

Abstract
Purpose:
Clinical relevance thresholds and laboratory methods are poorly defined for MET amplification, a targetable biomarker across malignancies.
Patients and Methods:
The utility of next-generation sequencing (NGS) in assessing MET copy number (CN) alterations was determined in >50,000 solid tumors. Using fluorescent in situ hybridization (FISH) as reference, we validated and optimized NGS analysis.
Results:
Incorporating read depth and focality analyses achieved 91% concordance, 97% sensitivity, and 89% specificity. Tumor heterogeneity, neoplastic cell proportions, and genomic focality affected MET amplification assessment. NGS methodology showed superiority in capturing overall amplification status in heterogeneous tumors and defining amplification focality among other genomic alterations. MET copy gains and amplifications were found in 408 samples across 23 malignancies. Total MET CN inversely correlated with amplified segment size. High-level/focal amplification was enriched in certain genomic subgroups and associated with targeted therapy response.
Conclusions:
Leveraging our integrated bioinformatic approach, targeted therapy benefit was observed across diverse MET amplification contexts.
Keywords: MET, amplification, FISH, next-generation sequencing
INTRODUCTION
MET proto-oncogene activation is implicated in the generation and progression of a variety of tumors, either as a primary driver or as a co-driver in the setting of acquired resistance to EGFR tyrosine kinase inhibitors (TKIs)(1). Located on the long arm of chromosome 7 (7q31), the MET gene encodes the tyrosine kinase receptor for hepatocyte growth factor known as HGFR or, more commonly, c-MET. Ligand binding results in receptor homodimerization and activation of mitogen activated protein kinase (MAPK) and other downstream pathways which regulate cellular functions, including cell proliferation, survival, migration, motility, invasion, angiogenesis, and epithelial-to-mesenchymal transition. While c-MET function is tightly regulated in its normal physiologic state, a diverse set of pathologic alterations may cause dysregulation and lead to oncogenesis. Mutations, rearrangements, gene amplification, and protein overexpression have functional implications that are biologically complex and highly variable. Different mutations may act through distinct mechanisms and result in variable transforming activities. Similarly, the degree of amplification and protein expression varies across tumors. Responses in clinical trials testing novel and selective c-MET inhibitors mirror this biological heterogeneity, further fueling efforts to better define MET as a predictive biomarker with robust therapeutic response (2).
Somatic alterations leading to c-MET-dependence are reported across several tumor types, including lung cancer (1), gastroesophageal cancer (3), and renal cell carcinoma (4). However, the most clinically recognized alterations are in non-small cell lung cancer (NSCLC). In this setting, two partially overlapping MET-altered states show promise in their targetability: MET exon 14 (MET ex14) skipping alterations and MET gene amplification. Juxtamembrane domain mutations disrupting the splice-sites flanking MET ex14 eliminate the exon and generate a constitutively active, degradation-resistant receptor (2). These alterations occur in 3–4% of NSCLC cases and are mutually exclusive with other lung cancer drivers (5–7). Dramatic and durable responses to c-MET inhibitors in clinical trials provided the evidence for the recent accelerated approval of capmatinib (8) and tepotinib (9) by the United States Food and Drug Administration (FDA) for patients with these alterations. Of note, a subset of tumors with MET ex14 skipping (~20%) also harbor MET copy number gains (CNGs)(10,11). It remains unclear if these tumors with MET ex14 skipping and MET amplification are driven solely by the ex14 skipping mutation or if the concurrent MET amplification increases the oncogenic potency of MET ex14 skipping alteration. Co-occurring MET amplification is also an acquired event in the context of EGFR TKI resistance (12–15); newer EGFR TKIs in combination with c-MET inhibitors has been explored in this setting, aiming to overcome c-MET bypass activation (15–18). Unfortunately, results have been inconsistent and partially confounded by variations surrounding the definition of MET amplification in preselection criteria. Finally, de novo MET amplification, in the absence of other oncogenic drivers, also has a low incidence in lung cancer, with few studies exploring its targetability. Preliminary data suggest that the strongest predictor of a true c-MET-driven tumor is high-level amplification (1,19). Crizotinib (20–22) and capmatinib (11) have clinical activity in patients with high-level MET amplification, with or without concurrent MET ex14. While neither drug has FDA approval for this indication, MET amplification has been designated as an emerging biomarker in the National Comprehensive Cancer Network guidelines (23).
There is currently no clear consensus about the optimal methodology or cutoff to define MET amplification clinically. Historically, MET copy number status has been determined using fluorescence in situ hybridization (FISH), and the incidence of CNG/amplification ranges from 0.7% to 21% (24,25), depending on the technique and cutoff. Early studies (19) defined amplification (high MET gene copy number) as a mean of ≥ 5 MET copies/cell. Alternatively, amplification has been measured as a ratio relative to a chromosomal enumeration probe (CEP7), providing distinction between focal MET amplification versus polysomy (extra copies of chromosome 7), the latter of which commonly occurs in NSCLC. Clinically, this distinction is relevant as restricted amplification is more likely to represent a true MET driver rather than a bystander event (1,26,27). Different cutoffs of MET:CEP7 ratios have been explored across studies to define amplification, including ratios of ≥ 1.8, ≥2.0 and >2.2, in combination with mean MET/cell ≥4 or 5 copies (19,22,24,28–30). More recently, a schema stratifying tumors into categories of low (ratio ≥1.8 to ≤2.2), intermediate (>2.2 to <5), or high (≥5) was used, showing that ratios ≥5 predict the most dramatic responses to crizotinib (22). However, while high amplifications are rare, the definition of a cutoff with the greatest predictive utility remains largely undefined.
With increased adoption of comprehensive next-generation sequencing (NGS) for routine assessment of patients with lung cancer and other malignancies, the capability to concurrently derive copy number alteration (CNA) data from these assays provides a cost-effective and less labor-intensive alternative to single-gene MET FISH assessment. While MET copy number assessment by NGS has been explored (31), there is a lack of defined assessment criteria and direct comparisons to FISH. Here, we explore the feasibility of assessing MET amplification by NGS in routine clinical practice. We correlate our data with concurrent FISH testing and examine the benefits and pitfalls of both. We further explore the overall landscape of genetic alterations identified in patients and review response data of c-MET targeted therapy.
METHODS
Case Selection.
After approval by the Memorial Sloan Kettering Cancer Center (New York, NY) institutional review board, molecular pathology service records from 2014 through 2021 were retrospectively reviewed. Three patient cohorts were examined in this study: landscape, FISH, and clinical cohorts.
For the MET amplification landscape cohort, 50,748 patients who had MSK-IMPACT NGS at our institution were queried for the presence of MET CNGs, with the goal to select cases with MET amplification, using our previously published criteria for the clinical assessment of HER2 amplification (32). Briefly, cases with MET CNGs with fold-change (fc) ≥1.5 and p<0.05 were selected, including those with focal gains and broad arm/chromosome-level gains. Although an fc of 2.0 was previously established as our cutoff for gene amplifications, 1.5 was used in the first exploration to account for potential sensitivity loss in low purity tumor samples. MET CNGs were further analyzed using Fraction and Allele-Specific Copy Number Estimates from the Tumor Sequencing (FACETS)(33) to assess tumor purity-adjusted MET integer copy number as previously described and detailed below. To focus on the molecular and clinical correlates of MET amplification, landscape cohort was further refined to include only those with MET amplification as described below. (34)
For the FISH cohort, NGS cases were retrospectively identified that either (1) also had FISH testing for MET as part of the clinical workup, (2) were part of the clinical validation for detection of CNA by NGS, including MET, or (3) had residual patient tissue remaining after NGS clinical testing that could be used for FISH analysis. This cohort included cases with no MET gains and cases with variable degrees of amplification to explore the correlation between NGS and FISH data across a broad range of MET copy states.
The treatment cohort consisted of patients who received c-MET targeted therapy, either alone or in conjunction with other therapies. Clinical characteristics, outcomes, and responses were examined (Supplementary Fig. 1).
NGS and copy number determination.
MSK-IMPACT is a hybridization-capture based NGS assay that interrogates all coding regions of 341–505 genes (35). The assay also captures >1,000 intergenic and intronic single-nucleotide polymorphisms (tiling probes), interspersed homogenously across the genome, aiding in the accurate assessment of genome-wide copy number changes. The probes target approximately 1.2 megabses (Mbs) of the human genome. Tumor testing was performed concurrently with patient-specific normal control (blood) to enable accurate determination of somatic variants and more comprehensive copy number analysis. Furthermore, to ensure sensitive detection of kinase fusions and alternative splicing variants, RNA-based fusion testing via Archer was performed in all treatment naïve tumors lacking mitogenic tumors on MSK-IMPACT, post-treatment tumors lacking a resistance mechanism on MSK-IMPACT, and any cases with kinase structural variants of unknown significance (36).
Two independent methods were used to determine copy number: read-depth and allele-specific copy number. The read-depth approach determines copy number based on the read-depth of each target region normalized to GC content and compared to normal control as previously described (32). This method assumes that increases or decreases in read counts of a particular region are proportional to CNGs or losses. To eliminate sample type-dependent coverage differences, we used formalin-fixed paraffin-embedded (FFPE) normal samples captured and sequenced with MSK-IMPACT. P-values were calculated to determine significant deviance from the null distribution and thereby provide statistical confidence to CNA reporting. Copy number changes are expressed as a fold change (fc) and interpreted as amplification (fc ≥2.0), CNG/borderline amplification (fc ≥1.5 but <2), or deletion (fc ≤−2.0). Changes with p-values <0.05 were considered significant. Cases with borderline amplification and low tumor content were flagged and reflexed to FISH analysis for further assessment, if material was available.
The allele-specific copy number approach uses the FACETS algorithm (33). This analysis employs matched patient normal control for comparison to determine CNGs, deletions, allele-specific amplifications, and copy neutral loss of heterozygosity. In contrast to the read-depth method which only uses read counts, FACETS includes single-nucleotide polymorphisms to provide allelic information. This combined approach increases the sensitivity and precision for detecting copy number aberrations, especially in low purity samples. Moreover, FACETS assesses tumor purity and ploidy, enabling integer copy number determination. In brief, integer copy number calls from FACETS were derived from modeling the expected values of total copy number log ratio (logR) and allele-specific log-odds-ratio data (logOR). Cases were classified as MET gains (copy numbers of 3–5) or MET amplifications (copy numbers ≥6), based on established criteria (33).
The FACETS algorithm uses segmentation analysis to determine the boundaries of the region of the genome exhibiting a copy number change. Using this information, we determined the length of the amplified region of chromosome 7 associated with the MET amplification and thereby determined whether amplification was focal or part of broad arm-level or chromosomal gains. Focality assessment was performed using length of the amplified region both as a continuous variable and as a binary threshold of 25 Mbs, one quarter the length of the long arm of chromosome 7.
FISH.
FISH analysis was performed on FFPE tissue sections (4 μm thickness) with tumor areas marked following standard protocols. A MET (7q31.2) FISH probe of approximately 800 kb was used in combination with a CEP7 (both from ZytoVision, Bremerhaven, Germany). Signal analysis was performed in combination with morphology correlation, and at least 100 interphase cells within the marked tumor area were evaluated and imaged using a Zeiss fluorescence microscope coupled with Metasystems ISIS software (Newton, MA). MET and CEP7 signals were counted and cases were initially classified as either amplified, high CNG, low CNG, or negative, as follows: For calling MET amplification, the ratio of MET:CEP7 must be >2 and average MET signals per cell must be >5, in which case the percentage of amplified cells was calculated. If the MET:CEP7 ratio was <2, the average MET signals per cell were used to determine high CNG (greater than 8 signals/cell), low CNG (4–8 signals/cell), or negative (less than 4 signals/cell). For cases which appeared heterogeneous, multiple areas across the tissue were examined, and the area with the highest mean MET copy number was used for stratification and heterogeneity was noted. Images of selected areas were captured for documentation.
Limit of Detection Analysis.
Serial dilutions of a formalin-fixed paraffin-embedded (FFPE) patient tumor sample were prepared by mixing the tumor DNA with appropriate amounts of the same patient’s normal control DNA derived from WBCs that were formalin-fixed and paraffin-embedded. The following dilutions were made: 100%, 50%, 25%, 12.5%, 6.25%, and 3.125%. These DNA mixtures were then sequenced to determine MET copy number using both read-depth and allele specific copy number (FACETS) methods.
Clinical outcomes.
This study was approved by the MSK Privacy Board. We reviewed pharmacy records of patients with NSCLC identified in the landscape cohort treated with one or more c-MET inhibitors from January 2014 through June 2021. Five patients with other tumor types were also treated with c-MET inhibitors during this time, for a total of 45 patients. Basic demographic information, tumor characteristics, treatment history, and radiographic and clinical response were collected from treated patients (clinical cohort). Patients with available baseline and at least one follow-up imaging study were evaluated by a radiologist (A.P.) according to RECIST v1.1 criteria. Overall response rate (ORR) was defined as the proportion of patients with a complete response (CR) or partial response (PR) as their best response to therapy; response confirmation by a second scan was not required. Time to treatment discontinuation (TTD) was defined as date of treatment initiation to date of treatment discontinuation. Patients were censored at their last known clinical follow-up if they continued treatment past the data lock of June 1, 2021.
Data availability.
The validation data generated in this study are publicly available in cBioPortal (https://www.cbioportal.org/study/summary?id=mixed_impact_subset_2022). Other data from the remaining cohort is available upon request to the corresponding author.
RESULTS
A total of 66,285 tumor samples from 50,748 patients of various histological subtypes underwent NGS by MSK-IMPACT; a standard clinical pipeline analysis identified 408 cases with MET CNGs or amplification (Supplementary Fig. 1). Prior to analysis of this larger cohort, we first concentrated on identifying a more focused diagnostic validation cohort to refine the existing MET amplification calling strategy of the standard pipeline using expanded bioinformatics analyses and FISH as the reference method.
For the validation cohort, 70 cases with sufficient material for MET FISH testing were selected: 48 with MET CNGs of various levels based on NGS and 22 without MET CNAs (Supplementary Table 1a and Supplementary Table 2). The cohort comprised 16 unique primary cancer types. The most common tumors were lung carcinoma (NSCLC and small cell lung cancer [SCLC]; 59%), followed by gastrointestinal (gastric, colorectal, biliary, pancreatic, and hepatocellular; 13%) carcinomas, renal cell carcinomas (7%), and glioblastoma (7%). NGS and FISH result comparisons were performed to establish concordance and further define criteria for calling amplification by NGS. NGS was evaluated for (a) its binary ability to determine the presence or absence of MET amplification and (b) its ability to quantitate MET copy number.
Binary MET amplification calling.
We sequentially tested three methods for determining MET amplification status in a binary (present or absent) fashion: (1) the standard read-depth method, (2) additional FACETS algorithm analysis, and (3) additional FACETS and focality analysis. The latter two were investigated to determine if the sensitivity and specificity could be improved. FISH analysis was considered the gold standard for comparison.
The first and simplest approach was to use a read-depth methodology, the current standard for calling amplification by MSK-IMPACT (Fig. 1a). MET amplification by NGS was defined as a MET fc ≥2.0 (p<0.05). This approach, however, showed low FISH concordance (81% concordance, 81% sensitivity, 82% specificity). This was primarily related to under detection in cases with relatively low tumor content and overcalling in the setting of polysomy, arm-level CNAs, and fragmented genomes (Supplementary Table 1b). Lowering the fc threshold did not significantly improve concordance (A threshold of fc ≥1.7 had 83% concordance, 94% sensitivity, and 74% specificity; a threshold of fc ≥1.5 had 80% concordance, 94% sensitivity, and 68% specificity).
Figure 1.
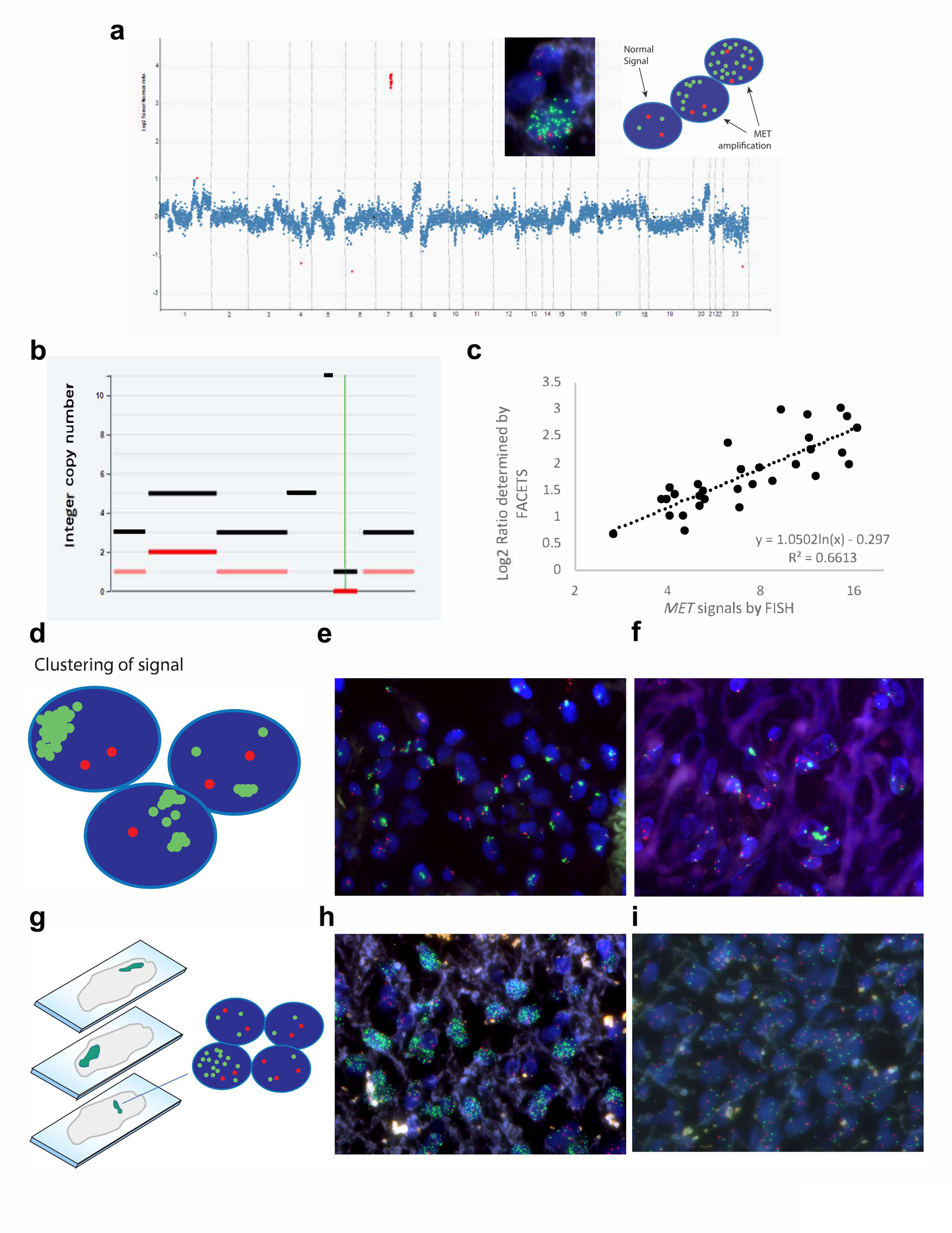
Comparison of MET amplification detection by fluorescence in situ hybridization (FISH) versus next generation sequencing. a Example copy number plot of a MET amplified case. The MET gene copy number, highlighted in red, is significantly higher than the rest of the copy number alterations, suggesting a biologically relevant focal amplification is driving oncogenesis. Insets show a representative FISH image and diagram demonstrating MET amplification. The normal FISH signal is two MET signals (green) and two CEP7 signals (red). When amplification of MET occurs, many green signals are observed, with an overall MET/CEP7 ratio ≥ 2, and some cells having more than 20 green signals. b Representative FACETS output demonstrating MET amplification at an integer copy number of eleven. c While there is good concordance when determining the binary call of whether MET is amplified, some discrepancy is observed with quantitative determination of MET copy number. When the signals are dispersed and evenly distributed, the signals can be accurately counted to give an assessment of MET copy number. Sometimes, however, difficulties arise. d-f MET signals can cluster, making it difficult to get an accurate count, especially when the number of signals per cell is greater than 20, and this can lead to an underestimation of the true MET copy number. g-i Other times, tumor heterogeneity can affect the count. While contiguous cells in a defined area are assessed, the areas selected for counting can be enriched for MET amplification, overestimating the true MET copy number. Both photographs are from the same tumor section,demonstrating one area with marked amplification (h), and another area without amplification (i).
The second approach additionally applied the FACETS algorithm to determine allele-specific copy number (Fig. 1b). FACETS was more sensitive for calling amplification than the read-depth method but exhibited a higher false positive rate (86% concordance, 100% sensitive and 74% specific, Supplementary Table 1c). Manual review of the copy number data suggested that a proportion of discordant calls were related to arm-level or whole chromosome 7 gains (polysomy) rather than MET specific amplification.
The third and most comprehensive approach introduced a focality criterion (<25 Mb threshold) to refine the algorithm. In conjunction with FACETS, this further improved agreement: 91% concordance, 97% sensitivity and 89% specificity (Supplementary Table 1d). Six discordant cases were encountered, all six cases had de novo low-level MET amplification called by FISH or NGS in the context of high tumor content (Supplementary Fig. 2). Five cases (NGS+/FISH−) exhibited low-level MET CNGs that met criteria for amplification by FACETS but were in a background of genomic fragmentation and variable partial gains and losses within regions of chromosome 7. This may have interfered with the accurate classification of the MET alteration with reference to the centromeric probe by FISH, or FACETS may overcall focal amplification due to the complexity of the overall genomic gains and losses. One other case (NGS−/FISH+) did not meet criteria for amplification because the amplified region exceeded our focality criteria (31 Mb).
Finally, FISH analysis exposed a complexity in MET amplified cases that highlights how accurate amplification assessment via FISH can be challenging, particularly in the setting of amplification acquired as a resistance mechanism to TKI therapy. MET signals displayed high heterogeneity across different regions of a tumor, predisposing to counting selection that favored nuclei with multiple signals and relying on narrow regions (50–100 selected nuclei in a single section; Fig. 1). Additionally, clustering and overlapping commonly precluded counting of individual signals, leading to under- or overestimation of amplification.
Quantitative MET amplification calling.
While concordance between number of MET signals by FISH and log2 ratio of total copy number divided by ploidy determined by FACETS was strong (R2=0.66), several cases deviated from the trendline (Fig. 1c). Deviations were primarily related to two main observations: (1) tight clustering of MET signals per cell often precluded accurate signal counting by FISH, particularly in samples with higher levels of CNG (Fig. 1d–f) and (2) high heterogeneity of signal distribution in which amplified cells clustered unevenly in different regions of a tumor (Fig. 1g–i). Using FACETS total copy number instead of the log2 ratio or MET:CEP7 ratios by FISH instead of total MET signals did not improve concordance (R2=0.47 and R2=0.42 respectively, Supplementary Fig. 3).
To further understand the analytical sensitivity of NGS, a limit of detection (LOD) analysis was performed (Supplemental Table 3) to determine tumor content requirements for accurate copy number assessment. Sequential dilutions of a tumor sample (~30% tumor content at baseline without dilution, with FISH demonstrating a MET copy number of 16.49 and a MET:CEP7 ratio of 8.36) were sequenced, and we compare the LOD of FACETS to read-depth methodology. MET amplification was detected using FACETS in the undiluted sample (integer copy number 11) and at 50% dilution (consistent with ~15% tumor content, integer copy number 12). Further dilutions did not show MET amplification, likely because FACETS can no longer accurately calculate tumor content below 20%. If using the threshold of MET fc ≥2.0 (p<0.05) as described above, read depth methodology was only able to call amplification in the undiluted sample, although increased fold change was also significant in the 50% dilution. Even though FACETS was able to call MET amplification at ~15% tumor content), its tumor percentage calculation becomes unreliable below 20%, and therefore we recommend 20% as a conservative limit of minimum tumor content for reliable copy number reporting.
Lastly, we explored the reliability of FACETS copy calls by estimating confidence intervals around the integer copy numbers. FACETS uses median log-ratio for each segment to estimate the integer copy number. The reliability of the estimate is a function of the number of single nucleotide polymorphisms and the variability of the log-ratio within that segment. Using these data from each sample, we can calculate a confidence interval around the estimate to provide some measure of reliability. To demonstrate this, we calculated estimates of confidence intervals for three cases with approximately 100, 50, and 20 copy numbers. For P-0003424-T01-IM5, the integer copy number for the segment that harbors MET is 105 with a 95% confidence interval (99, 117). For P-0048436-T01-IM6, the integer copy number is 49 with a 95% confidence interval (46, 51). For P-0000772-T01-IM3, the integer copy number is 20 with a 95% confidence interval (17, 22). These intervals clearly show that the confidence for these amplification calls is high.
MET landscape cohort analysis.
Having refined the criteria for MET amplification, the 408 cases with MET copy gains/amplification by standard NGS analysis were reanalyzed. Using FACETS, 50 (12%) had a MET copy number between 3–5 (MET gain) whereas 358 (88%) had a MET copy number ≥6 (MET amplification).
Since tumor purity can affect copy number analysis, we compared tumor purity as assessed by histologic review between those with MET gain and MET amplification. This was to ensure that MET gain was not due to low tumor purity in the case of occult amplification and that MET-amplified tumors were not enriched in cases with high tumor content. The proportion of cases with low tumor content did not differ between tumors with MET gain and MET amplification (13% vs. 20%, p=0.3, Fisher’s Exact Test, respectively), nor did median tumor purity (50% vs. 40%, p=0.09, Mann-Whitney U Test, respectively).
De novo and emergent MET amplification.
Focusing on tumors with MET amplification using the criteria above, we analyzed MET amplification frequency across tumor types (Fig. 2a). Notably, MET amplification was found most frequently in papillary renal cell carcinoma (8.4%), high-grade glioma (5.3%), and gastroesophageal junction adenocarcinoma (5.0%). MET amplification was also recurrently seen in NSCLC (1.9%) and was enriched in pleomorphic and sarcomatoid morphologies (8.5%; Fig. 2b).
Figure 2.
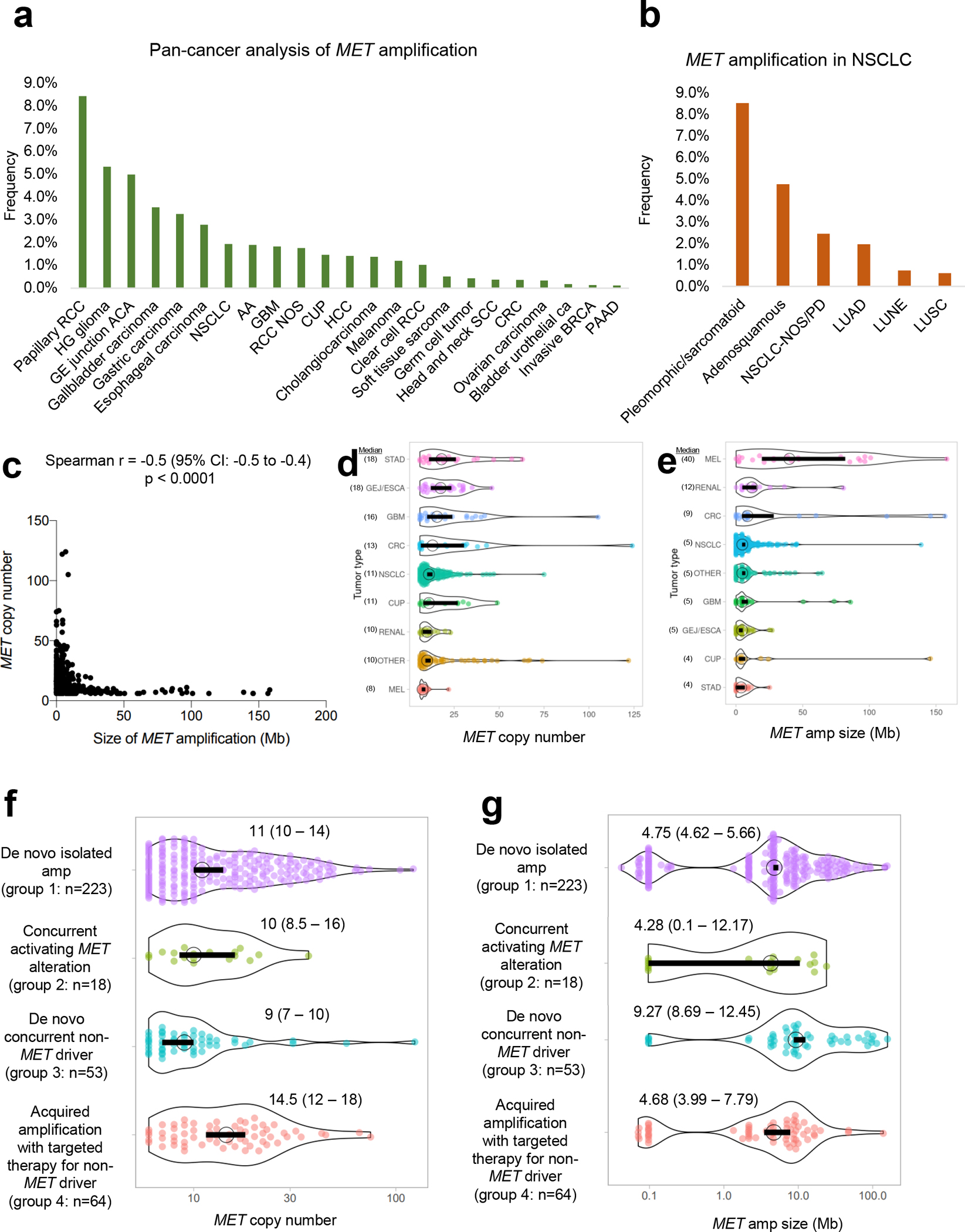
A: Frequency of MET amplification across solid tumors. RCC=renal cell carcinoma,HG=high grade, GE=gastroesophageal, NSCLC=non-small cell carcinoma,AA=anaplastic astrocytoma, GBM=glioblastoma multiforme, RCC=renal cellcarcinoma, NOS=not otherwise specified, CUP=carcinoma of unknown primary,HCC=hepatocellular carcinoma,SCC=squamous cell carcinoma, CRC=colorectal carcinoma, BRCA=breast carcinoma, PAAD=pancreatic adenocarcinoma. B: Frequency of MET amplification in NSCLC. NOS/PD=not otherwise specified, poorly differentiated, LUAD=lung adenocarcinoma, LUNE=large cell neuroendocrine carcinoma, LUSC=lung squamous cell carcinoma. C: Correlation of size of MET amplification vs. MET copy number. D and E: Landscape of MET copy number and MET amplification size across tumor types, respectively. MEL=melanoma, GEJ/ESCA=GE junction adenocarcinoma/esophageal adenocarcinoma, STAD=stomach adenocarcinoma. F andG: Distribution of MET copy number and MET amplification size according to four clinicopathologic groups. Median and 95% confidence intervals are provided.
Among NSCLCs, we separated MET amplification into four clinicopathologic cohorts: group 1, targeted therapy-naïve cases without established mitogenic drivers (“de novo isolated amplification”); group 2, cases with concurrent MET ex14 alterations or MET kinase fusions (“concurrent activating MET alteration”); group 3, targeted therapy-naïve cases with concurrent mitogenic non-MET drivers (“de novo concurrent non-MET driver”); group 4, mitogenic driver-positive tumors with acquired resistance to targeted therapy (“acquired amplification with targeted therapy for non-MET driver”). MET amplification was found de novo in 60% (n=84/140) of cases, with 51/84 (61%) cases occurring without another driver (group 1). A concurrent MET ex14 alteration was found in 18% (n=15/84) of cases and a concurrent MET fusion was found in 1% (n=1/84) of cases (group 2). The remaining 20% (n=17/84) fell into group 3 categorization with the following non-MET drivers: EGFR (n=7), KRAS (6), ERBB2 (n=2), ALK (n=1) and RET (n=1). Finally, 56/140 (40%) cases with MET amplification were associated with resistance to targeted therapy (group 4). Concurrent non-MET drivers included EGFR (n=50/56, 89%), ALK (n=3/56, 5%), RET (n=2/56, 4%) and KRAS G12C mutation (n=1/56, 2%).
In contrast, among non-lung cancers, MET amplification was found de novo in most cases (96%, n=210/218), with resistance to targeted therapy for a non-MET driver in the remaining minority (4%, n=8/218). In the first group, isolated MET amplification was found in 82% (n=172/210) of cases. A concurrent MET fusion was found in 1% (n=2/210). The remaining 17% (n=36/210) had a concurrent non-MET driver: ERBB2 amplification (n=10), KRAS mutations (9), BRAF mutations (7), FGFR2 fusions (2), NRAS mutations (2), BRAF fusions (2), EGFR mutation (1), FGFR3 mutation (1), RET fusion (1), and FGFR1 fusion (1). In the group with resistance to targeted therapy, concurrent non-MET drivers were BRAF mutations (n=3/8, 38%), ERBB2 amplification (n=3/8, 38%), FGFR2 fusion (n=1/8, 13%), and NTRK1 fusion (n=1/8, 13%).
Copy number versus focality.
MET amplification focality correlated with the degree of amplification and genomic focality (Fig. 2c). Cases with high amplification were more likely to have focal MET amplification; those with low amplification were associated with concurrent amplification of wider genomic regions extending beyond MET (Spearman rho= −0.5, 95%CI −0.5 to −0.4, p<0.0001, Fig. 2c). There was broad distribution in MET copy number and the size of the genomic region concurrently amplified for various tumor types, with gastric adenocarcinomas having the highest copy number and most focal MET amplification (Figs. 2d–e).
To further assess the dynamics of MET copy number and focality, the MET landscape cohort was subdivided into the four clinicopathologic cohorts described above (Fig. 3): group 1, n=223 (62%); group 2, n=18 (5%); group 3, n=53 (15%); group 4, n=64 (18%). Samples with CNGs were not included. MET copy number varied significantly amongst the different groups (p=0.002, Kruskal-Wallis Test). Groups 1 and 4 had higher MET copy number compared to group 3 (p=0.005 and p=0.002, respectively, Dunn’s Multiple Comparisons Test; Fig. 2f). Similarly, MET amplification also varied significantly among the different groups (p=0.0006 Kruskal-Wallis Test), with groups 1, 2, and 4 showing more focal MET amplification than group 3 (p=0.002, p=0.03, and p=0.002, respectively, Dunn’s Multiple Comparisons Test; Fig. 2g).
Figure 3.
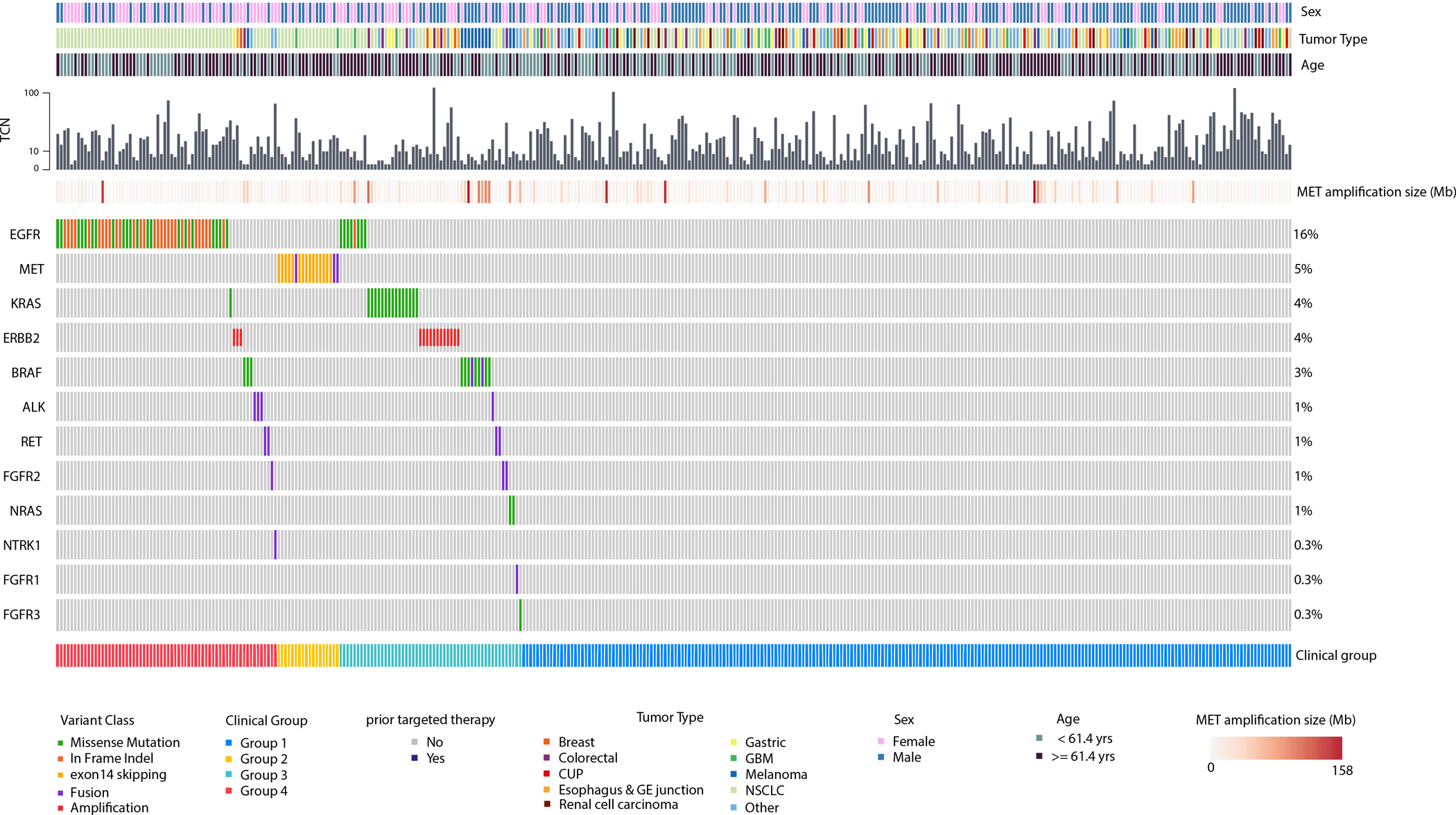
Oncoprint of MET amplified tumors. Group 1=de novo isolated MET amplification;Group 2=concurrent activating MET alteration; Group 3=de novo concurrent non-MET driver;Group 4=acquired amplification with targeted therapy for non-MET driver;TCN=total copynumber; CUP=cancer of unknown primary;GE=gastroesophageal;GBM=glioblastomamultiforme; NSCLC=non-small cell lung cancer; yrs=years; Mb=megabase.
Taken together, the findings suggest that high-level and focal MET amplification may be necessary to serve as a sole mitogenic driver (group 1) and mediate targeted therapy resistance (group 4). In contrast, de novo tumors with other oncogenic dependencies (group 3) may not require a similarly high level of amplification and focality, thus resulting in MET amplification with relatively lower copy number and broader gains.
Clinical response to c-MET inhibition.
Of the 408 patients in the landscape cohort, 45 patients received one or more c-MET inhibitors. Clinicopathologic features are summarized in Supplementary Table 4. Lung cancer (n=40, NSCLC) was the most common histology, followed by renal cell carcinoma (n=3), gastric adenocarcinoma (n=1), and intrahepatic cholangiocarcinoma (n=1). Crizotinib was most frequently used (n=22), followed by an investigational agent (n=13), cabozantinib (n=8), capmatinib (n=1), or tepotinib (n=1; Supplementary Table 5). In 27 cases, the c-MET inhibitor was combined with a non-c-MET TKI to address acquired resistance: osimertinib (n=24), selitrectinib (n=1), selpercatinib (n=1), and afatinib (n=1).
The best response to a c-MET inhibitor is shown in Figs. 4a–b for patients with measurable disease; those who received single-agent versus combination c-MET TKI therapy and concurrent non-MET drivers are indicated. The ORR was 25% (95% CI 3–71%, n=1/4) for isolated MET amplification (group 1), 67% (95% CI 39–86%, n=8/12) for concurrent MET ex 14 alteration (group 2, none had concurrent MET fusions), 25% (95% CI 3–71%, n=1/4) for de novo MET amplification and concurrent non-MET driver (group 3), and 44% (95% CI 25–66%, n=8/18) for acquired MET amplification after targeted therapy for non-MET driver (group 4). The median TTD for each group was 7.8, 8.3, 4.3 and 6.1 months, respectively (Fig. 4c). The duration of c-MET inhibition compared to other systemic therapies is shown per patient in Supplementary Fig. 4. In summary, clinical benefit appeared to be most enriched in patients with concurrent MET amplification and MET ex14 alteration. Response rates appeared more attenuated in de novo isolated MET amplification and MET amplification in the presence of a non-MET driver (either in the de novo or acquired resistance setting).
Figure 4.
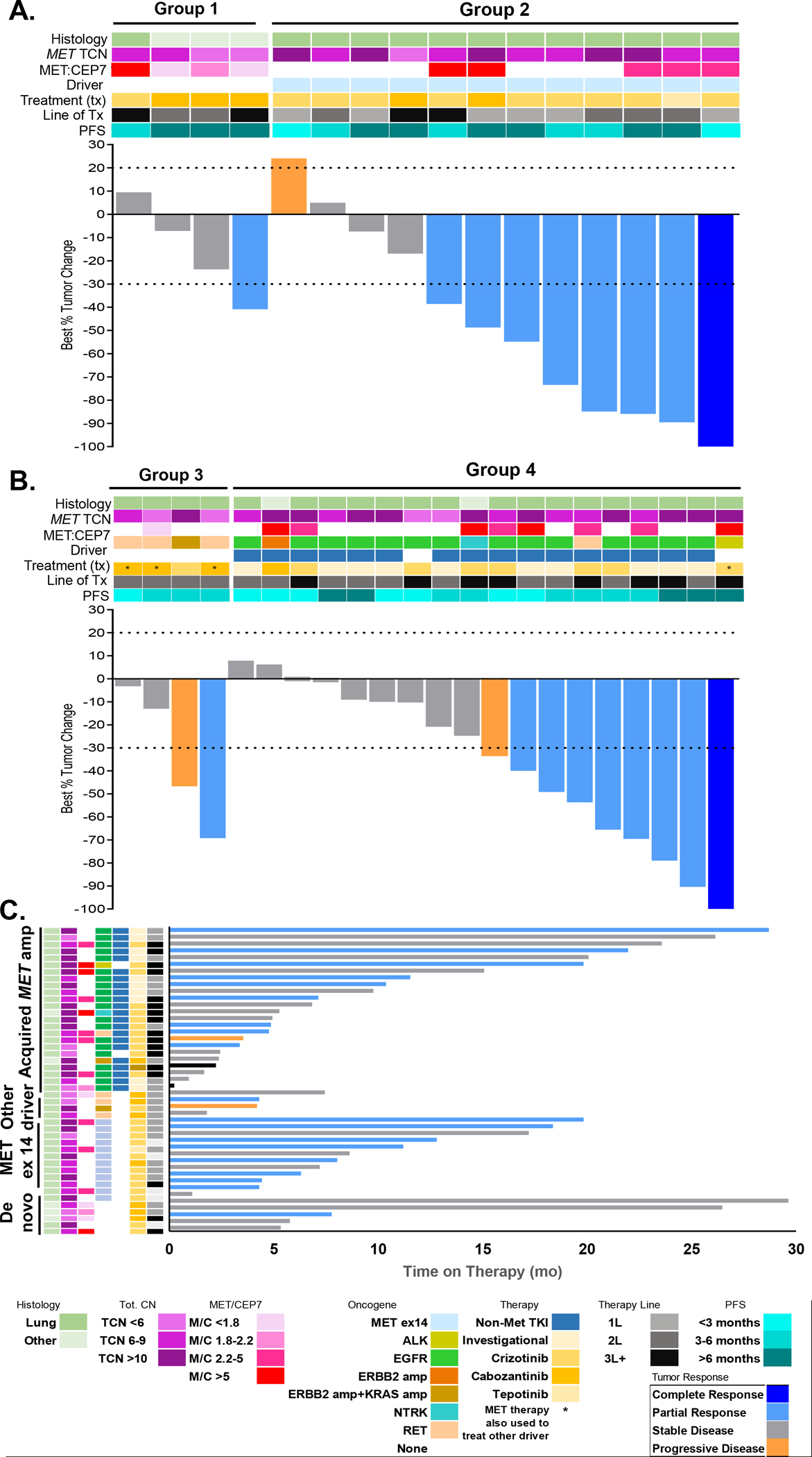
Clinical outcomes on MET-directed therapies. A and B: Waterfall plots demonstrating objective responses for RECIST-evaluable patients subgrouped into de novo isolated MET amplification (n=4; group 1), concurrent activating MET alterations (n=12), de novo with other concurrent driver (n=4), or MET amplification acquired due to resistance (n=18). If patients were treated with multiple sequential MET inhibitors, best response is shown. Dashed gray line indicates RECIST thresholds for progressive disease and partial response. C: Swimmers plot showing time on therapy in months. Tot. CN, total copy number; PFS, progression-free survival.
We then examined whether clinical benefit differences might be mediated by MET amplification level. MET copy number was stratified by <6 (n=7), 6–9 (n=15) and ≥10 copies (n=16). The ORR to c-MET inhibitors in these cohorts was 29% (95% CI 37–71%), 53% (95% CI 35–68%), and 50% (95% CI 28–72%), respectively; response rates appeared numerically higher in patients with 6 or more copies of MET. The ORR in these three cohorts is broken down into groups in Supplementary Table 6. Interestingly, all but one patient in this cohort met criteria for focal MET amplification (despite this not being a requirement for inclusion). The patient with broad MET amplification had concurrent MET ex 14 alteration and had a partial response to therapy. Furthermore, primary progression in any MET-amplified cancer regardless of amplification level was low at 5%.
DISCUSSION
MET amplification has become an increasingly relevant and targetable biomarker, both as a de novo driver and in the setting of acquired TKI resistance (37). While FISH has been historically considered the gold standard for assessment of CNG or gene amplifications, we exposed technical challenges with FISH assessment in this series. MET signal distribution was highly variable and signals clustered or overlapped, predisposing to counting selection and signal over- or underestimation, particularly when MET amplification was an acquired TKI resistance mechanism. Moreover, FISH uses one locus (the centromeric region) as the sole determinant for copy number enumeration without information on arm-level and broad genomic changes which may be critical for clinical interpretation. Importantly, the focal nature of MET gene amplification cannot be discerned by FISH.
By contrast, data from NGS-based approaches represent an average of a much larger number of cells from several levels (i.e. 250 ng of DNA represents ~38,000 diploid nuclei), allowing a more comprehensive assessment of MET status along with the ability to discern the focal nature of MET amplification, the landscape of gains and losses across chromosome 7 and other chromosomes. Interrogation of a larger number of tumor cells is important given the diversity of MET amplification inclusive states such as the heterogenous and potentially sub-clonal nature of MET amplification in acquired TKI resistance. This comprehensive analysis is contingent on assay design with adequate regional tiling and controls for tumor purity. Furthermore, with the increased need to identify several other actionable genetic alterations across cancer types, diagnostic algorithms have shifted in many contexts to incorporate all assessments into single comprehensive NGS assays (31). While many NGS-based assays can be validated to provide copy number analysis, there are important differences amongst these assays that can potentially influence the performance of copy number calling. In our study, we show that a large, hybrid-capture-based NGS panel enables a comprehensive characterization of MET copy number states. Future studies are needed to explore the performance of MET copy number analysis using other NGS assays, particularly those with amplicon-based panels that require less input.
Unfortunately, there are no established clinical cutoffs that define MET amplification by NGS. There are also no established guidelines for assessment or reporting of somatic CNAs by NGS as they exist for clinically relevant somatic sequence variants (38,39). Similarly, the paucity of FFPE reference standards for specific MET copy levels remains a barrier to standardization across different laboratories. CNA assessment using NGS may vary widely depending on the assay, the type of analysis, sample type, and sequencing quality. In addition, there are several biological phenomena that affect CNG detection using NGS data, including tumor content, allele copy number, and the presence of aneuploidy. We thus focused on ways to standardize algorithms for amplification detection that could be incorporated into updated guidelines.
Our validation of MET gene amplification by NGS mirrored methods used for HER2 (32). However, modifications had to be incorporated to account for the higher biologic complexity. First, addition of FACETS analysis more accurately determined the allele specific integer copy number corrected for tumor purity. This enabled higher sensitivity that was particularly useful in samples with low tumor content or in resistance samples exhibiting subclonal MET amplification. Second, a separate analysis of the genomic footprint of the amplified region in chromosome 7 was instituted to further assess MET amplification focality. In terms of the binary determination of MET amplified vs. non-amplified, our optimized NGS method showed high concordance with FISH, with discrepancies primarily associated in borderline categories. It is important to note, however, that the definition of focality assessment is not well defined. While we used 25 Mb here, consistent with one-quarter of the length of the chromosomal arm, additional rigorous studies are needed to clinically define focal amplification not only for MET amplification in NSCLC, but also for other genes and clinical contexts.
An important caveat is that for cases with low tumor content, quantitation of MET copy number and detection of amplification by NGS is less reliable, highlighting that, in selected cases, a combination of methods may represent the best approach. NGS can be limited in detecting CNG in specimens with low neoplastic cell percentages as we demonstrated in our LOD study. We specifically note that there are intrinsic technical limitations of our LOD study, in that the sample utilized had a high baseline amplification, raising the concerns for overestimation of our LOD across dilutions. However, through extensive studies of amplification for other markers such as HER2, MYC, and EGFR, which are also validated in our lab, the cutoff of 20% tumor also holds for lower levels of amplification allowing, at least, detection of gains that can be further confirmed by an alternate method. Tumor content below 20% with negative or borderline results represents an indication for reflex testing by FISH as an additional level of assessment.
Having established more optimized parameters for NGS assessment, we analyzed our existing data from >50,000 unique tumors sequenced by MSK-IMPACT. MET amplification was seen at an overall rate of approximately 0.7% which is in keeping with previously published data, including AACR (American Association for Cancer Research) GENIE (Genomics Evidence Neoplasia Information Exchange) cases (40). We demonstrate that the amplitude of the MET amplification correlates inversely with the overall size of the amplified region. Additionally, higher and more focal amplification was noted in cases where MET amplification was the only driver or a co-driver in the setting of TKI resistance, suggesting that the dynamics of amplification correlates with biological relevance and has potential treatment implications.
Finally, several observations were made in relation to MET amplification cutoffs as selection criterion for targeted therapy. The prevailing hypothesis was that higher amplification would associate with increased clinical benefit with c-MET inhibition. While we observed a similar trend in our NGS data, this did not meet statistical significance. In addition, durable responses were observed in MET amplified cancers with lower copy numbers by NGS (including those with FISH MET:CEP7 ratios <5 by reference testing), and the rate of primary progression in NGS-detected cases regardless of amplification level was extremely low. Therefore, we suggest that any degree of true MET amplification is reasonable to explore in clinical trials of c-MET targeted therapy when using an optimized NGS algorithm that incorporates expanded FACETS analyses or other analogous segmental analyses that adjust for tumor purity. While amplification focality correlates with the degree of amplification, the added predictive value of amplification focality to c-MET inhibition is unclear and warrants further study. Establishing only high-level amplification as a cutoff may disqualify patients who could benefit from targeted therapy. We continue, however, to recommend stratification by amplification level to prospectively elucidate the role of MET amplification as a quantitative variable as it relates to clinical benefit.
Our study highlights that, with proper validation, MET assessment by NGS is a suitable and potentially more informative approach than FISH in most cases across a variety of solid tumors. Understanding the technical considerations and biologic phenomena that impact performance is important. FISH analyzes only tumor cells but represents a narrower assessment that can be highly biased in the setting of acquired resistance. The overall assessment of MET, neighboring genomic regions, and focality is also limited. In contrast, NGS integrates data across the entire sample, determines integer copy number via a less biased computational method, and can more accurately determine the focal nature of MET amplification provided adequate tumor cellularity. Overall, this maximizes the capture of tumor heterogeneity by virtue of examining more tissue levels and defines the landscape for MET amplification and other genomic regions of interest. Given the high heterogeneity of MET as a biomarker, it is likely that unique cutoffs may be needed depending on the clinical scenario, and a combination of techniques may be required to comprehensively assess challenging cases. Our hope is that large prospective clinical trials further examining the clinical utility of multiple methods of copy number determination may elucidate objective thresholds to guide treatment.
Supplementary Material
Statement of translational relevance.
MET amplification is a targetable oncogenic driver in several tumor types, including lung cancer, gastroesophageal cancer, and renal cell carcinoma. Approved therapies exist for patients with MET-amplified tumors; however, the clinical utility of these treatments is limited by ambiguous diagnostic criteria and expensive non-generalizable testing methods that require valuable and limited tumor tissue. With the routine use of next-generation sequencing (NGS), sequencing data is being explored as a potential diagnostic method for evaluating MET amplification. We further refine NGS analysis strategies and compare the gold standard MET amplification detection method of fluorescence in-situ hybridization (FISH) with an NGS-based approach. Our results suggest that NGS methodology shows high sensitivity and specificity for identifying MET amplifications and support the use of this bioinformatic approach as an alternative to FISH in many MET amplification contexts.
Acknowledgements:
This was supported by the National Cancer Institute at the National Institutes of Health Cancer Center Support Grant [grant number P30 CA008748].
Footnotes
Competing interests: J.P.S. reports continuing medical education honoraria from Medscape. N.C. reports royalties from Wolters Kluwer for Pocket Oncology; and honoraria from MJH Life Sciences. M.L. reports consulting or advisory roles for Takeda Oncology, Janssen Pharmaceuticals. M.B. reports personal fees from Roche, grants from Illumina, grants from Grail, outside the submitted work; in addition, M.B. has a patent Systems and Methods for Detecting Cancer Via cfDNA Screening pending. A.D. reports HONORARIA/ADVISORY BOARDS: Ignyta, Genentech, Roche, MORE Health, AXIS, Loxo, Eli Lilly and Company, Bayer, AbbVie, EPG Health, Takeda, Ariad, Millenium, 14ner, Elevation Oncology, Harborside Nexus, TP Therapeutics, ArcherDX, Liberum, AstraZeneca, Monopteros, RV More, Pfizer, Novartis, Ology, Blueprint Medicines, EMD Serono, Amgen, Helsinn, Medendi, TouchIME, BeiGene, Repare RX, Janssen, BergenBio, Nuvalent, Entos, Hengrui Therapeutics, Merus, Treeline Bio, Exelixis, Chugai Pharmaceutical, Prelude, Tyra Biosciences, Remedica Ltd, Applied Pharmaceutical Science, Inc., Verastem, mBrace, and AXIS; ASSOCIATED RESEARCH PAID TO INSTITUTION: Pfizer, Exelixis, GlaxoSmithKlein, Teva, Taiho, PharmaMar; ROYALTIES: Wolters Kluwer; Other (Food/Beverage): Merck, Puma, Merus, Boehringer Ingelheim CME Honoraria: Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice, Axis, Peerview Institute, Paradigm Medical Communications, WebMD, MJH Life Sciences, AXIS, EPG Health, JNCC/Harborside. M.E.A. reports personal fees for ad-hoc board participation from AstraZeneca, Bristol Myers Squibb, Jansen Global services, speaker fees from Biocartis, Invivoscribe, RMEI Medical Education, Physician Education Resources, Peerview Institute for Medical Education, I3 Health Education on Point outside the submitted work. The other authors declare no potential conflicts of interest.
REFERENCES
- 1.Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J Thorac Oncol 2017;12(1):15–26 doi 10.1016/j.jtho.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch JP, Aebersold DM, Zimmer Y, Medová M. MET targeting: time for a rematch. Oncogene 2020;39(14):2845–62 doi 10.1038/s41388-020-1193-8. [DOI] [PubMed] [Google Scholar]
- 3.Lee HE, Kim MA, Lee HS, Jung EJ, Yang HK, Lee BL, et al. MET in gastric carcinomas: comparison between protein expression and gene copy number and impact on clinical outcome. Br J Cancer 2012;107(2):325–33 doi 10.1038/bjc.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schöffski P, Wozniak A, Escudier B, Rutkowski P, Anthoney A, Bauer S, et al. Crizotinib achieves long-lasting disease control in advanced papillary renal-cell carcinoma type 1 patients with MET mutations or amplification. EORTC 90101 CREATE trial. Eur J Cancer 2017;87:147–63 doi 10.1016/j.ejca.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015;5(8):850–9 doi 10.1158/2159-8290.Cd-15-0285. [DOI] [PubMed] [Google Scholar]
- 6.Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov 2017;7(6):596–609 doi 10.1158/2159-8290.Cd-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paik PK, Drilon A, Fan PD, Yu H, Rekhtman N, Ginsberg MS, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5(8):842–9 doi 10.1158/2159-8290.Cd-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Food and Drug Administration. FDA grants accelerated approval to capmatinib for metastatic non-small cell lung cancer. FDA news release, May 6, 2020. [cited June 2, 2020]. Available at https://www.fda.gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-capmatinib-metastatic-non-small-cell-lung-cancer.
- 9.Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC, et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. New England Journal of Medicine 2020;383(10):931–43 doi 10.1056/NEJMoa2004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol 2016;34(7):721–30 doi 10.1200/jco.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- 11.Wolf J, Seto T, Han J-Y, Reguart N, Garon EB, Groen HJM, et al. Capmatinib in MET Exon 14–Mutated or MET-Amplified Non–Small-Cell Lung Cancer. New England Journal of Medicine 2020;383(10):944–57 doi 10.1056/NEJMoa2002787. [DOI] [PubMed] [Google Scholar]
- 12.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316(5827):1039–43 doi 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 13.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007;104(52):20932–7 doi 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010;17(1):77–88 doi 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo R, Luo J, Chang J, Rekhtman N, Arcila M, Drilon A. MET-dependent solid tumours - molecular diagnosis and targeted therapy. Nat Rev Clin Oncol 2020. doi 10.1038/s41571-020-0377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng L, Kiedrowski LA, Ravera E, Cheng H, Halmos B. Response to Dual Crizotinib and Osimertinib Treatment in a Lung Cancer Patient with MET Amplification Detected by Liquid Biopsy Who Acquired Secondary Resistance to EGFR Tyrosine Kinase Inhibition. Journal of Thoracic Oncology 2018;13(9):e169–e72 doi 10.1016/j.jtho.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Zhu VW, Schrock AB, Ali SM, Ou SI. Differential response to a combination of full-dose osimertinib and crizotinib in a patient with EGFR-mutant non-small cell lung cancer and emergent MET amplification. Lung Cancer (Auckl) 2019;10:21–6 doi 10.2147/lctt.S190403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.York ER, Varella-Garcia M, Bang TJ, Aisner DL, Camidge DR. Tolerable and Effective Combination of Full-Dose Crizotinib and Osimertinib Targeting MET Amplification Sequentially Emerging after T790M Positivity in EGFR-Mutant Non-Small Cell Lung Cancer. J Thorac Oncol 2017;12(7):e85–e8 doi 10.1016/j.jtho.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 2009;27(10):1667–74 doi 10.1200/jco.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caparica R, Yen CT, Coudry R, Ou SI, Varella-Garcia M, Camidge DR, et al. Responses to Crizotinib Can Occur in High-Level MET-Amplified Non-Small Cell Lung Cancer Independent of MET Exon 14 Alterations. J Thorac Oncol 2017;12(1):141–4 doi 10.1016/j.jtho.2016.09.116. [DOI] [PubMed] [Google Scholar]
- 21.Ou S-HI, Kwak EL, Siwak-Tapp C, Dy J, Bergethon K, Clark JW, et al. Activity of Crizotinib (PF02341066), a Dual Mesenchymal-Epithelial Transition (MET) and Anaplastic Lymphoma Kinase (ALK) Inhibitor, in a Non-small Cell Lung Cancer Patient with De Novo MET Amplification. Journal of Thoracic Oncology 2011;6(5):942–6 doi 10.1097/JTO.0b013e31821528d3. [DOI] [PubMed] [Google Scholar]
- 22.Camidge DR, Otterson GA, Clark JW, Ignatius Ou SH, Weiss J, Ades S, et al. Crizotinib in Patients With MET-Amplified NSCLC. J Thorac Oncol 2021;16(6):1017–29 doi 10.1016/j.jtho.2021.02.010. [DOI] [PubMed] [Google Scholar]
- 23.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. In NCCN Guidelines. version 3.2022. accessed 6/19/2022. [Google Scholar]
- 24.Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba, II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama 2014;311(19):1998–2006 doi 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol 2008;3(4):331–9 doi 10.1097/JTO.0b013e318168d9d4. [DOI] [PubMed] [Google Scholar]
- 26.Go H, Jeon YK, Park HJ, Sung SW, Seo JW, Chung DH. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J Thorac Oncol 2010;5(3):305–13 doi 10.1097/JTO.0b013e3181ce3d1d. [DOI] [PubMed] [Google Scholar]
- 27.Kawakami H, Okamoto I, Okamoto W, Tanizaki J, Nakagawa K, Nishio K. Targeting MET Amplification as a New Oncogenic Driver. Cancers (Basel) 2014;6(3):1540–52 doi 10.3390/cancers6031540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casadevall D, Gimeno J, Clavé S, Taus Á, Pijuan L, Arumí M, et al. MET expression and copy number heterogeneity in nonsquamous non-small cell lung cancer (nsNSCLC). Oncotarget 2015;6(18):16215–26 doi 10.18632/oncotarget.3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watermann I, Schmitt B, Stellmacher F, Müller J, Gaber R, Kugler C, et al. Improved diagnostics targeting c-MET in non-small cell lung cancer: expression, amplification and activation? Diagn Pathol 2015;10:130 doi 10.1186/s13000-015-0362-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noonan SA, Berry L, Lu X, Gao D, Barón AE, Chesnut P, et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number-Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J Thorac Oncol 2016;11(8):1293–304 doi 10.1016/j.jtho.2016.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng LX, Jie GL, Li AN, Liu SY, Sun H, Zheng MM, et al. MET amplification identified by next-generation sequencing and its clinical relevance for MET inhibitors. Exp Hematol Oncol 2021;10(1):52 doi 10.1186/s40164-021-00245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross DS, Zehir A, Cheng DT, Benayed R, Nafa K, Hechtman JF, et al. Next-Generation Assessment of Human Epidermal Growth Factor Receptor 2 (ERBB2) Amplification Status: Clinical Validation in the Context of a Hybrid Capture-Based, Comprehensive Solid Tumor Genomic Profiling Assay. The Journal of molecular diagnostics : JMD 2017;19(2):244–54 doi 10.1016/j.jmoldx.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44(16):e131 doi 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat Commun 2021;12(1):3770 doi 10.1038/s41467-021-24109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin Cancer Res 2019;25(15):4712–22 doi 10.1158/1078-0432.CCR-19-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo R, Offin M, Brannon AR, Chang J, Chow A, Delasos L, et al. MET Exon 14-altered Lung Cancers and MET Inhibitor Resistance. Clin Cancer Res 2021;27(3):799–806 doi 10.1158/1078-0432.Ccr-20-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tack V, Spans L, Schuuring E, Keppens C, Zwaenepoel K, Pauwels P, et al. Describing the Reportable Range Is Important for Reliable Treatment Decisions: A Multiple Laboratory Study for Molecular Tumor Profiling Using Next-Generation Sequencing. The Journal of molecular diagnostics: JMD 2018;20(6):743–53 doi 10.1016/j.jmoldx.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Deans ZC, Costa JL, Cree I, Dequeker E, Edsjö A, Henderson S, et al. Integration of next-generation sequencing in clinical diagnostic molecular pathology laboratories for analysis of solid tumours; an expert opinion on behalf of IQN Path ASBL. Virchows Archiv : an international journal of pathology 2017;470(1):5–20 doi 10.1007/s00428-016-2025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7(8):818–31 doi 10.1158/2159-8290.Cd-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The validation data generated in this study are publicly available in cBioPortal (https://www.cbioportal.org/study/summary?id=mixed_impact_subset_2022). Other data from the remaining cohort is available upon request to the corresponding author.