

Abstract
The development of selective KRASG12C inhibitors that directly inhibit KRAS, an oncogene historically thought to be “undruggable”, represents a watershed moment in oncology and developmental therapeutics. Now, as KRAS-targeted therapy moves into its second phase, there is significant excitement and anticipation for durable disease control in tumor types where options remain limited, with clinical trials testing combination therapies, indirect pan-RAS/MAP kinase pathway inhibitors, and active-state RAS(on) inhibitors.
However, there is also reason for caution regarding the safety and tolerability of expanded RAS inhibition. This is evidenced by the intolerability of some combination therapies with selective KRASG12C inhibitors and foreshadowed by prior failures of combination therapies in other oncogene-driven tumors.
Herein, we review the landscape of and outlook for KRAS-targeted therapies. We specifically focus upon strategies to combat resistance to KRAS-targeted therapies, and discuss the possibility of off-target or unanticipated on-target effects that may limit clinical use.
Introduction
The RAS gene family consists of KRAS (kirsten rat sarcoma), HRAS (harvey rat sarcoma), and NRAS (neuroblastoma RAS viral oncogene homolog)(1,2). Together they are the most frequently mutated oncogenes in human cancer. KRAS is the predominant oncogenic driver in non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and pancreatic cancer (PDAC)(1). Further, NRAS is the second most common oncogenic driver in cutaneous melanoma after BRAF(1). Finally, HRAS is an emerging actionable oncogenic driver in a small subset of HPV (human papilloma virus)-negative squamous cell carcinoma of the head and neck(1). Historical attempts to target RAS directly, in particular KRAS, have been fraught with failure(1,3). Hence, clinical outcomes in RAS-driven tumors have been historically poorer than outcomes in tumors with non-RAS oncogenic drivers, when comparing within cancer types(4–6).
However, the long-considered “undruggability” of RAS has now been pierced with the development and proven clinical efficacy of the selective KRASG12C inhibitors sotorasib (AMG 510) and adagrasib (MRTX849)(7–9). Unfortunately, 1 year into FDA approval of sotorasib and 3 years into clinical use of selective KRASG12C inhibitiors, it is clear that although the development of these agents represents a watershed moment in cancer therapy, clinical efficacy as single agents is short-lived(7). Equally importantly, given the selectivity of current inhibitors to KRASG12C alone(10), there remains a substantial unmet need for novel targeting strategies that will expand the impact and improve the clinical outcomes for patients with RAS-driven tumors.
Herein, we review the current landscape of KRAS-targeted therapies including combination therapy and expanded RAS approaches, and discuss reasons for excitement and caution. We focus particularly on attempts to combat or overcome therapeutic resistance to currently available KRAS-directed therapy and avenues to expand the reach of direct RAS targeting. Finally, we discuss potential unanticipated difficulties with novel or combination therapies that warrant further investigation and may foreshadow hurdles to clinical translation.
Clinical efficacy and resistance to available direct KRAS inhibitors
Structurally, KRAS is a small, 21-kDa monomeric guanosine 5’-triphosphatase consisting of six beta strands and five alpha helices with a G-domain and a C-terminal membrane-targeting region. It belongs to the RAS protein family, which also includes HRAS and NRAS(11–13).
In non-malignant tissues, wild-type KRAS shifts between inactive GDP-bound RAS(off) state and active GTP-bound RAS(on) state. Activation and inactivation is regulated by a series of proteins called guanine nucleotide exchange factors (GEFs), such as Son-of-Sevenless (SOS1), which promote the formation of RAS(on) state by expelling GDP and allowing GTP to bind, and GTPase activating proteins (GAPs), such as Neurofibromin 1 (NF1) and RASA1/p120GAP, which accelerate conversion of GTP to GDP to promote return to the RAS(off) state(12,14,15). These proteins are in turn controlled by extracellular stimuli, such as epidermal growth factor (EGF) or interleukin 2 (IL-2), which act through cell-surface tyrosine-kinase receptors and are augmented by tyrosine phosphatase proteins such as src homology region 2 (SH2)-containing protein tyrosine phosphatase 2 (SHP2). Notably, localization of RAS, GEFs, and GAPs to the cell membrane is required for protein-protein interaction and is driven by post-translation lipid modifications on the RAS surface. HRAS relies solely upon the enzyme farneysyltransferase to acquire lipid modifications, whereas KRAS and NRAS can utilize either farnesyltransferase or geranylgeranyltransferase(12). Upon activation, KRAS communicates through a diverse array of effector pathways, including rapidly accelerated fibrosarcomoma (RAF)-mitogen-activated protein (MAP) kinase and phosphotidylinoside 3-kinase (PI3K) pathways to drive proliferative growth signals(12,14). This delicate machinery, of which KRAS serves as the intermediary, is a major highway connecting extracellular signals to intracellular response machinery, and loss of control of any component of this pathway is associated with uninhibited cellular growth and oncogenic transformation(14).
Oncogenic KRAS relies upon mutation-associated stabilization of the active GTP-bound RAS(on) state, with mutation-specific mechanisms including inhibition of GTP hydrolysis (class 1 mutation), enhanced nucleotide exchange (class 2 mutation), or both (class 3 mutation)(15), leading to constitutive downstream proliferative signaling. For nearly 40 years, direct KRAS targeting was largely fruitless. However, characterization of the switch II pocket of KRAS in the last decade led to the successful development by Shokat and colleagues of small molecules capable of covalently binding the acquired cysteine in KRASG12C to stabilize the inactive GDP-bound RAS(off) state and induce apoptotic cell death(16–19). With this achievement, there has been a renaissance in the development of targeted therapeutics against KRAS, leading to the groundbreaking FDA approval of sotorasib in lung adenocarcinoma(8).
With publication of CodeBreaK 100 and KRYSTAL-1 trials, two inactive-state RAS(off)-selective KRASG12C inhibitors, sotorasib and adagrasib, have now demonstrated clinical efficacy in patients with advanced cancer(7,8,20). These agents were very well tolerated as single agents, but, unfortunately, durable disease control is rare, with objective response rate (ORR) and median progression-free survival (PFS) in lung adenocarcinoma of ~40% and 6 months, respectively, and ORR and PFS in other advanced cancers, including colon and pancreatic adenocarcinoma, of only ~8% and 4 months, respectively(7,8).
Mechanisms of resistance are an area of intense study and appear to fall into two basic categories: primary and acquired(21). Acquired resistance reflects disease progression despite initial clinical benefit from therapy. Seminal clinical studies now suggest an expansive array of mechanisms, best studied in lung and colon adenocarcinoma, that overcome KRASG12C inhibition(22,23) and reactivate RAS-MAPK signaling. These include: on-target secondary resistance mutations within the switch II binding pocket that impact non-covalent drug binding (e.g., KRASR68X, KRASH95X, KRASY96X(22)); on-target secondary KRASnon-G12C mutations involving the trans allele (e.g., KRASG12X, KRASG13X, KRASQ61X)(22,23); off-target activating alterations that drive bypass MAPK signaling (e.g., NRASQ61X, ALK/RET fusions, BRAFV600E; MET amplification)(22,23); and histologic transformation to an alternative resistant histology (e.g., squamous transformation)(22).
Primary resistance, best defined as lack of clinical response despite on-target binding, is also suggested to be driven by multiple mechanisms, although not proven clinically. These include rapid adaptive feedback RTK-RAS-MAPK reactivation signaling upon exposure to drug or defects in host immune response. Adaptive oncogenic signaling, although difficult to evaluate clinically, is supported by pre-clinical evidence suggesting mechanisms including preferential transcription and formation of active GTP-bound KRASG12C from non-uniform cycling between active GTP-bound and inactive GDP-bound states, as driven by epidermal growth factor (EGF) and aurora kinase (AURKA)(24); persistent upstream RTK (e.g., EGFR, FGFR, HER2, c-MET) activity with signaling through alternative wild-type RAS forms, particularly in colorectal cancer(25,26); induction of epithelial-mesenchymal transition (EMT)(27); and, particularly in lung adenocarcinoma, disinhibition of cell-cycle transition by co-occurring alterations in CDKN2A(21,28). Further, defects in host immune response are hypothesized to be related to co-occurring genomic alterations or possibly host-specific intrinsic impairment(10,21,29). Indeed, administration of selective KRASG12C inhibitors to immunocompetent mouse models induced marked immune response within tumors including IFN signaling and infiltration by CD8+ T-cells, dendritic cells, and macrophages, and was associated with durable disease control(30). In contrast, immunodeficient mice showed initial tumor regression followed by rapid recurrence, suggesting the need for an intact host immune response to induce durable disease control. Modulators of immune response, particularly in lung adenocarcinoma, are co-occurring loss-of-function mutations in KEAP1, a regulator of oxidative damage response, or in STK11, a gene involved in diverse functions including metabolism and polarity, and which induces a cold immune microenvironment with decreased T-cell infiltration, possibly by decreasing positive regulators of inflammatory cytokines or down regulation of MHC class II expression, respectively(8,31).
Altogether, it is clear that although direct KRAS targeting via selective inhibition of the inactive-state RAS(off) state is a revolutionary step in cancer therapeutics, there remain significant unmet needs including the need to develop combination therapies to potentiate the effect of currently available inhibitors; the need to develop therapeutics that target mutations other than KRASG12C, which is found rarely in colon and pancreatic cancer; and/or the need to target KRAS in a mutation-independent manner. These key issues have spurred significant scientific investment and development of novel therapeutic strategies that have created much excitement and anticipation. However, these strategies evoke concern for possible off-target and unanticipated on-target effects that warrant discussion and investigation.
Role of KRAS in non-malignant tissues
To begin, understanding the role of KRAS in non-malignant tissues is increasingly important to predict possible unanticipated on-target effects of direct KRAS targeting. Indeed, the focus of scientific study since the initial identification of KRAS as a major driver of oncogenesis has been, unfortunately and understandably, to clarify its role in malignant tissues and identify avenues to oppose its constitutive activation. Hence, little is known to date about the normal function of KRAS in tissues such as muscle, colon, bone marrow, and liver, where notable toxicities have been seen from single-agent selective KRASG12C inhibitors(7,20,32) or from indirect pan-RAS inhibitors(33–35). In the lung, KRAS has been suggested to play a role in regulating the secretory, ciliated, and squamous differentiation of the epithelium(36). However, due to the scant literature on the role of KRAS in other mature tissue, understanding of the tissue-specific role of KRAS is extrapolated from the complex phenotypes seen in inherited RASopathies, a group of developmental syndromes that are universally associated with mutations in and hyperactivation of the RAS/MAPK pathway components. The most common of these is Neurofibromatosis type 1 (NF1) (OMIM 162200) and Noonan Syndrome (OMIM 163950), which together affect approximately one in 2,500 live births. NF1 involves loss-of-function lesions in Neurofibromin, a negative regulator of RAS, and presents with optic gliomas, short stature, café-au-lait macules, neurofibromas, learning disabilities, and predisposition to multiple cancers including malignant peripheral nerve sheath tumors, gastrointestinal stromal tumors, and leukemia(12,37). Similarly, Noonan syndrome, a genetically heterogeneous syndrome associated with germline mutations in PTPN11 (which encodes SHP2), SOS1, HRAS, KRAS, and other members of the RAS-MAPK pathway, is associated with diverse phenotypes including dysmorphic faces, infertility, congenital heart disease, bleeding diatheses, short stature, and increased risk of juvenile myelomonocytic leukemia(12,37). Together, these findings highlight the diverse role of KRAS across non-malignant tissue. Further, although small molecule inhibition is unlikely to produce dramatic phenotypes as found in RASopathies, these phenotypes hint at possible on-target side effects of non-selective KRAS (or pan-RAS) blockade and reinforce the need for caution and careful assessment in clinical studies.
Next generation of direct KRAS targeting: reasons for excitement and concern
Recognizing the limitations of single-agent inactive-state RAS(off)-selective KRASG12C inhibitors, multiple ongoing studies are testing the next generation of targeted KRAS therapies with hope of increased efficacy and durable disease control. These approaches include mutation-specific direct KRAS therapies or mutation-independent strategies, each with distinct advantages and disadvantages (Figure 1).
Figure 1:
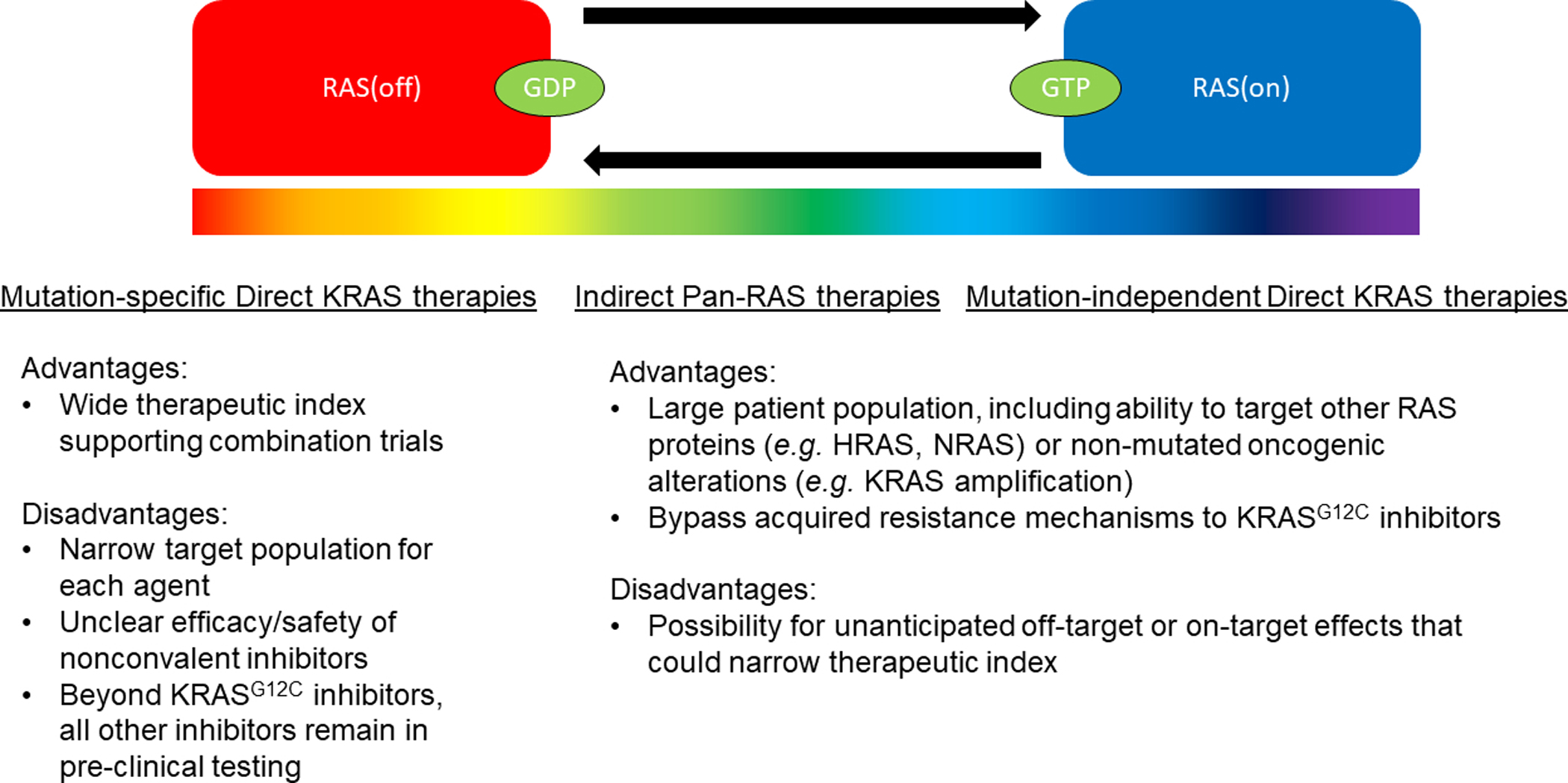
Spectrum of therapeutic approaches to target KRAS with focus upon advantages and disadvantages.
Other mutant-selective KRAS inhibitors
To begin, JNJ-74699157, formerly referred to as ARS-3248, is a selective KRASG12C inhibitor that covalently binds the acquired cysteine residue within the switch II pocket, like sotorasib and adagrasib. It demonstrated potent pre-clinical inhibition of downstream MAPK signaling(18,19,38). However, this drug showed significant toxicities in a recently published Phase I clinical trial, in sharp contrast to sotorasib and adagrasib(32). Specifically, six of 10 patients enrolled experienced treatment-emergent adverse events (TEAE) with increased blood creatinine phosphokinase (CPK) manifesting clinically as skeletal muscle weakness and myalgias that were considered dose-limiting toxicities (DLT) and prompted treatment interruption or discontinuation and eventually termination of the trial. Authors hypothesized that due to the selectivity of JNJ-74699157 for KRASG12C, toxicity is likely from off-target effects(32). However, as the mechanism of muscular toxicity is unknown and there is limited information about the role of KRAS in muscle tissue, additional validation of this hypothesis is needed. Similarly, the selective KRASG12C small molecule inhibitor LY3499446 advanced to clinical trials, but has now been terminated for an undisclosed toxicity(39). Thus, mutant-selective covalent inhibitors of KRASG12C have experienced both significant success and notable failure in clinical trials, and teasing out reasons for clinical intolerance should inform the development of 2nd generation mutant-selective inhibitors.
On the other hand, the recent identification of a noncovalent selective inhibitor (MRTX1133) of KRASG12D, the most common KRAS mutation, represents a beacon for targeted treatment for a patient population with historically poor outcomes and limited options, although this drug is still in early pre-clinical testing (40). Accordingly, the efficacy and safety of this molecule remain to be determined. Similarly, recent press releases by Revolution medicine discusses ongoing development of selective KRASG12C (RMC-6291), KRASG12D (RMC-9805), and KRASG13D (RMC-8839) using tri-complex technology (https://www.revmed.com/pipeline). However, at this time, very limited pre-clinical evidence to support efficacy and safety is available.
Combination therapies: Dual RTK-RAS-MAPK targeted therapies
Due to the excellent tolerability of sotorasib and adagrasib but rapid development of adaptive and acquired resistance, multiple clinical trials are now attempting to combine these agents with other targeted therapies to potentiate clinical efficacy and combat resistance. However, this path is fraught with both tales of success and difficulties, particularly synergistic toxicities, which temper expectations.
Notable combination therapy successes include dual inhibition of BRAF and MEK, both downstream mediators of KRAS within the MAPK pathway, in BRAF V600-driven metastatic melanoma(41) and of BRAF, MEK, and EGFR in metastatic colorectal adenocarcinoma(42) where combination therapy not only increased efficacy but also increased tolerance. In contrast, combination with MAPK and PI3K pathway inhibitors has repeatedly been associated with significant toxicities that have limited clinical translation(43,44).
Current ongoing clinical trials (CodeBreaK 101; KRYSTAL1; KRYSTAL 2) aim to combine either sotorasib or adagrasib with other RTK-RAS-MAPK/PI3K components including upstream targets (e.g., EGFR), tyrosine phosphatase proteins (e.g., SHP2), GEFs (e.g., SOS1), and downstream effectors (e.g., MEK, mTOR, and CDK4/6).
Specifically, combination trials with EGFR monoclonal antibodies, cetuximab and panitumumab, are ongoing in colorectal adenocarcinoma, and with the pan-ErbB inhibitor, afatinib, in lung adenocarcinoma. In colorectal cancer, the rationale was to target adaptive resistance via persistent EGFR signaling(26), an approach shown to be effective pre-clinically(10,24) in mouse xenograft models and is predicated on an already FDA-approved treatment(42,45). Excitingly, combination of adagrasib or sotorasib with EGFR inhibitors showed ORR of 30–40%(46,47) with minimal toxicity beyond expected anti-EGFR associated acneiform rash, in comparison to ~10% ORR seen in single-agent sotorasib and 22% for single-agent adagrasib. In contrast, the combination of sotorasib and afatinib in lung adenocarcinoma, where afatinib is approved but rarely used in EGFR-driven lung adenocarcinoma due to toxicity relative to osimertinib(48), showed moderate efficacy in a heavily pre-treated population, including those who had progressed on sotorasib monotherapy, but raised concerns for synergistic toxicity (grade ≥ 3 diarrhea: 21%; n=7)(49).
Inhibition of downstream targets of MAPK (MEK), PI3K signaling (mTOR), and cell cycle regulators (CDK4/6) to oppose feedback activation have shown synergy with sotorasib or adagrasib in mouse xenograft models(10,30). Thus, clinical protocols testing combinations in advanced cancer are ongoing. Recently reported early efficacy and safety data from combination of sotorasib and trametinib suggested synergistic activity with slightly increased ORR in colorectal cancer at 14%. However, as with afatinib, grade ≥ 3 toxicities were observed in 34% of patients with the most common being decrease in cardiac ejection fraction or increase in CPK(50). At this time, clinical efficacy and safety data for combining sotorasib with either everolimus or palbociclib has not been reported.
Finally, combination approaches of selective KRASG12C inhibitors and SHP2 and SOS1 inhibitors to oppose active/inactive state cycling and nucleotide exchange, respectively, have shown pre-clinical synergy(24,25,33,34,51) and are under active investigation. These include the combination of RMC-4630 (SHP2; Revolution Medicine; NCT04185883) with sotorasib and of TNO155 (SHP2; Novartis; NCT04330664) or BI 1701963 (SOS1; Boehringer Ingelheim; NCT04975256) with adagrasib. At this time, clinical efficacy(52) and safety data for combination therapies have not been reported. However, although these agents have been suggested to be selective to KRAS-addicted cells(34), due to their presence upstream of all RAS forms (HRAS, NRAS, KRAS), it remains to be seen if clinical exposure will have unanticipated on-target effects that will limit patient tolerance, as has been suggested by monotherapy studies where schedule changes to intermittent dosing have been required to improve tolerance(33–35,52).
Thus, targeted therapy combinations with inactive-state RAS(off) selective KRASG12C inhibitors is a dynamic space with multiple competing clinical protocols. Considering pre-clinical promise of synergy, there is substantial optimism about the ability of other agents to potentiate the effects of and overcome resistance to KRASG12C inhibitor monotherapy, which appears to now be borne out in colorectal cancer with cetuximab. However, as evidenced by combination therapy with afatinib or trametinib, regimens incorporating two small molecule inhibitors of MAPK and/or PI3K pathways are likely to show overlapping toxicities that may hinder dose titration and tolerance, as has been seen before(53). Finally, therapeutic index will be the primary concern for pan-RAS/MAPK therapies, which remains unknown at this time.
Combination therapies: Checkpoint inhibitors
Just prior to the rapid development of direct KRAS inhibitors, the oncology community saw an explosion of approvals for PD-1/PD-L1/CTLA-4 checkpoint inhibitors in nearly all advanced cancers. Although these agents are of limited use in colorectal or pancreatic cancer, approvals of checkpoint inhibitors changed treatment paradigms for and prolonged the survival of patients with advanced lung cancer, in particular those with KRAS-driven tumors(29,54,55). Indeed, significant excitement exists at the prospect of combining KRAS/MAPK inhibitors with immunotherapy to address primary resistance to monotherapy, with pre-clinical evidence suggesting that direct and indirect KRAS inhibitors can enhance tumor immunogenicity(30,51).
Notably, prior attempts to combine targeted therapy with immunotherapy in lung cancer were hindered by toxicity. Most famously, the combination of osimertinib, a 3rd generation EGFR tyrosine kinase inhibitor, and durvalumab, a PD-L1 checkpoint inhibitor, did not increase efficacy in a Phase Ib trial in EGFR-mutant lung adenocarcinoma when compared retrospectively to single-agent osimertinib(56), and 22% of patients developed interstitial lung disease leading to study suspension(57). However, unlike for sotorasib, the pre-clinical evidence to support the use of EGFR inhibitor and immunotherapy was sparse and was actually suggestive of lack of synergy as, interestingly, EGFR inhibitors enhance anti-tumor immunity through PD-L1 downregulation(58).
Thus, there is notable optimism that pre-clinical synergy will translate to durable disease control. Accordingly, sotorasib + atezolizumab, a PD-L1 antibody, is being tested in advanced lung cancer with KRASG12C as part of the Phase 1b CodeBreaK 101 trial, although no efficacy or safety data have been reported. Nevertheless, there is concern that concurrent or sequential exposure to sotorasib after checkpoint inhibitor therapy may generate severe-associated immune related adverse events(59), as suggested by case reports. Similarly, as part of the KRYSTAL-7, combination therapy with adagrasib + pembrolizumab is also now enrolling patients with advanced lung cancer with KRASG12C.
Indirect Pan-RAS inhibitors
As described above in the context of combination therapy approaches, oncogenic KRAS continues to rely upon upstream mediators, SHP2 and SOS1, for active/inactive state cycling and nucleotide exchange, respectively(24,60). Agents targeting these mediators has shown significant promise(24,34,38,60,61) and due to mutation-independent activity, have spurred excitement for a possible avenue to target all activating KRAS alterations.
Multiple SHP2 inhibitors are currently in development, including three, RMC-4630, TNO155, and JAB-3068, which have advanced to Phase II clinical trials. Nevertheless, evidence of efficacy and tolerability remains sparse. Preliminary reports from the Phase I dose-escalation study of RMC-4630 (NCT03634982) in advanced cancer, including refractory lung and colorectal cancer with KRASamp, KRASG12C, BRAFclass 3, or NF1LOF alterations demonstrated sustained inhibition of p-ERK and disease control rate (DCR) of 67% (n=12/18) among lung cancer patients with KRAS mutations(35), and, specifically in KRASG12C lung cancer, DCR of 75% (n=6/8). However, continuous dosing was associated with increased Grade ≥ 3 toxicities, particularly anemia and thrombocytopenia, which were ameliorated with intermittent dosing(35). Similarly, Phase I clinical data of TNO155 monotherapy (NCT03114319) in 118 heavily pre-treated advanced cancers, including lung and colorectal cancer, showed promising on-target inhibition on multiple different treatment schedules, as evidenced by change in DUSP6 expression,. However, no objective responses were observed at the time of that report. Further, Grade ≥ 3 cytopenias and diarrhea complicated use and as a result investigators reported that the optimal dosing schedule remains to be determined(52).
Similarly, clinical data regarding efficacy and safety of SOS1 inhibitors are limited. Currently, BI 1701963 is the only SOS1 inhibitor to reach the clinic (NCT94111458), enrolling patients with advanced cancer with KRAS mutations. Initial report on the first 31 patients to be treated identified maximal tolerated dose (MTD) due to two DLTs, Grade 4 thrombocytopenia and Grade 3 cardiomyopathy. Further, monotherapy demonstrated no objective responses(62).
Collectively, SHP2 and SOS1 are exciting targets against oncogenic RAS with strong pre-clinical rationale, both as monotherapies, and even more so in combinations(63–65). However, by their very nature, as indirect non-selective inhibitors of KRAS, there are concerns about unanticipated on-target activity, as evidenced by clear dose-associated cytopenias. It remains to be seen if this can be overcome by adjustments in treatment schedule.
Direct RAS(on) inhibitors
Finally, novel strategies to directly target active GTP-bound RAS(on) state of KRAS have recently been identified. BI-2852 (Boehringer Ingelheim) is a novel small-molecule nanomolar inhibitor that binds in the switch I/II pocket of KRAS and has been shown to block interaction of KRAS with SOS1 to impair nucleotide exchange and concurrently block interaction with the downstream effectors CRAF and PI3Kα leading to decreased p-ERK and cell proliferation(66). The switch I/II pocket is distinct from the site of KRASG12C inhibitor binding and present in both the active and inactive state of KRAS(66). However, due to concerns of indiscriminant RAS targeting of all RAS forms (HRAS, NRAS, KRAS), which has been shown to be embryonically lethal in mice(67), this compound remains in early phases of development with concerns regarding therapeutic index.
RMC-6236 (Revolution Medicine), a novel potent covalent KRAS inhibitor which binds an abundant chaperone protein, cyclophilin A, to generate a bi-complex(68,69). This bi-complex has then been shown to develop neofunction activity of binding KRAS to create an inhibitory tri-complex that sterically hinders the binding of downstream effectors to KRAS(68,69). Pre-clinical modeling has shown this tri-complex successfully inhibits GTP-bound RAS(on) state in a mutation-independent manner, including in cases of acquired resistance mutations from selective KRASG12C RAS(off) inhibitors(23). However, considering the ubiquitous nature of cyclophilin A, both intracellularly and extracellularly, it remains to be seen if not only tri-complex inhibition will have a safe therapeutic window between KRASWT and KRASMUT, but also if binary complexes of RMC-6236 and cyclophilin A will not have unanticipated off-target neofunctions or effects(70,71).
Discussion
Open optimism abounds as KRAS-targeted therapy moves into its second phase. Developmental pipelines are robust, with identification of novel therapeutic strategies to both overcome primary, adaptive, and acquired resistance to currently available mutant-specific inhibitors or to bypass them entirely with novel mutation-independent indirect pan-RAS or direct RAS(on) strategies.
Mutant-specific inhibitors, such as selective KRASG12C inhibitors, although narrow in scope, are exceedingly well tolerated and offer significant opportunity for combination regimens. However, careful attention will need to be paid to overlapping toxicities, particularly when utilizing other small-molecule inhibitors of the RTK/RAS/MAPK/PI3K pathways(53). Understandably, the “Achilles heel” of these inhibitors is their selectivity, and whether non-covalent inhibitors targeting other common oncogenic KRAS mutations, such as KRASG12D, will be successful remains to be seen.
In contrast, mutation-independent inhibitors of KRAS, either by indirect inhibition via SHP2 or SOS1 or by direct active state RAS(on) inhibition, offer broad opportunities to target all KRAS mutations, from common to uncommon, and other RAS forms such as HRAS or NRAS where no targeted agents are currently approved. However, by their very nature, this lack of selectivity enriches for unanticipated on-target effects from inhibition of wild-type RAS that warrants further study. Whether other approaches not discussed in detail in this review, such as cancer vaccines or proteolysis-targeted chimeras (PROTACs), will widen the therapeutic index of these drugs remains to be seen(72).
Thus, KRAS, a protein that only a short time ago was deemed “undruggable”, is now targetable thanks to the creativity and persistence of scientists and, as we enter the next phase of drug development, a vast new hope sits on the horizon for investigators and patients with KRAS-driven malignancies.
Footnotes
Conflict of interest: DSH receives research/grant funding from AbbVie, Adaptimmune, Adlai-Nortye, Amgen, Astra-Zeneca, Bayer, Bristol-Myers Squibb, Daiichi-Sankyo, Deciphera, Eisai, Endeavor, Erasca, F. Hoffmann-La Roche, Fate Therapeutics, Genentech, Genmab, Ignyta, Infinity, Kite, Kyowa Kirin, Lilly, LOXO, Merck, Medimmune, Mirati, Mologen, Navier, NCI-CTEP, Novartis, Numab, Pfizer, Pyramid Bio, SeaGen, Takeda, TCR2, Teckro, Turning Point Therapeutics, VM Oncology. Futher serves as in consulting/advisory role with Adaptimmune, Alpha Insights, Acuta, Alkermes, Amgen, Aumbiosciences, Axiom, Baxter, Bayer, Boxer Capital, BridgeBio, COR2ed, COG, Cowen, Ecor1, Gennao Bio, Genentech, Gilead, GLG, Group H, Guidepoint, HCW Precision, Immunogen, Infinity, Janssen, Liberium, MedaCorp, Medscape, Numab, Oncologia Brasil, Pfizer, Pharma Intelligence, POET Congress, Prime Oncology, RAIN, Seattle Genetics, ST Cube, Takeda, Tavistock, Trieza Therapeutics, Turning Point, WebMD,YingLing Pharma, Ziopharm. Finally, notes ownership interests in Molecular Match (Advisor), OncoResponse (Founder, Advisor), Telperian (Founder, Advisor).
References
- 1.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014;13(11):828–51 doi 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol 2018;15(11):709–20 doi 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- 4.Wiesweg M, Kasper S, Worm K, Herold T, Reis H, Sara L, et al. Impact of RAS mutation subtype on clinical outcome-a cross-entity comparison of patients with advanced non-small cell lung cancer and colorectal cancer. Oncogene 2019;38(16):2953–66 doi 10.1038/s41388-018-0634-0. [DOI] [PubMed] [Google Scholar]
- 5.Hayama T, Hashiguchi Y, Okamoto K, Okada Y, Ono K, Shimada R, et al. G12V and G12C mutations in the gene KRAS are associated with a poorer prognosis in primary colorectal cancer. Int J Colorectal Dis 2019;34(8):1491–6 doi 10.1007/s00384-019-03344-9. [DOI] [PubMed] [Google Scholar]
- 6.Nadal E, Chen G, Prensner JR, Shiratsuchi H, Sam C, Zhao L, et al. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J Thorac Oncol 2014;9(10):1513–22 doi 10.1097/JTO.0000000000000305. [DOI] [PubMed] [Google Scholar]
- 7.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020;383(13):1207–17 doi 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med 2021;384(25):2371–81 doi 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabari JK, Velcheti V, Shimizu K, Strickland MR, Heist RS, Singh M, et al. Activity of Adagrasib (MRTX849) in Brain Metastases: Preclinical Models and Clinical Data From Patients With KRASG12C-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res 2022. doi 10.1158/1078-0432.CCR-22-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10(1):54–71 doi 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos E, Nebreda AR. Structural and functional properties of ras proteins. FASEB J 1989;3(10):2151–63 doi 10.1096/fasebj.3.10.2666231. [DOI] [PubMed] [Google Scholar]
- 12.Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell 2017;170(1):17–33 doi 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simanshu DK, Morrison DK. A Structure is Worth a Thousand Words: New Insights for RAS and RAF Regulation. Cancer Discov 2022;12(4):899–912 doi 10.1158/2159-8290.CD-21-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasan N, Boyer JL, Herbst RS. A RAS renaissance: emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin Cancer Res 2014;20(15):3921–30 doi 10.1158/1078-0432.CCR-13-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson C, Burkhart DL, Haigis KM. Classification of KRAS-Activating Mutations and the Implications for Therapeutic Intervention. Cancer Discov 2022;12(4):913–23 doi 10.1158/2159-8290.CD-22-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 2016;15(11):771–85 doi 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- 17.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018;172(3):578–89 e17 doi 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov 2016;6(3):316–29 doi 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 19.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503(7477):548–51 doi 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ou SI, Janne PA, Leal TA, Rybkin II, Sabari JK, Barve MA, et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J Clin Oncol 2022:JCO2102752 doi 10.1200/JCO.21.02752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhave NS, Biter AB, Hong DS. Mechanisms of Resistance to KRAS(G12C)-Targeted Therapy. Cancer Discov 2021;11(6):1345–52 doi 10.1158/2159-8290.CD-20-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 2021;384(25):2382–93 doi 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov 2021;11(8):1913–22 doi 10.1158/2159-8290.CD-21-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020;577(7790):421–5 doi 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res 2020;26(7):1633–43 doi 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov 2020;10(8):1129–39 doi 10.1158/2159-8290.CD-20-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R, et al. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res 2020. doi 10.1158/1078-0432.CCR-20-2077. [DOI] [PubMed] [Google Scholar]
- 28.Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010;18(1):63–73 doi 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 29.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5(8):860–77 doi 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575(7781):217–23 doi 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 31.Ricciuti B, Arbour KC, Lin JJ, Vajdi A, Vokes N, Hong L, et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J Thorac Oncol 2022;17(3):399–410 doi 10.1016/j.jtho.2021.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Martin-Romano P, Cassier P, Johnson M, Haura E, Lenox L, et al. Phase I Study of JNJ-74699157 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. Oncologist 2022. doi 10.1093/oncolo/oyab080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savarese F, Gmachl M, Federico L, Trapani F, Gerlach D, Daniele J, et al. Vertical pathway inhibition with a SOS1:: KRAS inhibitor enhances the efficacy of KRAS G12C inhibitors, delays feedback resistance and demonstrates durable response. European Journal of Cancer 2020;138:S22. [Google Scholar]
- 34.Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov 2021;11(1):142–57 doi 10.1158/2159-8290.CD-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ou S, Koczywas M, Ulahannan S, Janne P, Pacheco J, Burris H, et al. A12 the SHP2 inhibitor RMC-4630 in patients with KRAS-mutant non-small cell lung cancer: preliminary evaluation of a first-in-man phase 1 clinical trial. Journal of Thoracic Oncology 2020;15(2):S15–S6. [Google Scholar]
- 36.Ogawa F, Walters MS, Shafquat A, O’Beirne SL, Kaner RJ, Mezey JG, et al. Role of KRAS in regulating normal human airway basal cell differentiation. Respir Res 2019;20(1):181 doi 10.1186/s12931-019-1129-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The RASopathy Family: Consequences of Germline Activation of the RAS/MAPK Pathway. Endocr Rev 2018;39(5):676–700 doi 10.1210/er.2017-00232. [DOI] [PubMed] [Google Scholar]
- 38.Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016;351(6273):604–8 doi 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palma G, Khurshid F, Lu K, Woodward B, Husain H. Selective KRAS G12C inhibitors in non-small cell lung cancer: chemistry, concurrent pathway alterations, and clinical outcomes. NPJ Precis Oncol 2021;5(1):98 doi 10.1038/s41698-021-00237-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J Med Chem 2022;65(4):3123–33 doi 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- 41.Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 2019;381(7):626–36 doi 10.1056/NEJMoa1904059. [DOI] [PubMed] [Google Scholar]
- 42.Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N Engl J Med 2019;381(17):1632–43 doi 10.1056/NEJMoa1908075. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 2012;18(8):2316–25 doi 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 44.Tolcher AW, Bendell JC, Papadopoulos KP, Burris HA 3rd, Patnaik A, Jones SF, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol 2015;26(1):58–64 doi 10.1093/annonc/mdu482. [DOI] [PubMed] [Google Scholar]
- 45.Qin S, Li J, Wang L, Xu J, Cheng Y, Bai Y, et al. Efficacy and Tolerability of First-Line Cetuximab Plus Leucovorin, Fluorouracil, and Oxaliplatin (FOLFOX-4) Versus FOLFOX-4 in Patients With RAS Wild-Type Metastatic Colorectal Cancer: The Open-Label, Randomized, Phase III TAILOR Trial. J Clin Oncol 2018;36(30):3031–9 doi 10.1200/JCO.2018.78.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fakih M, Falchook G, Hong D, Yaeger R, Chan E, Mather O, et al. 434P CodeBreaK 101 subprotocol H: Phase Ib study evaluating combination of sotorasib (Soto), a KRASG12C inhibitor, and panitumumab (PMab), an EGFR inhibitor, in advanced KRAS p. G12C-mutated colorectal cancer (CRC). Annals of Oncology 2021;32:S551. [Google Scholar]
- 47.Weiss J, Yaeger R, Johnson M, Spira A, Klempner S, Barve M, et al. LBA6 KRYSTAL-1: Adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Annals of Oncology 2021;32:S1294. [Google Scholar]
- 48.Yang Y, Liu Q, Cao L, Sun W, Gu X, Liu B, et al. Osimertinib versus afatinib in patients with T790M-positive, non-small-cell lung cancer and multiple central nervous system metastases after failure of initial EGFR-TKI treatment. BMC Pulm Med 2021;21(1):172 doi 10.1186/s12890-021-01539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gandara D, Marrone K, Govindan R, Skoulidis F, Durm G, Clarke J, et al. Abstract P05–02: A phase 1b study evaluating the combination of sotorasib, a KRASG12C inhibitor, and afatinib, a pan-ErbB tyrosine kinase inhibitor, in advanced KRAS p. G12C mutated non-small cell lung cancer (NSCLC). AACR; 2021. [Google Scholar]
- 50.Ramalingam S, Fakih M, Strickler J, Govindan R, Li BT, Goldberg S, et al. Abstract P05–01: A phase 1b study evaluating the safety and efficacy of sotorasib, a KRASG12C inhibitor, in combination with trametinib, a MEK inhibitor, in KRAS p. G12C-Mutated Solid Tumors. AACR; 2021. [Google Scholar]
- 51.Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 2021;218(1) doi 10.1084/jem.20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brana I, Shapiro G, Johnson ML, Yu HA, Robbrecht D, Tan DS-W, et al. Initial results from a dose finding study of TNO155, a SHP2 inhibitor, in adults with advanced solid tumors. J Clin Oncol 2021;39(suppl 15):3005. [Google Scholar]
- 53.Park SR, Davis M, Doroshow JH, Kummar S. Safety and feasibility of targeted agent combinations in solid tumours. Nat Rev Clin Oncol 2013;10(3):154–68 doi 10.1038/nrclinonc.2012.245. [DOI] [PubMed] [Google Scholar]
- 54.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(22):2078–92 doi 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 55.Negrao MV, Skoulidis F, Montesion M, Schulze K, Bara I, Shen V, et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J Immunother Cancer 2021;9(8) doi 10.1136/jitc-2021-002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(2):113–25 doi 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 57.Oxnard GR, Yang JC, Yu H, Kim SW, Saka H, Horn L, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol 2020;31(4):507–16 doi 10.1016/j.annonc.2020.01.013. [DOI] [PubMed] [Google Scholar]
- 58.Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol 2015;10(6):910–23 doi 10.1097/JTO.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 59.Begum P, Goldin RD, Possamai LA, Popat S. Severe Immune Checkpoint Inhibitor Hepatitis in KRAS G12C-Mutant NSCLC Potentially Triggered by Sotorasib: Case Report. JTO Clin Res Rep 2021;2(9):100213 doi 10.1016/j.jtocrr.2021.100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hofmann MH, Gerlach D, Misale S, Petronczki M, Kraut N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov 2022;12(4):924–37 doi 10.1158/2159-8290.CD-21-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 2018;20(9):1064–73 doi 10.1038/s41556-018-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson M, Gort E, Pant S, Lolkema M, Sebastian M, Scheffler M, et al. 524P A phase I, open-label, dose-escalation trial of BI 1701963 in patients (pts) with KRAS mutated solid tumours: A snapshot analysis. Annals of Oncology 2021;32:S591–S2. [Google Scholar]
- 63.Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin Cancer Res 2021;27(1):342–54 doi 10.1158/1078-0432.CCR-20-2718. [DOI] [PubMed] [Google Scholar]
- 64.Theard PL, Sheffels E, Sealover NE, Linke AJ, Pratico DJ, Kortum RL. Marked synergy by vertical inhibition of EGFR signaling in NSCLC spheroids shows SOS1 is a therapeutic target in EGFR-mutated cancer. Elife 2020;9 doi 10.7554/eLife.58204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Mohseni M, Grauel A, Diez JE, Guan W, Liang S, et al. SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci Rep 2021;11(1):1399 doi 10.1038/s41598-021-80999-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A 2019;116(32):15823–9 doi 10.1073/pnas.1904529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakamura K, Ichise H, Nakao K, Hatta T, Otani H, Sakagami H, et al. Partial functional overlap of the three ras genes in mouse embryonic development. Oncogene 2008;27(21):2961–8 doi 10.1038/sj.onc.1210956. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Z, Shokat KM. Bifunctional Small-Molecule Ligands of K-Ras Induce Its Association with Immunophilin Proteins. Angew Chem Int Ed Engl 2019;58(45):16314–9 doi 10.1002/anie.201910124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nichols RJ, Cregg J, Schulze CJ, Wang Z, Yang K, Jiang J, et al. A next generation tri-complex KRASG12C (ON) inhibitor directly targets the active, GTP-bound state of mutant RAS and may overcome resistance to KRASG12C (OFF) inhibition. AACR; 2021. [Google Scholar]
- 70.Nigro P, Pompilio G, Capogrossi MC. Cyclophilin A: a key player for human disease. Cell Death Dis 2013;4:e888 doi 10.1038/cddis.2013.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xue C, Sowden M, Berk BC. Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation. Arterioscler Thromb Vasc Biol 2017;37(6):1138–46 doi 10.1161/ATVBAHA.117.309212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nagasaka M, Potugari B, Nguyen A, Sukari A, Azmi AS, Ou SI. KRAS Inhibitors- yes but what next? Direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat Rev 2021;101:102309 doi 10.1016/j.ctrv.2021.102309. [DOI] [PubMed] [Google Scholar]