

Summary
Technological advances have made it feasible to collect multi-condition multi-omic time courses of cellular response to perturbation, but the complexity of these datasets impedes discovery due to challenges in data management, analysis, visualization, and interpretation. Here, we report a whole-cell mechanistic analysis of HL-60 cellular response to bendamustine. We integrate both enrichment and network analysis to show the progression of DNA damage and programmed cell death over time in molecular, pathway, and process-level detail using an interactive analysis framework for multi-omics data. Our framework, Mechanism of Action Generator Involving Network analysis (MAGINE), automates network construction and enrichment analysis across multiple samples and platforms, which can be integrated into our annotated gene-set network to combine the strengths of networks and ontology-driven analysis. Taken together, our work demonstrates how multi-omics integration can be used to explore signaling processes at various resolutions and demonstrates multi-pathway involvement beyond the canonical bendamustine mechanism.
Subject areas: Bioinformatics, Complex system biology, Systems biology, Data processing in systems biology
Graphical abstract
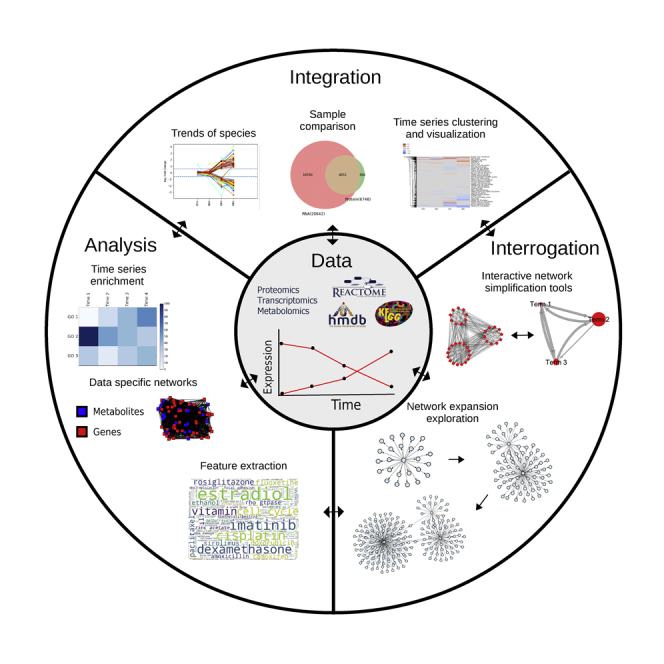
Highlights
-
•
Whole-cell analysis reveals how multiple cellular processes contribute to cell fate
-
•
MAGINE enables multi-scale mechanism exploration in multi-omics data
-
•
Annotated gene networks combine interaction networks and ontology-driven analysis
Bioinformatics; Complex system biology; Systems biology; Data processing in systems biology
Introduction
The cellular response to molecular perturbagens typically involves multiple cellular processes, such as gene expression modulation, changes in protein and metabolic activity, and in extreme cases, changes in DNA sequence or structure (Karran, 2001), all of which contribute differently to cellular phenotype (Hasin et al., 2017; Huang et al., 2017). Until recently, these processes were typically studied in isolation (Schenone et al., 2013), thus neglecting the wider cellular context for challenge-response mechanism outcome. The advent of technologies such as mass spectrometry (MS) and RNA sequencing (RNA-seq) has now enabled the measurement of biochemical interactions at molecular resolution for genomes, proteomes, and metabolomes that cover the whole cell (Aebersold and Mann, 2003; Toby et al., 2016; Wang et al., 2009). Recent work by our laboratories and others has already shown the potential of these kinds of datasets to gain a systems-level understanding of dynamic cellular response mechanisms to perturbations, with measurements that can easily number in the thousands to millions of data points, thus opening the door for the study of perturbation-response mechanisms at the whole-cell level (Gutierrez et al., 2018; Norris et al., 2017; Palmer et al., 2019).
Two major classes of analysis for multi-omics data are enrichment analysis and network analysis. Enrichment analysis is an established methodology that provides insights about relevant cellular processes by comparing multiple experimental conditions following cellular perturbations (e.g., drug treatments) (Subramanian et al., 2005; King et al., 2003). This approach can be used to identify cellular processes by identifying groups of genes or proteins that are enriched or depleted. Unfortunately, for the purposes of mechanistic exploration, these approaches often fail to provide insights into the specific molecular interactions that drive a specific cellular process. In addition, existing tools have not been designed to handle large multi-omics and multi-experiment exploration necessary to extract mechanistic hypotheses. For example, popular web-based enrichment analysis tools such as EnrichR (Chen et al., 2013) and Webgestalt (Wang et al., 2017) can process one sample at a time through their web interfaces, thus posing a limitation for data analysis for high-throughput multi-omics experiments, which can easily identify tens to hundreds of thousands of species with statistically significant changes in response to a challenge.
In contrast to enrichment-based analysis methods, network-based analysis maps biochemical species and their interactions (Goh et al., 2012). These networks can then be explicitly analyzed—e.g., using graph theoretic methods (Albert and Barabási, 2002)—to find paths or groups of relevant interactions between two or more network points. However, these become difficult to visualize and interrogate when the graph grows beyond a few tens of nodes, as seen in genome-wide networks whereby structural insights and process-level activity can be hard to ascertain (Longabaugh, 2012; Röttjers and Faust, 2018; Proulx-Giraldeau et al., 2017). Network tools, most notably Cytoscape (Shannon et al., 2003), partially address the need for network analysis in a biological context through the use of plug-ins, but their capabilities for multi-sample analysis are limited. Ingenuity Pathway Analysis (Krämer et al., 2014), a useful pathway analysis tool available for multi-omics data, can provide cellular process exploration, but its closed, proprietary nature limits extension by users to meet the needs of the field.
Enrichment and network analyses provide differing yet complementary insights into -omics datasets by providing process-focused and system-wide perspectives, respectively. Therefore, we hypothesize that concurrent exploration of biochemical networks and molecular enrichment for multi-sample, multi-omics data would facilitate mechanism exploration and shed insights about cellular response mechanisms. Although tools exist that can handle small -omics datasets (Zhou et al., 2019), their focus on point-and-click interfaces comes with a lack of flexibility and analysis capabilities required to explore large and complex, multi-time point or multi-sample datasets in the context of whole-cell mechanism exploration using untargeted methodologies. Therefore, there is an unmet need for tools to (1) integrate -omics datasets from multiple experimental modalities, (2) provide a platform where multiple analysis tools can be automated and used in tandem, and (3) enable human-guided mechanism exploration within a reproducible, shareable, extensible workflow environment (Marx, 2013).
To address these needs, we developed the Mechanism of Action Generator Involving Network analysis (MAGINE) to explore cellular response mechanisms from large and complex multi-condition, multi-omics datasets. MAGINE is a Python-based analysis framework that unifies and automates enrichment and network analyses onto a common platform under a high-level application programming interface (API), thereby enabling users to explore interactions across multiple cellular processes along with the molecular interactions that drive these processes using a minimal amount of code. We also introduce a new annotated gene-set network (AGN) methodology that facilitates exploration of biological processes at the whole-cell level. We have previously used MAGINE to identify putative contributions to the resistance mechanism to the anti-cancer drug cisplatin (Norris et al., 2017). To demonstrate the capabilities of MAGINE in detail, we explore the DNA-damage response (DDR) mechanism of HL-60 cells to bendamustine treatment. Bendamustine is a well-established DNA-damage agent used for cancer treatments in the clinic with an established consensus mechanism of action (Leoni and Hartley, 2011). Our analysis reveals detailed, systems-level, dynamic molecular mechanisms, which comprise thousands of biochemical interactions across multiple cellular processes. We present a multi-scale cellular response mechanism, at the enriched cellular process level, as well as specific interactions at the molecular level. Our results complement the traditionally accepted mechanisms for bendamustine-induced cell death, which comprise a few tens of molecular interactions (Leoni and Hartley, 2011; Cheson and Rummel, 2009). Finally, all our MAGINE-based analysis is documented using Jupyter notebooks, which offer a means to transparently report complete analyses, suitable for distribution across the scientific community to evaluate and expand on as desired.
Results
Multi-omics datasets to explore DNA-damage response mechanisms from a whole-cell perspective
To demonstrate the power of MAGINE, we decided to explore the DDR execution mechanism at the whole-cell level. We collected time course measurements of HL-60 cells treated with bendamustine, using multiple -omics modalities as shown schematically in Figure 1. Briefly, HL-60 cells were plated in triplicate and exposed to 100 μM bendamustine as described previously (Gutierrez et al., 2018). After introduction of drug, measurements were collected at 12 time points ranging from 30 s to 72 h across 6 experimental platforms, as shown in Figure 1A and Table 1. Sample preparation for transcriptomics, label-free proteomics, and metabolomics analyses was performed as previously described (Gutierrez et al., 2018). Stable isotope labeled aminoacids in cell culture (SILAC) and phosphorylation enriched SILAC (phSILAC) samples were prepared in a similar manner as reported (Norris et al., 2017), with the exception that SILAC samples were run by 1D chromatography rather than MudPIT.
Figure 1.

Number of biochemical species measured and found significant in bendamustine-treated HL-60 cells proteomic and transcriptomic datasets
(A) Number of species found significantly changed versus control (orange) and measured but not significantly changed (blue) for each experimental platform.
(B) Overlap between significantly changed species across platforms.
Table 1.
Experimentally measured platforms for bendamustine/HL-60 dataset by time point
| 30 s | 30 m | 01 h | 03 h | 06 h | 12 h | 18 h | 24 h | 36 h | 48 h | 60 h | 72 h | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNA-seq | – | – | – | – | – | – | – | – | ||||
| SILAC | – | – | – | – | – | – | – | |||||
| phSILAC | – | – | – | – | – | – | – | |||||
| Label-free | ||||||||||||
| HILIC | ||||||||||||
| RPLC |
In total, we detected 54,818 unique molecular species across all platforms, of which 14,426 were significantly changed versus control in at least one time point upon drug treatment. By platform, we saw 2,437 changed species versus control from phSILAC MS, 1,483 from label-free MS, 133 from SILAC MS, and 736 from RNA-seq; the remainder were from hydrophilic interaction liquid chromatograph (HILIC) and reverse phase liquid chromatography (RPLC) metabolomics platforms (Figure 1A). Early changes were detected at the phosphorylation level; phSILAC detected >700 such changes at the 30 s time point. Abundance changes for phosphorylated peptides were observed relatively evenly across all time points, whereas a gradual increase of significant differences versus control occurred over time in overall protein and RNA (Data S1). We next examined the overlap between significantly changed species across the experimental platforms (Figure 1B). The highest overlap among proteomic platforms was between phSILAC and label-free (621 species). Indeed, even within the protein platforms, a majority (>2,500) of measured species were unique to a single platform, whereas 688 species were measured in at least two platforms, and 22 species were measured in all three platforms. Between both RNA and at least one of the proteomic platforms (phSILAC, SILAC, and label-free), 88 species were significantly changed versus respective controls (Figure 1). For the metabolomic platforms (HILIC and RPLC), more than 9,700 species were unique to one of the two platforms, whereas 1,200 were common to both. Within-platform detection is generally reliable and repeatable (Gutierrez et al., 2018), thus the low overlap between significant species changes across platforms demonstrates the value of integrative, multi-platform analysis.
MAGINE: Data integration and mechanism exploration in Python
We wanted to explore the HL-60 response mechanism to bendamustine treatment by integrating and analyzing all -omics datasets and time points. To carry out this and other analyses, we developed MAGINE, a platform for -omics data integration and analysis in Python. MAGINE provides interactive analysis and exploration of multi-time point, multi-omics cellular perturbation data to identify and elucidate molecular species involved in differential cellular responses. MAGINE’s modular design allows custom workflows built around three concepts: data management, enrichment analysis, and network analysis. A typical MAGINE workflow, as we applied to the HL-60/bendamustine dataset, comprises three broad steps (covered in more detail later): first, data from multiple platforms are imported into the modeling framework as described in STAR Methods (Figure 2, top). Second, a data mining step is carried out to identify key biochemical interactions from curated networks (e.g., Reactome, HMDB, Biogrid, and/or Signor) as well as enrichment analysis through the Enrichr API to identify biological processes (Figure 2, middle). Third, the user interacts with the data to extract molecular-level networks and time course enrichment analysis and combine these into AGNs (Figure 2, bottom).
Figure 2.

MAGINE workflow applied to bendamustine-treated HL-60 cells proteomic and transcriptomic datasets
Multi-omics data were generated over multiple time points and experimental platforms. MAGINE was used to perform automated network analysis, combining prior knowledge from multiple databases, and automated enrichment analysis using Enrichr. MAGINE facilitates interactive exploration of datasets using molecular-level networks, enrichment analysis, and annotated gene-set networks.
The AGN is motivated by the desire to combine dynamic, high-level information about biological processes from enrichment analysis with biomolecule interactions extracted from molecular network databases. A combination of enrichment analysis and molecular network analysis results in a multi-scale mechanistic network that enables us to explore cellular response at multiple levels of resolution. At the coarse level, nodes represent biological processes and edges represent collective molecular interactions between nodes. This network can then be expanded into a fine-grained network, which enables exploration of chemical species and their interactions.
Because MAGINE has been developed in the Python programming language, users automatically accrue this ecosystem for scientific computing. MAGINE uses Jupyter Notebooks for documented, reproducible, extensible workflows; cytoscape.js (Franz et al., 2015) for network visualization; py2cytoscape for cytoscape session management (Ono et al., 2015); matplotlib for data visualization (Hunter, 2007); and igraph (Csardi and Nepusz, 2006), networkx (Hagberg et al., 2008), or graphviz (Ellson et al., 2001) for network analysis. In addition, networks can be exported using networkx (Hagberg et al., 2008), for further external manipulation. In what follows we carry out all the presented analysis with MAGINE, unless otherwise specified. Jupyter Notebooks are provided in the supplemental information.
Literature-derived DDR response network from multi-omics data
Our first step in the network analysis pipeline was to build a biochemical interaction network that maps all of our measured chemical species onto known biochemical interactions. Nodes in our network are biochemical species (i.e., gene, metabolite), with node attributes corresponding to the state (post translational modification, RNA level, protein expression), whereas edges corresponding to physical interactions (phosphorylation, ubiquitination, etc.). We require all edges to have an activating or inhibiting effect.
As described in STAR Methods and shown in Figure S1, we start with a list of seed species and grow the network by finding possible interconnecting molecular entities. We start by using significantly enriched or depleted proteins/genes across any time point or experimental platform and produce a network describing the physical relationships among those seeds. The resulting data-driven network (DDN) contains 21,511 biochemical species nodes and 528,905 interaction edges, out of 38,393 possible nodes and 995,599 possible edges (Figure 3A). We note that due to our choice of databases, the edges represent biochemical reactions (other databases may contain broader or indirect species relationships). As shown in Figure 3A, network coverage for measured species in the DDN was 83.3%, whereas coverage of significantly changed species throughout the time course for the DDN was 24.9%. Given the number of nodes and measurements, our data suggest that cellular DDR to bendamustine involved ∼5,000 nodes in the DDN.
Figure 3.

Network analysis of bendamustine-treated HL-60 cells dataset
(A) Data-driven network (DDN) containing 21,511 nodes and 528,905 edges. Yellow nodes highlight experimentally detected molecular species on left and significantly changed species on right.
(B) Manually curated molecular processes and proteins expected to be involved in the cellular response to bendamustine.
(C) Prior network constructed from the molecular species in (B). Seeds are shown in yellow, whereas seeds that are significantly changed in our dataset are shown in red.
(D) DDN with species highlighted in yellow for each of the molecular processes from (B).
(E) DDN with species highlighted in yellow for the same processes as (B); however, we expanded from our manually curated list and leveraged Reactome_2016 gene sets obtained from EnrichR.
We then asked how the literature-based DDR mechanism mapped to the DDN built from the multi-omics dataset. DDR for bendamustine comprises three main cellular processes, namely, DNA-damage repair, cell cycle arrest, and programmed cell death (Leoni and Hartley, 2011; Parikh et al., 2014; Kuruvilla et al., 2017; Leoni et al., 2008; Cheson and Rummel, 2009; Beeharry et al., 2012), leading us to develop a list of 71 biomolecules associated with these cellular processes, as shown in Figure 3B. We then built another interaction network, but this time we used these 71 species instead of all significantly changed proteins. This prior-knowledge network comprised 1,246 nodes and 9,123 edges, as shown in Figures 3C and 3D, representing 6% coverage of the DDN. Of these, 1,023 were measured in our dataset and 395 were significantly changed. This suggests that the interaction between the 71 key biomolecules involved with bendamustine’s DDR can require at least an order of magnitude more biochemical species to capture all their interactions. It also highlights the fact that guided studies of bendamustine response, which focus on such biomolecules based on prior knowledge, can give alternative perspectives on the mechanism to our unbiased approach due to guided studies’ more context-specific but less comprehensive overview of the global cellular response.
To broaden the set of genes from the manual literature search, we utilized enrichR gene sets to extract genes from Reactome_2016 in cell cycle, DNA damage, and programmed cell death. This resulted in 251, 124, and 76 genes, respectively. As shown in Figure 3E, the coverage of these terms is larger than our limited literature-compiled list. However, it is still two orders of magnitude smaller than the experimentally detected set of species described earlier. This highlights the complexity of the cell and demonstrates how a manual, limited literature search and a multi-omics approach produce vastly different results in size and coverage of the responses.
Time-dependent enrichment analysis of DDR to bendamustine
As shown in the previous section, network analysis can be useful to explore interactions among measured species, but due to the complexity of the network, we were not able to attain useful information about participating cellular processes in an unbiased manner. We therefore turned to enrichment analysis to identify relevant cellular processes in our dataset. We encoded a time-dependent enrichment analysis functionality to MAGINE and carried out enrichment-based analysis at each time point, as described in STAR Methods. Extracting meaningful knowledge from these large enrichment datasets requires iterative steps to filter, query, and visualize the results. We therefore integrated functions for these capabilities into MAGINE (shown in Notebook S3). Even after filtering, enrichment results can contain many redundant and closely related terms, which often detract from interpretation due to the added noise. We therefore developed a method called “enrichment compression” (described in STAR Methods and shown in Figure S2) that allows one to minimize similar or redundant enriched terms, while maximizing the overall information content of the remaining terms. We employed RNA-seq and all proteomics data to identify relevant cellular processes captured by over- or under-represented biomolecules, using MAGINEs enrichR function and the Reactome_2016 reference gene set. This resulted in 505 significantly enriched terms. We then required that a term be enriched in at least three time points, resulting in 155 terms. Finally, we applied enrichment compression to the 155 terms by ranking terms both by total gene number (providing broader terms) and by enrichment score (providing more specific terms), resulting in a compact 17-term (Figure 4B) and a more detailed 41-term summary (Figure 4A). The MAGINE enrichment compression step is reversible, thus providing transparency to the user for verification purposes and downstream analysis. Early response to DNA damage by bendamustine seems to elicit significant nuclear activity as shown in Figure 4A for enrichment score-based compression. For example, terms containing cellular processes such as Sumoylation, Nuclear Import, Nuclear Breakdown, and Heterochromatin Foci Formation are highly active at the 30 s, 1 h, and 6 h time points in the data. In addition, cell growth activity, represented by MTORC1-mediated Signaling and metabolic activity, represented by Proton-coupled Monocarboxylate Transport terms, also exhibits enriched activity (i.e., have higher detected relative copy number). These activities are further confirmed by the gene number-based enrichment (Figure 4B) where more general terms such as Glucose Transport and Proton-coupled Monocarboxylate Transport exhibit the highest activity.
Figure 4.

Enrichment analysis of bendamustine-treated HL-60 cells versus control over time
(A and B) Enrichment analysis yielded 84 significant terms with a large degree of redundancy, therefore we applied enrichment “compression,” which rank orders terms, and then iterates through each term, comparing it to all lower terms. Any lower-ranked term that is above a given threshold (too similar) is removed. Users can choose which information to rank the terms by, providing a larger number of specific terms, as shown in (A) by ranking by the combined score (enrichment), or fewer but more general terms by ranking by total number of genes per term, as shown in (B).
Nuclear transport and nucleus-related activities remain highly enriched at the 12–24 h time points, as shown by similarly enriched terms related to nuclear activity. This is not surprising given the extensive DNA damage that is taking place in the cell due to bendamustine. Terms such as Mitotic Anaphase and Resolution of Sister Chromatid Cohesion suggest that the cell attempts to engage in DNA duplication as shown in enrichment-based compression (Figure 4A). This is further supported by the observed enrichment in Cell Cycle, seen in the gene number-based compression (Figure 4B). At this stage, we also see enrichment of DNA Repair and Cellular Response to Stress terms, indicative of cellular activity in response to bendamustine-based DNA damage.
The enrichment score-based data exhibit enrichment in Mitotic Anaphase, G2/M Transition, G2/M Checkpoints, and Switching of Origins to a Post-replicative State, indicative of perturbations in the cell cycle at or around the G2/M transition. Specifically, G2/M transitions and checkpoints are indicated starting at 6 h post-exposure and persist out to 72 h (Figure 4A). In addition, homology-directed repair, which occurs only during S or G2, is enriched at 6, 12, and 24 h post-exposure. Taken together, these observations suggest that the stress imposed by DNA damage is inducing a cell cycle arrest in S or G2. Consistent with this observation, cyclin B levels were found to be stable when measured at 24, 36, 48, and 72 h post-exposure (Figures S7 and S8), suggesting abrogation of cell cycle as cyclin B levels normally oscillate. Also, PLK1 Thr210 phosphorylation, an indicator of progression from G2 to M, was detected in untreated cells at 6, 18, and 24 h. However, phospho-PLK1 was either not detected or was detected at greatly reduced levels in bendamustine-treated cells (Figure S9). Aberrant cyclin B levels coupled with the absence of active PLK1 support the enrichment terms that pointed toward perturbation of the cell cycle at G2/M and allow further interrogation of cell cycle disruption at specific checkpoints.
Emerging between 24 and 72 h, cellular activity has shifted away from nuclear and high metabolism activity toward programmed cell death execution. Apoptosis is highly enriched, highlighting the execution of programmed cell death. This is accompanied by TP53 depletion (i.e., lower relative copy number), likely in DNA repair, as well as by depletion of metabolic activity. This is further confirmed in the gene number-based data (Figure 4B), where programmed cell death is notably enriched. These observations are supported by direct measurement of active caspase 3/7 at 24 h and decreased viability at 48 h post-exposure (Figure S10). Although we also see enrichment in Gene Expression, Cell Cycle, and FGFR2 Alternative Splicing, it is harder to interpret the implications of these enrichment terms given the data.
Taken together, the data suggest early nuclear import and nucleus-involved responses, likely due to the ongoing DNA damage by bendamustine and the cellular engagement of the DNA repair machinery. Multiple other processes accompany this response, notably metabolism-related processes that appear enriched throughout the cell response. Although the data suggest activity at checkpoint proteins such as TP53 prevalent at multiple time points, we note that HL-60 is TP53 deficient (Wolf and Rotter, 1985) and we thus interpret these in the context of biochemical interactions relevant for DDR response. Thus, using MAGINE we can both direct analysis at known responses of bendamustine and explore processes with a less biased, data-driven way. We can cycle through terms, experimental data, and networks to come up with descriptive explanations of the data.
Biological processes in DNA-damage response to bendamustine
Next, we asked whether we could place the cellular processes identified in the time-dependent enrichment analysis from the previous section within a dynamic, molecular context. To attain this goal, we introduce the concept of an AGN to explore biological processes and explore molecular interactions across and within biological processes (Figure 5). Formally, an AGN is a molecular network constructed from the merger of molecular networks and annotated gene sets (described in supplemental information and shown in Figure S5). As shown in Figure 4, the AGN can be visualized over time, where each node size is adjusted based on the enrichment score, providing a high-level representation of the dynamics of signal flow, with animation if desired (Data S5). We present the dynamics of bendamustine-induced DDR in HL-60 cells for five time points where data were collected across all -omics techniques.
Figure 5.

Annotated gene-set network (AGN), showing significant changes of gene ontology terms in bendamustine-treated HL-60 cells versus control over time
The size of each node corresponds to the enrichment of said term, and the edge width corresponds to the number of edge pairs between each node at the given time point. Early response (30 s) is dominated by glucose transport; by 12 h post-treatment, DNA repair is at its peak size, whereas cell cycle is the most significant term; by 72 h programmed cell death is the most significant term.
At 30 s, the ontology terms with the most significant change involve Proton-coupled Monocarboxylate Transport, Glucose Transport, Cell Cycle, and Gene Expression. Proteins in the Proton-coupled monocarboxylate transport term include SLC16A1, SLC16A3, SLC16A7, and BSG. Closer inspection of the molecular species relevant for each term shows enriched activity in nuclear pore proteins (e.g., NUP188, NUP153), POLA1 (a DNA polymerase), RBL1 (regulates entry into cell division), and CARM1 (involved in DNA packaging). Inspection of the edge interactions shows that gene expression activity is connected with the terms Cellular Responses to Stress, Immune System, Disease, Cell Cycle, and Glucose Transport. At first glance these terms appear broad and uninformative. We therefore carried out term decompression and found, for example, that the Cell Cycle term comprises nuclear pore complex disassembly, initiation of nuclear envelope reformation, and nuclear envelope reassembly, pointing to high import/export nuclear activity likely due to the presence of bendamustine.
Some notably enriched proteins among these interactions include MINK1 (negative regulator of Ras-related Rap2 signaling), RB1 (a key regulator of entry into cell division), MAPK1 (a key member of the eponymous MAPK pathway), and many members of the NUP family of proteins. Taken together, these data suggest that at 30 s following drug treatment, the cell is engaging in a stress response (as evidenced by the components present in Disease, Immune System, and Glucose Transport terms), but the effects of DNA damage are already evident, as shown by increased protein copy number involved in processes such as Nuclear Pore Complex Disassembly.
At the 1-h mark, we see a slight increase in DNA repair, involving processes such as Nucleotide Excision Repair and PCNA-dependent Base Excision Repair. The main molecular contributors are MDC1, ATRIP, ERCC3, EP300, POLD3, RPA3, and RPS27A. These proteins are all involved in various forms of DNA damage, DNA repair, polymerase activity, and ubiquitination. The activity in Glucose Transport and Gene Expression terms also increases, but the activity is, again, comparable to that at 30 s with high activity in the NUP family of proteins.
At 6 h, Cell Cycle becomes the dominant term, followed by Signaling by Rho GTP-ases and Gene Expression. Among Cell Cycle sub-terms, Activation of nima kinases nek9, nek6, and nek7 is the most enriched, involving decrease in phosphorylation of NEK7, NEK9, and CDK1 and RNA decreases of CCNB1 and PLK1. The Depolymerisation of the nuclear lamina term also appears significant, with phosphorylation increases in PRKCB and decrease of CCNB1, CDK1, EMD, LMNA, LMNB1, and TMPO. We also note increases in TP53BP1, indicating some activity in cell cycle checkpoints and possibly programmed cell death, despite the fact that HL-60 cells do not express TP53. Taken together, these results suggest that activity toward cell cycle execution remains sustained, accompanied by significant remodeling of the nuclear envelope, and some activity surrounding cell cycle checkpoints seems to emerge.
At 12 h, the Cell Cycle term remains the most enriched, but we also start to see more interaction pairs between terms, as indicated by the thicker lines that connect each node. At this stage, however, we see enrichment in proteins such as NBN and TOPBP1, related to DDR as well as DNA-damage repair. In addition, we see high activity of WEE1, a negative regulator of the G2 to M cell cycle transition. We also note high activity of DYNC1H1, indicative of increased transport in the cell. The Glucose Transport term exhibits marked depletion of NUP210 proteins, likely indicative of nuclear pore instability as DDR progresses.
At 24 h, the cell cycle term remains dominant but a marked increase in enriched interactions across terms is observed. We observe enrichment in MCM6, RANBP2, and MCM3, all related to DNA replication, and notably NDC1, involved directly in nuclear pore activity. The highest interaction among terms is that between Cell Cycle and Disease (590 enriched interacting species), followed closely by Gene Expression (557 enriched interacting species). Notable proteins in the Disease term include NDC1 and SUPT16H (transcription initiation). In the Gene Expression term we note increased copy number in PSMB1 (proteasome subunit beta 1, a component of the 20 s proteasome), NDC1, KHSRP (possible mRNA transport activity), and SF3A3 (pre-mRNA splicing protein).
Finally, at 72 h, we see a significant shift in the enriched terms. We note that this time point has less data than the other time points in the AGN, which could affect the number of observed interactions, and we therefore only explore it for general enrichment trends. The dominant term is Programmed Cell Death, followed by Gene Expression and Cell Cycle. Interestingly, we do not see increased abundance of key proteins associated with programmed cell death at the 72 h point, but we do see, e.g., BAX (key protein in mitochondrial outer membrane permeabilization) and RIPK1 (key protein in necroptosis regulation) at the 24 h time point.
Taken together, these data suggest that following DNA damage, the cell continues to carry out significant cell cycle activity. Activity in DNA damage and DNA repair emerges early on until a Programmed Cell Death takes over toward the end. We also note that non-apoptotic cell death processes were enriched at the 24-h mark, suggesting a possibility of alternate modes of cell death.
Molecular interactions that play key roles in DNA-damage response to bendamustine
As shown in the previous section, the Cell Cycle enrichment term was prominent across all time points. In fact, the term Cell Cycle was the overall highest compressed enriched term across all datasets. Decompressing this high-level term, we saw that “nuclear”-related terms were highly enriched (Figure 6) and that Nuclear Pore Complex (NPC) Disassembly was the highest enriched term not involved in transport activity. We generated a molecular heatmap from the NPC member proteins as shown in Figure 6B, and a molecular subnetwork as shown in Figure 6C.
Figure 6.

Using MAGINE to explore the nuclear pore complex
MAGINE provides tools to switch perspectives between broad terms, measurements, and molecular networks.
(A) Identification of terms with nuclear in name.
(B) Filtered experimental data to visualize trends of species responsible for enrichment of nuclear pore complex disassembly (from A).
(C) We then used these proteins to construct a network, where the node size corresponds to the absolute value of the maximum fold change and color corresponds to maximum fold change in our dataset.
Most of the molecules in the NPC Disassembly term exhibit decreased expression, suggesting that these proteins are present in lower abundances than expected. We note that at the 30 s time point, NUP214 exhibits a low expression value. This protein is typically involved in nucleocytoplasmic transport and is a structural constituent of the nuclear pore. NUP35 and RANBP2 are also depleted. The latter is a protein involved in sumoylation and is thought to bind various possible proteins and DNA to assist in transport across the nuclear envelope. These early time points suggest that early activity targets NPC stability.
Subsequent molecular enrichment does not exhibit major changes until the 12 and 24 h time points, where we see that NUP210 and NUP98 are significantly depleted, whereas RANBP2 expression is slightly increased. The depletion of NUP210 is suggestive of successful disassembly of the nuclear pore complex as this nucleoporin is essential for NPC assembly, fusion, and structural integrity. NUP98 typically works in tandem with NUP96 to coordinate bidirectional transport across the NPC, and its depletion further suggests dismantling of the NPC. Taken together, the time course enrichment analysis suggests that the NPC continues to exhibit some function until final dismantling takes place at later time points.
We wanted to further explore how molecular interactions could regulate NPC dismantling. The molecular interaction network (Figure 6C) exhibits a highly connected network, with CDK1 playing a central role. We see significant phosphorylation activity in NUP210, NUP214, NUP153, NUP107, NUP35, NUP98, and RANBP2. Since CDK1 is an upstream regulator of these proteins, their depletion could be a direct result of CDK1 deactivation. We provide further examples in the supplemental information showing how we can extract other relevant subnetworks to DDR such as CASP3 activity based on downstream targets.
Thus, using MAGINE we were able to start with a broad, known biological process (cell cycle), dive into terms that are more specific, extract and visualize the measurements of the species involved, and then view the interactions among those molecular species, all in a human-driven exploratory cycle. The tool enables custom workflows, dynamic analysis, and is all shareable and transparent.
Discussion
Multi-omics measurements provide larger cellular coverage than traditional in vitro/in-cell measurements (western blots or ELISA), and identification of unexpected significantly changed biochemical species is common. MAGINE enables users to elucidate these findings through exploration of the molecular data and their biological network context, which is often a slow step. Our results show the complexity in interpreting multi-omics data, as the response to bendamustine involves multiple genes and cellular pathways. Single time point measurements do not capture an entire mechanism, with some events occurring early (phosphorylation events of DNA repair proteins) and some later (changes in cell cycle proteins, cleavage events of apoptotic CASP3). Interactive exploration of these data enables users to piece together the mechanism and design further experiments for validation and follow-up exploration.
Perhaps the most biologically interesting finding, but intuitively obvious, is the vast cellular response to DNA-damaging agents. In this study we observed thousands of molecular changes, across multiple molecular types (RNA, phosposites, protein expression) and across different time points. Our approach allows opportunity to explore this large-scale dataset in a piece-wise, exploratory manner. As technology advances, we see an increase in both the resolution of depth at a single time point and the ability to measure more time points of the response, meaning ultimately more and more data to sift through. Rather than depending on spreadsheets exploration (of nearly 100 individual spreadsheets across time point and molecular type measurement) or limited-scope custom tools, we built MAGINE in Python, allowing instant access to the large ecosystem of Python libraries.
MAGINE enables users to construct custom analysis protocols in Jupyter Notebooks that are both reproducible and transferable. Existing software such as Biojupies (Torre et al., 2018) and PaDua (Ressa et al., 2019) has demonstrated the power of this approach on RNA-seq and phospho-proteomics analysis. We expect the use of Jupyter Notebooks to increase and see MAGINE as highly complementary to such pipelines.
As highlighted in our introduction, the multi-omics tool space has many options, but few of them can natively handle multi-time point, multi-platform datasets. We believe the tools most closely aligned with MAGINE’s scope and goals are Cytoscape (Shannon et al., 2003), Metascape (Zhou et al., 2019), and Enrichr (Chen et al., 2013), thus we provide a detailed comparison in Table S1. In addition to the aforementioned capabilities, we believe MAGINE is unique in the multi-omics analysis space in that it is both open source and easily scriptable into documented, extensible Jupyter Notebook workflows. We believe this provides the optimum balance of flexibility, transparency, and ease of use. MAGINE’s design allows for extension to other -omics tools, e.g., Mummichog (Li et al., 2013), to automate time series analysis (in progress).
In summary, MAGINE enables users to easily switch between enrichment analysis, networks, and experimental data within an iterative, interactive framework. This allows users to vary resolutions (molecular/biological process, static/dynamic), depending on the question at hand, and generate new hypotheses. As improvements and falling costs with -omic data generation allow multiple sample acquisitions per perturbation, we envision the need for integrative tools such as MAGINE increasing significantly.
Limitations of the study
MAGINE is built as a Python toolkit, requiring the user to program, limiting its scope to Python-fluent scientists. The tool requires the data to be in a specific format, requiring additional steps before use. The tool also provides results directly back from outside databases, requiring contextualization and interpretation that cannot be automated away. Thus, the results we provide could be different from results others might distill from the same information. Finally, MAGINE was initially designed for human databases. Most of its parts are organism agnostic (data exploration, network tools, etc.); however, generation of networks and enrichment analysis rely on and are thus limited by outside database resources.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Caspase 3 | Cell Signaling | CST9661 |
| Caspase 9 | Cell Signaling | CST9502 |
| GAPDH | Cell Signaling | CST2118 |
| PARP | Cell Signaling | CST9532 |
| PLK1 | Abcam | ab17056 |
| phosphorylated PLK1 | Abcam | ab133442 |
| anti-mouse HRP-conjugated secondary antibody | Cell Signaling | 7075 |
| Anti-rabbit HRP-conjugated secondary antibody | Cell Signaling | 7074 |
| Chemicals, peptides, and recombinant proteins | ||
| Nonidet P40 (NP40) containing protease and phosphatase inhibitors | ThermoFisher Scientific | 78446 |
| Isocove’s Modified Dulbecco’s Medium | ThermoFisher Scientific | 12440054 |
| Heat-inactivated fetal bovine serum | Atlanta Biologicals | S11150 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | G7573 |
| Apo-ONE homogeneous caspase-3/7 assay | Promega | G7792 |
| LumiGLO Kit | Cell Signaling | 5067501 |
| Deposited data | ||
| Processed data | This paper |
https://github.com/LoLab-VU/MAGINE_Supplement_notebooks/blob/master/Data/bendamustine_data.csv.gz https://doi.org/10.5281/zenodo.6639374 |
| Experimental models: Cell lines | ||
| Human: HL-60 | ATCC | CCL-240 |
| Software and algorithms | ||
| MAGINE | This paper | https://github.com/LoLab-VU/MAGINE |
| MAGINE docker image | This paper | docker pull lolab/magine-complete |
| MAGINE notebooks for this study | This paper | https://github.com/LoLab-VU/MAGINE_Supplement_notebooks |
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contact, Carlos F. Lopez, clopez@altoslabs.com.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
∗Cell culture and viability assays
HL-60 cells were obtained from ATCC and were cultured in Isocove’s Modified Dulbecco’s Medium (12440054, ThermoFisher Scientific) with 10% v/v heat-inactivated fetal bovine serum (FBS, S11150, Atlanta Biologicals). Cells were maintained at 37°C with 5% CO2 atmosphere. Bendamustine was re-suspended in DMF to a final concentration of 18.6 mM and diluted in media prior to intoxication of cells with 100 uM compound. A DMF vehicle was included in all experiments. Viability was assessed using CellTiter-Glo Luminescent Cell Viability Assay (G7573, Promega), according to manufacturer’s protocol with the exception of diluting 6-fold in tissue culture grade PBS. Apoptosis was measured using the Apo-ONE homogeneous caspase-3/7 assay (G7792, Promega) to quantify active caspase-3/7 levels. For viability and apoptosis assays, each condition was measured in triplicate and averaged. Relative viability and apoptosis were calculated by normalizing to a vehicle treated control.
∗Western blots
Cells were lysed in 50 mM Tris Cl (pH 8), 150 mM NaCl, 1% Nonidet P40 (NP40) containing protease and phosphatase inhibitors (78446, ThermoFisher Scientific) and then resolved on 12% SDS/PAGE gels. Immunoblots were probed with antibodies against caspase 3 (CST9661, Cell Signaling), caspase 9 (CST9502, Cell Signaling), GAPDH (CST2118, Cell Signaling), PARP (CST9532, Cell Signaling), PLK1 (ab17056, Abcam), or phosphorylated PLK1 (ab133442, Abcam). Binding of an anti-mouse (7075, Cell Signaling) or an anti-rabbit (7074, Cell Signaling) HRP-conjugated secondary antibody was detected with the LumiGLO Kit (Cell Signaling) according to the manufacturer’s instructions.
Method details
MAGINE: A framework to explore cellular response mechanisms using multi-omics
MAGINE is implemented in Python, a suitable language for a biological exploration platform due to its ease of use, large user base, and integration with over 200,000 packages in the Python Package Index. The platform has been tested on Windows, Mac OS, and Linux. It can be used within the Python console, interactive Jupyter Notebooks, or within data processing and analysis pipelines. We envisage the majority of users to be best served through the Jupyter notebook option, and thus describe that in the most detail, including tutorial notebooks (supplemental information).
MAGINE comprises three main modules (Figure S6): data management and visualization, enrichment analysis, and network analysis. Each module can be used independently, but data can be easily shared across modules. We present a brief summary of each module below, followed by an applied case study.
Data analysis module
The data management module handles data storage, access, and analysis. MAGINE utilizes the pandas (McKinney, 2010) library to provide database-like capabilities for -omics data querying. Data are loaded using a tabular, comma-separated values (CSV) file, with one measurement per row (shown in supplemental information). MAGINE stores these in an ExperimentalData class, which provides a simple, high-level interface to access, filter, and search these data as needed. This module also provides visualization capabilities, which include sample comparisons, time-series clustering, and species differential expression trends over time (Figure S6). A summary of these methods is shown in Table S3, and example usage can be found online or in Notebook S1.
Enrichment module
The enrichment analysis module identifies over- or under-represented biochemical species that share common annotation (e.g., biological processes or molecular functions) compared to random background sets (Huang et al., 2009). MAGINE leverages the capabilities of EnrichR, which includes over 120 gene set “libraries” (Chen et al., 2013; Kuleshov et al., 2016). For analysis, users provide one or more lists of genes, which can be manually constructed or created by the ExperimentalData class (e.g., all enriched genes, species detected on a specific experimental platform, or filtered by time point). Currently, analysis of metabolomics is not supported by the enrichment module, and thus was not used for the bendamustine dataset enrichment analysis. MAGINE automates analysis through EnrichR, as shown in Figure S3. A single command is provided to query EnrichR across all time points and data platforms present in a dataset. The results are stored in an EnrichmentResult class, which includes methods to further query, filter, and visualize enrichment terms. Terms can be compared, ranked, grouped, and visualized with built-in methods (e.g., time-series heat map, word cloud). Genes corresponding to each term can be extracted and used to subset ExperimentalData or create subgraphs.
Even through the use of multiple databases, traditional enrichment analyses can yield terms of varied granularity, ranging from very broad (e.g., “biological process”), to highly specific (e.g., “cysteine-type endopeptidase inhibitor activity involved in apoptotic process”), within a single results output. The problem is exacerbated due to each gene mapping to multiple terms, thus introducing term redundancy, increasing the total number of explorable terms and hindering human interpretation. To address these issues, we developed an ontology compression method to aggregate terms based on gene content similarity (described in supplemental information). This significantly reduces enrichment term redundancy, which in turn greatly aids human interpretation. For example, on the bendamustine dataset analyzed herein, 84 terms from traditional enrichment analysis of our data can be compressed to 17 terms, an 80% reduction (see results). A summary of functions available in the enrichment module is shown in Table S4, and example usage can be found online or in Notebook S3).
Network module
MAGINE’s network module allows users to build, query, and visualize molecular and gene annotation networks. A summary of the network module’s methods is shown in Table S5, and example usage can be found online or in Notebook S2. The network module utilizes connectivity information from multiple databases, including KEGG (Ogata et al., 1999; Kanehisa et al., 2016), SIGNOR (Perfetto et al., 2016), Reactome Functional Interactions (Wu et al., 2010), BioGrid (Chatr-Aryamontri et al., 2017; Stark, 2006), and HMDB (Wishart et al., 2009; Wishart et al., 2007). The graphs underlying these databases are merged into a single network, which can be used to perform queries (e.g., find paths between nodes, apply clustering methods) or construct context-specific networks based on a user-provided seed species list. Seed nodes can be obtained from various sources: significantly changed species, mutational evidence, literature review, or manual curation. The module iterates through the databases and expands the network by adding edges and nodes based on connectivity to the seed species in those databases (Figure S1). The resulting networks are a subset of the background network, focused around the specific molecular seed species. These networks can be large (>20,000 nodes and >100,000 edges). MAGINE users can use the Subgraph class to generate subnetworks (Table S5). For example, these functions enable users to find paths between species, or find neighbors (upstream or downstream) of nodes of interest. Options are provided to limit the network expansion, such as setting a maximum distance from specific nodes.
Additionally, we introduce a method to create a coarse-grained network from gene sets, which we refer to as an Annotated Gene-set Network (AGN). The AGN is motivated by the desire to combine dynamic, high-level information about biological processes from enrichment analysis with inter-process communication provided by molecular networks. This results in a coarse-grained network, where nodes are biological process terms and the edges are connections between the sets of nodes in the molecular network. This can be expanded into a fine-grained network, which contains the chemical species and their connections, thus enabling multi-resolution exploration. MAGINE’s network module also provides various tools for network visualization, allowing users to overlay data or update network attributes. Users can visualize networks in Jupyter notebooks using cytoscape.js (Franz et al., 2015), modify a cytoscape session via py2cytoscape (Ono et al., 2015), or create a sequence of figures of network activity through matplotlib (Hunter, 2007), igraph (Csardi and Nepusz, 2006), or graphviz (Ellson et al., 2001). Networks can be exported using networkx (Hagberg et al., 2008), for further external manipulation.
Enrichment term aggregation
MAGINE contains a method, remove_redundancy, for reducing the number of gene enrichment terms by aggregating redundant terms. First, it calculates the ratio of the sizes of the intersection and union (Jaccard index (Gilbert, 1972)) between genes within all term pairs. It then ranks all terms based on either their combined score (from EnrichR), number of genes in the term, or p-value. Starting from the highest ranked, it compares all lower ranked terms and removes them if their similarity is above a user-defined threshold, as demonstrated in Figure S2. This allows the user to minimize the number of total terms while maintaining a level of information content that preserves total information.
Annotated gene set network construction
Annotated gene set networks start with a set of annotation terms, which can be selected based on expert knowledge, rank of enrichment, all compressed terms, or any other criteria. We first extract the set of nodes from a molecular network identified with the selected annotation terms. From there, we search through all possible combinations of pairs between the terms. For example, if term 1 has genes (A, B, C) and term 2 has (D, E), we count the number of edges from the possible sets ((A, D), (A, E), (B, D), (B, E), (C, D), (C, E)) that are found in the network edges. We then do the reverse (term 2 to term 1). If a node is in both sets, we consider the edges that connect the other term, not edges that are within the term. This is demonstrated in Figure S5.
Quantification and statistical analysis
Control versus triplicate for each time point were considered for significant differences. Fold change was calculated using Protalizer (Heaven et al., 2018), Progenesis (Qi et al., 2012), or cuffdiff (Trapnell et al., 2013) (for proteomics, metabolomics, and RNAseq respectively). Significance was determined based on an Benjamini-Hochberg corrected p-value of 0.05 and an absolute fold change of 1.5. These values were choose based on previously published cut-offs (Norris et al., 2017).
Acknowledgments
We thank Randi Gant-Branum, Lauren D. Palmer, D. Borden Lacy, and Eric P. Skaar for data generation and feedback. We also thank Oscar Ortega, Michael Kochen, Lisa Poole-Pino, and Nate Braman for input and ideas, and Michael Ripperger for discussions regarding visualization tools. Research was sponsored by the U.S. Army Research Office and the Defense Advanced Research Projects Agency and was accomplished under Cooperative Agreement no. W911 NF-14-2-0022. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Army Research Office, DARPA, or the US Government. The US Government is authorized to reproduce and distribute reprints for government purposes notwithstanding any copyright notation herein. This work was supported in part by the National Science Foundation grant MCB-1411482 (C.F.L., J.C.P). This work was also supported in part by the National Institutes of Health cooperative agreement 1U01CA215845-01 (C.F.L.) and by the National Institutes of Health grant T32-CA119925 (J.C.P).
Author contributions
J.C.P., A.L.R.L., L.A.H., D.B.G., N.M., M.A.F., T.T., S.D.S., J.L.N., J.A.M., R.M.C., J.P.W., and C.F.L. conceived the study. D.B.G., N.M., M.A.F., T.T., S.D.S., J.L.N., J.A.M., R.M.C., and J.P.W. performed experiments and generated all the data shown in the manuscript. J.C.P., A.L.R.L., L.A.H., D.B.G., N.M., M.A.F., T.T., S.D.S., J.L.N., J.A.M., R.M.C., J.P.W., and C.F.L. interpreted the data and wrote the manuscript. J.C.P. wrote the software.
Declaration of interests
The authors declare no competing interests.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105341.
Supplemental information
The area of each node is proportional to the enrichment of that ontology term at the indicated time point. The thickness of edges is related to the number of edges connecting the molecular nodes underlying the two term nodes (not time-dependent).
Data and code availability
-
1.
MAGINE is released as open source software under the GNU General Public License, version 3.
-
2.
All original code has been deposited at github.com/LoLab-VU/Magine and is publicly available as of the date of publication.
-
3.
Full documentation can be found at magine.readthedocs.io.
-
4.
All Jupyter notebooks are available as part of the supplement, and are also available online at https://github.com/LoLab-VU/MAGINE_Supplement_notebooks.
-
5.
All data are available as part of the supplement and are also available online at https://github.com/LoLab-VU/MAGINE_Supplement_notebooks.
-
6.
Any additional information required to reproduce this work is available from the lead contact (clopez@altoslabs.com).
References
- Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Albert R., Barabási A.L. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002;74:47–97. [Google Scholar]
- Beeharry N., Rattner J.B., Bellacosa A., Smith M.R., Yen T.J. Dose dependent effects on cell cycle checkpoints and DNA repair by bendamustine. PLoS One. 2012;7:e40342. doi: 10.1371/journal.pone.0040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N.K., O'Donnell L., Oster S., Theesfeld C., Sellam A., et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–D379. doi: 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheson B.D., Rummel M.J. Bendamustine: rebirth of an old drug. J. Clin. Oncol. 2009;27:1492–1501. doi: 10.1200/JCO.2008.18.7252. [DOI] [PubMed] [Google Scholar]
- Csardi G., Nepusz T. The igraph software package for complex network research. InterJ. Complex Syst. 2006;1695:1–9. [Google Scholar]
- Ellson J., Gansner E., Koutsofios L., North S.C., Woodhull G. In: International Symposium on Graph Drawing. Mutzel Petra, Jünger Michael., editors. Springer; 2001. Graphviz–open source graph drawing tools; pp. 483–484. [Google Scholar]
- Franz M., Lopes C.T., Huck G., Dong Y., Sumer O., Bader G.D. Cytoscape. js: a graph theory library for visualisation and analysis. Bioinformatics. 2015;32:309–311. doi: 10.1093/bioinformatics/btv557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert G. Distance between sets. Nature. 1972;239:174. [Google Scholar]
- Goh W.W.B., Lee Y.H., Chung M., Wong L. How advancement in biological network analysis methods empowers proteomics. Proteomics. 2012;12:550–563. doi: 10.1002/pmic.201100321. [DOI] [PubMed] [Google Scholar]
- Gutierrez D.B., Gant-Branum R.L., Romer C.E., Farrow M.A., Allen J.L., Dahal N., Nei Y.W., Codreanu S.G., Jordan A.T., Palmer L.D., et al. An integrated, high-throughput strategy for multiomic systems level analysis. J. Proteome Res. 2018;17:3396–3408. doi: 10.1021/acs.jproteome.8b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg A., Swart P., S Chult D. United States; 2008. Exploring network structure, dynamics, and function using networkx.https://www.osti.gov/servlets/purl/960616 [Google Scholar]
- Hasin Y., Seldin M., Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:1–15. doi: 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaven M.R., Cobbs A.L., Nei Y.W., Gutierrez D.B., Herren A.W., Gunawardena H.P., Caprioli R.M., Norris J.L. Micro-data-independent acquisition for high-throughput proteomics and sensitive peptide mass spectrum identification. Anal. Chem. 2018;90:8905–8911. doi: 10.1021/acs.analchem.8b01026. [DOI] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Chaudhary K., Garmire L.X. More is better: recent progress in multi-omics data integration methods. Front. Genet. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P. Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis. 2001;22:1931–1937. doi: 10.1093/carcin/22.12.1931. [DOI] [PubMed] [Google Scholar]
- King O.D., Foulger R.E., Dwight S.S., White J.V., Roth F.P. Predicting gene function from patterns of annotation. Genome Res. 2003;13:896–904. doi: 10.1101/gr.440803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla J., Savona M., Baz R., Mau-Sorensen P.M., Gabrail N., Garzon R., Stone R., Wang M., Savoie L., Martin P., et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood. 2017;129:3175–3183. doi: 10.1182/blood-2016-11-750174. [DOI] [PubMed] [Google Scholar]
- Leoni L.M., Hartley J.A. Mechanism of action: the unique pattern of bendamustine-induced cytotoxicity. Semin. Hematol. 2011;48:S12–S23. doi: 10.1053/j.seminhematol.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Leoni L.M., Bailey B., Reifert J., Bendall H.H., Zeller R.W., Corbeil J., Elliott G., Niemeyer C.C. Bendamustine (Treanda) displays a distinct pattern of cytotoxicity and unique mechanistic features compared with other alkylating agents. Clin. Cancer Res. 2008;14:309–317. doi: 10.1158/1078-0432.CCR-07-1061. [DOI] [PubMed] [Google Scholar]
- Li S., Park Y., Duraisingham S., Strobel F.H., Khan N., Soltow Q.A., Jones D.P., Pulendran B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013;9:e1003123. doi: 10.1371/journal.pcbi.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longabaugh W.J.R. Combing the hairball with BioFabric: a new approach for visualization of large networks. BMC Bioinf. 2012;13:275–316. doi: 10.1186/1471-2105-13-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V. The big challenges of big data. Nature. 2013;498:255–260. doi: 10.1038/498255a. [DOI] [PubMed] [Google Scholar]
- McKinney W. 2010. Data Structures for Statistical Computing in Python. [Google Scholar]
- Norris J.L., Farrow M.A., Gutierrez D.B., Palmer L.D., Muszynski N., Sherrod S.D., Pino J.C., Allen J.L., Spraggins J.M., Lubbock A.L.R., et al. Integrated, high-throughput, multiomics platform enables data-driven construction of cellular responses and reveals global drug mechanisms of action. J. Proteome Res. 2017;16:1364–1375. doi: 10.1021/acs.jproteome.6b01004. [DOI] [PubMed] [Google Scholar]
- Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K., Muetze T., Kolishovski G., Shannon P., Demchak B. CyREST: turbocharging cytoscape access for external tools via a RESTful API. F1000Research. 2015;4:478. doi: 10.12688/f1000research.6767.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer L.D., Jordan A.T., Maloney K.N., Farrow M.A., Gutierrez D.B., Gant-Branum R., Burns W.J., Romer C.E., Tsui T., Allen J.L., et al. Zinc intoxication induces ferroptosis in A549 human lung cells. Metallomics. 2019;11:982–993. doi: 10.1039/c8mt00360b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh K., Cang S., Sekhri A., Liu D. Selective inhibitors of nuclear export (SINE)–a novel class of anti-cancer agents. J. Hematol. Oncol. 2014;7:1–8. doi: 10.1186/s13045-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetto L., Briganti L., Calderone A., Cerquone Perpetuini A., Iannuccelli M., Langone F., Licata L., Marinkovic M., Mattioni A., Pavlidou T., et al. SIGNOR: a database of causal relationships between biological entities. Nucleic Acids Res. 2016;44:D548–D554. doi: 10.1093/nar/gkv1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx-Giraldeau F., Rademaker T.J., François P. Untangling the hairball: fitness-based asymptotic reduction of biological networks. Biophys. J. 2017;113:1893–1906. doi: 10.1016/j.bpj.2017.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi D., Brownridge P., Xia D., Mackay K., Gonzalez-Galarza F.F., Kenyani J., Harman V., Beynon R.J., Jones A.R. A software toolkit and interface for performing stable isotope labeling and top3 quantification using progenesis LC-MS. OMICS. 2012;16:489–495. doi: 10.1089/omi.2012.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressa A., Fitzpatrick M., van den Toorn H., Heck A.J.R., Altelaar M. PaDuA: a Python library for high-throughput (Phospho)proteomics data analysis. J. Proteome Res. 2019;18:576–584. doi: 10.1021/acs.jproteome.8b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röttjers L., Faust K. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol. Rev. 2018;42:761–780. doi: 10.1093/femsre/fuy030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenone M., Dančík V., Wagner B.K., Clemons P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013;9:232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toby T.K., Fornelli L., Kelleher N.L. Progress in top-down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem. 2016;9:499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre D., Lachmann A., Ma’ayan A. BioJupies: automated generation of interactive notebooks for RNA-seq data analysis in the cloud. Cell Syst. 2018;7:556–561.e3. doi: 10.1016/j.cels.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. https://www.nature.com/articles/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., Knox C., Guo A.C., Eisner R., Young N., Gautam B., Hau D.D., Psychogios N., Dong E., Bouatra S., et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:603–610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., Tzur D., Knox C., Eisner R., Guo A.C., Young N., Cheng D., Jewell K., Arndt D., Sawhney S., et al. HMDB: the human metabolome database. Nucleic Acids Res. 2007;35:521–526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D., Rotter V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc. Natl. Acad. Sci. USA. 1985;82:790–794. doi: 10.1073/pnas.82.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Feng X., Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523–1610. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The area of each node is proportional to the enrichment of that ontology term at the indicated time point. The thickness of edges is related to the number of edges connecting the molecular nodes underlying the two term nodes (not time-dependent).
Data Availability Statement
-
1.
MAGINE is released as open source software under the GNU General Public License, version 3.
-
2.
All original code has been deposited at github.com/LoLab-VU/Magine and is publicly available as of the date of publication.
-
3.
Full documentation can be found at magine.readthedocs.io.
-
4.
All Jupyter notebooks are available as part of the supplement, and are also available online at https://github.com/LoLab-VU/MAGINE_Supplement_notebooks.
-
5.
All data are available as part of the supplement and are also available online at https://github.com/LoLab-VU/MAGINE_Supplement_notebooks.
-
6.
Any additional information required to reproduce this work is available from the lead contact (clopez@altoslabs.com).