

Abstract
Lipidomics studies suffer from analytical and annotation challenges because of the great structural similarity of many of the lipid species. To improve lipid characterization and annotation capabilities beyond those afforded by traditional mass spectrometry (MS)-based methods, multidimensional separation methods such as those integrating liquid chromatography, ion mobility spectrometry, collision-induced dissociation and MS (LC-IMS-CID-MS) may be used. Although LC-IMS-CID-MS and other multidimensional methods offer valuable hydrophobicity, structural and mass information, the files are also complex and difficult to assess. Thus, the development of software tools to rapidly process and facilitate confident lipid annotations is essential. In this Protocol Extension, we use the freely available, vendor-neutral and open-source software Skyline to process and annotate multidimensional lipidomic data. Although Skyline (https://skyline.ms/skyline.url) was established for targeted processing of LC-MS-based proteomics data, it has since been extended such that it can be used to analyze small-molecule data as well as data containing the IMS dimension. This protocol uses Skyline’s recently expanded capabilities, including small-molecule spectral libraries, indexed retention time and ion mobility filtering, and provides a step-by-step description for importing data, predicting retention times, validating lipid annotations, exporting results and editing our manually validated 500+ lipid library. Although the time required to complete the steps outlined here varies on the basis of multiple factors such as dataset size and familiarity with Skyline, this protocol takes ~5.5 h to complete when annotations are rigorously verified for maximum confidence.
Introduction
As interest in lipidomic studies continues to grow because of their linkages with xenobiotic exposures and various diseases, including cardiovascular disease and metabolic disorders, the demand for innovative data analysis and processing techniques has greatly increased1–5. Lipids are a broad class of biomolecules that perform essential roles in various cellular processes including molecular signaling, energy storage and the formation of cellular membranes. On the basis of these functions, lipids have become targets for novel diagnostics and treatments6–8. Although research efforts to characterize and understand the lipidome are expanding, these studies are currently challenged by the many lipid isomers present in complex sample types. Lipid isomers have the same elemental composition but differences in structural arrangements such as variation in head group and fatty acyl tail composition, connectivity (sn-position) and double bond position and orientation. This isomeric problem along with their extensive concentration range in biological and environmental samples often complicates the identification of unique lipid species9,10.
To address the challenges related to assigning unique lipid species to detected features for more comprehensive lipidomic characterization, powerful analytical tools beyond traditional mass spectrometry (MS)-based methods have been used, including derivatization methods11, Paternò-Büchi reactions12, ozonolysis-induced dissociation13,14, electron impact excitation of ions from organics15 and ultraviolet photochemical detection16,17. In addition, multidimensional separation methods such as liquid chromatography (LC), ion mobility spectrometry (IMS), collision-induced dissociation (CID) and MS (LC-IMS-CID-MS) are becoming increasingly common techniques for advanced lipidomic evaluations. The IMS size-based separation enhances selectivity and separations of lipid isomers, in addition to pinpointing different lipid and biomolecular classes to increase confidence in the lipid annotations9,18–22. Furthermore, the collision cross section (CCS) values measured with IMS platforms provide a direct correlation to the ion’s gas-phase size. Thus, multidimensional LC-IMS-CID-MS measurements contain valuable feature-specific information such as lipid hydrophobicity, structure, mass and fragments. However, these datasets are large in both complexity and file size, causing manual feature assignment to be extremely time consuming and feasible for only a small number of targets within small sample sets19. Developing software capable of processing these complex data files, while facilitating accurate feature annotation, is crucial10.
Recently, the freely available and open-source software tool Skyline was adapted to process IMS data from Agilent, Waters, Bruker, SCIEX and Thermo. An evaluation of LC-IMS-CID-MS peptide data demonstrated the capabilities of both IMS and Skyline for enabling enhanced proteomic data collection and analysis. Specifically, Skyline’s IMS filtering function facilitated rapid LC-CID-MS and LC-IMS-CID-MS data comparisons in which the resulting calibration curves showed improved linearity and lower detection limits with the additional IMS separation19. Although Skyline has traditionally been used for proteomic analyses23–25, it has recently been expanded to support small-molecule data processing26. Thus, in this work, we have combined these advancements to use Skyline as a tool for LC-IMS-CID-MS lipidomic data processing and annotation, which has been shown to provide hundreds of confident lipid annotations in complex sample matrices27. This protocol describes the step-by-step procedure for using Skyline and many of its unique features to analyze LC-IMS-CID-MS lipidomic data. Although IMS enhances the selectivity, separation capability and annotation confidence of lipidomic analyses27, untargeted lipidomic analyses using high-resolution MS (HRMS) without the IMS dimension are more common because of the accessibility of LC-HRMS instruments and corresponding data analysis software platforms. However, the steps described are widely applicable to any LC-HRMS/MS lipidomic data, because Skyline disregards the IMS-specific functions when data without this dimension are imported. LC-HRMS/MS lipidomics data from complex matrices are still highly complex, and manual validation of lipid annotations by using all available analytical dimensions (LC, MS1 and tandem mass spectrometry (MS/MS)) is crucial; thus, this protocol is applicable because this is readily achieved by using Skyline.
This LC-IMS-CID-MS lipidomics protocol begins with Skyline setup and data import, followed by indexed retention time (iRT) calibration, annotation validation and library editing and, finally, results report export (Fig. 1). Some familiarity with Skyline is highly recommended, and detailed, hands-on tutorials can be found at https://skyline.ms/tutorials.url. The downloadable lipid library used in this protocol was built from human plasma lipidomic data and manually validated on the basis of stringent criteria (Supplementary Table 5) including mass measurement accuracy, drift time aligned fragments, subclass-specific retention time, lipid presence in multiple samples, CCS database matching and lipid subclass CCS versus mass-to-charge ratio (m/z) trendlines27, which previous studies have noted to be informative for structural annotation of unknowns9,18. Although the library was optimized for human plasma lipidomic analyses, it can be readily applied to any sample type27. Additional sample-specific libraries (e.g., additional human and animal biofluids, as well as plant and animal tissues) will also be shared via Panorama in the future.
Fig. 1 |. Lipidomic LC-IMS-CID-MS protocol overview.
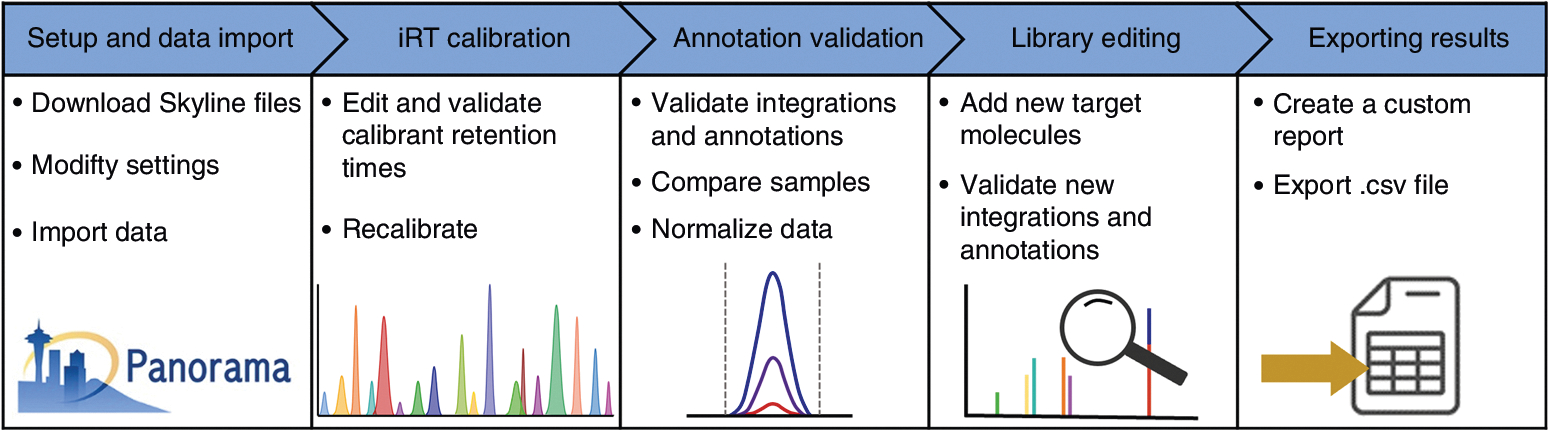
This protocol includes the Skyline setup and data import, iRT calibration, annotation validation, library editing and how to export the results report.
The specific lipid library used in this protocol includes 854 lipid targets and 6,149 transitions (i.e., total precursor and fragment m/z values targeted by Skyline). However, because some lipids are observed in both positive and negative mode and with multiple adducts, the library correlates to 516 unique lipids spanning multiple classes from five of the eight lipid categories as defined by LIPID MAPS28 (Fig. 2). Each lipid in the library has stored information on its class or subclass, common name28, molecular formula, m/z for one or more adducts, fragments and neutral losses, CCS and iRT. A custom small-molecule spectral library is also included and can be viewed directly in Skyline to compare the user-observed lipid MS/MS spectra to the spectra used to generate the library27. Although the library was built from experimental data, the transition lists were primarily generated and modified by using LipidCreator, a free and open-source tool integrated with Skyline and designed to create targeted lipidomics assays but used here to build transition lists29. The fragmentation patterns observed in our spectra were validated via comparisons to literature and in silico spectra when available29–31.
Fig. 2 |. Summary of library lipids.
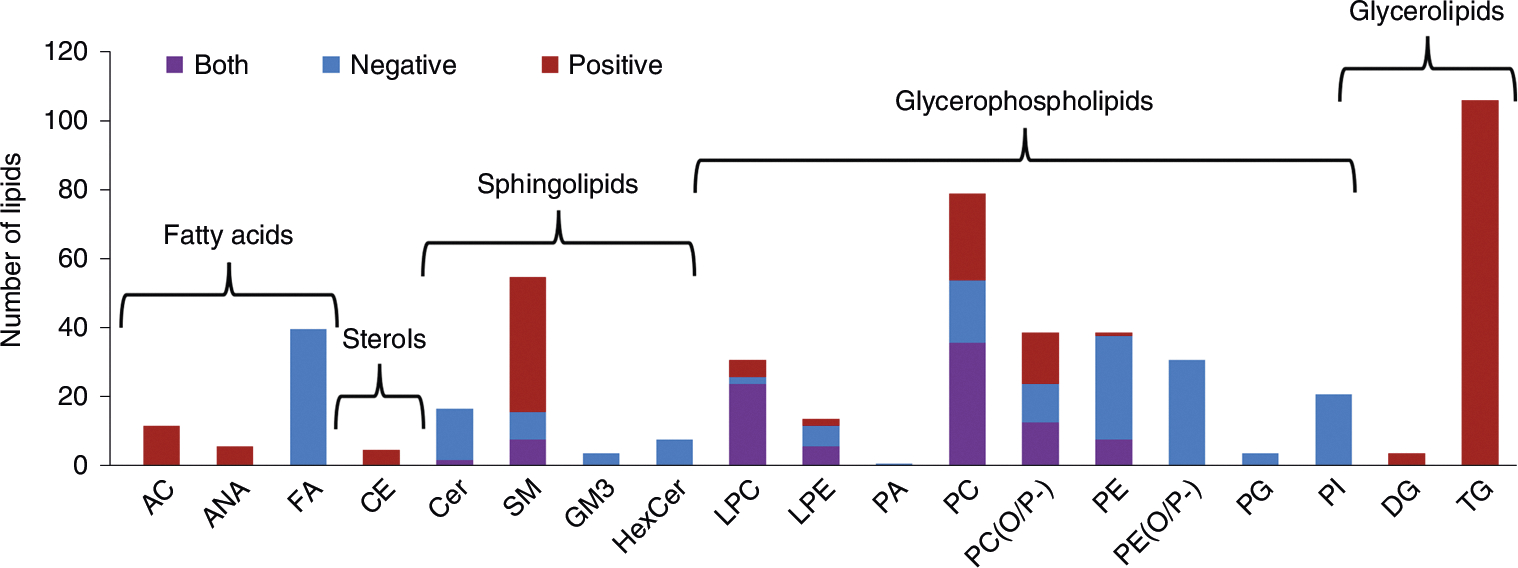
The number of lipids in the library used for this protocol graphed per class or subclass. Lipid categories are also labeled, and the bars are colored on the basis of the ionization mode(s) each lipid was detected in, with positive mode only illustrated with red, negative mode only with blue and both modes with purple. AC, acylcarnitine; ANA, anandamide; CE, cholesterol ester; Cer, ceramide; DG, diacylglycerol; FA, fatty acid; GM3, ganglioside; HexCer, hexose ceramide; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; PA, phosphatidic acid; PC, phosphatidylcholine; PC(O/P-), alkyl/alkenyl ether (plasmalogen) phosphatidylcholine; PE, phosphatidylethanolamine; PE(O/P-), alkyl/alkenyl ether (plasmalogen) phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; SM, sphingomyelin; TG, triacylglycerol.
An additional Skyline feature that we adapted for small molecules and used in this protocol is iRT, which standardizes and predicts molecule retention times to account for run time or retention time shifts27,32. In each ionization mode, we assign a set of 20 endogenous reference lipids that span the LC gradient iRT values between 0 and 100 to calibrate the iRT calculator. Reference lipids were chosen on the basis of their span across the gradient and their presence in multiple sample types. The iRT calculator used in this protocol was previously applied to experimental plasma samples and validated for both the 38-min LC gradient used here and a shortened 14-min gradient, with averages of 2% and 3% difference, respectively, between the predicted and observed retention times27. All remaining lipids in the library are assigned an iRT value on the basis of their retention times relative to these calibrant lipids. In subsequent datasets, the user simply verifies the retention times of the calibrant lipids, and the retention times of the remaining ~800 targets are predicted by using a locally weighted scatterplot smoothing (LOESS) model. Importantly, the default reference lipids can be exchanged with other lipids from the library if they are not present or easily identified in a given dataset, and new internal standards can be added as iRT landmarks, all while retaining the prediction functionality. Finally, lipid CCS values have been calculated within Skyline from experimental data and compared to existing standards, literature or database values when available. All CCS values in our library had observed differences of ≤2% from reference values27,33. Because CCS values are molecular descriptors unique to each ion, these values are stored in the spectral library rather than the variable IMS output values21,22. Skyline then converts the stored CCS values to drift time for drift tube IMS data or other IMS output values, such as inverse reduced mobility (1/K0), depending on the IMS platform and user-defined instrument tolerances19. Prior calibration of data files by using vendor software accounts for differences in drift times (or other IMS output values) due to variable experimental parameters such as temperature24,27. This drift time range then determines the window of signal for chromatogram extraction. This process filters out interferences at both the MS and MS/MS levels and inherently increases the confidence of the lipid annotations, specific examples of which are shown throughout the protocol19,27,33. Although this protocol is not focused on absolute quantification of lipids, it can be easily expanded to do so by using the Skyline small-molecule quantification tutorial (https://skyline.ms/tutorial_small_quant.url) as a reference. Briefly, users are able to define matched and surrogate internal standards, create linear or nonlinear calibration curves, define and calculate figures of merit (i.e., limits of detection and quantitation) and export the concentration of each target. Skyline also supports relative quantification, because analyte peak areas can be normalized to either the total ion current or equalized medians to export relative abundances. Additional examples and screenshots corresponding to important steps of the protocol can be found in Supplementary Figs. 1–17.
Comparison with other methods
Because of the limited number of lipid data–processing software tools that support IMS data, manual assignment of lipids for LC-IMS-CID-MS data is commonly used to generate confident annotations. However, manual analysis is extremely time consuming and feasible for only a small number of lipid targets and samples. When considering other automated software tools, one major distinction of this protocol is that Skyline is free and vendor neutral, compared to commercially available software such as LipidSearch (Thermo Scientific), Lipidyzer (SCIEX), LipidAnnotator (Agilent) and SimLipid (PREMIER Biosoft). Of the many freely available software tools for lipid data processing and annotation, such as Liquid34, MS-DIAL35, LipidIMMS36, LipidMatch Flow37, Lipid Data Analyzer 238 and Greazy/LipidLama39, only LipidIMMS and MS-DIAL are compatible with IMS data8,35,36. Although these tools take database and in silico CCS values into account for feature identification and scoring, ion mobility filtering before feature assignment is desirable to filter out common interferences in the MS and MS/MS dimensions. Although many of these software packages can process and annotate large datasets rapidly by using extensive databases, the lipidomics community has called for more stringent validation of results, higher confidence in the level of lipid speciation reported and sample-specific searches, all of which are covered by this protocol8,10,27. In addition, the Skyline interface allows users to not only validate their results but control the peak integration and annotations as needed to ensure consistent and confident annotations.
Limitations
Skyline is designed as an inherently targeted data analysis platform; therefore, while untargeted data are being analyzed, a target list is necessary, and no feature-finding or other untargeted searches are included in this workflow. Furthermore, only annotated lipids are included in the library, although it is common in metabolomic and lipidomic analyses to report unknown features and/or perform statistical analyses to determine those worth identifying with further studies. The target list is, however, fully editable, and Skyline can be used as a complementary tool with feature finding–based workflows for validation of results. Beginners in lipidomics should refer to additional resources to understand small-molecule data-annotation confidence40–43 and the general limitations of lipidomics research8,10,20,43,44. Here, annotation confidence refers to both the identification level based on the collected evidence40,41 and the validation of the identification, because peak-picking algorithms are flawed, and small-molecule researchers have not yet adopted a false discovery rate to define the reliability of automated annotations42–44. In addition, the protocol will not illustrate all of the Skyline software features that may be useful for lipidomic data processing, such as group comparisons or the use of heavy labeled internal standards for absolute quantitation26. Information and tutorials on how to use additional Skyline software functions can be found at https://skyline.ms/tutorials.url. The MS/MS spectral library in this protocol was also generated from experimental data collected by using CID with ramped collision energy and an all-ions data-independent acquisition scheme (Supplementary Methods). Thus, the fragmentation pattern and fragment ion intensities may differ for spectra collected by using different collision energies, isolation schemes such as data-dependent acquisition or fragmentation methods such as high-energy collision dissociation. This protocol can be applied to any HRMS data; however, annotation confidence may be limited without the LC, IMS and MS/MS dimensions. The iRT calculator built for this protocol was calibrated with a commonly used lipidomics LC method (Supplementary Methods) and reversed-phase C18 column. The retention time prediction performance should not be affected by the gradient time27, but prediction performance may be limited or not applicable when different mobile phases and LC gradient methods are applied. Prediction performance may also be limited if many of the iRT calibrant lipids are not detected; however, the calibrant lipids can be re-defined by the user in this case. The iRT calculator cannot be used for other LC separation modes such as hydrophilic interaction LC/normal-phase LC or for shotgun lipidomics or direct injection-IMS-CID-MS data. Not all acyl chains were able to be confidently annotated when building the library; thus, some lipids are given only as their sum composition (i.e., total number of fatty acyl carbons and double bonds). We feel these limitations are minor and will not affect the importance of this protocol.
Materials
Equipment
PC running Windows XP or later
Skyline version 21.2+ (https://skyline.ms/skyline.url)
Data files. Skyline supports data from six major instrument vendors (Agilent, Bruker, Sciex, Waters, Thermo and Shimadzu) as well as any mzML, mz5 and mzXML files26. This protocol minimally recommends untargeted lipidomic data collected on an LC-HRMS/MS or IMS-MS platform. For best results, untargeted lipidomic data collected on an LC-IMS-CID-MS platform is encouraged, because the library was built from data collected by using this platform (Supplementary Methods). However, the IMS functionalities are compatible with any platform on which CCS values are calculated, including drift tube IMS, trapped IMS and traveling wave IMS instruments from Agilent, Bruker and Waters19. In addition, field asymmetric IMS data from Thermo can be processed, but CCS values would need to be replaced with compensation voltage values. IMS data must be calibrated before import by using vendor software to allow Skyline to convert between CCS and empirically measured values such as drift time. IMS data can be viewed in Skyline without calibration. However, the ion mobility filtering feature, which is based on the stored CCS values of each lipid, will not function.
Procedure
Skyline setup and data import ● Timing varies (2 min plus 1–5 min per sample)
-
1
Download the positive and negative mode calibration Skyline documents and zip files from Panorama (https://panoramaweb.org/baker-lipid-ims.url) or Zenodo (https://zenodo.org/record/6374209#.Yplt7hPML9E).
-
2
Open Skyline and ensure that the small molecule interface is selected.
-
3Perform either option A or B, depending on whether the iRTs are applicable to your LC method.
- iRTs applicable
- Click ‘Open File’ and select either ‘1_Plasma_Lipid_Library_Positive_iRT_Calibration.sky.zip’ or ‘1_Plasma_Lipid_Library_Negative_iRT_Calibration.sky.zip’.
- Click ‘Open’.
- iRTs not applicable
- Click ‘Open File’ and select either ‘2_Plasma_Lipid_Library_Positive.sky.zip’ or ‘2_Plasma_Lipid_Library_Negative.sky.zip’.
- Select ‘Settings’ → ‘Molecule Settings’ → ‘Prediction’ and change the ‘Retention Time Predictor’ field to ‘None’.
-
4
Select ‘View’ → ‘Spectral Library’ at any time to view the full spectral library, as displayed in Fig. 3, for the ionization mode in use.
-
5
(Optional, Steps 5–8) Editing the MS settings for various mass spectrometers. Select ‘Settings’ → ‘Transition Settings’ → ‘Full Scan’.
-
6
In the ‘MS1 filtering’ section, change the ‘Precursor mass analyzer’ from time-of-flight (TOF) to the mass analyzer used to collect your data. Edit the appropriate setting for your mass analyzer, such as ‘Resolving power’ or ‘Mass accuracy’. The original settings are appropriate for a TOF instrument with a resolving power between 20,000 and 30,000 (Supplementary Methods). It is recommended for small molecules that the ‘Isotope peaks included’ field is set to ‘Count’ and the ‘Peaks’ field is set to ‘3’.
-
7
In the ‘MS/MS filtering’ section, change the ‘Acquisition method’, ‘Product mass analyzer’, the appropriate setting for your product mass analyzer such as ‘Resolving power’ or ‘Mass accuracy’ and ‘Isolation scheme’ settings to that of your MS/MS method. The original settings are appropriate for a TOF instrument with a resolving power between 20,000 and 30,000 using a data-independent acquisition method with an All Ions isolation scheme (i.e., alternating high and low energy with no precursor selection) (Supplementary Methods).
-
8
Ensure that ‘Use high-selectivity extraction’ is checked to extract a single-resolution width around the target m/z, which reduces interference in complex sample matrices.
-
9
(Optional, Steps 9 and 10) Editing the IMS settings for various ion mobility spectrometers. Select ‘Settings’ → ‘Transition Settings’ → ‘Ion Mobility’. Ensure that the ‘Ion mobility library’ field is set to ‘None’ and that ‘Use spectral library ion mobility values when present’ is checked.
-
10
Change the ‘Resolving power’ field from 50 to the resolving power of your instrument. The ‘Window type’ field can also be changed to either ‘Linear’ or ‘Fixed’ depending on the IMS platform used, in which case IMS drift time or inverse mobility widths or fixed width should be inputted on the basis of instrument parameters rather than resolving power.
▲CRITICAL STEP Setting the resolving power or width value(s) determines the width of the window in which signal will be extracted in the mobility dimension for each lipid. Edit these settings if the drift time ranges appear to be too narrow or broad when reviewing the data.
-
11
Select ‘Edit’ → ‘Expand All’ → ‘Molecules’ or Ctrl + D to view the m/z value(s) for each lipid.
-
12
Select ‘File’ → ‘Import’ → ‘Results’ and click ‘OK’. Select all the LC-MS/MS or calibrated LC-IMS-MS/MS data files to be analyzed and select ‘Open’.
Fig. 3 |. Skyline spectral library explorer view.
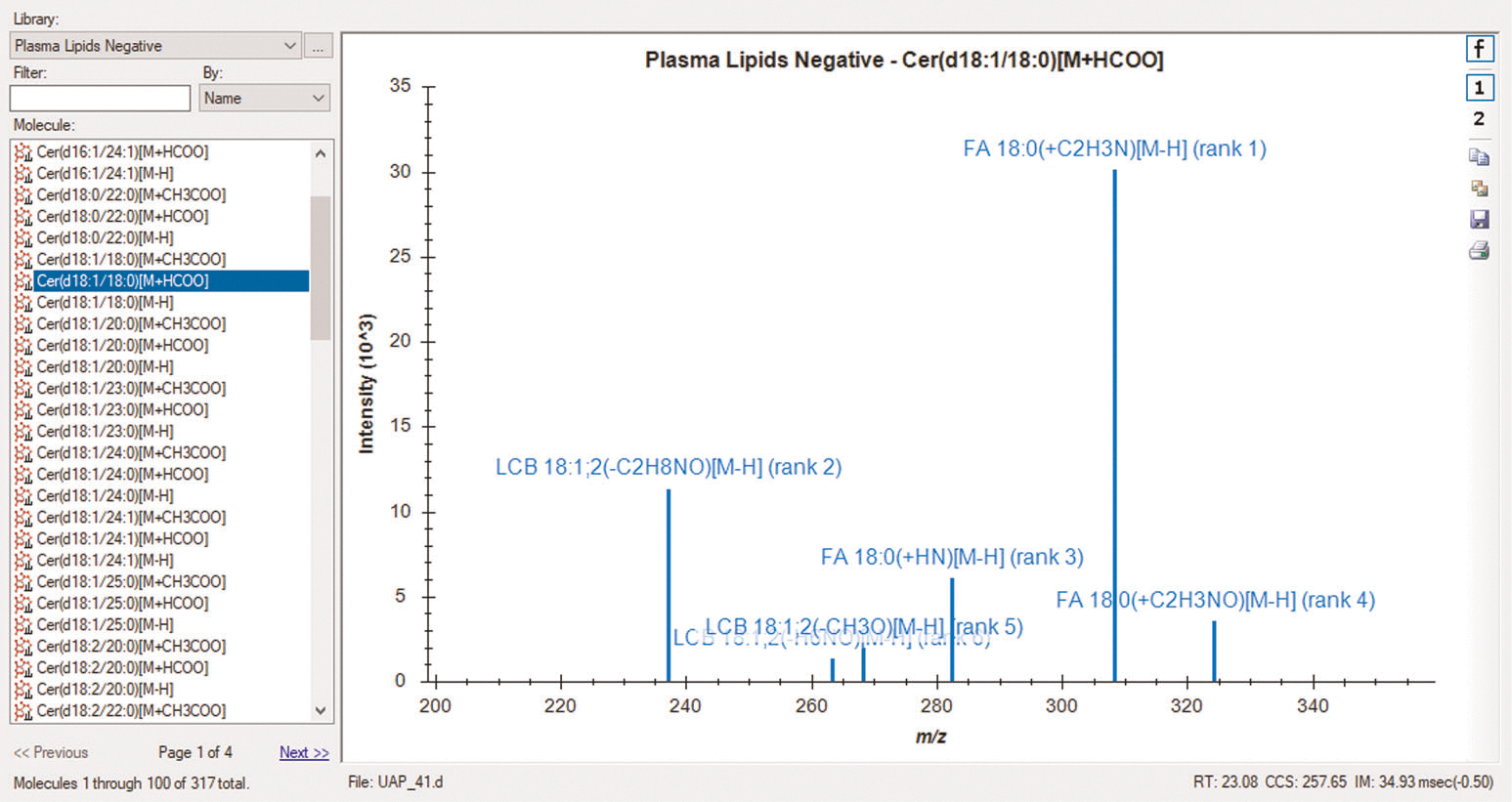
Skyline library MS/MS spectrum for Cer(d18:1/18:0) [M+HCOO]− from plasma lipid data collected in negative ionization mode. Fragment ions are labeled with names, adducts and intensity rankings. The precursor retention time (RT), CCS and drift time (IM) are also displayed in the bottom right corner. The spectral libraries can be viewed before and after importing data.
iRT calibration ● Timing 20 min
-
13
Select each of the 20 lipids under either the ‘Positive Plasma iRT Lipid Calibrants’ or ‘Negative Plasma iRT Lipid Calibrants’ list in the ‘Targets’ pane of the main window depending on the ionization mode of the selected document to view the extracted chromatograms and verify that the proper peak is selected. A predicted retention time will automatically appear on the chromatogram, which will shift if any calibrant peaks are edited.
? TROUBLESHOOTING
-
14
For lipids with multiple adducts, select the lipid name to view the overlayed chromatograms or select the precursor m/z to view the extracted ion chromatogram for an individual adduct and its transitions. The mass error values displayed above each peak should be within the tolerance of the instrument used for data collection (Supplementary Fig. 6).
-
15
Click the candidate precursor peak apex on the chromatogram to view the MS1 spectrum at that time point (Supplementary Fig. 7). Verify that the candidate peak is at the correct m/z and not part of the isotopic distribution of another lipid. The isotope dot product displayed next to the target m/z in the ‘Targets’ pane gives a measure of the similarity of the observed versus expected isotopic distribution from 0 to 1, where 1 is the highest degree of similarity. For example, the first eluting peak for TG(54:4) in Fig. 4 actually arises from the second isotopic peak of TG(54:5) at a lower m/z; thus, it is critical to verify both the MS1 and MS/MS spectra for each candidate peak.
? TROUBLESHOOTING
-
16
Click the Show 2D spectrum icon to change the plot to a 3D spectrum with drift time. Verify that the signal is within the horizontal violet range. Only signal within this drift time range is extracted when drift time filtering is in use (Supplementary Fig. 7).
? TROUBLESHOOTING
-
17
For calibrant lipids with transitions, select ‘View’ → ‘Library Match’ to view the MS/MS library spectra. A legend, which can be viewed by right-clicking on a chromatogram and selecting ‘Legend’, may be useful in comparing transitions (Supplementary Fig. 8). Alternatively, select ‘Edit’ → ‘Expand All’ → ‘Precursors’ and click each transition to highlight the corresponding chromatographic trace. The dot product (dotp) displayed next to the target m/z in the ‘Targets’ pane gives a measure of the similarity of the observed MS/MS spectra and the library match spectra from 0 to 1, where 1 is the highest degree of similarity45. If a similar CID method was used (Supplementary Methods), the dotp should be near 1; however, the dotp threshold will vary depending on the data quality, instrument used, fragmentation method applied and additional factors. Because the spectral library was created from experimental data rather than neat chemical standards, dotp values of 1 are not anticipated, and users should identify a threshold for their given dataset. Furthermore, the fragment ion ranks given in parentheses next to each transition in the ‘Targets’ pane should closely align with the ranks from the library match, which are listed in brackets next to the transitions as well as above each ion in the ‘Library Match’ window.
-
18
Click the peak apex of any fragment chromatogram trace to view the MS/MS spectrum at that time point.
-
19
Click the Show 2D spectrum icon to change the plot to a 3D spectrum with drift time. Verify that the signal is within the horizontal violet range. Note that high-energy drift time offsets will lower the precursor drift time range by 0.5 ms for all transitions. This accounts for the change in fragment ion drift time caused by the collision cell voltage giving smaller fragment ions a higher velocity than larger precursor ions.
-
20
The calibrant lipids are listed in order of increasing retention time, shown for positive mode in Fig. 4. Ensure that the integrated peaks from the imported data follow this pattern. If any calibrant lipids were not detected at a sufficient abundance or are unable to be distinguished from other peaks, the iRT calibrants can be replaced with any other lipids in the library (see Steps 52–56). The retention time of the peak apex is the data point used for calibration rather than the entire retention time window of a given peak; therefore, if peak broadening occurs, such as is the case for TG(54:4) in Fig. 4, probably due to multiple co-eluting isomers of various 54:4 fatty acyl compositions, it should not affect the prediction accuracy. If peak broadening skews the selected retention time and affects prediction accuracy, this iRT calibrant should be removed or replaced (see Steps 52–56).
▲CRITICAL STEP The performance of the retention time prediction calculator will depend on the accuracy of the observed calibrant retention times. The calibrant lipids were selected on the basis of their spread along the gradient as well as high signal-to-noise ratios and ease of identification in multiple lipidomic datasets. However, each peak should still be carefully validated to avoid miscalibration. Only the peak apex is used in calibration, making it unnecessary to fine-tune integration boundaries as long as the correct peak apex is captured. If any calibrant lipids are not present or cannot be readily identified, follow Steps 52–56 to replace the iRT calibrants with preferred targets.
-
21
(Optional) Viewing iRT instead of retention time. Right-click on the chromatogram and select either ‘Show Positive Plasma Lipid iRT Score’ or ‘Show Negative Plasma Lipid iRT Score’.
-
22
If the integration boundaries need to be edited, either click and drag beneath the x-axis to select the correct peak or click the retention time and mass error label above the correct peak. If the retention time reproducibility between samples or replicates is high, the integration boundaries can be synchronized to apply the same integration boundaries to each sample. To do this, right-click on the chromatogram and select ‘Synchronize Integration’. Deselect any sample boxes in the ‘Synchronized Integration’ window that should not be synchronized. In the ‘Align To’ dropdown, select ‘None’ to synchronize the integration to any sample as edited, a sample name to align to a specific sample or ‘Retention Time Calculator’ to align to the iRT predictions. For tuning integration boundaries, it is also possible to click and drag the dashed integration boundary lines in the graph. If one or more of the calibrant lipid(s) were not detected, right-click the chromatogram and select ‘Remove Peak’.
-
23
Review the generated regression and the retention times of the calibrant lipids across each sample. Select ‘View’ → ‘Retention Times’ → ‘Replicate Comparison’ to view a plot of the retention times of the selected lipid across each sample.
-
24
Right-click in the ‘Retention Times – Replicate Comparison’ window and select ‘Value’ → ‘Retention Time’ to see a line plot of only the peak apex retention times.
-
25
If any retention times do not match the validated peak, and synchronized integration is not being used, right-click on the validated chromatogram and select ‘Apply Peak to All’. If the selected peak remains incorrect, manually edit the integration boundaries as described in Step 22. Repeat this for each calibrant lipid until the correct peaks are chosen for each of the 20 calibrant lipids.
-
26
Select ‘View’ → ‘Retention Times’ → ‘Regression’ → ‘Score to Run’ to view the iRT calibration regression.
-
27
Right-click in the ‘Retention Times – Score to Run Regression’ window and select ‘Regression Method’ → ‘Loess’.
Fig. 4 |. Precursor chromatogram for positive-mode iRT calibrants.
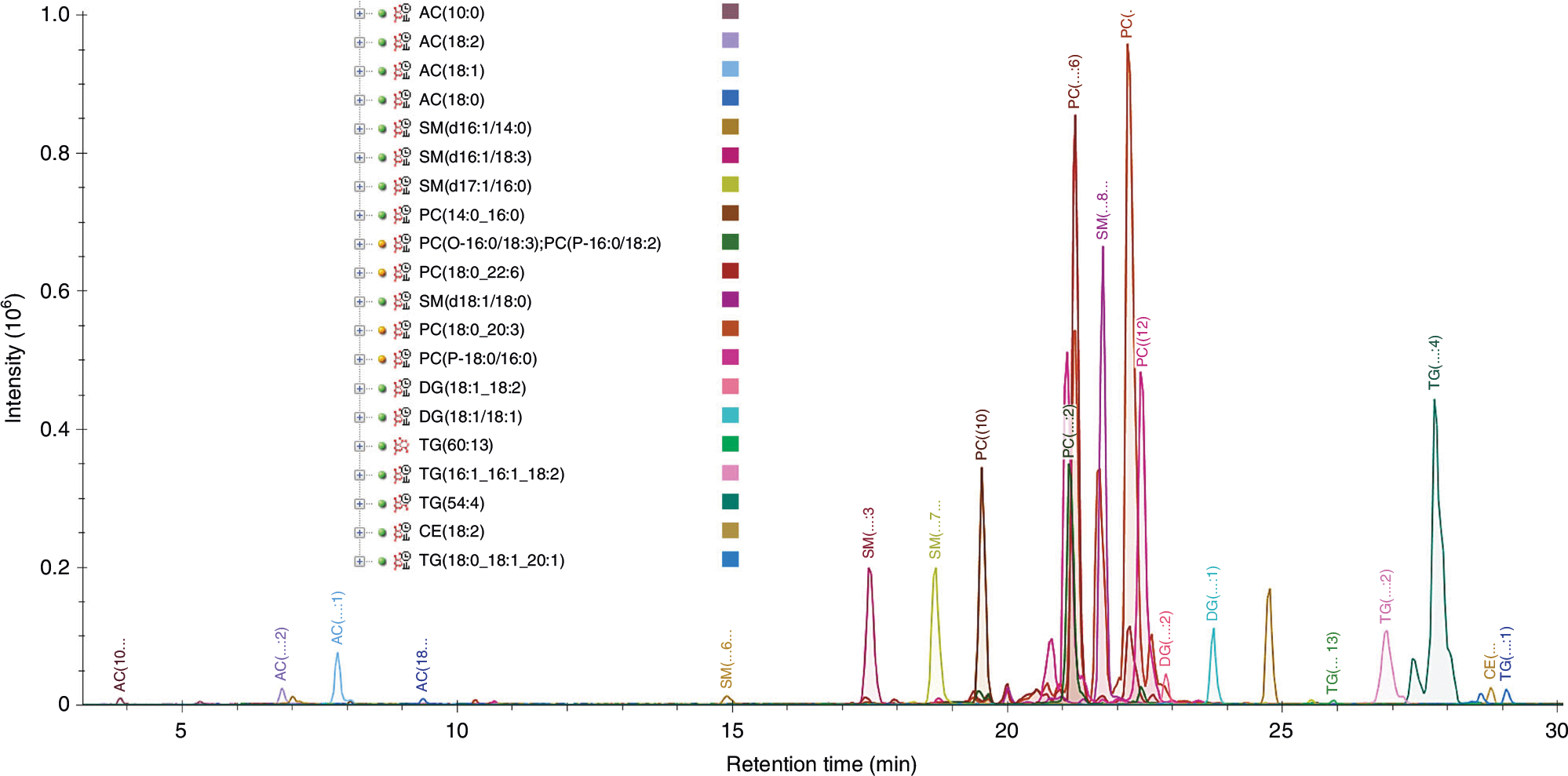
The overlayed extracted precursor lipid iRT calibrant chromatograms for positive ionization mode from a test plasma sample are displayed. This is viewed in Skyline by selecting the ‘Positive Plasma Lipid iRT Calibrants’ molecule list name in the ‘Targets’ pane. Tip: Holding the shift key and pressing the down-arrow key advances the selection through the peptides in the list, highlighting each in red on the overlayed chromatogram graph.
Annotation validation ● Timing varies (~5 h plus 1–5 min per sample)
-
28
Select ‘File’ → ‘Import’ → ‘Document’ and select either ‘2_Plasma_Lipid_Library_Positive.sky.zip’ or ‘2_Plasma_Lipid_Library_Negative.sky.zip’.
-
29
Select ‘Edit’ → ‘Expand All’ → ‘Molecules’ or Ctrl + D to view the m/z value(s) for each lipid. Lipids are sorted by subclass and then by increasing m/z within each subclass.
-
30
Select ‘Edit’ → ‘Manage Results’, then select all data files and select ‘Reimport’ → ‘OK’.
-
31
Validate each lipid target in at least one sample (Fig. 5). To view more than one sample at a time, select ‘View’ → ‘Arrange Graphs’ and select ‘Tiled’, ‘Column’ or ‘Row’ for a small sample set, or ‘Grouped’ for a large sample set.
-
32
For lipids with multiple adducts, select the lipid name to view the overlaid chromatograms or select each precursor m/z to view the extracted ion chromatogram for an individual adduct and its transitions. The mass error values displayed above each peak should be within the tolerance of the instrument used to collect the data. Candidate peaks should be within the yellow 2-min window surrounding the predicted retention time if iRT calibration was successful (Supplementary Fig. 6).
-
33
Click the candidate precursor peak apex on the chromatogram to view the MS1 spectrum at that time point. The isotope dot product displayed next to the target m/z in the ‘Targets’ pane gives a measure of the similarity of the observed versus expected isotopic distribution from 0 to 1, where 1 is the highest degree of similarity. Verify that the candidate peak is at the correct m/z and not part of the isotopic distribution of another lipid, an example of which is demonstrated in Fig. 6.
-
34
Click the Show 2D spectrum icon to change the plot to a 3D spectrum with drift time (Fig. 6). Verify that the signal is within the horizontal violet range. Only signal within this drift time range is extracted when drift time filtering is in use.
-
35
For lipids with transitions, select ‘View’ → ‘Library Match’ to view the MS/MS library spectra (Fig. 3). The dotp displayed next to the target m/z in the ‘Targets’ pane gives a measure of the similarity of the observed MS/MS spectra and the library match spectra from 0 to 1, where 1 is the highest degree of similarity45. If a similar CID method was used (Supplementary Methods), the dotp should be near 1; however, the dotp threshold will vary depending on the data quality, instrument used, fragmentation method applied and additional factors. Because the spectral library was created from experimental data rather than neat chemical standards, dotp values of 1 are not anticipated, and users should identify a threshold for their given dataset. Furthermore, the fragment ion ranks given in parentheses next to each transition in the ‘Targets’ pane should closely align with the ranks from the library match, which are listed in brackets next to the transitions as well as above each ion in the ‘Library Match’ window.
-
36
Click the peak apex of any fragment chromatogram trace to view the MS/MS spectrum at that time point. Tip: You can select ‘View’ → ‘Transitions’ → ‘Split Graph’ to separate precursor and fragment chromatograms into different panes with different y-axis scales.
-
37
Click the Show 2D spectrum icon to change the plot to a 3D spectrum with drift time. Verify that the signal is within the horizontal violet range (Fig. 7). Note that high-energy drift time offsets will lower the precursor horizontal violet drift time range by 0.5 ms for all transitions. This accounts for the change in fragment ion drift time caused by the collision cell voltage giving smaller fragment ions a higher velocity than larger precursor ions.
-
38
If the original peak is incorrect, and the integration boundaries need to be edited, either click and drag beneath the x-axis to select the correct peak or click the retention time and mass error label above the correct peak. If the retention time reproducibility between samples or replicates is high, the integration boundaries can be synchronized to apply the same integration boundaries to each sample. To do this, right-click on the chromatogram and select ‘Synchronize Integration’. Deselect any sample boxes in the ‘Synchronized Integration’ window that should not be synchronized. In the ‘Align To’ dropdown, select ‘None’ to synchronize the integration to any sample as edited, a sample name to align to a specific sample or ‘Retention Time Calculator’ to align to the iRT predictions. For tuning the integration boundaries, it is also possible to click and drag the dashed integration boundaries in the graph. If synchronized integration is not being used, right-click on the chromatogram with the corrected boundaries and select ‘Apply Peak to All’, or manually edit the integration boundaries in the remaining samples.
-
39
Remove lipids that are not of interest or not present in any sample by selecting the lipid name(s) in the target list and pressing ‘Delete’.
-
40
To ensure consistency between samples, review the replicate comparison windows and correct any outliers. To compare retention times, select ‘View’ → ‘Retention Times’ → ‘Replicate Comparison’ (Fig. 8).
-
41
To compare peak areas, select ‘View’ → ‘Peak Areas’ → ‘Replicate Comparison’. Peak areas will likely vary across samples; thus, peak area percentages may be a more helpful comparison. To view the peak area percentages, right-click on the ‘Peak Area – Replicate Comparison’ window and select ‘Normalized to’ → ‘Total’ (Fig. 8). To view normalized peak areas for relative quantification, right-click on the ‘Peak Area – Replicate Comparison’ window and select ‘Normalized to’ → ‘Equalize Medians’ or ‘Total Ion Current’. Tip: To view precursor ions separately from fragment ions, select ‘View’ → ‘Transitions’ → ‘Split Graph’.
-
42
To compare mass errors, select ‘View’ → ‘Mass Errors’ → ‘Replicate Comparison’.
-
43
Select any outliers directly in the replicate comparison window by clicking the bars or x-axis label to jump to the outlying lipid and sample, and then review the extracted ion chromatogram.
-
44
Review the regression generated by plotting the observed retention time versus the iRT (Fig. 9) to identify any remaining outliers. Select ‘View’ → ‘Retention Times’ → ‘Regression’ → ‘Score to Run’.
-
45
Right-click in the ‘Retention Times – Score to Run Regression’ window and select ‘Regression Method’ → ‘Loess’.
? TROUBLESHOOTING
-
46
Move and click the mouse cursor over any outlier points directly on the plot to examine the outlying lipid extracted ion chromatogram(s).
-
47
Right-click in the ‘Retention Times – Score to Run Regression’ window and select ‘Plot’ → ‘Residuals’ to evaluate the residuals.
Fig. 5 |. Annotation validation.
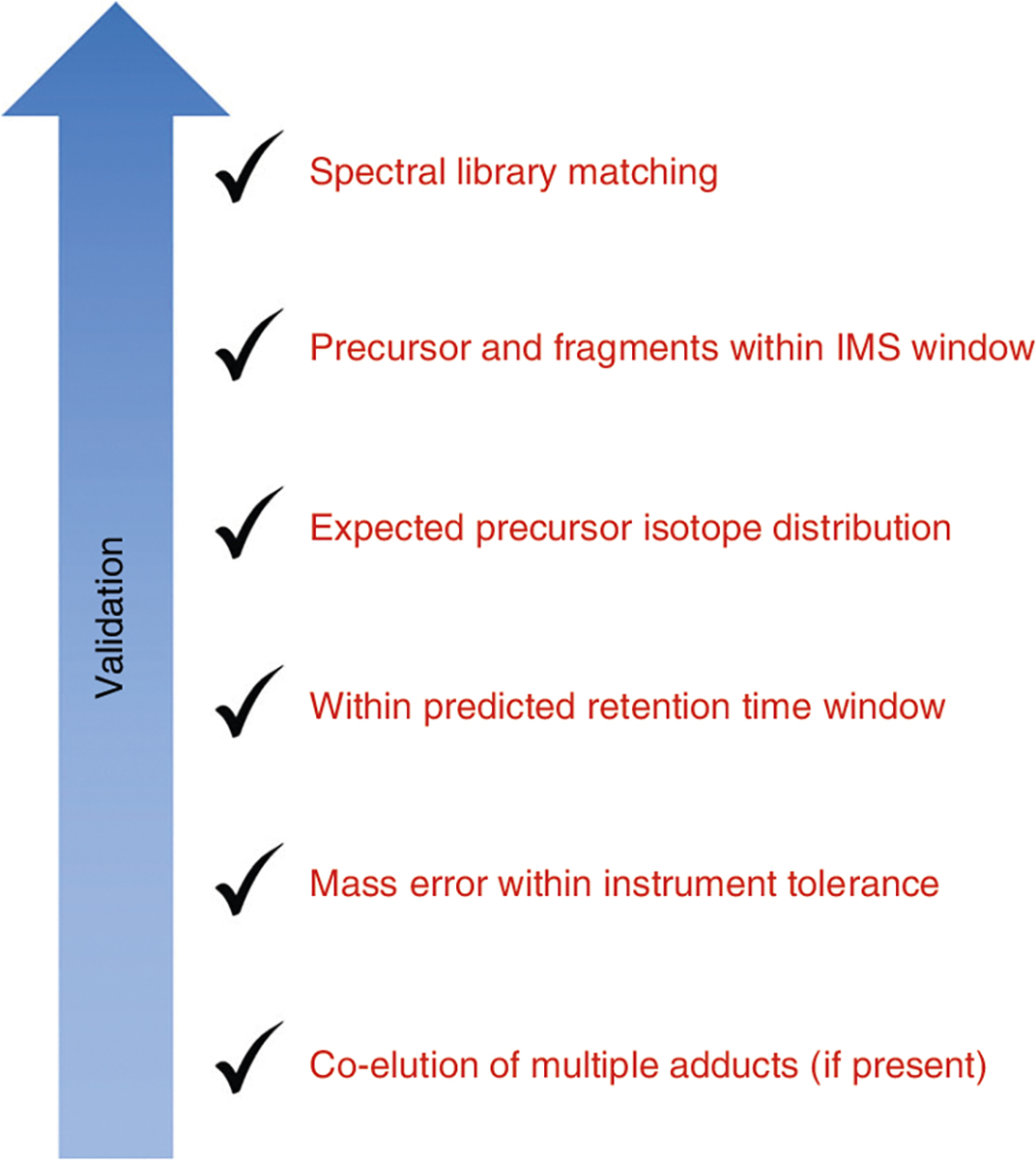
Criteria for validating lipid annotations for this workflow. The specifics of these criteria will vary depending on the instrument used and the similarity of the method to that used for the library creation (Supplementary Methods). Expectations for validated lipids using this procedure would be detection and co-elution of all adducts listed; precursor mass error <5 ppm; fragment mass error <10 ppm; retention time within ±1 min of the predicted retention time; presence of the precursor, [M+1] and [M+2] signals within the drift time filtering window with a resolving power of 50; and fragment ranks closely matching the library match ranks.
Fig. 6 |. Annotation validation example.
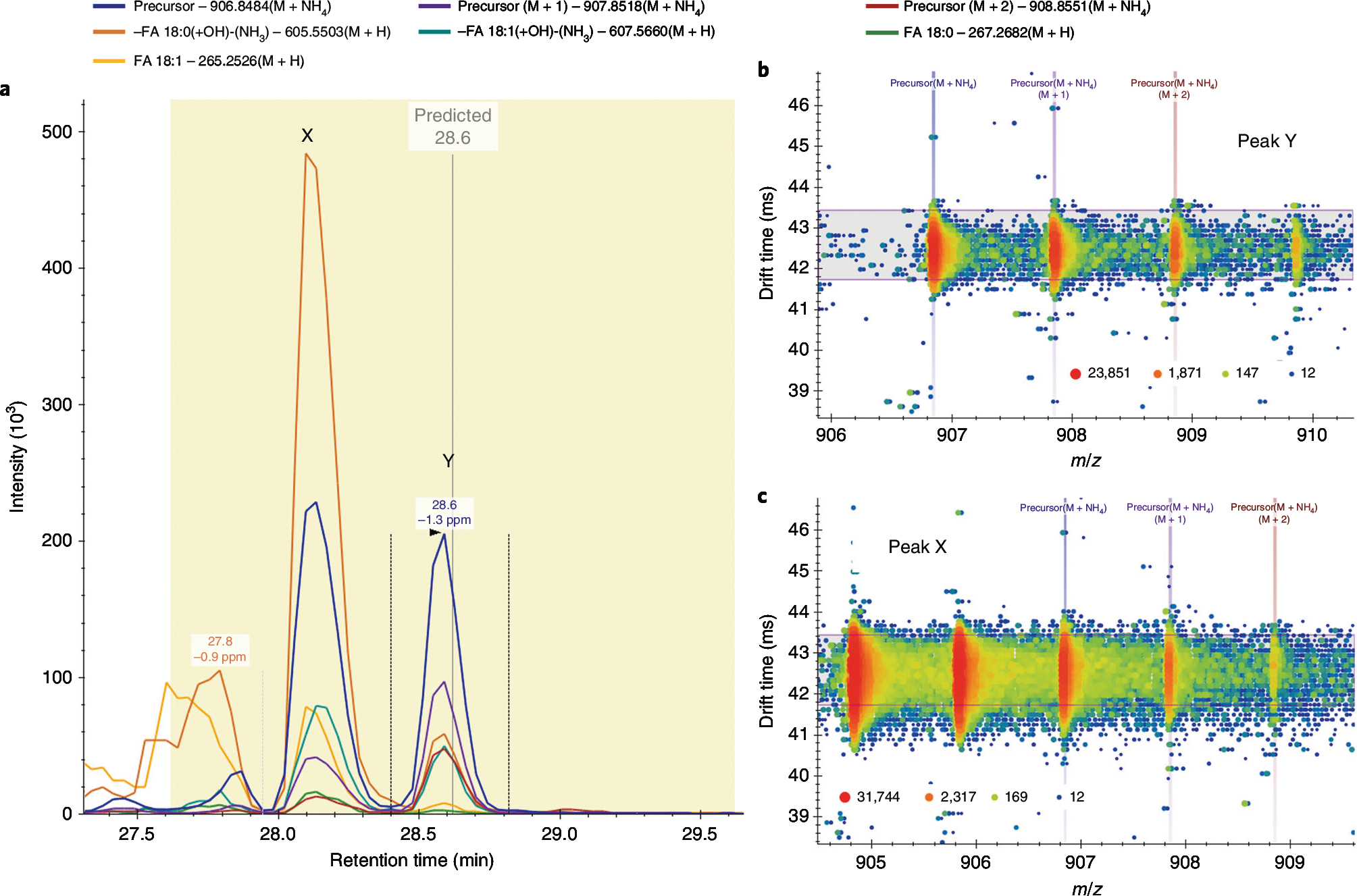
Validation of the TG(18:0_18:0_18:1) annotation. a, The precursor and fragment signals extracted for TG(18:0_18:0_18:1) [M+NH4]+. The vertical gray line shows the predicted retention time (28.6 min) with a yellow 2-min window. The two candidate peaks within this window are labeled X (28.2 min) and Y (28.6 min). The vertical lines around peak Y are the integration boundaries. b,c, The precursor drift spectra at the apex of peaks Y (b) and X (c), where the horizontal violet range shows the filter imposed by the lipid’s CCS of 329.37 Å2. Only signal within this range is extracted for a. Signal intensity is displayed as a heat map from blue (low intensity) to red (high intensity). The signal from peak X is directly related to the precursor [M+2] signal for the lipid TG(18:0_18:1_18:1) at a lower m/z. Therefore, peak Y is validated as the correct peak for TG(18:0_18:0_18:1).
Fig. 7 |. Lipid MS/MS spectra example.
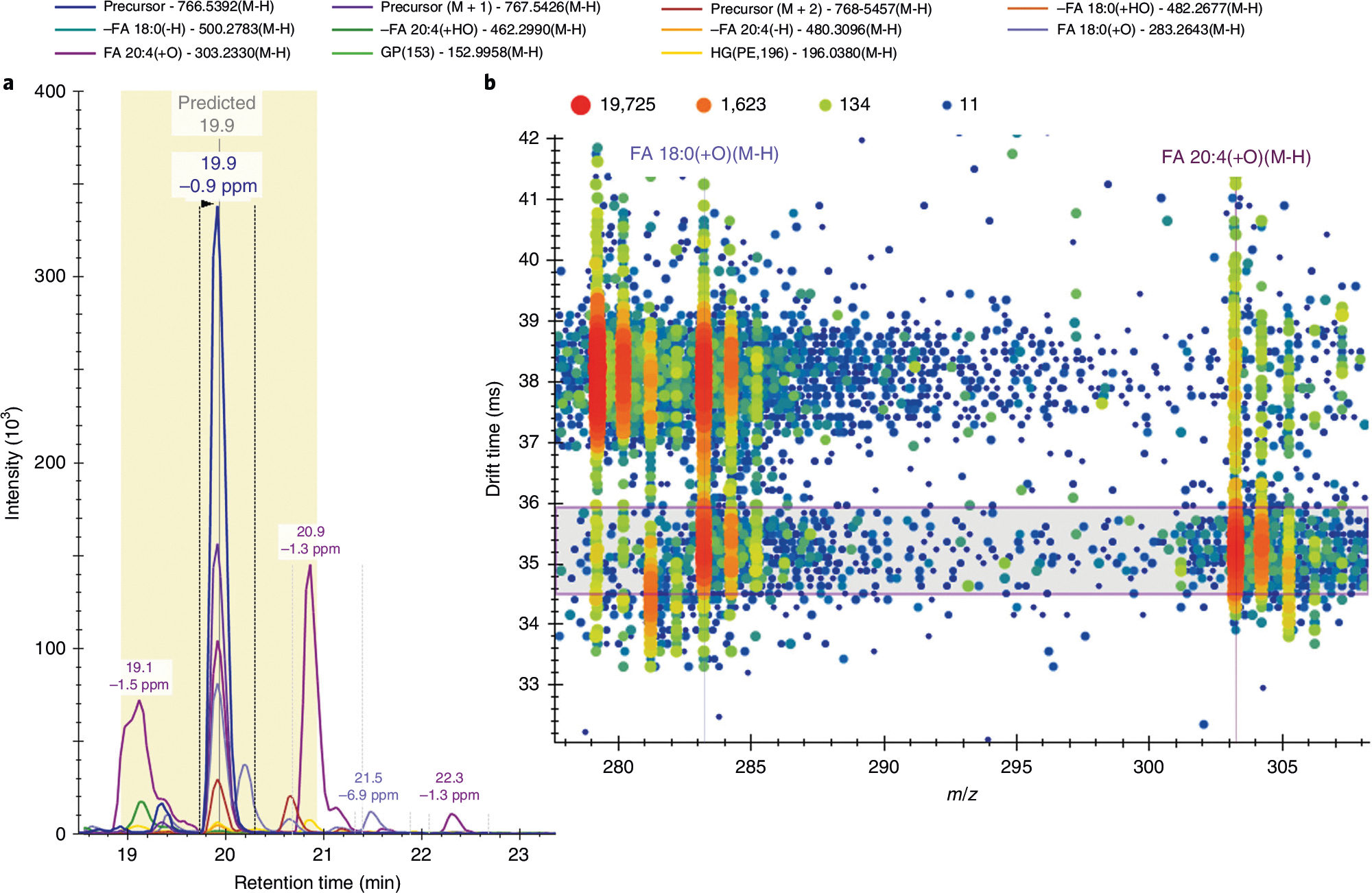
Skyline negative-mode MS/MS spectra for PE(18:0_20:4). a, Illustration of the precursor and fragment signals extracted for PE(18:0_20:4) [M-H]−. The vertical gray line illustrates the predicted retention time (19.9 min) with a yellow 2-min window. The vertical lines around the peak at 19.9 min are the integration boundaries. b, MS/MS drift spectra at 19.9 min for the m/z range of 278–308 showcase the two main fragment ions used to identify the fatty acyl chain composition of this lipid. Signal intensity is displayed as a heat map from blue (low intensity) to red (high intensity). The horizontal violet range shows the filter imposed by the lipid’s CCS of 275.2 Å2, with a high-energy-drift-time offset of −0.5 ms. Without drift time filtering, at least two visible ions would contribute to the extracted fragment intensities, and the fatty acyl chain composition would be more challenging to identify given the retention time overlap of many lipid species with various fatty acyl chains. For example, the FA 18:0(+O)[M-H] ion at ~35.4 ms is a fragment of the lipid of interest PE(18:0_20:4); however, the ion at ~38 ms is a fragment of the co-eluting lipid PC(18:0_18:2).
Fig. 8 |. Skyline replicate comparisons.
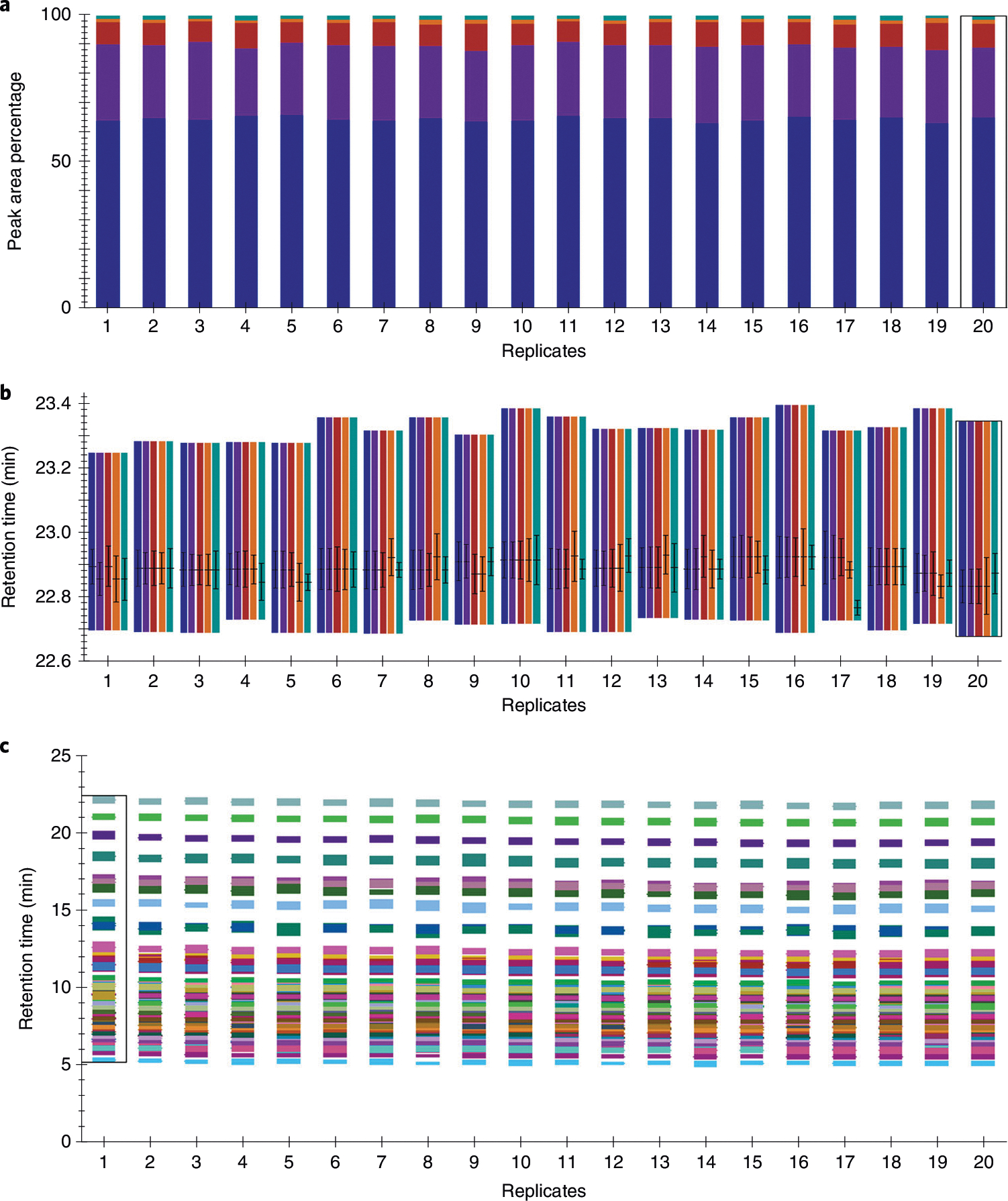
a,b, Skyline replicate comparison examples for DG(18:1_18:2) in positive ionization mode including the normalized peak areas to confirm integration and total peak area percentage reproducibility (a) and retention times (b), where the line indicates the retention time of the peak apex and the whiskers indicate the full width at half maximum centered around the apex, to confirm retention time reproducibility across 20 test control plasma samples. The colored bars in a and b indicate precursor (blue, purple and red for precursor, M+1 and M+2, respectively) and fragment (orange and teal for M-FA 18:1(-H)-(H2O+NH3) and M-FA 18:2(-H)-(H2O+NH3), respectively) signals. c, Illustration of the replicate comparison of retention times for all lipids within the fatty acyl lipid category in negative ionization mode across 20 test control plasma samples, where each color signifies a single lipid. The black rectangle on each graph signifies which sample is currently in view in the main chromatogram window (sample 20 in a and b, and sample 1 in c). Other samples can be selected within the ‘Retention Times – Replicate Comparison’ window, within the ‘Replicates’ dropdown menu within the ‘Targets’ pane or by using the sample tabs.
Fig. 9 |. iRT performance assessment.
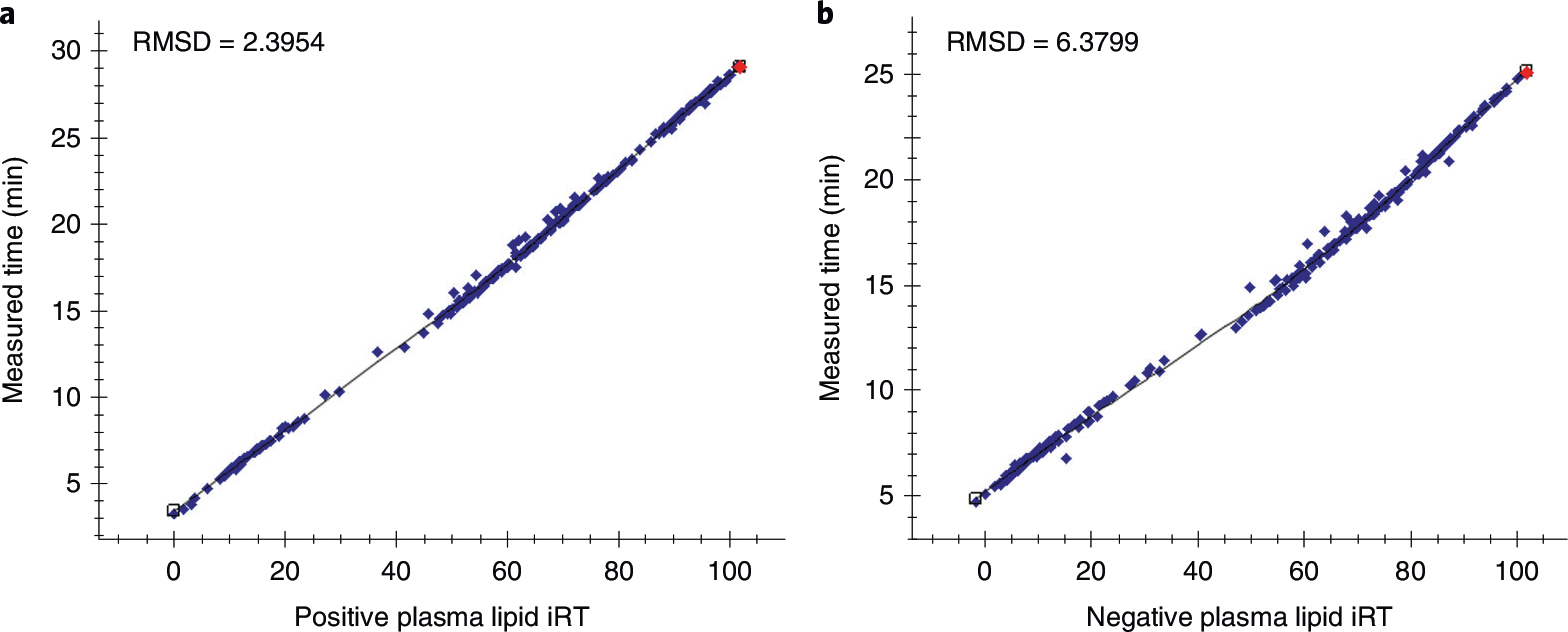
a,b, Measured retention times of each lipid plotted as a function of iRT for positive ionization mode (a) and negative ionization mode (b) using LOESS regressions for one representative sample of a test dataset. The same LC method was used for the calibration and test datasets, but on secondary columns and instruments. The red dot signifies which lipid is currently in view in the main chromatogram window. Other lipids can be selected within the ‘Retention Times – Score to Run Regression’ window or the ‘Targets’ pane. RMSD, root mean square deviation.
(Optional) Library and iRT editing ● Timing varies (5 min plus 1–5 min per sample)
-
48
To search for additional targets that are not included in the library such as internal standards or lipids of interest, create29 or use an existing transition list containing at least a molecule list name, precursor name, precursor m/z, charge or precursor formula and adduct.
-
49Import the new transition list by using one of the following options:
- Import a .csv file with proper headings (Supplementary Table 1) by selecting ‘File’ → ‘Import’ → ‘Transition List’.
- Paste a transition list with proper headings by right-clicking at the bottom of the ‘Targets’ pane and selecting ‘Paste’.
? TROUBLESHOOTING
-
50
Re-import results data by selecting ‘Edit’ → ‘Manage Results’, then select all of the data files and select ‘Re-import’ and then ‘OK’.
-
51
Validate or remove additional targets by repeating Steps 32–37.
-
52
(Optional Steps 52–56) Adding new targets to iRT calculator for retention time prediction in future datasets and editing the iRT reference lipids. Select ‘Settings’ → ‘Molecule Settings’ → ‘Prediction’.
-
53
Click the calculator icon and select ‘Edit Current’.
-
54
At the bottom of the ‘Edit iRT Calculator’ window, select ‘Add’ → ‘Results’. The number of added molecules will be displayed at the top of the ‘Add iRT Molecules’ window.
-
55
To change the iRT calibrant lipids from the default to a set of internal standards or preferred endogenous lipids, select ‘Choose Standards’ from the ‘Edit iRT Calculator’ window. Select your set of calibrants from the standard molecule list.
-
56
Click ‘OK’ in all open windows to save and close.
Exporting results ● Timing 5 min
-
57
Select ‘File’ → ‘Export’ → ‘Report’.
-
58
Select any of the reports under ‘Main’ and select ‘Preview’ to preview the export file and determine which report contains the information of interest. The report named ‘Transition Results’ is the recommended default report, because it contains each lipid name, subclass, precursor and product m/z and charge, retention time, area, background and peak rank for each sample (‘replicate’).
-
59
To incorporate additional information to the ‘Transition Results’ report, select ‘Edit List’ and then select ‘Transition Results’ and ‘Edit’. Use the hierarchical list to select additional columns or click the binocular icon to search, and then select the checkbox next to the items of interest.
-
60
To remove default information, use the hierarchical list or click the binocular icon to search, and select the checkbox to uncheck items. Additional items of interest may include total ion current area, CCS, mass error ppm, predicted result retention time and normalized area. To set the normalization method before export for relative quantification, select ‘Settings’ → ‘Molecule Settings’ → ‘Quantification’, and then select an option (‘Total Ion Current’ or ‘Equalize Medians’) from the ‘Normalization method’ dropdown.
-
61
Click ‘OK’ in the ‘Edit Report’ and ‘Manage Reports’ windows.
-
62
Select ‘Export’ to export a .csv file. Disregard the iRT calibrant lipids in subsequent analyses, because they are repeated in the target list within their respective classes.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
|
| |||
| 13 | No predicted retention time is displayed | Viewing predicted retention times is not enabled, or an incorrect calculator was chosen | Right-click a chromatogram and select ‘Predicted Retention Times’. Ensure that the correct calculator is in use by selecting ‘Settings’ → ‘Molecule Settings’ → ‘Prediction’ and selecting the proper calculator from the ‘Retention time predictor’ dropdown. Save your document, and then close and reopen the Skyline window |
| 15 | No horizonal violet box is displayed, indicating drift time filtering is not in use | Data files are not calibrated, or ion mobility settings are incorrect | Calibrate data files by using vendor software and re-import. Ensure that the ‘Transition Settings – Ion Mobility’ tab is appropriately configured for your instrument and verify that ‘Use spectral library ion mobility values when present’ is checked |
| 16 | Cannot open MS1 or MS/ MS data | Data files have been moved or renamed | Ensure that raw data file names have not been changed. Move the raw data files back to their original file location or to the document parent folder to be locally accessed. Use ‘File’ → ‘Open containing folder’ to locate the parent folder quickly |
| 45 | The ‘Retention Times – Score to Run Regression’ window is blank | The wrong iRT calculator is selected | Right-click in ‘Retention Times – Score to Run Regression’ window and click ‘Calculator’. Select the proper iRT calculator from the list for the ionization mode you are viewing |
| 49 | The [M+1] and [M+2] peaks of added molecules are missing | Transition settings were not reapplied | Select ‘Settings’ → ‘Transition Settings’ → ‘Full-Scan’. Change the ‘Isotope peaks included’ field to ‘None’ and click ‘OK’, then navigate back to the ‘Full-Scan’ settings, change the ‘Isotope peaks included’ field back to ‘Count’ and click ‘OK’ |
Timing
The time required to perform the steps outlined in the protocol depend on the PC characteristics, dataset size, instrument and method used to collect the data, user familiarity with Skyline and lipid data and number of lipids of interest. Software-dependent steps will generally take seconds to minutes, while user-dependent steps such as annotation validation will be more time consuming. Although Skyline supports analyzing both ionization modes in one document, the libraries were built separately to avoid analyzing an excessive number of targets and data files in one document. Therefore, these steps should be repeated for both ionization modes (if applicable).
Steps 1–12, Skyline setup and data import: varies (2 min plus 1–5 min per sample)
Steps 13–27, iRT calibration: 20 min
Steps 28–47, annotation validation: varies depending on the aforementioned factors (~5 h plus 1–5 min per sample)
Steps 48–56, library editing: varies (5 min plus 1–5 min per sample)
Steps 57–62, exporting results: 5 min
Anticipated results
The workflow presented here allows rapid and targeted processing of multidimensional data containing hundreds of lipids based on precursor m/z, MS/MS spectral library matching, iRT retention time predictions and IMS filtering27. The exported lipid abundances can be further analyzed with data visualization software and statistical and pathway mapping tools to provide biological insights27,46. The library contains 516 unique lipids across multiple classes and subclasses from five of the eight lipid categories28. Although the library was optimized for human plasma lipidomics data analysis, it can be easily edited to fit the user’s interests and has been used to uniquely identify 370 lipids from bronchoalveolar lavage fluid, 359 lipids from zebrafish tissue and 269 lipids from fruit fly tissue samples (Supplementary Table 1) without inserting additional lipid targets. Utilization of Skyline’s iRT feature should control for run-to-run retention time shifts and give accurate retention time predictions for data collected by using the same or similar LC methods. Retention time predictions were found to be within +/− 1 min of the observed retention time for each lipid in the test data collected on a secondary column and instrument, with an average percentage difference between observed and predicted retention times of 2.5% in both ionization modes (Supplementary Table 2). Drift time filtering based on lipid CCS values further aided in annotating the detected lipids by increasing lipid identification confidence and improving the signal-to-noise ratio for the targeted species19. Although this protocol is not focused on lipid quantification, additional steps can be taken to use Skyline’s small-molecule quantification features to give quantitative values for the annotated lipid species rather than relative abundances.
Data availability
All associated library and data files are publicly available at Panorama Public (https://panoramaweb.org/baker-lipid-ims.url) and Zenodo (https://zenodo.org/record/6374209#.YpzPMxPMJJU) 47.
Code availability
Skyline source code is freely available at https://skyline.ms/source.url.
Supplementary Material
Acknowledgements
Portions of this research were supported by grants from the NIH National Institute of Environmental Health Sciences (P30 ES025128, P42 ES027704 and P42 ES031009), NIH National Institute of General Medicine Sciences (R24 GM141156, P41 GM103533 and T32 GM133366), a cooperative agreement with the United States Environmental Protection Agency (STAR RD 84003201) and startup funds from North Carolina State University. In addition, most of the LC-IMS-CID-MS measurements were made in the Molecular Education, Technology, and Research Innovation Center (METRIC) at North Carolina State University.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-022-00714-6.
Peer review information Nature Protocols thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Soppert J, Lehrke M, Marx N, Jankowski J & Noels H Lipoproteins and lipids in cardiovascular disease: from mechanistic insights to therapeutic targeting. Adv. Drug Deliv. Rev. 159, 4–33 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Maradonna F & Carnevali O Lipid metabolism alteration by endocrine disruptors in animal models: an overview. Front. Endocrinol. 9, 654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niemann R The effects of xenobiotics on hepatic lipid and lipo-protein metabolism. Exp. Pathol. 39, 213–232 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Miao H et al. Lipidomics biomarkers of diet-induced hyperlipidemia and its treatment with Poria cocos. J. Agric. Food Chem. 64, 969–979 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Perla FM, Prelati M, Lavorato M, Visicchio D & Anania C The role of lipid and lipoprotein metabolism in non-alcoholic fatty liver disease. Children 4, 46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wenk MR The emerging field of lipidomics. Nat. Rev. Drug Discov. 4, 594–610 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Rolim AEH, Henrique-Araújo R, Ferraz EG, de Araújo Alves Dultra, F. K. & Fernandez, L. G. Lipidomics in the study of lipid metabolism: current perspectives in the omic sciences. Gene 554, 131–139 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Züllig T, Trötzmüller M & Köfeler HC Lipidomics from sample preparation to data analysis: a primer. Anal. Bioanal. Chem. 412, 2191–2209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leaptrot KL, May JC, Dodds JN & Mclean JA Ion mobility conformational lipid atlas for high confidence lipidomics. Nat. Commun. 10, 985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liebisch G et al. Lipidomics needs more standardization. Nat. Metab. 1, 745–747 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Yang K, Dilthey BG & Gross RW Identification and quantitation of fatty acid double bond positional isomers: a shotgun lipidomics approach using charge-switch derivatization. Anal. Chem. 85, 9742–9750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma X & Xia Y Pinpointing double bonds in lipids by Paternò-Büchi reactions and mass spectrometry. Angew. Chem. Int. Ed. Engl. 53, 2592–2596 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Brown SHJ, Mitchell TW & Blanksby SJ Analysis of unsaturated lipids by ozone-induced dissociation. Biochim. Biophys. Acta 1811, 807–817 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Thomas MC, Mitchell TW & Blanksby SJ Ozonolysis of phospholipid double bonds during electrospray ionization: a new tool for structure determination. J. Am. Chem. Soc. 128, 58–59 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Campbell JL & Baba T Near-complete structural characterization of phosphatidylcholines using electron impact excitation of ions from organics. Anal. Chem. 87, 5837–5845 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Klein DR & Brodbelt JS Structural characterization of phosphatidylcholines using 193 nm ultraviolet photodissociation mass spectrometry. Anal. Chem. 89, 1516–1522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan E, Nguyen CQN, Shiea C & Reid GE Detailed structural characterization of sphingolipids via 193 nm ultraviolet photodissociation and ultra high resolution tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 28, 1406–1419 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Kyle JE et al. Uncovering biologically significant lipid isomers with liquid chromatography, ion mobility spectrometry and mass spectrometry. Analyst 141, 1649–1659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maclean BX et al. Using Skyline to analyze data-containing liquid chromatography, ion mobility spectrometry, and mass spectrometry dimensions. J. Am. Soc. Mass Spectrom. 29, 2182–2188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hancock SE, Poad BLJ, Batarseh A, Abbott SK & Mitchell TW Advances and unresolved challenges in the structural characterization of isomeric lipids. Anal. Biochem. 524, 45–55 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Paglia G, Kliman M, Claude E, Geromanos S & Astarita G Applications of ion-mobility mass spectrometry for lipid analysis. Anal. Bioanal. Chem. 407, 4995–5007 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Groessl M, Graf S & Knochenmuss R High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids. Analyst 140, 6904–6911 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Egertson JD, MacLean B, Johnson R, Xuan Y & MacCoss MJ Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 10, 887–903 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maclean B et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pino LK et al. The Skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 39, 229–244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams KJ et al. Skyline for small molecules: a unifying software package for quantitative metabolomics. J. Proteome Res 19, 1447–1458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirkwood KI et al. Development and application of multidimensional lipid libraries to investigate lipidomic dysregulation related to smoke inhalation injury severity. J. Proteome Res 21, 232–242 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahy E et al. A comprehensive classification system for lipids. J. Lipid Res. 46, 839–862 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Peng B et al. LipidCreator workbench to probe the lipidomic landscape. Nat. Commun. 11, 2057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kind T et al. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 10, 755–758 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy R Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids (Royal Society of Chemistry, 2015). [Google Scholar]
- 32.Escher C et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Picache JA et al. Collision cross section compendium to annotate and predict multi-omic compound identities. Chem. Sci. 10, 983–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kyle JE et al. LIQUID: an-open source software for identifying lipids in LC-MS/MS-based lipidomics data. Bioinformatics 33, 1744–1746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsugawa H et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Z et al. LipidIMMS Analyzer: integrating multi-dimensional information to support lipid identification in ion mobility—mass spectrometry based lipidomics. Bioinformatics 35, 698–700 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Koelmel JP et al. LipidMatch: an automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinforma. 18, 331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartler J et al. Deciphering lipid structures based on platform-independent decision rules. Nat. Methods 14, 1171–1174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kochen MA et al. Greazy: open-source software for automated phospholipid tandem mass spectrometry identification. Anal. Chem. 88, 5733–5741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sumner LW et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3, 211–221 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blaženović I, Kind T, Ji J & Fiehn O Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites 8, 1–23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Köfeler HC et al. Quality control requirements for the correct annotation of lipidomics data. Nat. Commun. 12, 4771 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koelmel JP, Ulmer CZ, Jones CM, Yost RA & Bowden JA Common cases of improper lipid annotation using high-resolution tandem mass spectrometry data and corresponding limitations in biological interpretation. Biochim. Biophys. Acta 1862, 766–770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rampler E et al. Recurrent topics in mass spectrometry-based metabolomics and lipidomics—standardization, coverage, and throughput. Anal. Chem. 93, 519–545 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frewen BE et al. Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal. Chem. 78, 5678–5684 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Odenkirk MT et al. Combining micropunch histology and multidimensional lipidomic measurements for in-depth tissue mapping. ACS Meas. Sci. Au 2, 67–75 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirkwood KI et al. Utilizing Skyline to analyze lipidomics data containing liquid chromatography, ion mobility spectrometry and mass spectrometry dimensions. Zenodo; https://zenodo.org/record/6374209#.YpzYhBPMJJU (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All associated library and data files are publicly available at Panorama Public (https://panoramaweb.org/baker-lipid-ims.url) and Zenodo (https://zenodo.org/record/6374209#.YpzPMxPMJJU) 47.