

Abstract
Blood-brain barrier (BBB) damage is closely related to morbidity and mortality in patients with traumatic brain injury (TBI). Inhibition of VEGF effectively protects BBB integrity in clinical ischemic stroke. Protecting BBB integrity, reducing brain edema and alleviating post-TBI secondary brain injury are key to a favorable patient prognosis. MMP-9 affects BBB integrity by destroying the tight junction of vascular endothelial cells and inhibiting the transport and enzymatic systems. The present study aimed to examine the possible interplay between VEGF and MMP-9 in TBI. A TBI model was established in 87 male Sprague-Dawley rats. Reverse transcription-quantitative PCR, western blotting, wet-dry brain edema assessment, TUNEL and Fluoro-Jade C staining were performed to analyze the brain tissue samples of the rats. The results showed that compared with in the Sham group rats, the mRNA and protein expression levels of VEGF and MMP-9 were increased at 24 h post-TBI. After bevacizumab treatment, BBB permeability and nerve cell apoptosis were markedly reduced. In conclusion, the present study revealed a potential role for TBI-associated VEGF and MMP-9 upregulation in BBB disruption and nerve damage post-TBI.
Keywords: VEGF, MMP-9, blood-brain barrier, apoptosis, traumatic brain injury
Introduction
Traumatic brain injury (TBI) can be caused by anything from a blow to the head to penetrating brain damage induced by external forces (1). TBI has a global annual incidence of >294/100,000, and is more common in older adolescents (15-19 years) and the elderly (≥65 years) (2). Notably, TBI is mainly caused by falling injuries, blows and car accidents, and often results in impairment of consciousness, movement, sensation, language, vision, hearing and memory, and even death (3). Secondary brain injury after TBI occurs as a result of cerebral blood vessel and brain parenchymal damage, and involves oxidative stress, inflammatory response, excitatory toxicity, imbalanced calcium homeostasis, increased vascular permeability, blood-brain barrier (BBB) damage, brain edema and other pathological processes (4). BBB damage can aggravate cerebral edema, disrupt ion balance and induce immune cell infiltration, leading to cell death (5). To the best of our knowledge, the specific pathway involved in early brain injury and BBB permeability changes post-TBI remains unclear.
The BBB separates the blood from the brain, and mostly comprises vascular endothelial cells, pericytes, astrocytes and basement membrane (6). In healthy individuals, BBB integrity heavily depends on the capability of the aforementioned cells to prevent blood-derived factors and immune cells from entering the brain tissue while maintaining the highly restricted environment of the brain (7). BBB breakdown allows excess water to accumulate in the brain, leading to edema, or swelling (8). Regulatory molecules, including MMPs, also induce BBB damage by affecting the transport system of vascular endothelial cells, leading to cerebral edema (9).
As zinc-dependent proteolytic enzymes, MMPs represent multifunctional endopeptidases with several functions under physiological and pathological conditions. In the brain, MMPs are essential for tissue generation, neural network remodeling and BBB integrity, and constitute the main proteases involved in extracellular matrix degradation (10). MMP-9 is a subtype of MMPs, also referred to as gelatin enzyme B, which is closely related to changes in BBB permeability (11). Various cell types contribute to MMP-9 synthesis and secretion, including endothelial cells, glial cells, neurons, mesenchymal cells, T cells, macrophages, neutrophils and eosinophils (12). The upregulation of MMPs following brain damage leads to enhanced BBB permeability and mediates cerebral edema formation (10). It has been suggested that VEGF inhibition in early cerebral ischemia may serve an anti-cerebral ischemia role partially via MMPs (13).
VEGF is a growth factor that induces angiogenesis and increases vascular permeability, which can indirectly promote the development of brain edema and tightly regulate angiogenesis (14). VEGF inhibition has been reported to attenuate vascular permeability and reduce vasogenic edema in acute cerebral ischemia (15). It has been demonstrated that blocking VEGF with bevacizumab can attenuate the brain injury-induced disruption of tight junction proteins (16) and block the increase of brain edema in the rat brain (17). In addition, although MMP-9 upregulation enhances the destruction of tight junction proteins, whether MMP-9-induced damage to tight junction proteins is related to VEGF upregulation in TBI remains unclear. The present study aimed to investigate whether VEGF affects BBB integrity by controlling MMP-9 secretion, in order to provide additional evidence for the application of VEGF inhibitors in the treatment of TBI.
Materials and methods
Study design and grouping
Two distinct assays were performed (Fig. 1). Experiment 1 assessed the time courses of VEGF and MMP-9 post-TBI. A total of 36 rats underwent randomization into the Sham, TBI 12 h, TBI 24 h, TBI 48 h, TBI 72 h and TBI 7 days groups (n=6/group). After rats were sacrificed and tissues were collected, reverse transcription-quantitative PCR (RT-qPCR) and western blotting (WB) were performed to measure the mRNA and protein expression levels of VEGF and MMP-9, respectively (Fig. 1B). Brain tissue samples were collected around the injured area. WB was performed on the tissue collected from the front of the damaged area and tissue from the rear was used for RT-qPCR (Fig. 1A).
Figure 1.

TBI model establishment and study design. (A) Brain tissue specimens collected around the injured area in the TBI group or at the same location in Sham rats were examined via RT-qPCR, western blotting, TUNEL and FJC staining assays. (B) Schematic diagram of Experiment 1. VEGF and MMP-9 expression levels were detected in the brain tissue surrounding the injured area after TBI, to determine the time point of maximum expression change for the second experiment. (C) Schematic diagram of Experiment 2. Effects and potential mechanisms of the VEGF/MMP-9 pathway were assessed post-TBI. TBI, traumatic brain injury; FJC, Fluoro-Jade C; RT-qPCR, reverse transcription-quantitative PCR; Beva, bevacizumab; SD, Sprague-Dawley.
In Experiment 2, 48 rats were randomly assigned to the Sham, TBI, TBI + Vehicle and TBI + bevacizumab (TBI + Beva) groups (n=12/group), to assess the roles of VEGF and MMP-9 in TBI brain injury. A total of 24 h after TBI, which was performed based on Experiment 1, animal euthanasia was performed, followed by the collection of injured brain tissues. Specimens from six rats were assessed by WB of VEGF, MMP-9, occludin and collagen-IV protein expression, and TUNEL and Fluoro-Jade C (FJC) staining of neuronal apoptosis and necrosis. Tissue from the front of the damaged area was used for WB, and tissue from the rear was used for TUNEL and FJC assays (Fig. 1A). The remaining six rats per group were selected for analyzing brain edema. In addition, six rats per group were randomly selected for neurological score assessment, which was performed 24 h after TBI and before euthanasia (Fig. 1C). All experiments strictly used the blinded method.
Animals
A total of 87 male Sprague-Dawley rats (weight, 300-350 g; age, 8 weeks) were provided by JOINN Laboratories (China) Co., Ltd., including 84 that were analyzed (one rat in Experiment 1 and two rats in Experiment 2 died during anesthesia or modeling). Rats were maintained under a 12-h light/dark cycle with constant temperature (25˚C) and humidity (50%), as well as free access to food and drinking water. All experimental protocols were approved by the Animal Ethics and Welfare Committee of Zhangjiagang TCM Hospital Affiliated to Nanjing University of Chinese Medicine (approval no. 2020-14-1; Zhangjiagang, China) and conformed to the guidelines on the care and use of animals outlined by the National Institutes of Health (18).
The rats were sacrificed upon reaching humane endpoints. The reasons for the death of rats during anesthesia or modeling were likely caused by: i) Intraperitoneal injection of sodium pentobarbital into the internal organs; ii) shock due to excessive bleeding in the sagittal sinus when the bone window was opened; and iii) heart rate and breathing not recovered after the 40-g weight was dropped. The humane endpoints of the study included dyspnea, cyanosis, persistent convulsions and severe hypothermia that could not be recovered by warming measures. Death in rats was verified as heart rate and breathing arrest.
Establishment of the TBI rat model
Experimental TBI in rats was established as described in a previous report (19). Rats underwent intraperitoneal anesthesia with 1% sodium pentobarbital at 40 mg/kg followed by fixation onto a stereotaxic apparatus (Shanghai Yuyan Instruments Co., Ltd.).
A bone drill was used to make a 5-mm diameter parietal window at the right of the midline and behind the coronal suture, keeping the dura intact. Next, a 4-mm diameter and 5-mm height copper weight was placed in the bone window. This was followed by dropping a 40-g steel rod from a height of 25 cm onto the copper weight to cause trauma. A short pause in the heart rate and breathing of the rats indicated successful modeling. After disinfection and suturing, the rats were allowed to recover in a warm room. Rats in the Sham group were also submitted to the aforementioned procedure, without dropping the steel rod. Important parameters were monitored during modeling, including heart rate, respiration, body temperature and body weight. These parameters were monitored at 9 a.m. every day after the TBI model was established until the rats were sacrificed at the indicated time points (12, 24, 48, 72 h and 7 days after TBI).
Drug injection
In the TBI + Beva group, bevacizumab (10 mg/kg, in saline solution; Selleck Chemicals) was intraperitoneally injected once immediately after TBI (16,20). The TBI + Vehicle group animals were intraperitoneally administered equal amounts of saline.
Tissue collection and sectioning
Sodium pentobarbital (1%, 40 mg/kg) anesthesia was performed at the indicated time points post-injury (12, 24, 48, 72 h and 7 days after TBI). The animals underwent perfusion with 200 ml 0.9% normal saline via the heart before being sacrificed by the intraperitoneal injection of sodium pentobarbital (2%; 150 mg/kg). Cortical specimens around the injury area (3 mm from the edge of the injury site in the TBI group or the same location in Sham animals) were obtained and placed on ice. A portion of the specimens from experiment 1 and experiment 2 underwent snap freezing and storage at -80˚C for WB and RT-qPCR, whereas the remaining samples from experiment 2 were fixed with 4% formalin overnight at room temperature, embedded in paraffin and cut into 5-µm sections using a paraffin slicer (SLEE medical GmbH) for TUNEL and FJC staining. Two blinded pathologists extracted and selected the tissue samples.
RT-qPCR
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was used to extract total RNA from the brain tissue around the injury. Total RNA (1 µg) underwent RT. qPCR was carried out on a QuantStudio™ Dx RT-PCR Instrument (Thermo Fisher Scientific, Inc.) with SYBR™ Green Master Mix (Thermo Fisher Scientific, Inc.). Thermocycling conditions were as follows: Denaturation at 95˚C for 2 min; followed by 40 amplification cycles at 95˚C for 15 sec, 60˚C for 15 sec and 72˚C for 60 sec; final extension at 72˚C for 10 min, and hold at 4˚C. The 2-∆∆Cq method was utilized to analyze data that were normalized to GAPDH expression (21). The assays were performed in triplicate. The sequences of primers used for RT-qPCR are presented in Table I.
Table I.
Primer sequences for quantitative PCR.
| Primer name | Primer sequence | Product size, bp |
|---|---|---|
| VEGF | F: 5'-ACGGGCCTCTGAAACCATGAA-3' | 121 |
| R: 5'-TTTCTGCTCCCCTTCTGTCGT-3' | ||
| MMP-9 | F: 5'-GCCGGGAACGTATCTGGAAA-3' | 177 |
| R: 5'-GGTTGTGGAAACTCACACGC-3' | ||
| GAPDH | F: 5'-TGTGAACGGATTTGGCCGTA-3' | 208 |
| R: 5'-GATGGTGATGGGTTTCCCGT-3' |
F, forward; R, reverse.
WB
WB was performed based on a previous method (22). Brain tissue homogenization was performed in tissue protein extraction reagent (CoWin Biosciences) containing protease inhibitors on ice for 20 min. The homogenates were cleared by centrifugation at 12,000 x g for 20 min at 4˚C. Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.) was utilized for protein quantification. Equal amounts of total protein (30 µg/lane) were separated by 10% SDS-PAGE and transferred onto a PVDF membrane (MilliporeSigma). Membranes were blocked with QuickBlock™ Blocking Buffer (Beyotime Institute of Biotechnology) at room temperature for 1 h. Subsequently, membranes were incubated overnight at 4˚C with primary antibodies (Table II) and then with goat anti-rabbit (dilution, 1:5,000; cat. no. 65-6120; Invitrogen; Thermo Fisher Scientific, Inc.) or anti-mouse IgG-HRP (dilution, 1:5,000; cat. no. 62-6520; Invitrogen; Thermo Fisher Scientific, Inc.) at room temperature for 1 h. Finally, Immobilon™ Western Chemiluminescent HRP Substrate (MilliporeSigma) was used to detect immunoblots and an imaging system (Bio-Rad Laboratories, Inc.) was used for visualization. ImageJ (version 1.8.0; National Institutes of Health) was utilized for semi-quantification.
Table II.
Primary antibodies used for western blotting.
| Antibody against | Catalogue number | Supplier | Host species | Dilution |
|---|---|---|---|---|
| VEGF | ab214424 | Abcam | Rabbit monoclonal | 1:1,000 |
| MMP-9 | ab76003 | Abcam | Rabbit monoclonal | 1:3,000 |
| Occludin | sc-271842 | Santa Cruz Biotechnology, Inc. | Mouse monoclonal | 1:500 |
| Collagen-Ⅳ | ab6586 | Abcam | Rabbit polyclonal | 1:500 |
| GAPDH | PA1-987 | Invitrogen; Thermo Fisher Scientific, Inc. | Rabbit polyclonal | 1:10,000 |
Neurological score assessment
Neurological score evaluation was performed 24 h post-TBI using the modified Garcia test (23,24). The scoring system consisted of seven components: i) Spontaneous activity; ii) axial sensation; iii) vibrissae proprioception; iv) symmetry of limb movement; v) lateral turning; vi) forelimb outstretching; and vii) climbing. Every subtest was scored from 0 to 3, with a maximum score of 21. The higher the score, the lower the nerve damage.
TUNEL assay
The TUNEL assay was performed to measure neuronal apoptosis using a kit purchased from Beyotime Institute of Biotechnology, according to the manufacturer's instructions. After dewaxing in xylene for 5 min twice, anhydrous ethanol for 5 min, 90% ethanol for 2 min, 70% ethanol for 2 min and distilled H2O for 2 min (all at room temperature), paraffin-embedded sections were treated for 20 min with DNase-free proteinase K (20 µg/ml) at 37˚C. Subsequently, the sections were incubated for 1 h TUNEL working solution at 37˚C in the dark. DAPI Fluoromount-G™ (Shanghai Yeasen Biotechnology Co., Ltd.) was utilized for counterstaining for 10 min at room temperature before observation under a fluorescence microscope (Olympus Corporation). The apoptotic index was determined as (TUNEL-positive cells)/(total cells) x100.
FJC staining
Necrosis was measured via FJC staining using a kit from Biosensis Pty Ltd., according to the manufacturer's instructions. After dewaxing in xylene for 5 min twice, anhydrous ethanol for 5 min, 90% ethanol for 2 min, 70% ethanol for 2 min and distilled H2O for 2 min (all at room temperature), paraffin-embedded sections were transferred to solution B (1:9, potassium permanganate:distilled water) and incubated for 10 min at room temperature. Subsequently, the sections were incubated with solution C (1:9, FJC solution:distilled water) for 30 min at room temperature in the dark and washed with distilled water. After drying at 60˚C for 10 min, specimens underwent soaking in xylene for 5 min at room temperature. Finally, sealing was performed using neutral balsam (Shanghai Yeasen Biotechnology Co., Ltd.) before observation under a fluorescence microscope (Olympus Corporation) to count the FJC-positive cells.
Brain edema assessment
Brain edema assessment was performed using the wet-dry method (25). After the rat brains were collected, they were divided into ipsilateral and contralateral sides, and immediately weighed to assess the wet weights. Subsequently, brain tissues were dried at 100˚C for 24 h and dry weights were obtained. The percentage of water content in the brains was quantified as [(wet weight-dry weight)/wet weight] x100.
Statistical analysis
GraphPad Prism 8.0 (GraphPad Software, Inc.) was used for statistical analysis. Data distribution was analyzed using the Shapiro-Wilk test and all data were normally distributed except for the neurological score data. The neurological score is shown as the median and interquartile range, and other data are presented as the mean ± standard deviation. One-way ANOVA followed by Dunnett's post hoc test was performed to compare experimental groups with the Sham group in Experiment 1. One-way ANOVA followed by Tukey's post hoc test was performed to compare the groups in Experiment 2. Kruskal-Wallis test followed by Dunn's post hoc test was used to analyze the neurological scores. P<0.05 was considered to indicate a statistically significant difference.
Results
VEGF and MMP-9 expression in the rat brains after TBI
The mRNA and protein expression levels of VEGF and MMP-9 were assessed in the Sham group, and at 12, 24, 48 and 72 h and 7 days after TBI by RT-qPCR and WB, respectively (Fig. 2). The mRNA and protein expression levels of VEGF peaked at 24 h after TBI (P<0.001; Fig. 2A and C). The changes in the mRNA and protein expression levels of MMP-9 exhibited an increase detected 12 h after TBI and peaking at 24 h (P<0.001; Fig. 2B and D). Notably, 24 h (P<0.001) and 48 h (P<0.01) after TBI, the expression levels of VEGF and MMP-9 were significantly increased compared with those in the Sham group. The time point of 24 h after TBI, which was associated with the maximum expression of VEGF and MMP-9, was selected to investigate the effects of bevacizumab.
Figure 2.

mRNA and protein expression levels of VEGF and MMP-9 post-TBI. mRNA expression levels of (A) VEGF and (B) MMP-9 relative to GAPDH. Amplification plots and melting curves of VEGF and MMP-9 levels were used to confirm the specificity of reverse transcription-quantitative PCR amplification. Western blotting was performed to assess (C) VEGF and (D) MMP-9 protein expression levels in the Sham group, and at 12, 24, 48 and 72 h and 7 days after TBI. Data were normalized to the Sham group. Statistical analysis was performed using one-way ANOVA with Dunnett's post hoc test. n=6/group. *P<0.05, **P<0.01, ***P<0.001 vs. Sham group. TBI, traumatic brain injury.
Effects of bevacizumab on VEGF and MMP-9 expression after TBI
Bevacizumab was injected following TBI. Compared with in the Sham group, the protein expression levels of VEGF and MMP-9 were markedly increased in the TBI group (P<0.001; Fig. 3A and B), which was consistent with the results of Experiment 1. Their levels in the TBI and TBI + Vehicle groups were comparable. Furthermore, the expression levels of VEGF and MMP-9 in the TBI + Beva group were significantly lower than those in the TBI + Vehicle group (P<0.01; Fig. 3A and B). These results indicated that bevacizumab administration may reduce the expression of VEGF and MMP-9 after TBI.
Figure 3.

Effects of bevacizumab on the protein expression levels of VEGF, MMP-9, occludin and collagen-IV, brain edema and neurological score at 24 h post-TBI. Protein expression levels of (A) VEGF, (B) MMP-9, (C) occludin and (D) collagen-IV, and (E) brain water content and (F) neurological scores were assessed to determine the effects of bevacizumab at 24 h post-TBI. Protein levels were normalized to levels in the Sham group. Statistical analyses of western blotting and brain edema were performed using one-way ANOVA with Tukey's test post hoc. Statistical analyses of neurological scores were performed using Kruskal-Wallis test with post hoc Dunn's test. n=6/group. ***P<0.001 vs. Sham group; &&P<0.01, &&&P<0.001 vs. TBI + Vehicle group. TBI, traumatic brain injury; TBI + Beva, TBI + bevacizumab.
BBB integrity in rats with TBI upon bevacizumab administration
The expression levels of the tight junction proteins occludin and collagen-IV were assessed as indicators of BBB degradation (Fig. 3C and D). The expression levels of occludin and collagen-IV were significantly decreased in the TBI group compared with those in the Sham group (P<0.001). Their levels in the TBI and TBI + Vehicle groups were comparable. Furthermore, the TBI + Beva group exhibited significantly increased expression levels of occludin (P<0.001) and collagen-IV (P<0.01) compared with in the TBI + Vehicle group, indicating that BBB damage was ameliorated after bevacizumab administration. Additionally, brain edema was assessed using the wet-dry method (Fig. 3E). Compared with in the Sham group, brain water levels were increased on the ipsilateral side as a result of TBI (P<0.001). Bevacizumab injection significantly reduced brain edema in the damaged hemisphere after TBI compared with in the TBI + Vehicle group (P<0.01). Brain edema on the ipsilateral side of injury was generally higher than that on the contralateral side. However, brain edema on the contralateral side showed no significant changes among the Sham group and all of the TBI groups (P>0.05).
Neurological scores in TBI rats following bevacizumab administration
The modified Garcia test was used to reflect neurological scores at 24 h post-TBI (Fig. 3F). Neurological scores were significantly decreased in the TBI group compared with those in the Sham group (P<0.001). The scores in the TBI and TBI + Vehicle groups were comparable. Neurological scores in the TBI + Beva group were markedly improved in comparison with the TBI + Vehicle group (P<0.01).
Neuronal apoptosis and necrosis in rats with TBI after bevacizumab intervention
The TUNEL assay (Fig. 4A and C) and FJC staining (Fig. 4B and D) were performed to assess neuronal apoptosis and necrosis, respectively. The TBI group exhibited significantly increased neuronal apoptosis compared with in the Sham group (P<0.001; Fig. 4C), whereas it was comparable to the TBI + Vehicle group (P>0.05). Neuronal apoptosis was significantly decreased in the TBI + Beva group compared with that in the TBI + Vehicle group (P<0.01; Fig. 4C). Neuronal necrosis was significantly increased in the TBI group in comparison with the Sham group (P<0.001), whereas it was significantly decreased in the TBI + Beva group (P<0.001; Fig. 4D), which was consistent with the results of TUNEL assay.
Figure 4.

Effects of bevacizumab on neuronal apoptosis and necrosis 24 h after TBI. (A) Representative TUNEL assay images showing TUNEL-positive (green) apoptotic cells and DAPI-stained (blue) nuclei. (B) Representative FJC staining assay images showing cell necrosis. (C) Apoptotic and (D) necrotic cells. Scale bar, 50 µm. One-way ANOVA with Tukey's post hoc test was utilized for group comparisons n=6/group. ***P<0.001 vs. Sham group; &&P<0.01, &&&P<0.001 vs. TBI + Vehicle group. TBI, traumatic brain injury; TBI + Beva, TBI + bevacizumab; FJC, Fluoro-Jade C.
Discussion
VEGF is an important factor that induces vascular permeability, and contributes to BBB destruction and brain edema (26). In the present study, the effects of VEGF inhibition on BBB protection and neuroprotection were investigated in a rat model of TBI. The results showed that the mRNA and protein expression levels of VEGF were increased post-TBI and reached their maximum level at 24 h post-TBI. Bevacizumab, also known as Avastin, can bind to VEGF and block its biological activity and is commonly used as a clinical antitumor agent (27). Bevacizumab has been reported to serve an important role in brain tumor-related brain edema, particularly in the treatment of brain radiation necrosis (28,29). However, it remains unclear as to whether there is an effect on TBI-induced brain edema. In the present study, bevacizumab was administered immediately after TBI to inhibit the expression of VEGF and to investigate whether the inhibition of VEGF ameliorated BBB damage by reducing the expression of MMP-9. The present results demonstrated that after bevacizumab injection, the expression levels of VEGF were decreased compared with those in the TBI group, whereas brain edema was ameliorated. These findings indicated that VEGF may promote cerebral edema in the TBI rat model. Moreover, the inhibition of VEGF by bevacizumab may improve brain edema after TBI.
The occurrence of brain edema after TBI and BBB dysfunction are directly correlated (30). Previous studies have shown that the degradation of the tight junction protein occludin has a critical function in the loss of BBB integrity (31,32). Collagen-IV represents the main constituent of the basement membrane of the BBB and its degradation may lead to defects in basement membrane function, which may serve an essential role in the pathogenetic mechanism of cerebral damage (33,34). The present study investigated the BBB integrity by measuring the levels of occludin and collagen-IV. The protein expression levels of occludin and collagen-IV were decreased following TBI compared with those in the Sham group, with reduced integrity of the BBB. However, following inhibition of VEGF by bevacizumab, the expression levels of occludin and collagen-IV were increased compared with those in the TBI group. The injection of bevacizumab may increase the levels of tight junction proteins and possibly repair the basement membrane of the BBB after TBI, as also suggested by the improvement in brain edema.
Previous findings have suggested that MMPs can damage tight junctions and the basement membrane in the BBB, aggravating brain edema (35). Our previous study also confirmed that MMP-9 is upregulated in a rat model of TBI and the specific MMP-9 suppressor SB-3CT can reverse the increase in MMP-9 and brain edema, and reduce nerve apoptosis and necrosis (36). The present study used bevacizumab to inhibit VEGF following TBI, which led to the downregulation of MMP-9, upregulation of occludin and collagen-IV, and improved BBB integrity compared with in the TBI group.
Damage to the BBB can aggravate brain edema, disrupt ion balance and induce immune cell infiltration, leading to cell death (7,37). In the present study, TUNEL and FJC staining were used to assess apoptosis and necrosis in the brain tissue surrounding the TBI area, respectively. The present results showed increased levels of TUNEL- and FJC-positive cells with increasing VEGF and MMP-9 levels post-TBI. Upon bevacizumab administration, the levels of TUNEL- and FJC-positive cells were significantly lower than those in the TBI group, suggesting that bevacizumab treatment may be helpful in improving secondary nerve injury after TBI.
The mechanism of TBI leading to secondary brain injury is very complex. As aforementioned, VEGF was revealed to be significantly increased in the brain tissue surrounding the TBI area, resulting in a corresponding increase in MMP-9 levels. MMP-9 increases brain water content by regulating BBB permeability, leads to neuron necrosis and apoptosis, and causes neurological dysfunction (36). In the present study, inhibition of VEGF with bevacizumab reduced TBI-induced brain edema and may inhibit secondary brain injury; this effect may partly be regulated by occludin and collagen-IV through the MMP-9 pathway (Fig. 5). This result indicated that VEGF suppression in early TBI may have great potential in the treatment of secondary brain injury post-TBI. The possible mechanism of action was explored in the present study to a certain extent in terms of improved brain edema after TBI.
Figure 5.
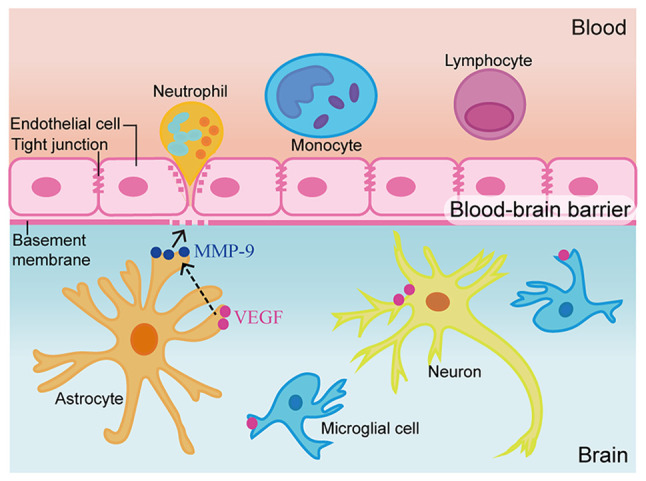
Schematic diagram of the possible mechanism by which VEGF affects secondary brain injury post-traumatic brain injury via MMP-9. After TBI, VEGF and MMP-9 levels in the brain tissue surrounding the injured area were increased. By decreasing occludin and collagen-IV levels, blood-brain barrier permeability may be enhanced, leading to an increase in water content, and further resulting in apoptosis and necrosis. Inhibition of VEGF by bevacizumab may reverse this damage.
The present study has some limitations. Firstly, since female rats have estrus and menstrual cycles, which can add an additional variable to scientific experiments and data analysis, only male rats were used in the present study. The effect of this will be considered in future studies. Secondly, the present study only used animal experiments to investigate the effect of bevacizumab on the expression of VEGF/MMP-9 after TBI, as well as on brain edema and neurological function, whereas the specific mechanism of action was not verified in vitro. Future studies will be performed to investigate the mechanism of action of bevacizumab in the treatment of brain edema after TBI in vitro. Thirdly, the half-life of bevacizumab is estimated to be 20 days and there is a potential risk of it affecting wound healing (38); therefore, further validation studies are needed to assess the clinical application of bevacizumab in patients with TBI. The present study explored the effect of bevacizumab on brain edema from the perspective of BBB integrity in an animal model of TBI, with the aim of providing experimental guidance for the possible future clinical application of bevacizumab in the treatment of TBI.
In conclusion, VEGF was upregulated after TBI in rats, which damaged BBB integrity by activating MMP-9 and may aggravate secondary brain injury. Additionally, inhibition of VEGF exerted neuroprotective effects. The present findings indicated that VEGF may represent a critical target for the prevention and control of secondary brain injury after TBI.
Acknowledgements
Not applicable.
Funding Statement
Funding: The present study was funded by the Zhangjiagang Health Youth Science and Technology Project (grant no. ZJGQNKJ202011), the Zhangjiagang Science and Technology Support Project (grant nos. ZKS2020, ZKS2029) and the Suzhou Science and Technology Development Project (grant nos. SKJY2021001 and SKJY2021003).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
BD and HS contributed to study conception and design. MW, YG, LJ, MZ and HG performed experiments. MW and YG drafted the manuscript. HS and YG generated figures and performed data analysis. HS and BD confirm the authenticity of all the raw data. MW and BD revised the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
All experiments involving animals received ethical approval from the Animal Ethics and Welfare Committee of Zhangjiagang TCM Hospital Affiliated to Nanjing University of Chinese Medicine (approval no. 2020-14-1). No human subjects were assessed in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Georges A, M Das J. Traumatic Brain Injury. In: StatPearls [Internet]. StatPearls Publishing, Treasure Island, FL, 2021. [Google Scholar]
- 2.Nguyen R, Fiest KM, McChesney J, Kwon CS, Jette N, Frolkis AD, Atta C, Mah S, Dhaliwal H, Reid A, et al. The International incidence of traumatic brain injury: A systematic review and meta-analysis. Can J Neurol Sci. 2016;43:774–785. doi: 10.1017/cjn.2016.290. [DOI] [PubMed] [Google Scholar]
- 3.Capizzi A, Woo J, Verduzco-Gutierrez M. Traumatic brain injury: An overview of epidemiology, pathophysiology and medical management. Med Clin North Am. 2020;104:213–238. doi: 10.1016/j.mcna.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Sivandzade F, Alqahtani F, Cucullo L. Traumatic brain injury and blood-brain barrier (BBB): Underlying pathophysiological mechanisms and the influence of cigarette smoking as a premorbid condition. Int J Mol Sci. 2020;21(2721) doi: 10.3390/ijms21082721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang Y, Gao S, Wang X, Cao Y, Lu J, Chen S, Lenahan C, Zhang JH, Shao A, Zhang J. Programmed Cell Deaths and Potential Crosstalk With Blood-Brain Barrier Dysfunction After Hemorrhagic Stroke. Front Cell Neurosci. 2020;14(68) doi: 10.3389/fncel.2020.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7(a020412) doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhowmick S, D'Mello V, Caruso D, Wallerstein A, Abdul-Muneer PM. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp Neurol. 2019;317:260–270. doi: 10.1016/j.expneurol.2019.03.014. [DOI] [PubMed] [Google Scholar]
- 8.Jha RM, Kochanek PM, Simard JM. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology. 2019;145(Pt B):230–246. doi: 10.1016/j.neuropharm.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blixt J, Svensson M, Gunnarson E, Wanecek M. Aquaporins and blood-brain barrier permeability in early edema development after traumatic brain injury. Brain Res. 2015;1611:18–28. doi: 10.1016/j.brainres.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Rempe RG, Hartz AMS, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36:1481–1507. doi: 10.1177/0271678X16655551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, An Q, Wang T, Gao S, Zhou G. Autophagy- and MMP-2/9-mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-increased Blood-brain barrier permeability during early reperfusion in stroke. Neuroscience. 2018;377:126–137. doi: 10.1016/j.neuroscience.2018.02.035. [DOI] [PubMed] [Google Scholar]
- 12.Dang B, Duan X, Wang Z, He W, Chen G. A therapeutic target of cerebral hemorrhagic stroke: Matrix metalloproteinase-9. Curr Drug Targets. 2017;18:1358–1366. doi: 10.2174/1389450118666170427151657. [DOI] [PubMed] [Google Scholar]
- 13.Zhang HT, Zhang P, Gao Y, Li CL, Wang HJ, Chen LC, Feng Y, Li RY, Li YL, Jiang CL. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol Med Rep. 2017;15:57–64. doi: 10.3892/mmr.2016.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma C, Zhou J, Xu X, Wang L, Qin S, Hu C, Nie L, Tu Y. The construction of a radiation-induced brain injury model and preliminary study on the effect of human recombinant endostatin in treating radiation-induced brain injury. Med Sci Monit. 2019;25:9392–9401. doi: 10.12659/MSM.917537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura R, Nakase H, Tamaki R, Sakaki T. Vascular endothelial growth factor antagonist reduces brain edema formation and venous infarction. Stroke. 2005;36:1259–1263. doi: 10.1161/01.STR.0000165925.20413.14. [DOI] [PubMed] [Google Scholar]
- 16.Deng Z, Zhou L, Wang Y, Liao S, Huang Y, Shan Y, Tan S, Zeng Q, Peng L, Huang H, Lu Z. Astrocyte-derived VEGF increases cerebral microvascular permeability under high salt conditions. Aging (Albany NY) 2020;12:11781–11793. doi: 10.18632/aging.103348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pishko GL, Muldoon LL, Pagel MA, Schwartz DL, Neuwelt EA. Vascular endothelial growth factor blockade alters magnetic resonance imaging biomarkers of vascular function and decreases barrier permeability in a rat model of lung cancer brain metastasis. Fluids Barriers CNS. 2015;12(5) doi: 10.1186/2045-8118-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. National Research Council (US) Institute for Laboratory Animal Research: Guide for the Care and Use of Laboratory Animals. National Academies Press (US), Washington, DC, 1996. [PubMed] [Google Scholar]
- 19.Gao F, Li D, Rui Q, Ni H, Liu H, Jiang F, Tao L, Gao R, Dang B. Annexin A7 levels increase in rats with traumatic brain injury and promote secondary brain injury. Front Neurosci. 2018;12(357) doi: 10.3389/fnins.2018.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu CS, Wang ZF, Dai LM, Chu SH, Gong LL, Yang MH, Li ZQ. Induction of proline-rich tyrosine kinase 2 activation-mediated C6 glioma cell invasion after anti-vascular endothelial growth factor therapy. J Transl Med. 2014;12(148) doi: 10.1186/1479-5876-12-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Gong Y, Wu M, Shen J, Tang J, Li J, Xu J, Dang B, Chen G. Inhibition of the NKCC1/NF-κB signaling pathway decreases inflammation and improves brain edema and nerve cell apoptosis in an SBI rat model. Front Mol Neurosci. 2021;14(641993) doi: 10.3389/fnmol.2021.641993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng D, Wang B, Wang L, Abraham N, Tao K, Huang L, Shi W, Dong Y, Qu Y. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J Pineal Res. 2017;62(e12395) doi: 10.1111/jpi.12395. [DOI] [PubMed] [Google Scholar]
- 24.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 25.Gong Y, Wu M, Gao F, Shi M, Gu H, Gao R, Dang BQ, Chen G. Inhibition of the pSPAK/pNKCC1 signaling pathway protects the bloodbrain barrier and reduces neuronal apoptosis in a rat model of surgical brain injury. Mol Med Rep. 2021;24(717) doi: 10.3892/mmr.2021.12356. [DOI] [PubMed] [Google Scholar]
- 26.Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen Nv, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, Chinot OL. Bevacizumab (Avastin(R)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86(102017) doi: 10.1016/j.ctrv.2020.102017. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang H, Shi S, Yuan Z, Chang JY. Bevacizumab treatment for radiation brain necrosis: Mechanism, efficacy and issues. Mol Cancer. 2019;18(21) doi: 10.1186/s12943-019-0950-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voss M, Wenger KJ, Fokas E, Forster MT, Steinbach JP, Ronellenfitsch MW. Single-shot bevacizumab for cerebral radiation injury. BMC Neurol. 2021;21(77) doi: 10.1186/s12883-021-02103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu J, Li X, Yin J, Hu Y, Gu Y, Pan S. Glycocalyx degradation leads to blood-brain barrier dysfunction and brain edema after asphyxia cardiac arrest in rats. J Cereb Blood Flow Metab. 2018;38:1979–1992. doi: 10.1177/0271678X17726062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim KA, Kim D, Kim JH, Shin YJ, Kim ES, Akram M, Kim EH, Majid A, Baek SH, Bae ON. Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS. 2020;17(21) doi: 10.1186/s12987-020-00182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Li X, Qiao S, Yang D, Li Z, Xu J, Li W, Su L, Liu W. Occludin degradation makes brain microvascular endothelial cells more vulnerable to reperfusion injury in vitro. J Neurochem. 2021;156:352–366. doi: 10.1111/jnc.15102. [DOI] [PubMed] [Google Scholar]
- 33.Lee WH, Warrington JP, Sonntag WE, Lee YW. Irradiation alters MMP-2/TIMP-2 system and collagen type IV degradation in brain. Int J Radiat Oncol Biol Phys. 2012;82:1559–1566. doi: 10.1016/j.ijrobp.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cottarelli A, Corada M, Beznoussenko GV, Mironov AA, Globisch MA, Biswas S, Huang H, Dimberg A, Magnusson PU, Agalliu D, et al. Fgfbp1 promotes blood-brain barrier development by regulating collagen IV deposition and maintaining Wnt/β-catenin signaling. Development. 2020;147(dev185140) doi: 10.1242/dev.185140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rempe RG, Hartz AMS, Soldner ELB, Sokola BS, Alluri SR, Abner EL, Kryscio RJ, Pekcec A, Schlichtiger J, Bauer B. Matrix metalloproteinase-mediated blood-brain barrier dysfunction in epilepsy. J Neurosci. 2018;38:4301–4315. doi: 10.1523/JNEUROSCI.2751-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu MY, Gao F, Yang XM, Qin X, Chen GZ, Li D, Dang BQ, Chen G. Matrix metalloproteinase-9 regulates the blood brain barrier via the hedgehog pathway in a rat model of traumatic brain injury. Brain Res. 2020;1727(146553) doi: 10.1016/j.brainres.2019.146553. [DOI] [PubMed] [Google Scholar]
- 37.Alluri H, Wiggins-Dohlvik K, Davis ML, Huang JH, Tharakan B. Blood-brain barrier dysfunction following traumatic brain injury. Metab Brain Dis. 2015;30:1093–1104. doi: 10.1007/s11011-015-9651-7. [DOI] [PubMed] [Google Scholar]
- 38.Bergsland E, Dickler MN. Maximizing the potential of bevacizumab in cancer treatment. Oncologist. 2004;9 (Suppl 1):S36–S42. doi: 10.1634/theoncologist.9-suppl_1-36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.