

Abstract
Rifamycin antibiotics are a valuable class of antimicrobials for treating infections by mycobacteria and other persistent bacteria owing to their potent bactericidal activity against replicating and non‐replicating pathogens. However, the clinical utility of rifamycins against Mycobacterium abscessus is seriously compromised by a novel resistance mechanism, namely, rifamycin inactivation by ADP‐ribosylation. Using a structure‐based approach, we rationally redesign rifamycins through strategic modification of the ansa‐chain to block ADP‐ribosylation while preserving on‐target activity. Validated by a combination of biochemical, structural, and microbiological studies, the most potent analogs overcome ADP‐ribosylation, restored their intrinsic low nanomolar activity and demonstrated significant in vivo antibacterial efficacy. Further optimization by tuning drug disposition properties afforded a preclinical candidate with remarkable potency and an outstanding pharmacokinetic profile.
Keywords: Antibiotic Resistance, Antibiotics, Drug Design, Mycobacterium Abscessus, Rifamycin
The first antibiotics with low nanomolar activity against the multidrug‐resistant Mycobacterium abscessus are achieved by rational redesign of rifamycins. Structure‐based modification on the rifamycin ansa‐chain, together with microbiological, biochemical, biophysical and in vivo characterizations, provides novel candidates that are 300‐fold more active than rifampicin, completely bypass the resistance, and possess promising pharmacokinetic profiles.
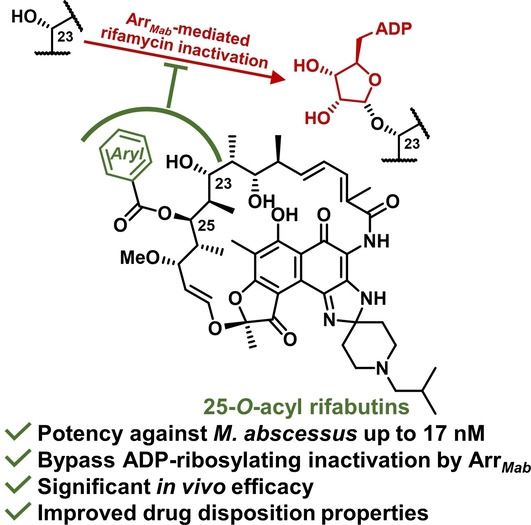
Introduction
Antimicrobial resistance (AMR) is a significant and ever‐increasing burden to public health with roughly 2–5 million annual deaths attributed to AMR worldwide. [1] Among the resistance mechanisms adopted by microorganisms, enzymatic inactivation of antibiotics is among the earliest identified, tracing back to 1940 when the first β‐lactamase was reported to “destroy penicillin”. [2] While microorganisms can degrade antibiotics by hydrolysis or redox transformation, modification of antibiotics via group transfer represents the most chemically diverse drug inactivation mechanism.[ 3 , 4 ] Many antibiotics including aminoglycosides, macrolides, and lincosamides are prone to group‐transfer inactivation via transformations such as acylation, phosphorylation, and glycosylation. Within this family of antibiotic‐modifying chemistry, ADP‐ribosylation of the rifamycins represents a particularly novel and noteworthy member.
The natural product‐derived rifamycin antibiotics function as inhibitors of bacterial transcription by allosteric binding to bacterial DNA‐dependent RNA polymerases (RNAPs) preventing elongation of the nascent RNA and exhibit exceptional sterilizing activity against Mycobacterium tuberculosis and many other pathogens.[ 5 , 6 ] With nanomolar antibacterial activity, rifamycins are specifically known for their clinical value in the treatment of persistent bacterial and mycobacterial infections due to their remarkable potency against bacterial persisters, often in biofilms and under dormant states, that are highly tolerant to most antibiotics. However, rifamycin drugs are clinically ineffective against Mycobacterium abscessus, an emerging nontuberculous mycobacteria that causes an often fatal pulmonary infection with no reliable treatment options since M. abscessus is intrinsically drug resistant to virtually all antibacterial classes. [7] The rifamycin resistance in M. abscessus is caused by a group‐transfer inactivation mechanism via a rifamycin ADP‐ribosyltransferase (Arr). [7] With NAD+ as the donor, Arr catalyzes the formation of the ADP‐ribosyl‐oxocarbenium intermediate and transfers this intermediate regioselectively to C23‐OH on the rifamycin polyketide ansa‐chain. This, in turn, prevents binding to the bacterial RNAP and significantly reduces the rifamycin potency (Figure 1).[ 7 , 8 , 9 , 10 ] As a consequence of this, for example, the semi‐synthetic rifamycin drug rifabutin, which exhibits low nanomolar activity against M. tuberculosis and many gram‐positive pathogens, displays only modest micromolar activity against M. abscessus.[ 11 , 12 ] Arr in M. abscessus and some other bacteria are by far the only known ADP‐ribosyltransferases that target small molecules, while most bacterial ADP‐ribosyltransferases are protein toxins that function through post‐translational ADP‐ribosylation of host proteins and are key virulence factors in Corynebacterium diphtheriae, Vibrio cholerae, Bordetalla pertussis, and Clostridrium botulinum. [13] Therefore, rifamycin ADP‐ribosylation represents an unprecedented and novel mechanism of antimicrobial resistance. [14]
Figure 1.
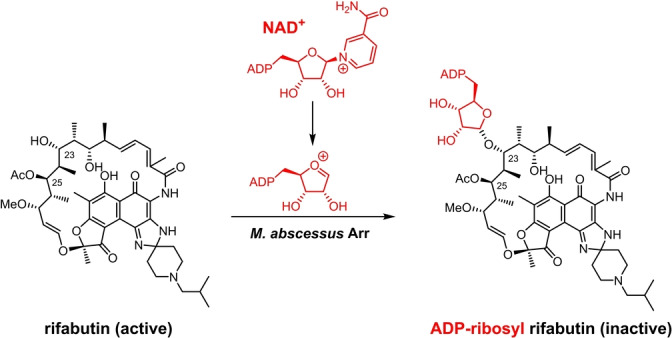
Rifabutin is inactivated in M. abscessus. Arr of M. abscessus catalyzes the formation of ADP‐ribosyl‐oxocarbenium intermediate from NAD+ and the consecutive ADP‐ribosylation on C23‐OH.
As a means to circumvent such pathogenic resistance, we hypothesized that rifamycins could be rationally modified to prevent Arr‐mediated ADP‐ribosylation at the C23 position while maintaining bacterial RNAP target engagement. To determine possible modification sites on rifamycins, we investigated the rifamycin binding mode in mycobacterial RNAP using the 3D structure of M. tuberculosis RNAP (RNAP Mtb ) in complex with rifampicin (PDB: 5UHB, Figure S1), which shares a 97 % sequence identity to the M. abscessus RNAP (RNAP Mab ) rifamycin binding site. Around the inactivation position C23‐OH, C21‐OH and C22‐CH3 are closely enveloped by the binding pocket while C24‐CH3 and C25‐OAc face larger spaces in the pocket. This led us to consider the C24 and C25 positions as two possible sites for modification, given they do not appear to compromise ligand binding. We reasoned that the C25 position was a suitable position for derivatization since C25‐OH, generated through acetyl hydrolysis, [15] could serve as a handle for chemical modifications.
The modification of rifamycin C25 position was previously explored. In the work by Combrink and co‐workers, various carbamates were installed on 3‐morpholino rifamycin S to evade ADP‐ribosylation and increase potency against Mycobacterium smegmatis and M. abscessus.[ 16 , 17 , 18 ] Two C‐25 malonates from 3‐morpholino rifamycin S were also reported by us [7] and other groups [19] to exhibit improved activity against a panel of different bacteria including M. abscessus. However, most of the reported C25‐modified analogs were demonstrated with only micromolar activity, which is not better than current anti‐M. abscessus drugs such as amikacin. All the reported C‐25 modifications were based on 3‐morpholino rifamycin S as a simplified scaffold, while there is no report on the derivatization of marketed rifamycin drugs including rifampicin and rifabutin, which is rewarding and will benefit from the already optimized properties including superb activity, favorable pharmacokinetics (PK), and lowered toxicity. Furthermore, since modifications on the rifamycin ansa‐chain simultaneously likely affect the binding to both Arr and RNAP, the balance of structural change to disturb interactions with Arr while maintaining RNAP engagement can be subtle. A detailed structure‐based analysis that can provide comprehensive rationale and guidance for the modification and structural optimization is valuable, but still absent.
Results and Discussion
To explore whether C25 modification affects the binding of rifamycins against RNAP Mab , molecular docking studies were conducted on a homology model of RNAP Mab that uses the rifamycin‐bounded RNAP Mtb structure 5UHB as the template. The binding of an archetypical ligand 25‐O‐benzoyl rifabutin (5 a, Figure 2A), in which a bulkier benzoyl moiety replaced the C25 acetyl group, was investigated and demonstrated a highly consistent binding mode compared to unmodified rifabutin in the conserved rifamycin binding site (Figure S2).
Figure 2.

Homology‐model‐guided rifamycin design depicts C‐25 modification blocking interaction with Arr Mab . A) The structure of the archetypical compound 25‐O‐benzoyl rifabutin (5 a), where the C25 acetyl group of rifabutin is replaced by a benzoyl group. B) The overall structure of Arr Mab homology model complexed with the ADP‐ribosyl‐oxocarbenium intermediate (blue) and 5 a (cyan). C) Proposed binding modes of rifabutin (magenta) and 5 a (cyan) to the Arr Mab homology model. The distance between C23‐OH and oxocarbenium‐C1′ is measured and shown as a purple dashed line.
To determine whether C25 modification on rifamycins can disturb the interaction with M. abscessus Arr (Arr Mab ), a homology model of Arr Mab was generated based on the Arr of M. smegmatis (64 % sequence identity, PDB: 2HW2). The ADP‐ribosyl‐oxocarbenium intermediate was positioned in this model and the complex was further optimized using molecular dynamics simulations (Figure 2B). A molecular docking analysis of 5 a and rifabutin using this Arr Mab model suggested that in contrast to rifabutin, 5 a formed a catalytically incompetent complex due to the increase in the distance between C23‐OH and NAD+‐C1′ from 4.9 Å (rifabutin) to 7.1 Å (5 a) (Figure 2C). With these results, we hypothesized that rifamycin analogs with modifications on the C25 position could be well accommodated by RNAP but were unable to undergo ADP‐ribosylation by Arr, thereby restoring their high potency against M. abscessus.
To experimentally validate this hypothesis, we synthesized a series of C‐25 substituted rifamycin analogs containing a range of sterically varied alkyl, aryl, and heteroaryl esters (Scheme 1). Rifabutin was selected as the template since it is the most potent rifamycin against M. abscessus, [7] has the lowest P450 induction potential and most favorable PK profile among clinically approved rifamycin antibiotics. [20] Prior to initiating modifications to C25, the C21,23‐diol was protected rendering an acetonide‐containing rifabutin 2. Carefully controlled methanolysis of 2 afforded the deacetylated intermediate 3 using potassium carbonate to minimize competitive lactam opening due to the electron‐withdrawing naphthoquinone core, which enhances the reactivity of the amide linkage. [15] Acylation of the newly liberated C25‐OH proved to be extremely challenging. The steric hindrance from the neighboring acetonide group prevented acylation with less‐reactive reagents; under stronger acylation conditions, on the other hand, the nucleophilic spiroimidazopiperidine N‐3 amine also reacted and lowered the regioselectivity. After extensive experimentation (Table S1), we found that the regioselective esterification on C25‐OH could only be achieved using a large excess of acid anhydride or mixed anhydride, formed in situ from acids and pivaloyl chloride. The acetonide group was successfully cleaved using CSA in methanol to yield final rifabutin analogs. For the most sterically demanding substrate, the less hindered 25‐O‐desacetyl rifabutin 6 [15] was directly acylated using acid anhydrides to generate desired products after careful separation. The structures of all the final products are verified by 1D and 2D NMR. 1H‐1H NOESY spectra were acquired for analogs 5 e, 5 g, and 5 i (see Table 1) as representatives to ensure the configurations of the chiral centers, especially C25, are maintained throughout the synthesis (see Supporting Information).
Scheme 1.
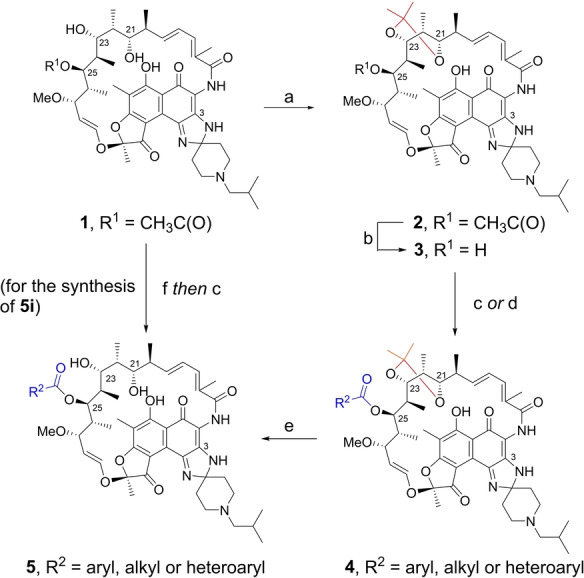
Synthesis of 25‐O‐acyl rifabutin analogs: a) 2,2‐dimethoxypropane, CSA, acetone, room temperature, 2 h, 68 %; b) K2CO3, MeOH, 50 °C, 48 h, 59 %; c) RC(O)OC(O)R, DMAP, 1,2‐dichloroethane, room temperature or 50 °C, 72–96 h; d) RCOOH, pivaloyl chloride, triethylamine, DMAP, DCM, 0 °C to room temperature, 4 h; e) CSA, MeOH, room temperature, 0.5 h; f) NaOH, ZnCl2, MeOH, room temperature, overnight, 71 %. For 5 a–5 h and 5 j–5 o, the yields were 7–72 % over two steps (c or d, and then e); For 5 i, the yield was 6 % with procedure c from 25‐O‐desacetyl rifabutin 6. CSA=camphorsulfonic acid.
Table 1.
Activity of the synthetic rifabutin analogs against WT and Δarr M. abscessus.[a]
|
compound |
R2 (see Scheme 1) |
WT[b] Mab MIC |
Δarr Mab MIC |
MIC ratio[c] |
|---|---|---|---|---|
|
clarithromycin |
|
1450 |
1800 |
0.7 |
|
rifampicin |
– |
5300 |
260 |
20.4 |
|
rifabutin |
Me |
1150 |
31 |
39.3 |
|
5 a |
C6H5‐ |
53 |
60 |
0.9 |
|
5 b |
o‐MeC6H4‐ |
43 |
64 |
0.7 |
|
5 c |
m‐MeC6H4‐ |
75 |
65 |
1.2 |
|
5 d |
p‐MeC6H4‐ |
200 |
110 |
1.8 |
|
5 e |
o‐FC6H4‐ |
60 |
58 |
1.0 |
|
5 f |
o‐ClC6H4‐ |
43 |
49 |
0.9 |
|
5 g |
o‐OMeC6H4‐ |
30 |
30 |
1.0 |
|
5 h |
o‐CF3C6H4‐ |
100 |
120 |
0.8 |
|
5 i |
o‐PhC6H4‐ |
260 |
140 |
1.8 |
|
5 j |
m‐FC6H4‐ |
55 |
90 |
0.6 |
|
5 k |
2‐Me‐butyryl |
1100 |
28 |
39.3 |
|
5 l |
2‐Et‐butyryl |
1150 |
49 |
23.5 |
|
5 m |
3‐Pyridyl |
24 |
26 |
0.9 |
|
5 n |
5‐Pyrimidyl |
18 |
25 |
0.72 |
|
5 o |
2‐Thiazolyl |
17 |
17 |
1.0 |
[a] All MIC values are given in nM. [b] M. abscessus ATCC 19977. [c] MIC ratio=WT MIC/Δarr strain MIC.
To reveal the impact of C‐25 modifications on the potency of the synthetic rifabutin analogs, the minimum inhibitory concentration that results in 90 % growth inhibition (MIC) of these analogs against M. abscessus was determined (Table 1). A wild‐type (WT) M. abscessus ATCC 19977 and an isogenic arr‐deleted (Δarr) M. abscessus strain were used in parallel to assess the resistance phenotype. Clarithromycin, rifampicin and rifabutin were included as controls. Compound 5 a containing a 25‐O‐benzyol group had an MIC of 53 nM, which is 20 and 100 times lower than rifabutin and rifampicin, respectively. More importantly, 5 a was equally potent against WT and Δarr M. abscessus strains, meaning that this compound was no longer inactivated by Arr. Compounds with ortho‐, meta‐ and para‐methyl benzoates (5 b–5 d) demonstrated that ortho‐substitution was preferred. Further investigation on different ortho‐substituted C‐25 benzoates clearly delineated the impact of steric bulkiness on the activity: small groups (F, Cl and OMe 5 e–5 g) could be appended without compromising the activity, while bulkier groups (CF3 and Ph 5 h and 5 i) caused 2‐ and 5‐fold loss of potency, respectively, compared to 5 a. Analog 5 j with the small F on the meta position of C25‐benzoate also retained the activity. On the other hand, analogs 5 k and 5 l with small alkyl groups display only modest micromolar MIC values against WT M. abscessus. Potent activity is only achieved against the isogenic Δarr deletion strain indicating 5 k and 5 l are inactivated by Arr. Furthermore, compounds 5 a, 5 b, and 5 j were shown to maintain potent activity against a panel of drug‐resistant M. abscessus clinical isolates (Table S2). Altogether, these results demonstrate C‐25 modification of rifabutin as a viable strategy to significantly increase the potency and effectively block rifamycin inactivation by ADP‐ribosylation.
In the following activity screening using a rifamycin‐resistant M. abscessus mutant RFB‐R1 carrying an RNAP point mutation, [7] analogs 5 a, 5 b, and 5 j lost detectable activity (Table S3), suggesting their on‐target activity through RNAP inhibition. To further understand how the C‐25 modification on rifabutin affects binding to RNAP, the crystal structure of a representative compound 5 a complexed with RNAP Mtb was solved at a resolution of 3.9 Å [26] (see Table S4 for the data‐collection and refinement statistics). The three‐dimensional structure reveals that 5 a adopts a highly similar binding mode to rifampicin in the same binding site (Figure S3) and preserves all the essential hydrogen bonds from C1‐O, C8‐OH, C21‐OH and C23‐OH (Figure 3A). A noteworthy difference in the binding of 5 a is that the C‐25 benzoate forms a unique π‐stacking interaction with Phe439, with the two phenyl rings demonstrating a “close parallel displaced” geometry (Figure 3B). These results indicate that the molecular target RNAP can accommodate the bulky C‐25 modification by forming novel ligand‐target interactions. Interestingly, a small cleft formed by Phe439 and Arg173 was found to envelop the C‐25 benzoate of 5 a (Figure 3C), which was not observed in the structure of the previous RNAP‐rifampicin complex. This cleft may account for the lower potency of analogs with phenyl‐ and trifluoromethyl‐substituted benzoates since these extremely large substituents cannot fit in this small cleft, thereby decreasing the overall binding affinity. Analogs 5 b–5 g were proposed to bind to RNAP with highly similar modes to 5 a, evaluated by molecular docking studies using the RNAP Mab homology model mentioned above (Figure S4).
Figure 3.

A) The active site structure of 5 a (orange) bound to RNAP Mtb . Residues involved in important hydrogen bonds are shown as cyan sticks. H‐bonds are shown as purple dashed lines. B) C‐25 benzoate (C25‐OBz) of 5 a (orange) and F439 (cyan) form a π‐stacking interaction. C) C‐25 benzoate of 5 a (orange) is positioned in a cleft formed by F439 and R173. The cleft is demonstrated in blue.
To biochemically validate the effect of C‐25 modifications on ADP‐ribosylation, we cloned, expressed, and purified recombinant Arr Mab and developed a liquid chromatography mass spectrometry (LC‐MS) enzymatic assay to quantify the rifamycins and ADP‐ribosylated adducts. Upon incubation with Arr Mab and NAD+, rifampicin and rifabutin were completely converted in 40 minutes (Figure 4 and Figure S5) and the new peaks were confirmed as ADP‐ribosyl adducts (m/z=694.90 for ADP‐ribosyl rifabutin, see Table S5 for MS identifications for all the LC signals). By contrast, no transformation was observed for 5 a and 5 b which do not exhibit a loss of activity against WT M. abscessus with a functioning antibiotic‐inactivating Arr (Figure 4 and Figure S5). Meanwhile, the small‐alkyl substituted analogs 5 k and 5 l, with considerable MIC shifts against WT and Δarr M. abscessus strains, were fully converted through ADP‐ribosylation (Figure S5). These results further demonstrate that 25‐O‐benzoyl rifabutin compounds cannot be modified by Arr Mab .
Figure 4.
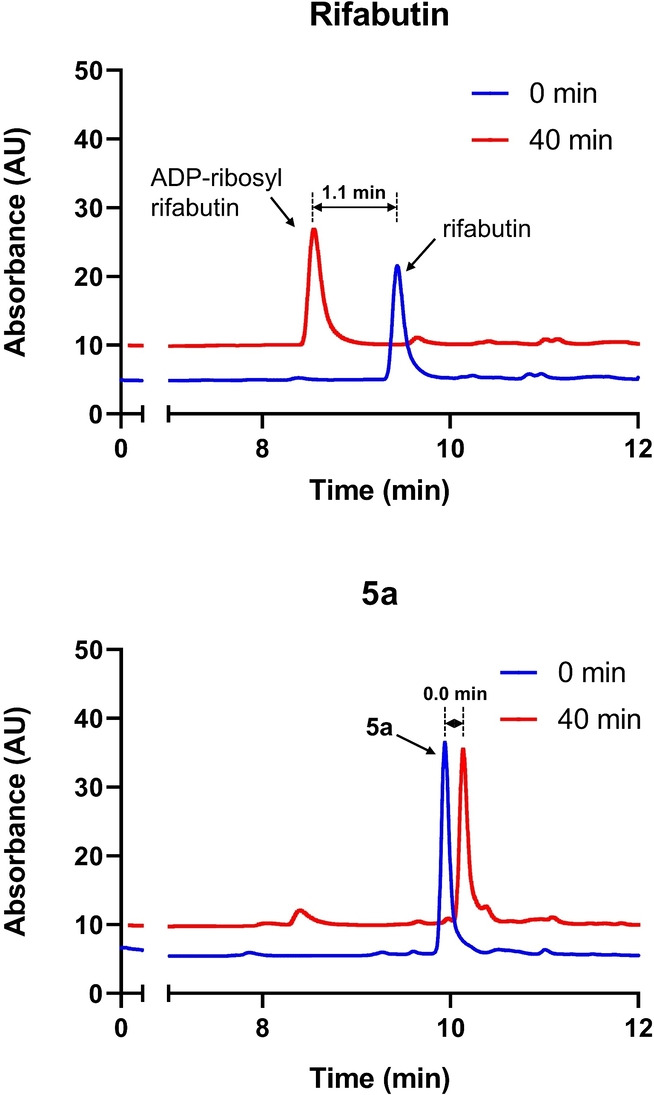
In vitro characterization of ADP‐ribosylation of rifamycins using overexpressed Arr Mab . All the peaks were identified by MS (Table S5). The retention time difference of the peaks is labelled. Rifabutin was fully converted into the ADP‐ribosyl adduct upon incubation in a 40‐minute time course. By contrast, no Arr Mab ‐catalyzed transformation was observed for the synthetic compound 5 a under the same incubation condition.
The promising in vitro activity of the rifabutin analogs led to the investigation of their in vivo PK properties. Candidates 5 b and 5 j were administered intraveneously (i.v.) and orally (p.o.) to CD1 mice to assess their PK parameters and determine the optimal dosing regimen for future efficacy studies (Table 2). Both compounds exhibited an improved volume of distribution (V d) and reduced clearance (CL) relative to rifabutin, resulting in a prolonged half‐life (t 1/2) and greater in vivo drug exposure as measured by the area‐under‐the‐curve (AUC) in the concentration‐time curve. C25‐desacetylation by esterases is a major metabolic pathway of rifamycin drugs.[ 21 , 22 ] To assess whether candidates 5 b and 5 j were also prone to enzymatic ester hydrolysis, we evaluated the amount of 25‐O‐desacetyl rifabutin 6 in mouse plasma after intraveneous and oral administration. Both 5 b and 5 j exhibited high metabolic stability to potential hydrolysis in either plasma or gut/liver, as less than 0.2 % of 6 was detected for both candidates in both administration routes (Table 2). Interestingly, hydrolysis of rifabutin C‐25 acetate was not obvious either in this mouse model with only 0.4 % of 6 detected in both administration routes. Whether the rifabutin scaffold can provide additional metabolic stability against rifamycin C25‐desacylation remains to be determined.
Table 2.
Important PK parameters of rifabutin and selected analogs.[a]
|
compound |
rifabutin |
5 b |
5 j |
5 m |
|---|---|---|---|---|
|
WT MIC [nM] |
1150 |
43 |
55 |
24 |
|
V d [L kg−1] |
3.51 |
1.1 |
1.9 |
1.6 |
|
t 1/2 [h] |
4.0 |
10.3 |
11.7 |
6.8 |
|
CL [mL/(kg×min)] |
10.3 |
1.5 |
2.7 |
3.0 |
|
C‐25 deacylation i.v. [%] |
0.4 |
n.d. |
n.d. |
0.2 |
|
C‐25 deacylation p.o. [%][c] |
0.4 |
0.2 |
n.d. |
0.01 |
|
PPB [% bound] |
96.24 |
99.99 |
99.98 |
97.95 |
|
unbound fraction [%] |
3.76 |
0.01 |
0.02 |
2.05 |
|
fAUC/MIC[b] |
0.22 |
0.04 |
0.06 |
28.2 |
[a] See Table S6 for the complete data and errors. [b] fAUC/MIC was measured based on p.o. doses of 10 mg kg−1. [c] For both administrative routes, deacylation %=AUC 6 /AUCcompound×100 %, n.d.: undetectable.
With the favorable PK profiles in hand, we next sought to characterize the in vivo efficacy of the candidate using an infected mouse model. [23] Compound 5 j, with the PK parameters validated, was selected as the candidate. 5 j, clarithromycin, rifabutin, and vehicle were orally administered once daily to M. abscessus‐infected mice for 10 consecutive days, and then the lung and spleen bacterial load was assessed. The efficacy of a drug was defined as a statistically significant reduction of colony‐forming unit (CFU) in a study group relative to the vehicle control at the end of the experiment (Figure 5). In this model, candidate 5 j significantly reduced the bacterial load in lung by 10‐fold and achieved in vivo bactericidal activity in a similar level to the positive control clarithromycin, which is a widely used anti‐M. abscessus drug. The comparable efficacy of 5 j and clarithromycin was also reflected in the spleen CFU reduction. However, 5 j did not exhibit improved efficacy relative to rifabutin, as both compounds reduced lung and spleen CFU counts to a similar extent.
Figure 5.
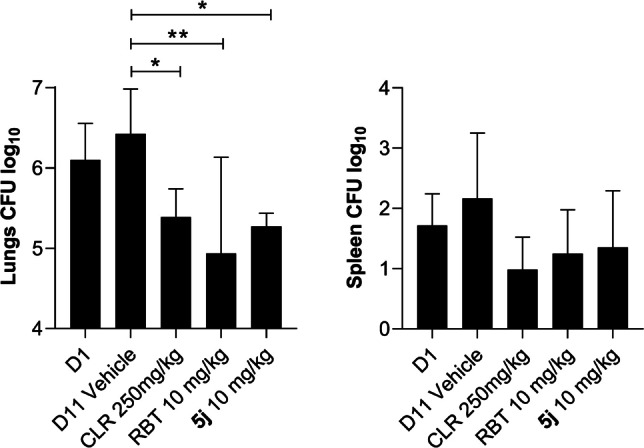
Analog 5 j exhibited bactericidal in vivo efficacy at 10 mg kg−1. Animals infected with M. abscessus underwent drug treatment for 10 consecutive days. Drugs were administered once daily to groups of 6 mice per study group. At 11 days postinfection, organ homogenates were plated on agar to determine the bacterial load. Results were analyzed using one‐way analysis of variance (ANOVA) multicomparison and Dunnett's posttest. *, P<0.05; **, P<0.01; ***, P<0.001. D1: Day‐1. D11: Day‐11. CLR: clarithromycin. RBT: rifabutin.
It was unexpected that the promising properties of 5 j, including in vitro nanomolar antibacterial activity and good in vivo PK properties, did not translate into improved in vivo efficacy compared to rifabutin. Therefore, we were prompted to look for possible causes. A further investigation of the pharmacokinetic/pharmacodynamic (PK/PD) profiles of the candidates led to plasma protein binding (PPB) as a likely limiting factor. Both 5 b and 5 j were highly protein bound as measured in a PPB assay and resulted in a significantly lower fraction of unbound drug in plasma than rifabutin (126‐ and 380‐fold, Table 2). Given that the free concentration of antimycobacterial drugs correlates well with their in vivo efficacy, [24] we selected the ratio of the area under the unbound drug concentration‐time profile to MIC (fAUC/MIC) as the PK/PD index driving efficacy, [25] and explored rifabutin analogs with higher fAUC/MIC in the following compound optimization.
In the characterization of the synthetic rifabutin analogs, it was discovered that switching C25‐acetate to benzoates increased the lipophilicity of the molecule, which is reflected by a longer retention time on LC. We therefore suspected that the lipophilicity of the analogs contributed to their PPB. To further optimize the candidate aimed at reducing the lipophilicity of C‐25 substituents while maintaining favorable potency, the phenyl group on 5 a was replaced with bioisosteric heterocycles to generate analogs with 3‐pyridyl (5 m), 5‐pyrimidyl (5 n) and 2‐thiazolyl (5 o) groups. All the heterocyclic analogs were found to be even more potent with MICs as low as 17 nM and not susceptible to ADP‐ribosylation (Table 1). In the PK characterization, the candidate compound 5 m not only maintained a favorable V d, low clearance, and high stability against C‐25 ester cleavage but also had an at least 100‐fold increase in the unbound fraction in plasma. The drastically increased plasma free fraction of 5 m, in combination with its low MIC, led to an astonishing fAUC/MIC value of 28.2, 128 times higher than rifabutin (Table 2). Taken together, the results delivered 5 m as a promising candidate with an excellent potency and superior PK properties.
Conclusion
In conclusion, we have rationally redesigned rifabutin to restore the low nanomolar antimycobacterial activity of rifamycin antibiotics and extend their clinical utility against intrinsically multidrug‐resistant M. abscessus by evading a novel rifamycin resistance mechanism. Structure‐based derivatization at the C‐25 position of rifabutin afforded analogs that are more than a hundred‐fold more potent than the widely used rifampicin and are no longer susceptible to the primary rifamycin resistance in M. abscessus through ADP‐ribosylation. X‐ray crystallography and molecular docking studies suggest that additional ligand‐target interactions contribute to the favored on‐target activity. The ability to overcome Arr‐mediated resistance was validated using an in vitro biochemical assay to directly detect ADP‐ribosylation, with results congruent with the observed microbiological activity using WT and Δarr M. abscessus strains. One representative compound also demonstrated strong in vivo efficacy comparable to the anti‐M. abscessus drug clarithromycin. In a further stage of modification, three heterocyclic C‐25 ester analogs were strategically designed based on compounds’ free fraction in plasma, an important driving factor. Compound 5 m emerged as an exemplary candidate with potent in vitro antibacterial activity, outstanding drug disposition and greatly improved in vivo PK properties. Given this promising profile, characterization of the in vivo efficacy of 5 m is ongoing in our labs and will be revealed soon.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The NMR analysis in this research was supported by Grant 1S10OD021536 from National Institute of General Medical Sciences. The ESI‐MS measurements were performed at Mass Spectrometry Laboratory of Department of Chemistry at the University of Minnesota. We thank the in vivo and analytical chemistry teams of the Center of Discovery and Innovation for their excellent technical assistance. We are grateful to Wei Chang Huang (Taichung Veterans General Hospital, Taichung, Taiwan) for providing M. abscessus Bamboo and to Sung Jae Shin (Department of Microbiology, Yonsei University College of Medicine, Seoul, South Korea) and Won‐Jung Koh (Division of Pulmonary and Critical Care Medicine, Samsung Medical Center, Seoul, South Korea) for providing M. abscessus K21. Research reported in this work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Numbers R01AI132374 (to T.D.), U19AI142731 (to T.D., J.F.S., R.H.E., and V.D.) and S10 OD023524 (to V.D.). We thank the Stanford Synchrotron Radiation Lightsource for beamline access and support with X‐ray data collection. The structural biology work also was supported by National Institutes of Health grant GM041376 to R.H.E.
T. Lan, U. S. Ganapathy, S. Sharma, Y.-M. Ahn, M. Zimmerman, V. Molodtsov, P. Hegde, M. Gengenbacher, R. H. Ebright, V. Dartois, J. S. Freundlich, T. Dick, C. C. Aldrich, Angew. Chem. Int. Ed. 2022, 61, e202211498; Angew. Chem. 2022, 134, e202211498.
Contributor Information
Prof. Dr. Thomas Dick, Email: thomas.dick.cdi@gmail.com.
Prof. Dr. Courtney C. Aldrich, Email: aldri015@umn.edu.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Murray C. J. L., Ikuta K. S., Sharara F., Swetschinski L., Robles Aguilar G., Gray A., Han C., Bisignano C., Rao P., Wool E., Johnson S. C., Browne A. J., Chipeta M. G., Fell F., Hackett S., Haines-Woodhouse G., Kashef Hamadani B. H., Kumaran E. A. P., McManigal B., Agarwal R., Akech S., Albertson S., Amuasi J., Andrews J., Aravkin A., Ashley E., Bailey F., Baker S., Basnyat B., Bekker A., Bender R., Bethou A., Bielicki J., Boonkasidecha S., Bukosia J., Carvalheiro C., Castañeda-Orjuela C., Chansamouth V., Chaurasia S., Chiurchiù S., Chowdhury F., Cook A. J., Cooper B., Cressey T. R., Criollo-Mora E., Cunningham M., Darboe S., Day N. P. J., De Luca M., Dokova K., Dramowski A., Dunachie S. J., Eckmanns T., Eibach D., Emami A., Feasey N., Fisher-Pearson N., Forrest K., Garrett D., Gastmeier P., Giref A. Z., Greer R. C., Gupta V., Haller S., Haselbeck A., Hay S. I., Holm M., Hopkins S., Iregbu K. C., Jacobs J., Jarovsky D., Javanmardi F., Khorana M., Kissoon N., Kobeissi E., Kostyanev T., Krapp F., Krumkamp R., Kumar A., Kyu H. H., Lim C., Limmathurotsakul D., Loftus M. J., Lunn M., Ma J., Mturi N., Munera-Huertas T., Musicha P., Mussi-Pinhata M. M., Nakamura T., Nanavati R., Nangia S., Newton P., Ngoun C., Novotney A., Nwakanma D., Obiero C. W., Olivas-Martinez A., Olliaro P., Ooko E., Ortiz-Brizuela E., Peleg A. Y., Perrone C., Plakkal N., Ponce-de-Leon A., Raad M., Ramdin T., Riddell A., Roberts T., Robotham J. V., Roca A., Rudd K. E., Russell N., Schnall J., Scott J. A. G., Shivamallappa M., Sifuentes-Osornio J., Steenkeste N., Stewardson A. J., Stoeva T., Tasak N., Thaiprakong A., Thwaites G., Turner C., Turner P., van Doorn H. R., Velaphi S., Vongpradith A., Vu H., Walsh T., Waner S., Wangrangsimakul T., Wozniak T., Zheng P., Sartorius B., Lopez A. D., Stergachis A., Moore C., Dolecek C., Naghavi M., Lancet 2022, 399, 629–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abraham E. P., Chain E., Nature 1940, 146, 837–837. [Google Scholar]
- 3. Wright G. D., Adv. Drug Delivery Rev. 2005, 57, 1451–1470. [DOI] [PubMed] [Google Scholar]
- 4. Awasthi D., Freundlich J. S., Trends Microbiol. 2017, 25, 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rothstein D. M., Cold Spring Harbor Perspect. Med. 2016, 6, a027011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarathy J. P., Via L. E., Weiner D., Blanc L., Boshoff H., Eugenin E. A., C. E. Barry 3rd , Dartois V. A., Antimicrob. Agents Chemother. 2018, 62, e02266-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ganapathy U. S., Lan T., Krastel P., Lindman M., Zimmerman M. D., Ho H., Sarathy J. P., Evans J. C., Dartois V., Aldrich C. C., Dick T., Antimicrob. Agents Chemother. 2021, 65, e00978-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rominski A., Roditscheff A., Selchow P., Böttger E. C., Sander P., J. Antimicrob. Chemother. 2017, 72, 376–384. [DOI] [PubMed] [Google Scholar]
- 9. Schäfle D., Selchow P., Borer B., Meuli M., Rominski A., Schulthess B., Sander P., Antimicrob. Agents Chemother. 2021, 65, e02215-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baysarowich J., Koteva K., Hughes D. W., Ejim L., Griffiths E., Zhang K., Junop M., Wright G. D., Proc. Natl. Acad. Sci. USA 2008, 105, 4886–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chew K. L., Octavia S., Go J., Ng S., Tang Y. E., Soh P., Yong J., Jureen R., Lin R. T. P., Yeoh S. F., Teo J., J. Antimicrob. Chemother. 2021, 76, 973–978. [DOI] [PubMed] [Google Scholar]
- 12. Aziz D. B., Low J. L., Wu M.-L., Gengenbacher M., Teo J. W. P., Dartois V., Dick T., Antimicrob. Agents Chemother. 2017, 61, e00155-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simon N. C., Aktories K., Barbieri J. T., Nat. Rev. Microbiol. 2014, 12, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wencewicz T. A., J. Mol. Biol. 2019, 431, 3370–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kump W., Bickel H., Helv. Chim. Acta 1973, 56, 2323–2347. [DOI] [PubMed] [Google Scholar]
- 16. Combrink K. D., Denton D. A., Harran S., Ma Z., Chapo K., Yan D., Bonventre E., Roche E. D., Doyle T. B., Robertson G. T., Lynch A. S., Bioorg. Med. Chem. Lett. 2007, 17, 522–526. [DOI] [PubMed] [Google Scholar]
- 17. Combrink K. D., Ramos A. R., Spring S., Schmidl S., Elizondo K., Morin P., De Jesus B., Maurer F. P., Bioorg. Med. Chem. Lett. 2019, 29, 2112–2115. [DOI] [PubMed] [Google Scholar]
- 18. Paulowski L., Beckham K. S. H., Johansen M. D., Berneking L., Van N., Degefu Y., Staack S., Sotomayor F. V., Asar L., Rohde H., Aldridge B. B., Aepfelbacher M., Parret A., Wilmanns M., Kremer L., Combrink K., Maurer F. P., PNAS Nexus 2022, 1, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wehrli W., Zimmermann W., Kump W., Tosch W., Vischer W., Zak O., J. Antibiot. 1987, 40, 1733–1739. [DOI] [PubMed] [Google Scholar]
- 20. Crabol Y., Catherinot E., Veziris N., Jullien V., Lortholary O., J. Antimicrob. Chemother. 2016, 71, 1759–1771. [DOI] [PubMed] [Google Scholar]
- 21. Jamis-Dow C. A., Katki A. G., Collins J. M., Klecker R. W., Xenobiotica 1997, 27, 1015–1024. [DOI] [PubMed] [Google Scholar]
- 22. Aristoff P. A., Garcia G. A., Kirchhoff P. D., Showalter H. D., Tuberculosis 2010, 90, 94–118. [DOI] [PubMed] [Google Scholar]
- 23. Dick T., Shin S. J., Koh W. J., Dartois V., Gengenbacher M., Antimicrob. Agents Chemother. 2020, 64, e01943-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lakshminarayana S. B., Huat T. B., Ho P. C., Manjunatha U. H., Dartois V., Dick T., Rao S. P., J. Antimicrob. Chemother. 2015, 70, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jayaram R., Gaonkar S., Kaur P., Suresh B. L., Mahesh B. N., Jayashree R., Nandi V., Bharat S., Shandil R. K., Kantharaj E., Balasubramanian V., Antimicrob. Agents Chemother. 2003, 47, 2118–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The RNAP Mtb -5 a co-crystal structure has been deposited in the Protein Data Bank. PDB code: 7U22.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.