ribosomal genes were excluded only from the k-means clustering (step 2.D as described in Martin et al., 2019 (33)) (Table S17).
Cell annotation
Using the gene module analysis described earlier, we identified highly variable genes and explored their expression across different clusters. Clusters were annotated by comparing gene expression patterns with profiles reported in prior literature.
For stromal cell clusters, EC expressed multiple identifying markers like PECAM1, VWF, CLDN5, and EMCN and lymphatics could be identified with TFF3, LYVE1, and PROX1. PvC were identified by combination of subset specific markers for and shared expression of contractile genes like ACTA2, TAGLN, MYL9 and TPM2. PvC subset specific genes included RGS5, COX4I2 and HIGD1B for pericytes and DES and ACTG2 for SM. Identifying fibroblast markers included those listed in the main text, PDGFRA, SPON1, and MMP2, but also DCN, FBLN1, LUM, COL1A2, RARRES2, CTGF.
A cluster (#13) with contaminating epithelial cells was identified by the high expression of multiple keratin genes including KRT17 and KRT19. Contaminating macrophages were identified in cluster 6 by expression of CD45 (PTPRC), C1QB, C1QA, C1QC, and MARCO. Cluster 18 and 16 were excluded due to high mitochondrial gene content and hemoglobin genes, respectively. The annotation process for fibroblast subsets is described in the text.
Histological staining
Multiplexed IHC was performed according to the protocol developed by (31) with some modifications. Slides were baked at 37°C overnight, deparaffinized in xylene, then rehydrated. Antigen retrieval was done in citrate buffer (pH 6 or 9) (Dako, S2367 or 2369) at 95°C for 30 minutes, followed by incubation in 3% hydrogen peroxide for 15 minutes, then blocked using serum-free protein block solution (Dako, X0909) before adding primary antibody for 1 hour at room temperature or overnight at 4°C. The primary antibody was detected using a secondary antibody conjugated to horseradish peroxidase followed by chromogenic revelation using 3-amino-9-ethylcarbazole (AEC) (Vector laboratories, SK4200). Slides were counterstained with hematoxylin (Sigma-Aldrich, HHS32–1L) and mounted with a glycerol-based mounting medium (Dako, C0563). Then the same slides were bleached and re-stained as previously described. Antibodies sources can be found in Table S5. Masson’s trichrome staining was performed by the Biorepository and Pathology core at the Icahn School of Medicine at Mount Sinai.
Mass Cytometry by Time Of Flight (CyTOF)
Samples were processed to a single cell suspension according to the tissue processing protocol listed earlier. Cell viability staining was achieved with Rh103 staining for 20 minutes at 37°C followed by staining with the CyTOF antibodies listed in Table S5. Acquisition of the samples was performed by the Human Immune Monitoring Center at Mount Sinai. All analysis of the CyTOF samples, including the creation of viSNE plots, was done using the Cytobank platform (https://www.cytobank.org/).
ECM and immunomodulatory gene expression profiles
Gene lists were sourced from (67) and (68) for ECM and immunomodulatory genes, respectively. Selected genes had to meet a mean expression threshold of 1 UMI per 2000 UMIs in 2% of cells in at least 1 cluster and meet a minimum 3-fold expression change between at least two clusters. Selected genes for display were further refined by qualitative analysis.
Gene module analysis
Gene correlation modules were generated using a similar method as previously described in (33). Briefly, cells are downsampled to 2000 total UMIs and highly variable genes are isolated. A gene-gene correlation matrix for the isolated gene set is computed for each sample over the cell population(s) of interest and correlation matrices are averaged following a Fisher Z-transformation. Applying the inverse transformation then results in the best-estimate correlation coefficients of gene-gene interactions across the dataset. Genes are clustered into modules using complete linkage hierarchical clustering over correlation distance. Ribosomal, mitochondrial, HLA and immunoglobulin genes were removed from the analysis prior to creation of gene modules as these genes were not of interest in this study, reflected patient genomic variability or were heavily influenced by contaminating plasma cells.
Acquisition of TCGA dataset and histological subtypes
The TCGA lung adenocarcinoma (LUAD) RNAseq data was downloaded using the GDCquery and GDCdownload functions from the TCGAbiolinks R package. GDCquery options included project=”TCGA-LUAD”, data.category=”Transcriptome Profiling”, data.type=”Gene Expression Quantification”, workflow.type=”HTSeq – FPKM”, experimental.strategy=”RNA-Seq”, and legacy=F. Whole exome sequencing data was downloaded using the GDCquery_Maf function with arguments tumor=”TCGA-LUAD” and pipelines=”mutect2”. Clinical data was downloaded using the GDCquery_clinic function with arguments project=”TCGA-LUAD” and type=”clinical”.
The dominant histological subtype for each TCGA tumor was sourced from (46).
CAF gene signatures and LCAM in bulk analysis
Our initial gene list was acquired from gene module analysis, as described above. To define cell type specific gene signatures, we first excluded genes well expressed in non-fibroblast lineages, such as EC and PvC within our dataset. Next, we utilized the (32) and (23) datasets to exclude genes found in epithelial and immune cells. For each dataset we performed in house clustering and identified stromal clusters, then excluded any genes from our signatures if they showed higher expression in the non-stromal clusters. We then compared the expression of the genes between fibroblast subsets and only kept genes if they showed high expression in the cluster of interest relative to other clusters. Due to their similarity, FAP+ CAF and FAP+αSMA+ CAF were treated as one group and due to ADH1B+ CAF similarity to adjacent fibroblast clusters they were not contrasted with alv. fib. and PI16+ fib. Signatures were further refined by manually checking that expression was consistently enriched in the cell type of interest across at least 3 patients.
Bulk RNA samples were scored in the following method; mitochondrial genes, hemoglobins, and Ig variants genes were removed from the data tables. Next genes were converted to a percent expression of the total reads and regularized with a constant value, 1e-8, added to each gene and log transformed. Z scores were then calculated for each gene across all samples. Finally, the overall cell type score, is calculated by taking the average of all genes within the signature. A complete list containing the signature and removed genes can be found in Table S17.
The derivation of LCAMhigh or LCAMlow scores is described in (32). In short, the cell types associated with each state were averaged to create an LCAMhigh or LCAMlow score. The difference between LCAMhigh and LCAMlow was the final LCAM score. All signatures are calculated using only tumor samples, with sample ID ending in ‘−01A’, and signatures were z-scored before graphing or other analysis. TCGA patients with their corresponding signatures scores can be found in Table S7.
External scRNAseq dataset analysis
Based on our scRNAseq data we defined separate gene signatures for; pan-fibroblast cells, perivascular cells, alveolar fib, mesothelial fib., MYH11+αSMA+ CAF, ADH1B+ CAF, FAP+ CAF and FAP+αSMA+ CAF (Table S17). Fibroblasts were isolated by selecting those above our pan-fibroblast score threshold and below the perivascular cell score threshold (Figure S6A). Then, alv. fib., meso. fib., and MYH11+αSMA+ CAF were identified and removed from subsequent plots (Figure S6A). The remaining cells were then compared against the ADH1B+, FAP+, FAP+αSMA+ CAF gene signatures (Figure S6B-D). The datasets can be found at the following locations; Lambrechts et al., 2018 (23) – ArrayExpress under accessions E-MTAB-6149 and E-MTAB-6653, Wu et al., 2021 (26) – Gene Expression Omnibus database accession code GSE13190712, GSE99254, Kim et al., 2020 (24) – Gene Expression Omnibus database accession code GSE131907, Laughney et al., 2020 (25) - Gene Expression Omnibus database accession code GSE123904.
Histology analysis and overlays
All histology analysis was performed using the open-source image analysis QuPath software (QuPath-0.2.3, https://qupath.github.io/)(69) and ImageJ/Fiji(70,71).
To create overlayed images, scans exported from QuPath as OME.TIFF then imported into ImageJ using the BioFormats plugin(72). Alignment was done using the “Linear Stack Alignment with SIFT” plugin(73). The AEC and hematoxylin stains were extracted from individual scans using “colour deconvolution” and colored as desired.
Quantifying T cell infiltration and inter-patient CAF heterogeneity
Cropped scans were imported into a newly created project in QuPath and were aligned using the “interactive image alignment” plugin. Alignment information was saved using QuPath_script_1. Tumor and stroma regions were defined by applying the “create cytokine annotation” function on the keratin scans. The stroma and tumor annotations were transferred onto the aligned CD3, CD8, αSMA, ADH1B, and FAP scans with QuPath_script_2. On the CD3 and CD8 scans, positive cell detection was used to count the CD3+ and CD8+ cells. We manually removed debris spots (which appear positive for any marker) to avoid false positives. Distance to the tumor and stroma annotations was calculated using the “distance to annotation 2D” option and the measurements were exported as raw data to be analyzed in R. For figure 4E the stroma and tumor annotations were tiled using QuPath function “Create Tiles” and αSMA, ADH1B, and FAP scans the positively stained area were calculated using the QuPath training classifier.
Quantifying T cell infiltration and intra-patient CAF heterogeneity
For the intra-patient analysis, we created a separate QuPath project with all the desired scans to analyze. The images were cropped and exported and then overlays were generated using an ImageJ script with the following steps:
Deconvolution (hematoxylin, AEC, residual)
Alignment on hematoxylin images
Creation of transformation matrix then application on AEC images
Threshold to remove background and then ”Stack of Images”
The composite image was transferred back to QuPath for further analysis. Adjacent tissue regions on slides were excluded from the analysis and in the region to analyze we used the “train pixel classifier” to annotate Tumor nests versus Stroma; while αSMA, FAP and ADH1B areas were annotated using “QuPath train classifier”. Dedicated script automated the tiling and quantification and resulted in 3 data files; cell information including staining intensity and distances to annotations, tile annotation parameters, and annotation measurements such as ADH1B stained area within an annotation. The resulting measurements were exported and analyzed in R.
Quantifying MYH11+αSMA+ CAF boundary enrichment
MYH11+αSMA+ CAF score (Figures 5C, 5D) approximates the enrichment of these cells at the tumor nest boundary (<10μm) relative to their distal background density (20–30μm). Iterating over tiles, we counted the number of MYH11+αSMA+ double positive cells in each distance bin of 1μm. The proximal value was defined as the quantile, 0.75, of the number of cells in the bins within distance <10μm. The distal value was similarly defined as the quantile, 0.75, of the number of cells in the bins with distances between 20–30μm. The MYH11+αSMA+ CAF score was defined as log2(proximal/distal). The 0.75 percentile was selected to maximize the sensitivity of detecting robust high-density regions.
Quantifying FAP+αSMA+ CAF correlation with collagens IV, XI and XII
After the images were cropped and exported, overlays were generated using an ImageJ script as described above. The composite image was transferred to QuPath software for further analysis. The train pixel classifier was used to annotate Tumor nests versus Stroma. In the stromal annotation, the Train pixel classifier was used to annotate FAP+ αSMA-, αSMA+, collagen IV+, collagen XI+ and collagen XII+ regions. Dedicated script automated the tiling and quantification and resulted in a data file containing per tile annotation measurements for each marker. The resulting measurements were exported and analyzed in R.
Quantifying FAP+αSMA+ CAF tumor islet coverage
Each tumor IHC image was divided into 1500µm tiles using QuPath, in each tile the fraction of tumor nest surface covered by FAP+αSMA+ CAF was estimated. Then, using all tiles the average FAP+αSMA+ tumor islet coverage was calculated for each patient.
Supplementary Material
SIGNIFICANCE.
The cellular and molecular programs driving T cell marginalization in solid tumors remain unclear. Here, we describe two CAF populations associated with T cell exclusion in human lung tumors. We demonstrate the importance of pairing molecular and spatial analysis of the tumor microenvironment, a prerequisite to develop new strategies targeting T cell-excluding CAF.
ACKNOWLEDGMENTS
The authors thank the important contributions of patients who participated in this study. This project was supported by Genentech, Inc. and carried out in collaboration with the Fondation ARC pour la recherche sur le cancer. The computational work was supported by the Scientific Computing at the Icahn School of Medicine at Mount Sinai and the Office of Research Infrastructure of the National Institutes of Health under award number S10OD026880. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This manuscript was edited at Life Science Editors and the cartoon illustrations were created using BioRender.com. We thank the Mount Sinai Flow Cytometry core, Human Immune Monitoring Center and the Biorepository and Pathology core for their support. We thank Sarah Lagha and Anne-Sophie Tedesco, Agathe Seguin-Givelet and Isabelle Sauret for their contribution to obtain and study additional FFPE NSCLC samples from Institut Mutualiste Montsouris. We thank Eliane Piaggio, Ana-Maria Lennon-Duménil, and Olivier Lantz for carefully reading and commenting the manuscript.
Footnotes
DECLARATION OF INTERESTS
Research support for this work was provided in part by Genentech, Inc. The authors declare no other competing financial interests.
Data availability
Sequencing data is available on GEO at GSE183219. Code is available on our Github website: https://github.com/effiken/Grout_et_al. Raw histology data are available at https://scdissector.org/grout/.
REFERENCES
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–86. [DOI] [PubMed] [Google Scholar]
- 2.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. New Engl J Medicine 2015;373:1627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019;18:197–218. [DOI] [PubMed] [Google Scholar]
- 4.Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature 2017;541:321–30. [DOI] [PubMed] [Google Scholar]
- 5.Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean M-C, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest 2012;122:899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol 2009;9:618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bajénoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, et al. Stromal Cell Networks Regulate Lymphocyte Entry, Migration, and Territoriality in Lymph Nodes. Immunity 2006;25:989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, et al. The Conduit System Transports Soluble Antigens from the Afferent Lymph to Resident Dendritic Cells in the T Cell Area of the Lymph Node. Immunity 2005;22:19–29. [DOI] [PubMed] [Google Scholar]
- 11.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein–α. Science 2010;330:827–30. [DOI] [PubMed] [Google Scholar]
- 12.Ziani L, Chouaib S, Thiery J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front Immunol 2018;9:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, et al. Single-cell analysis reveals diverse stromal subsets associated with immune evasion in triple-negative breast cancer. Biorxiv 2020;2020.06.04.135327.
- 14.Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Reports 2020;31:107701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res 2020;80:1846–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc National Acad Sci 2013;110:20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofheinz R-D, al-Batran S-E, Hartmann F, Hartung G, Jäger D, Renner C, et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer. Oncol Res Treat 2003;26:44–8. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol 2021;1–13. [DOI] [PMC free article] [PubMed]
- 20.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Medicine 2017;214:579–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov 2020;10:1330–51. [DOI] [PubMed] [Google Scholar]
- 22.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 2019;9:1102–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018;24:1277–89. [DOI] [PubMed] [Google Scholar]
- 24.Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 2020;11:2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée V-P, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 2020;26:259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu F, Fan J, He Y, Xiong A, Yu J, Li Y, et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun 2021;12:2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z, Zhou L, Liu L, Hou Y, Xiong M, Yang Y, et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat Commun 2020;11:5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 2020;10:232–53. [DOI] [PubMed] [Google Scholar]
- 29.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017;171:1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021;39:866–882.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Remark R, Merghoub T, Grabe N, Litjens G, Damotte D, Wolchok JD, et al. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci Immunol 2016;1:aaf6925–aaf6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021;39:1594–1609.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019;178:1493–1508.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillich A, Zhang F, Farmer CG, Travaglini KJ, Tan SY, Gu M, et al. Capillary cell-type specialization in the alveolus. Nature 2020;586:785–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corselli M, Chen C-W, Sun B, Yap S, Rubin JP, Péault B. The Tunica Adventitia of Human Arteries and Veins As a Source of Mesenchymal Stem Cells. Stem Cells Dev 2012;21:1299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020;587:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, et al. Cross-tissue organization of the fibroblast lineage. Nature 2021;593:575–9. [DOI] [PubMed] [Google Scholar]
- 38.Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer‐associated fibroblasts and their markers. Int J Cancer 2020;146:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker C, Rutten F, Yuan X, Pass H, Mew DM, Everitt J. Wilms’ tumor suppressor gene expression in rat and human mesothelioma. Cancer Res 1994;54:3101–6. [PubMed] [Google Scholar]
- 40.Fletcher AL, Acton SE, Knoblich K. Lymph node fibroblastic reticular cells in health and disease. Nat Rev Immunol 2015;15:350–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerdidani D, Aerakis E, Verrou K-M, Angelidis I, Douka K, Maniou M-A, et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J Exp Med 2022;219:e20210815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang VW. Collagen, stiffness, and adhesion: the evolutionary basis of vertebrate mechanobiology. Mol Biol Cell 2020;31:1823–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haruyama N, Sreenath TL, Suzuki S, Yao X, Wang Z, Wang Y, et al. Genetic evidence for key roles of decorin and biglycan in dentin mineralization. Matrix Biol 2009;28:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park J, Ivey MJ, Deana Y, Riggsbee KL, Sörensen E, Schwabl V, et al. The Tcf21 lineage constitutes the lung lipofibroblast population. Am J Physiol-lung C 2019;316:L872–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mecham RP. Elastin in lung development and disease pathogenesis. Matrix Biol 2018;73:6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warth A, Muley T, Meister M, Stenzinger A, Thomas M, Schirmacher P, et al. The Novel Histologic International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society Classification System of Lung Adenocarcinoma Is a Stage-Independent Predictor of Survival. J Clin Oncol 2012;30:1438–46. [DOI] [PubMed] [Google Scholar]
- 48.Fletcher AL, Turley SJ. Who am I? (re‐)Defining fibroblast identity and immunological function in the age of bioinformatics. Immunol Rev 2021;302:5–9. [DOI] [PubMed] [Google Scholar]
- 49.Chihara T, Suzu S, Hassan R, Chutiwitoonchai N, Hiyoshi M, Motoyoshi K, et al. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ 2010;17:1917–27. [DOI] [PubMed] [Google Scholar]
- 50.Menzies-Gow A, Ying S, Sabroe I, Stubbs VL, Soler D, Williams TJ, et al. Eotaxin (CCL11) and Eotaxin-2 (CCL24) Induce Recruitment of Eosinophils, Basophils, Neutrophils, and Macrophages As Well As Features of Early- and Late-Phase Allergic Reactions Following Cutaneous Injection in Human Atopic and Nonatopic Volunteers. J Immunol 2002;169:2712–8. [DOI] [PubMed] [Google Scholar]
- 51.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 1990;347:669–71. [DOI] [PubMed] [Google Scholar]
- 52.Taub D, Conlon K, Lloyd A, Oppenheim J, Kelvin D. Preferential migration of activated CD4+ and CD8+ T cells in response to MIP-1 alpha and MIP-1 beta. Science 1993;260:355–8. [DOI] [PubMed] [Google Scholar]
- 53.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell–dendritic cell interaction. Nature 2006;440:890–5. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y-J, Soumelis V, Watanabe N, Ito T, Wang Y-H, Malefyt R de W, et al. TSLP: An Epithelial Cell Cytokine that Regulates T Cell Differentiation by Conditioning Dendritic Cell Maturation. Annu Rev Immunol 2007;25:193–219. [DOI] [PubMed] [Google Scholar]
- 55.Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005;8:369–80. [DOI] [PubMed] [Google Scholar]
- 56.Hegde PS, Karanikas V, Evers S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res 2016;22:1865–74. [DOI] [PubMed] [Google Scholar]
- 57.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554:538–43. [DOI] [PubMed] [Google Scholar]
- 58.Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol Res 2015;3:1082–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 60.Chakravarthy A, Khan L, Bensler NP, Bose P, Carvalho DDD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun 2018;9:4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 2015;21:524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sekiguchi R, Yamada KM. Chapter Four Basement Membranes in Development and Disease. Curr Top Dev Biol 2018;130:143–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalluri R Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer 2003;3:422–33. [DOI] [PubMed] [Google Scholar]
- 64.Vázquez-Villa F, García-Ocaña M, Galván JA, García-Martínez J, García-Pravia C, Menéndez-Rodríguez P, et al. COL11A1/(pro)collagen 11A1 expression is a remarkable biomarker of human invasive carcinoma-associated stromal cells and carcinoma progression. Tumor Biol 2015;36:2213–22. [DOI] [PubMed] [Google Scholar]
- 65.Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387:1540–50. [DOI] [PubMed] [Google Scholar]
- 66.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020;20:174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife 2014;3:e01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Kloppman E, Lizio M, et al. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat Commun 2015;6:7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep-uk 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Linkert M, Rueden CT, Allan C, Burel J-M, Moore W, Patterson A, et al. Metadata matters: access to image data in the real world. J Cell Biology 2010;189:777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lowe DG. Distinctive Image Features from Scale-Invariant Keypoints. Int J Comput Vision 2004;60:91–110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data is available on GEO at GSE183219. Code is available on our Github website: https://github.com/effiken/Grout_et_al. Raw histology data are available at https://scdissector.org/grout/.