

Abstract
Functional and structural decline of the neuromuscular system is a recognized cause of decreased strength, impaired performance of daily living activities, and loss of independence in the elderly. However, in mammals, including humans, age-related loss of strength is greater than loss of muscle mass, so the underlying mechanisms remain only partially understood. This review focuses on the mechanisms underlying impaired skeletal muscle function with aging, including external calcium-dependent skeletal muscle contraction; increased voltage-sensitive calcium channel Cav1.1 β1a-subunit and junctional face protein JP-45 and decreased Cav1.1 (α1) expression, and the potential role of these and other recently discovered molecules of the muscle T-tubule/sarcoplasmic reticulum junction in excitation-contraction uncoupling. We also examined neural influences and trophic factors, particularly insulin-like growth factor-I (IGF-1). Better insight into the triad proteins’ involvement in muscle ECC and nerve/muscle interactions and regulation will lead to more rational interventions to delay or prevent muscle weakness with aging. The focus of this review is on the proteins mediating excitation-contraction coupling (ECC) and their expression and regulation in humans and rodent models of skeletal muscle functional decline with aging. Age-dependent changes in proteins other than those related to ECC, muscle composition, clinical assessment and interventions, have been extensively reviewed recently [1-3].
Keywords: Skeletal muscle, aging, sarcopentia, insulin-like growth factor 1, excitation-contraction coupling, denervation
DECREASED MUSCLE SPECIFIC FORCE IN MAMMALIAN SPECIES, INCLUDING HUMAN
The decline in muscular strength with age is caused largely by a loss of total muscle mass - but also a disproportionate loss of strength. Some studies in humans directly relate this diminished strength to muscle atrophy [4], while others find that it is greater than the decrease in muscle mass [5]. For example, the decline in normalized force (force/muscle mass, Nm/kg) in the knee extensors with aging has been found to follow a curvilinear relationship, starting at about 40 years and declining by about 28% from 40-49 to 70-79 years [5]. In vitro studies of single human muscle fiber contractility also reveal a decrease in specific force (force/cross-sectional area) with age [6]. Therefore, the intrinsic force-generating capacity of the skeletal muscle per contractile unit may be impaired in aging mammals, including humans. Postulated mechanisms include alterations to the excitation-contraction coupling (ECC) process [7-9] and decreased actin-myosin cross-bridge stability [10,11].
EXCITATION-CONTRACTION UNCOUPLING
The transduction of changes in sarcolemmal potential to elevated intracellular calcium concentration is a key event that precedes muscle contraction [12] (see Fig. 1). Electromechanical transduction in muscle cells requires the participation of the dihydropyridine receptor (DHPR) [13] located at the sarcolemmal transverse T-tubule. The DHPR is a multimeric voltage-gated L-type Ca2+ channel (dihydropyridine-sensitive). Cav1.1 (α1 subunit) activation evokes Ca2+ release from an intracellular store (sarcoplasmic reticulum, SR) through ryanodine-sensitive calcium channels (RyR1) into the myoplasm [14]. The functional consequence of the reduced number, function, or interaction of these receptors is reduced intracellular calcium mobilization and force development [15]. Calcium binds to troponin C, which by interaction with troponin I, T and tropomyosin, leads to crosslinkages between actin and myosin and sliding of thin-on-thick filaments to produce force [16]. Uncoupling of the excitation-contraction machinery is a major factor in age-dependent decline in the force- generating capacity of individual cells [17].
Fig. (1).
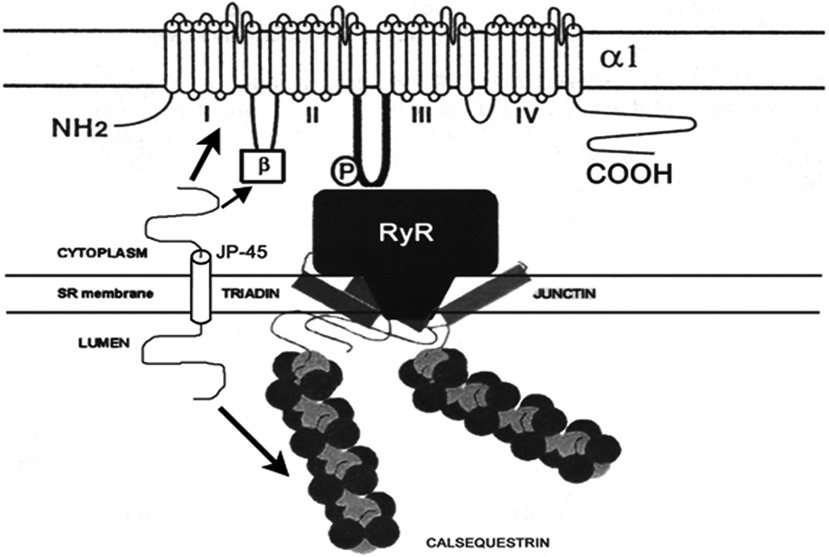
Organization of the triad junction emphasizing Cav1.1/DHPRα1 subunit, JP-45 and RyR interactions. JP-45 interacts with the Cav1.1/DHPR α1 and β1a subunits and calsequestrin (arrows). The ryanodine receptor (RyR) isoform-1 interacts with junction and triadin. Other triadic proteins such as calmodulin, FKBP, and protein kinases, have been omitted for clarity. Adapted from [168] and [169].
Aging muscle fibers exhibit less specific force than those from young-adult or middle-aged animals but similar endurance and recovery from fatigue [18-20]. Whether excitation-contraction uncoupling (ECU) results from altered neural control of muscle gene expression is not known. However, a series of studies support this concept. First, denervation results in a significant decrease in DHPR functional expression and alterations in ECC in skeletal muscle from adult rats [21]. Second, nerve crush leads to reduced levels of mRNA-encoding DHPR subunits and RyR1 in muscle [22], and studies show that both DHPR and RyR1 expression depend on skeletal muscle innervation [23,24]. Third, during development, DHPR mRNA levels change in relation to fiber innervation [25]. Fourth, myotube depolarization triggers the appearance of (+)-[3H]PN 200-110 binding sites [26]. Finally, exercise and chronic stimulation in vivo increase DHPR expression in homogenates of soleus and extensor digitorum longus (EDL) muscles [27,28]. Thus, fiber-type composition, DHPR and RyR1, and ECC seem to depend on nerve stimulation and muscle activity.
We are starting to understand how nerve stimulation of muscle activity influences muscle phenotype and the specific sarcolemmal-nuclear signaling pathways involved in muscle gene expression at different ages. Increasing evidence points to a decline in neural influence on skeletal muscle at later ages [29], leading to changes in muscle composition that result in ECU [11].
CHANGES IN SKELETAL MUSCLE INNERVATION AND PLASTICITY OF THE NEUROMUSCULAR JUNCTION WITH AGING INFLUENCE ECC
Muscle weakness in aging mammals may result from primary neural or muscular etiological factors or a combination [30]. Experimental muscle denervation leads to loss in absolute and specific force [31,32]. Although denervation contributes to the functional impairment of skeletal muscle with aging [33], its prevalence in human and animal models of aging remains to be determined.
Some studies have focused on the mechanisms underlying neuromuscular impairments in old age. Several aspects have been investigated: the phenomenon known as ECU [7,34], which leads to a decline in muscle specific force (force normalized to a cross-sectional area) [35]; the loss in muscle mass associated with a decrease in muscle fibers as well as fiber atrophy [36,37]; changes in fiber type [29, 38-40]; decreased maximal isometric force and slower sliding speed of actin on myosin [41,42]; and impaired recovery after eccentric contraction [43,44]. Identifying the triggers of these changes remains elusive. Some suggestions include decreased muscle loading [45], oxidative damage [46,47], age-dependent decrease in IGF-1 expression or tissue sensitivity [48-50], and decline in satellite cell proliferation [51].
Interaction between skeletal muscle and neuron is crucial to the capacity of both to survive and function throughout life. Thus, muscle atrophy and weakness may result from primary neural or muscular etiological factors or a combination. Growing evidence supports a role for the nervous system in age-related structural and functional alterations in skeletal muscle [52]. The number of motor neurons in the lumbosacral spinal cord of humans has been shown to decrease after the age of 60, and the number of large and intermediate-sized myelinated axon fibers decreases with age in the ventral roots with no change in small fiber numbers [30, 53,54].
Motor units decrease with motor neurons, as measured with electromyography in humans and in situ calculation in rats. As with motor neuron fibers, the loss of motor units seems to be greatest among the largest and fastest. A decline in the number and size of anterior horn cells in the cervical and lumbosacral spinal cord and cytons in motor neuron columns in the lumbar spinal cord in humans with age has been reported [55]. These studies found fewer large and intermediate-diameter cytons, which are the largest and fastest motor neurons [56,57]. In fact, aged motor units exhibit increased amplitude and duration of action potentials, supporting the idea that those remaining grow larger [33, 58]. Morphological evidence of this process can be found in the muscle. Fiber loss and atrophy with age is greatest among fast type-2 fibers, a finding that agrees with the loss of large and intermediate-sized motor neuron fibers and large motor units. Fiber type “grouping” has been found in human muscle with age, indicating a denervation/re-innervation process [30]. More direct evidence of a slow denervation process with aging is provided by the increased prevalence of old muscle fibers staining positive for neural cell adhesion molecule [59].
Overall fiber loss and a preferential decrease in type-2 fiber number and size in mixed fiber-type muscles, such as the vastus lateralis, is observed with aging (for a review see [30]). However, all lower limb muscles may not respond similarly to aging. The tibialis anterior, a predominantly type-2 muscle, has been shown to decrease, with compensatory hypertrophy in the remaining fibers to maintain overall muscle size (Lexell, unpublished results). Conversely, a recent report documents preferential atrophy of type-2 fibers in biceps brachii, an upper limb muscle, but not reduced numbers. This finding is consistent with clinical studies showing better preservation of upper limb muscle function with age [11].
Several groups have reported skeletal muscle denervation and reinervation and motor unit remodeling or loss in aging rodents or humans [57, 60-65]. Motor-unit remodeling leads to changes in fiber-type composition [66]. During development, muscle fiber-type phenotype is determined by interactions with subpopulations of ventral spinal cord motor neurons that activate contraction at different rates, ranging from 10 (slow fibers) to 100 (fast-fatigue resistant) or 150 Hz (fast-fatigue sensitive, limb muscles) [67-69]. Age-related motor-unit remodeling appears to involve denervation of fast muscle fibers with re-innervation by axonal sprouting from slow fibers [36, 58, 70, 71]. When denervation outpaces reinnervation, a population of muscle fibers becomes atrophic and is functionally excluded. Although denervation contributes to skeletal muscle atrophy and functional impairment with aging [33], its time course and prevalence in human and animal models of aging remain to be determined. Urbancheck et al. (2001) analyzed the contribution of denervation to deficits in specific force in skeletal muscle in 27-29-month (old) compared with 3-month (young) rats [59]. Contraction force recordings together with muscle immunostaining for NCAM (neural cell adhesion molecule), a marker of fiber denervation [72,73], showed a significantly higher number of denervated fibers in old rats. The area of denervated fibers detected by positive staining with NCAM antibodies accounts for a significant fraction of the decline in specific force [59].
We hypothesized that denervation in aging skeletal muscle is more extensive than predicted by standard functional and structural assays and asked whether it is a fully or partially developed process. To address these two questions, we combined electrophysiological and immunohisto-chemical assays to detect the expression of tetrodotoxin (TTX)-resistant sodium channels (Nav1.5) in flexor digitorum brevis (FDB) muscles from young-adult and senescent mice. The FDB muscle was selected for its fast fiber-type composition (~70% type IIx, 13% IIa, and 17% type I) [74] and because the shortness of the fibers makes them suitable for patch-clamp recordings [75].
Two sodium channel isoforms are expressed in skeletal muscle, the TTX-sensitive Nav1.4 and the TTX-resistant Nav1.5. Both were originally isolated from rat skeletal muscle and denominated SkM1 [76] and SkM2 [77], respectively. To determine the status of denervation of individual fibers from adult and senescent mice, we took advantage of the following properties of the Nav1.5 channel: (1) its expression after denervation but absence in innervated adult muscle; (2) its early increase in expression, recorded 24 h after denervation in hindlimb muscles [78]; and (3) its relative insensitivity to TTX [77, 79-81].
Sodium current density measured with the macropatch cell-attached technique did not show significant differences between FDB fibers from young and old mice. The TTX dose-response curve, using the whole cell voltage-clamp technique, showed three populations of fibers in senescent mice, one similar to fibers from young mice (TTX-sensitive), another similar to fibers from experimentally denervated muscle (TTX-resistance), and a third intermediate group. Partially and fully denervated fibers constituted approximately 50% of the total number of fibers tested, which agrees with the percent of fibers shown to be positive for the Nav1.5 channel by specific immunostaining [75]. These results confirmed our hypothesis that muscle denervation is more extensive than that reported using more classical techniques.
Recovery from denervation implies nerve sprouting and re-innervation by the same or neighboring motor units. Different methods of inducing transient nerve injury and recovery have been employed with contrasting results. Slower regeneration and reinnervation in aged compared to young motor endplates was recorded in response to crush injury of the peripheral nerve [52, 82]. The difference in the time needed to recover was attributed to a transient failure in the spatiotemporal relationship between Schwann cells, axons, and the postsynaptic acetylcholine receptor regions during reinnervation in aged rats [82]; that is, nerve/muscle interactions contribute significantly to impaired recovery after nerve injury in the aged.
However, in apparent contrast, a comparable capacity for regeneration has been shown in muscles from very old compared to young rats [83]. Effects of age on muscle regeneration were studied by injecting the local anesthetic, bupivacaine, in fast-twitch muscles. It induced similar muscle fiber damage and reduced the mean tetanic tension in fast-twitch muscles from young adult (4-month) and old (32- and 34-month) rats. The same authors investigated muscle regeneration using heterochronic transplantation of nerve-intact EDL, a fast-twitch muscle. EDL muscles from 4- or 32-month-old rats were cross-transplanted in place of the same muscle in 4-month-old hosts. As a control, contralateral muscles were autotransplanted back into the donors. After 60 days, the old-into-young muscle transplants regenerated as successfully as the young-into-young autotransplants. Lack of nerve damage provided favorable conditions for muscle regeneration, together with an age-related effect of the local environment on the transplants [83].
As evidence of the importance of neural factors in nerve regeneration, the same group reported that when axons are allowed to regenerate in an endoneurial environment, there is no evidence of age-related impairment in muscle reinnervation [84]. Therefore, although old muscle can regenerate as successfully as young muscle, an intact nerve supply seems critical to recovery, together with less clearly defined factors associated with the local environment. We believe IGF-1 secretion and signaling is vital for the protection of nerve and muscle from age-related degeneration.
Neural alterations occur at the ventral spinal cord motor neuron, peripheral nerve, and neuromuscular junction in aging mammals. Age-related changes have been documented in neuronal soma size [56, 85] and number [55, 57, 65] in the spinal cord and in peripheral nerve in tibialis nerves of mice aged 6-33 months [53], including accumulation of collagen in the perineurium and lipid droplets in the perineurial cells, together with an increase in macrophages and mast cells. From 6 to 12 months, numbers of Schwann cells associated with myelinated fibers (MF) decrease slightly in parallel with an increase in their internodal length, but then increase in older nerves in parallel with a greater incidence of demyelination and remyelination. The reported unmyelinated axon (UA) to myelinated fiber (UA/MF) ratio is about 2 until 12 months, decreasing to 1.6 by 27 months. In older mice, the loss of nerve fibers involves UA (50% loss at 27-33 months) more than MF (35%). In aged nerves, wide incisures and infolded or outfolded myelin loops are frequent, resulting in an increased irregularity in the morphology of fibers along the internodes [53].
In summary, adult mouse nerves (12-20 month) show several features of progressive degeneration, whereas general nerve disorganization and marked fiber loss occur from 20 months on [53]. The deterioration of myelin sheaths during aging may be due to decreased expression of the major myelin proteins (P0, PMP22, MBP). Axonal atrophy, frequently seen in aged nerves, may be explained by reduced expression and axonal transport of cytoskeletal proteins in the peripheral nerve [54]. The incidence and severity of the age-related peripheral nerve changes seem to depend on the animal’s genetic background. Thus, histological examination conducted on isolated sciatic nerves and brachial plexuses revealed more pronounced axonal degeneration and remyelination in B6C3F1 and C3H than in C57BL mice [86]. Impaired nerve regeneration in animals and humans has been correlated with diminished anterograde and retrograde axonal transport [87], and retardation in the slow axonal transport of cytoskeletal elements during maturation and aging has been reported [88,89]. This reduced axonal transport could account for the inability of the motor neuron in old mice to expand the field of innervation in response to partial denervation [90].
Alterations of the neuromuscular junction in association with aging have been attributed to its “instability” [91]. The process of neuromuscular synapse formation and activity-dependent editing of neuromuscular synaptic connections is better understood [92] than the events leading to denervation in aging mammals. Apparently, after synapse formation, the terminals of the same axon, described as a cartel, exhibit heterogeneity in terms of acetylcholine release, which may contribute to nerve terminal selection in the developmental transition from innervation of each muscle fiber by multiple nerve endings to the adult one-on-one pattern. Activity plays a crucial role in synapse elimination during this period (for a review see [92]). These concepts prompt the interesting hypothesis that senescent mammals retain a similar mechanism for eliminating neuromuscular synapse. The level of physical activity among the elderly is highly variable and considered important for successful neuromuscular function. Endurance exercise modulates the neuromuscular junction of C57BL/6NNia aging mice [93]. When synaptic terminals occupying motor endplates in adult rats were electrically silenced by the sodium channel blocker tetrodotoxin or the acetylcholine receptor blocker α-bungarotoxin, regenerating axons that were both inactive and synaptically ineffective frequently displaced them. This study concludes that neither evoked nor spontaneous activation of acetylcholine receptors is required for competitive re-occupation of neuromuscular synaptic sites by regenerating motor axons in adult rats [94].
Experimental denervation of skeletal muscle from aging rodents leads to a series of changes, such as re-orientation of costameres (rib-like structures formed by dystrophin and β-dystroglycan) [95,96], proliferation of triadic membranes [97], decrease in charge movement (functional expression of the DHPR voltage sensor), and alterations in the SR calcium-release channel [9, 15, 21 98, 99]. The molecular substrate for these alterations is only partially understood. We hypothesize that age-related denervation may induce these structural and functional changes in mammalian, including human, muscle. Costameric proteins transmit mechanical lateral forces and provide structural integrity when mechanically loaded muscle fibers contract [100]. Muscle activity and muscle agrin, two orders of magnitude lower than the effective concentration of neural agrin, regulate the organization of cytoskeletal proteins in skeletal muscle fibers [95]. It would be interesting to explore these molecular changes in aging muscle and examine the potential beneficial effect of muscle agrin on costamere structure and force development. The studies reported above strongly involve neural alterations in the onset and progression of age-related decline in skeletal muscle function.
TROPHIC FACTORS REGULATE SPINAL CORD MOTOR NEURON STRUCTURE AND FUNCTION AND ECC
Target-derived neurotrophic factors, including the neurotrophin, and nerve growth factor have a well-established role in regulating survival of developing neurons in the peripheral and central nervous systems [101,102]. Some other studies point to a continued role for target-derived trophic factors in the plasticity of adult and aged neurons [103,104]. A series of studies suggest a role for neurotrophins, at least, in the adult neuromuscular system. Neural activity appears to contribute significantly to the trophic interactions between nerve and muscle at the adult neuromuscular junction. Neurotrophins regulate the development of synaptic function [105], and participate in activity-induced modification of synaptic transmission [106]. Potentiation of synaptic efficacy by brain-derived neurotrophic factor is facilitated by presynaptic depolarization at developing neuromuscular synapses [107,108]. Using a nerve/muscle co-culture in which neurotrophin-4 (NT-4) is overexpressed in a subpopulation of postsynaptic myocytes, presynaptic potentiation was restricted to synapses on myocytes overexpressing NT-4. Nearby synapses formed by the same neuron on control myocytes were not affected [109]. Furthermore, the production of endogenous NT-4 messenger RNA in rat skeletal muscle was regulated by muscle activity; the amount of NT-4 mRNA decreased after blocking neuromuscular transmission with alpha-bungarotoxin and increased during postnatal development and after electrical stimulation. Finally, NT-4 may mediate the effects of exercise and electrical stimulation on neuromuscular performance [110]. Thus, muscle-derived NT-4 appears to act as an activity-dependent, muscle-derived neurotrophic signal for the growth and remodeling of the adult neuromuscular junction.
These investigations of the complex role of neural activity in regulating nerve-target interactions have not extended to the aging neuromuscular junction. However, a close correlation between altered ligand-receptor expression(s) and axonal/terminal aberrations in senescence supports a role for neurotrophin signaling in age-related degeneration of cutaneous innervation [111]. An age-related decrease in target neurotrophin expression, notably NT3 and NT4, correlated with site-specific loss of sensory terminals combined with aberrant growth of regenerating/sprouting axons into new target fields [111].
The role of IGF-1 and related binding proteins in neural control of aging skeletal muscle ECC and fiber-type composition in mammals is under investigation. Systemic overexpression of human IGF-1 cDNA in transgenic mice resulted in IGF-1 overexpression in a broad range of visceral organs and increased concentrations in serum [112]. These mice exhibited increased body weight but only a modest improvement in muscle mass.
Because of the possible confounding effects of systemic expression, Coleman et al. targeted IGF-1 overexpression specifically to striated muscle [113] using a myogenic expression vector containing regulatory elements from both the 5′- and 3′-flanking regions of the avian skeletal α-actin gene. IGF-1 overexpression in cultured muscle cells causes precocious alignment and fusion of myoblasts into terminally differentiated myotubes and elevated levels of myogenic basic helix-loop-helix factors, intermediate filament, and contractile protein mRNA [113]. Transgenic mice carrying a single copy of the hybrid skeletal α-actin/hIGF-1 transgene had hIGF-1 mRNA levels that were approximately half those of the endogenous murine skeletal α-actin gene on a per-allele basis but conferred substantial tissue-specific overexpression without elevating serum levels of IGF-1. This localized, muscle-specific overexpression of human IGF-1 caused significant hypertrophy of myofibers, suggesting that IGF-1 is a more potent myogenic stimulus when derived from sustained autocrine/paracrine release than when administrated exogenously. Similar hypertrophy has been observed in response to simple intramuscular injections of IGF-1 in adult rats [114].
Effects of IGF-1 on muscle in aging animals have also been investigated. In old mice, muscle-specific overexpression of IGF-1 preserves skeletal muscle force and DHPR expression [8, 115], while viral-mediated, muscle-specific expression prevents age-related loss of type-IIB fibers [116]. There is evidence that the capacity of IGF-1 to induce muscle hypertrophy declines in adult and senescent mice [117]. However, its effects on fiber specific force are sustained until late ages [118], suggesting that the pathways it uses to control fiber size and to generate force diverge. Overexpression of the mIGF-1 isoform, corresponding to the human IGF-1Ea gene, resulted in sustained mouse muscle hypertrophy and regenerative capacity throughout life (Musaro et al., 2001), indicating that this muscle-specific splice variant of the IGF-1 gene plays a different role in muscle molecular composition and function than the other IGF-1 splice variants.
We tested the hypothesis that target-derived IGF-1 prevents alterations in neuromuscular innervation in aging mammals [29]. We used senescent wild-type mice as a model of deficient IGF-1 secretion and signaling and S1S2 transgenic mice to investigate the role sustained IGF-1 overexpression in striated muscle plays in neuromuscular innervation. Analysis of the nerve terminal in EDL muscles from senescent mice showed that sustained overexpression of IGF-1 in skeletal muscle partially or completely reversed the decrease in cholinesterase-stained zones (CSZ) exhibiting nerve terminal branching, number of nerve branches at the CSZ, and nerve branch points. Target-derived IGF-1 also prevented age-related decreases in the postterminal α-bungarotoxin immunostained area. Postsynaptic folds were fewer and longer as shown by electron microscopy.
Transgenic overexpression of IGF-1 in skeletal muscle may also prevent the switch in muscle fiber-type composition recorded in senescent mice. The use of the S1S2 IGF-1 transgenic mouse model allowed us to provide morphological evidence for the role of target-derived IGF-1 in spinal cord motor neurons in senescent mice. The main conclusion of this study was that muscle IGF-1 prevents age-dependent changes in nerve terminal and neuromuscular junction, influencing muscle fiber-type composition and, potentially, muscle function [17, 115, 116].
The role of IGF-1 in motor neuron survival has been examined during embryonic or postnatal life [119] as well as in spinal cord pathology [120-122]. For example, in young rodents, IGF-1 expression is upregulated in Schwann cells and astrocytes following spinal cord and peripheral nerve injury, while IGF-binding protein 6 is strongly upregulated in the injured motor neurons [123]. In regions of muscle enriched with neuromuscular junctions, IGF-II was strongly upregulated in satellite and possibly glial cells during recovery from sciatic nerve crush [124] while IGF-1 showed less significant changes. In young animals, systemic administration of IGF-1 decreases lesion-induced motor neuron cell death and promotes muscle reinnervation [125]. It also promotes neurogenesis and synaptogenesis in diverse areas of the central nervous system, such as the hypocampal dentate gyrus during postnatal development [126], and increases proliferation of granule cell progenitors [127].
These studies suggest that IGF-1 might have beneficial effects on spinal cord motor neurons from senescent mammals. However, transgenic overexpression of IGF-1 in the central nervous system does not improve ECC or neuromuscular performance in the mouse [127,128]. In contrast to localized motor neuron expression, widespread IGF-1 may be deleterious for neuronal function or muscle innervation [128].
During embryonic and postnatal development, specific sets of CNS neurons show high levels of IGF-1 receptor gene expression combined with IGF-1 expression, while in hippocampal and cortical neurons, receptor and IGF-1 expression are localized in different cell groups [129]. These expression patterns suggest that IGF-1 exerts autocrine and paracrine effects in the CNS in addition to its previously described paracrine (muscle-derived) actions on spinal cord motor neurons. While these mechanisms contribute undoubtedly to the development of the appropriate neuronal phenotype and probably to its maintenance in adulthood, its involvement in aging processes remains substantially untested. Despite these uncertainties, an age-related decline in neuronal as well as muscle-derived IGF-1 combined with altered IGF-1 resistance through reduced expression or sensitivity of the receptor may contribute to the atrophy or death of motor and other CNS neurons in aging mammals. Through the previously described mechanisms, these changes may trigger a cascade of events leading to decreased skeletal muscle gene transcription.
IGF-1 REGULATES SKELETAL MUSCLE ECC
IGF-1 may affect functional interactions between nerve and muscle by regulating transcription of the Cav1.1 gene [130]. Although the Cav1.1 subunit is critical to ECC, the basic mechanisms regulating its gene expression are unknown. To understand them, we isolated and sequenced the 1.2-kb 5' flanking-region fragment immediately upstream of the mouse DHPRα1S gene [131]. Luciferase reporter constructs driven by different promoter regions of that gene were used for transient transfection assays in muscle C2C12 cells. We found that three regions, corresponding to the CREB, GATA-2, and SOX-5 consensus sequences within this flanking region, are important for DHPRα1S gene transcription, and antisense oligonucleotides against them significantly reduced charge movement in C2C12 cells [131]. This study demonstrates that the transcription factors CREB, GATA-2, and SOX-5 play a significant role in the expression of skeletal muscle DHPRα1S.
Whether IGF-1 regulates these transcription factors and subsequent expression of the Cav1.1 gene is not known. Using an approach similar to that described above [131], we investigated the effects of IGF-1 on various promoter deletion/luciferase reporter constructs. They were transfected into C2C12 cells, and IGF-I effects were measured by recording luciferase activity. IGF-I significantly enhanced DHPRα1S transcription, carrying the CREB binding site but not in CREB core binding site mutants. A gel mobility shift assay using a double-stranded oligonucleotide for the CREB site in the promoter region and competition experiments with excess unlabeled or mutated promoter oligonucleotide and unlabeled consensus CREB oligonucleotide indicate that IGF-1 induces CREB binding to the DHPRα1S promoter. We prevented IGF-1 from mediating enhanced charge movement by incubating the cells with antisense but not sense oligonucleotides against CREB. These preliminary results support the conclusion that IGF-1 regulates Cav1.1 transcription in muscle cells by acting on the CREB element of the promoter [130]. Confirming these results in skeletal muscle will be important as well as determining whether IGF-1/CREB signaling and the signaling pathway linking IGF-1R to CREB activation is preserved in aging mammals. We hypothesize that these effects are mediated by the direct action of IGF-1 on muscle cells, perhaps via activation of satellite cells (Barton-Davis et al., 1998), but may involve neuronal access to muscle-derived IGF-1.
Muscle IGF-1 is known to have trophic effects on motor neurons [29], so its overexpression is effective in delaying or preventing the deleterious effects of aging in both tissues. Since age-related decline in muscle function stems partly from motor neuron loss, we created a tetanus toxin fragment-C (TTC) fusion protein to target IGF-1 to motor neurons. IGF-1-TTC was shown to retain IGF-1 activity as indicated by [3H]thymidine incorporation into L6 myoblasts. Spinal cord motor neurons effectively bound and internalized the IGF-1-TTC in vitro. Similarly, IGF-1-TTC injected into skeletal muscles was taken up and transported back to the spinal cord in vivo, a process that could be prevented by denervation of the injected muscles. Three monthly IGF-1-TTC injections into muscles of aging mice did not increase muscle weight or fiber size but significantly increased single fiber specific force over aged controls injected with saline, IGF-1, or TTC. None of the injections changed muscle fiber-type composition, but neuromuscular junction postterminals were larger and more complex in muscle fibers injected with IGF-1-TTC compared to the other groups, suggesting preservation of muscle fiber innervation. This work demonstrates that induced overexpression of IGF-1 in spinal cord motor neurons of aging mice prevents muscle fiber specific force decline, a hallmark of aging skeletal muscle [132].
EXTERNAL Ca2+-DEPENDENT CONTRACTION IN AGING SKELETAL MUSCLE
We have demonstrated that a population of fast muscle fibers from aging mice depends on external Ca2+ to maintain tetanic force during repeated contractions [133]. We hypothesized that age-related denervation in muscle fibers plays a role in initiating this contractile deficit and that preventing denervation by IGF-1 overexpression would prevent external Ca2+-dependent contraction in aging mice, which was true. To determine whether IGF-1 overexpression affects muscle or nerve, aging mice were injected with a tetanus toxin fragment-C (TTC) fusion protein that targets IGF-1 to spinal cord motor neurons, and this treatment prevented external Ca2+-dependent contraction. We also showed that injections of the IGF-1-TTC fusion protein prevented age-related alterations to the nerve terminals at the neuromuscular junctions. We conclude that the slow, age-related denervation of fast muscle fibers is responsible for dependence on external Ca2+ to maintain tetanic force in a population of muscle fibers from senescent mice [134].
More recently, we examined the role of extracellular Ca2+, voltage-induced influx of external Ca2+ ions, SR Ca2+ depletion during repeated contractions, store-operated Ca2+ entry (SOCE), SR ultrastructure, SR subdomain localization of the ryanodine receptor, and sarcolemmal excitability in muscle force decline with aging. These experiments demonstrated that external Ca2+, but not Ca2+ influx, is needed to maintain fiber force with repeated electrical stimulation. Decline in fiber force is associated with depressed SR Ca2+ release. SR Ca2+ depletion, SOCE, and the putative segregated Ca2+ release store do not play a significant role in external Ca2+-dependent contraction. Note that a significant number of action potentials fail in senescent mouse muscle fibers subjected to a high stimulation frequency. These results indicate that failure to generate action potentials accounts for decreased intracellular Ca2+ mobilization and tetanic force in aging muscle exposed to a Ca2+-free medium [135].
THE SR JUNCTIONAL FACE MEMBRANE PROTEIN JP-45 PLAYS A ROLE IN SKELETAL MUSCLE ECU WITH AGING
JP-45 has been reported exclusively in skeletal muscle, and its expression decreases with age [136]. It colocalizes with the Ca2+-release channel (the ryanodine receptor) and interacts with calsequestrin and the skeletal muscle Cav1.1 [137]. We identified the JP-45 domains and the Cav1.1 involved in this interaction and investigated the functional effect of JP-45 on ECC. Its cytoplasmic domain, comprising residues 1-80, interacts with two distinct and functionally relevant domains of DHPRα1 subunit, the I-II loop and the C-terminal region. Interaction with the I-II loop occurs through the loop’s α-interacting domain. A DHPR subunit, β1α, also interacts with the cytosolic domain of JP-45, drasically reducing the interaction between JP-45 and the I-II loop (Fig. 1).
The functional effect of JP-45 on DHPRα1 subunit activity was assessed by investigating charge movement in differentiated C2C12 myotubes after overexpressing or depleting JP-45. Overexpression decreased peak charge-movement and shifted VQ1/2 to a more negative potential (−10 mV). Depletion decreased both the amount of DHPRα1 subunit and peak charge-movements. These results demonstrated that JP-45 is important for functional expression of voltage-dependent Ca2+ channels [137].
Another recent study demonstrates that deleting the gene that encodes JP-45 results in decreased muscle strength in young mice by decreasing functional expression of the Cav1.1 subunit, the molecule that couples membrane depolarization and calcium release from the SR. These results point to JP-45 as one of the molecules involved in the development or maintenance of skeletal muscle strength [138]. Whether JP-45 is modulated by neural activity and/or trophic factors is unknown.
In the last decade, a series of triad proteins have been identified, including mitsugumin-29 [139,140], junctophilin [141], SRP-27/TRIC-A [142-144], and junctate/hambug [145]. However, their role in ECC is only partially understood [146], and nerve-dependent regulation of their expression is unknown.
INCREASED CAvB1A EXPRESSION WITH AGING CONTRIBUTES SKELETAL MUSCLE WEAKNESS
Loss of specific force in old age [35, 147] is characterized in part by a deficit in Ca2+ release following depolarization [7, 148]. Excitation-contration uncoupling is not a result of decreased Ca2+ stores or RyR release function [148], and therefore may be caused by alterations in the functionality and expression of DHPR and its subunits with aging. The primary DHPR subunit in skeletal muscle is Cav1.1, previously known as DHPRα1s [149]. Cav1.1 is a large transmembrane protein, which contains both the Ca2+ conducting pore and the voltage sensing S4 domain. Auxiliary subunits (α2/δ, β1a and γ) bind Cav1.1 to make up DHPR (for review, see [150]), with the most widely studied being the cytosolic Cavβ1a subunit. Cavβ1a, a muscle specific member of the Cavβ family of proteins, binds to a region of the I-II intracellular loop of Cav1.1 known as the alpha interaction domain (AID) [151]. Cavβ1a is classically described by its role in chaperoning Cav1.1 to the plasma membrane and regulating L-type Ca2+ current [152-155]. Most notably, E-C coupling cannot occur without Cavβ1a [152]. Cavβ1a binds to charged residues on RyR [156] and neutralization of these residues reduces ECC, suggesting a direct interaction with RyR. The correct organization of Cav1.1 into tetrads within the t-tubule membrane is also a specific function of the Cavβ1a isoform [157].
Although classically known for augmenting the expression and function of Cav1 subfamily of calcium channels the Cavβ family of subunits may contribute to the down-regulation of Cav1 as well. A family of Ras-related G-proteins (RGKs) mediates the down-regulation of several Cav1 isoforms in a Cavβ dependent manner [158]. Additionally, the previously uncharacterized SH3 domain of Cavβ was shown to bind dynamin and mediate endocytosis of Cav1.2 [159]. As previous studies have shown that the Cav1.1 subunit declines in old rodents [128, 160, 161] and this causes an impairment of ECC [160], we wanted to investigate what effects aging had on Cavβ1a expression, as this subunit is also critical for ECC.
Western blot analysis shows a substantial increase of Cavβ1a expression over the full lifespan of FVB mice [162]. To examine the specific effects of Cavβ1a overexpression, a Cavβ1a -YFP plasmid was electroporated in vivo into young animals. The resulting increase in expression of Cavβ1a corresponded to decline of Cav1.1 over the same time period. YFP fluorescence, used as a measure of Cavβ1a -YFP expression in individual fibers, also showed an inverse relationship with charge movement, measured using the whole-cell patch-clamp technique. Specific force was significantly reduced in young Cavβ1a -YFP electroporated muscle fibers compared to sham-electroporated, age-matched controls. siRNA interference of Cavβ1a in young muscles reduced charge movement, while charge movement in old was restored to young control levels. These studies imply Cavβ1a serves as both a positive and negative regulator of Cav1.1 expression, and that endogenous overexpression of Cavβ1a during old age may play a roloe in the loss of specific force [162].
CONCLUDING COMMENTS
Age-related declines in the neuromuscular system are a recognized cause of impaired physical performance and loss of independence in the elderly. Epidemiological data associate these changes with increased risk of morbidity, disability, and mortality in the elderly [163-166].
We argue for the importance of neural factors in age-related impairment of mammalian skeletal muscle structure and function. Decreased local production of IGF-1 and/or neurotrophins and tissue resistance to these factors through altered receptor expression or responsiveness may result in loss and atrophy of spinal cord motor neurons. In fact, declining motor neuron function may be more extensive than that predicted by structural assays. Preliminary data support the concept that reduced IGF-1 synthesis may cause the failure of an IGF-mediated pathway to decrease CREB phosphorylation. In turn, reduced CREB phosphorylation may result in reduced DHPRα1S transcription, ECU, and decreased muscle force.
The characterization of a number of triad proteins is shedding light on the molecular signaling involved in excitation-gene expression and ECC [167]. The role of neural factors in regulating the expression and function of these newly identified triad proteins is a necessary focus of research in the coming years. We hypothesize that neural factors (autocrine trophic factors, nerve activity and connectivity) play a vital role in preventing age-related ECU. Based on this hypothesis, we predict that interventions aimed at counteracting nerve loss will play an important part in ameliorating the loss of force exhibited in animal models of aging as well as in elderly humans.
ACKNOWLEDGMENTS
Results reported in this article were obtained with the support of the National Institutes of Health/National Institute on Aging (AG15820, AG13934) and Muscular Dystrophy Association of America’s grants to Osvaldo Delbono and the Wake Forest University Claude D. Pepper Older Americans Independence Center (P30-AG21332).
Footnotes
DISCLOSURE
It should be noted that the author has previously published much of the material covered in this review article in a book chapter titled 'Excitation-Contraction Coupling Regulation in Aging Skeletal Muscle' in the book titled 'Sarcopenia — Age-Related Muscle Wasting and Weakness'. Springer Science+Business Media B.V. ISBN: 978-90-481-9712-5. 113-134, 2011.
REFERENCES
- [1].Lang T, Streeper T, Cawthon P, Baldwin K, Taaffe DR, Harris TB. Sarcopenia: etiology, clinical consequences, intervention, and assessment. Osteoporos Int 2010; 21(4): 543–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Henderson GC, Irving BA, Nair KS. Potential application of essential amino acid supplementation to treat sarcopenia in elderly people. J Clin Endocrinol Metab 2009; 94(5): 1524–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Doran P, Donoghue P, O'Connell K, Gannon J, Ohlendieck K. Proteomics of skeletal muscle aging. Proteomics 2009; 9(4): 989–1003. [DOI] [PubMed] [Google Scholar]
- [4].Kent-Braun JA, Ng AV. Specific strength and voluntary muscle activation in young and elderly women and men. J Appl Physiol 1999; 87(1): 22–9. [DOI] [PubMed] [Google Scholar]
- [5].Lynch NA, Metter EJ, Lindle RS, Fozard JL, Tobin JD, Roy TA, et al. Muscle quality. I. Age-associated differences between arm and leg muscle groups. J Appl Physiol 1999; 86(1): 188–94. [DOI] [PubMed] [Google Scholar]
- [6].Frontera WR, Hughes VA, Fielding RA, Fiatarone MA, Evans WJ, Roubenoff R. Aging of skeletal muscle: a 12-yr longitudinal study. J Appl Physiol 2000; 88(4): 1321–6. [DOI] [PubMed] [Google Scholar]
- [7].Delbono O, O'Rourke KS, Ettinger WH. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J Membr Biol 1995; 148(3): 211–22. [DOI] [PubMed] [Google Scholar]
- [8].Renganathan M, Messi ML, Delbono O. Overexpression of IGF-1 exclusively in skeletal muscle prevents age-related decline in the number of dihydropyridine receptors. J Biol Chem 1998; 273(44): 28845–51. [DOI] [PubMed] [Google Scholar]
- [9].Wang Z-M, Messi ML, Delbono O. L-type Ca2+ channel charge movement and intracellular Ca2+ in skeletal muscle fibers from aging mice. Biophys J 2000; 78: 1947–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lowe DA, Thomas DD, Thompson LV. Force generation, but not myosin ATPase activity, declines with age in rat muscle fibers. Am J Physiol Cell Physiol 2002; 283(1): C187–92. [DOI] [PubMed] [Google Scholar]
- [11].Payne AM, Delbono O. Neurogenesis of excitation-contraction uncoupling in aging skeletal muscle. Exerc Sport Sci Rev 2004; 32(1): 36–40. [DOI] [PubMed] [Google Scholar]
- [12].Dulhunty AF. Excitation-contraction coupling from the 1950s into the new millennium. Clin Exp Pharmacol Physiol 2006; 33(9): 763–72. [DOI] [PubMed] [Google Scholar]
- [13].Schneider MF, Chandler WK. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature 1973; 242(5395): 244–6. [DOI] [PubMed] [Google Scholar]
- [14].Tanabe T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature 1990; 346: 567–9. [DOI] [PubMed] [Google Scholar]
- [15].Delbono O, Renganathan M, Messi ML. Excitation-Ca2+ release-contraction coupling in single aged human skeletal muscle fiber. Muscle Nerve Suppl 1997; 5: S88–92. [DOI] [PubMed] [Google Scholar]
- [16].Loeser RF, Delbono O. The musculoskeletal and joint system. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, Eds. Hazzard's Geriatric Medicine and Gerontology. Sixth ed. New York: McGraw-Hill; 2009; pp. 1355–68. [Google Scholar]
- [17].Delbono O. Molecular mechanisms and therapeutics of the deficit in specific force in ageing skeletal muscle. Biogerontology 2002; 3(5): 265–70. [DOI] [PubMed] [Google Scholar]
- [18].Gonzalez E, Messi ML, Delbono O. The specific force of single intact extensor digitorum longus and soleus mouse muscle fibers declines with aging. J Membr Biol 2000; 178(3): 175–83. [DOI] [PubMed] [Google Scholar]
- [19].González E, Delbono O. Age-dependent fatigue in single intact fast- and slow-fibers from mouse EDL and soleus skeletal muscles. Mech Ageing Dev 2001; 122: 1019–32. [DOI] [PubMed] [Google Scholar]
- [20].González E, Delbono O. Recovery from fatigue in fast and slow single intact skeletal muscle fibers from aging mouse. Muscle Nerve 2001; 24: 1219–24. [DOI] [PubMed] [Google Scholar]
- [21].Delbono O. Calcium current activation and charge movement in denervated mammalian skeletal muscle fibres. J Physiol (Lond) 1992; 451:187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ray A, Kyselovic J, Leddy JJ, Wigle J, Jasmin BJ, Tuana BS. Regulation of dihydropyridine and ryanodine receptor gene expression in skeletal muscle. Role of nerve, protein kinase C and cAMP pathways. J Biol Chem 1995; 270: 25837–44. [DOI] [PubMed] [Google Scholar]
- [23].Kyselovic J, Leddy JJ, Ray A, Wigle J, Tuana BS. Temporal differences in the induction of dihydropyridine receptor subunits and ryanodine receptors during skeletal muscle development. J Biol Chem 1994; 269:21770–7. [PubMed] [Google Scholar]
- [24].Pereon Y, Sorrentino V, Dettbarn C, Noireaud J, Palade P. Dihydropyridine receptor and ryanodine receptor gene expression in long-term denervated rat muscles. Biochem Biophys Res Commun 1997; 240: 612–7. [DOI] [PubMed] [Google Scholar]
- [25].Chaudari N, Beam KG. mRNA for cardiac calcium channel is expressed during development of skeletal muscle. Dev Biol 1993; 155: 507–15. [DOI] [PubMed] [Google Scholar]
- [26].Pauwels PJ, Van Assouw HP, Leysen JE. Depolarization of chick myotubes triggers the appearance of (+)-[3H]PN 200-110-Binding Sites. Mol Pharmacol 1987; 32: 785–91. [PubMed] [Google Scholar]
- [27].Saborido A, Molano F, Moro G, Megias A. Regulation of dihydropyridine receptor levels in skeletal and cardiac muscle by exercise training. Pflug Archiv- Eur J Physiol 1995; 429: 364–9. [DOI] [PubMed] [Google Scholar]
- [28].Pereon Y, Navarro J, Hamilton M, Booth FW, Palade P. Chronic stimulation differentially modulates expression of mRNA for dihydropyridine receptor isoforms in rat fast twitch skeletal muscle. Biochem Biophys Res Commun 1997; 235: 217–22. [DOI] [PubMed] [Google Scholar]
- [29].Messi ML, Delbono O. Target-derived trophic effect on skeletal muscle innervation in senescent mice. J Neurosci 2003; 23(4): 1351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Delbono O. Neural control of aging skeletal muscle. Aging Cell 2003; 2: 21–9. [DOI] [PubMed] [Google Scholar]
- [31].Finol HJ, Lewis DM, Owens R. The effects of denervation on contractile properties or rat skeletal muscle. J Physiol 1981; 319: 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dulhunty AF, Gage PW. Excitation-contraction coupling and charge movement in denervated rat extensor digitorum longus and soleus muscles. J Physiol 1985; 358: 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Larsson L, Ansved T. Effects of ageing on the motor unit. Prog Neurobiol 1995; 45(5): 397–458. [DOI] [PubMed] [Google Scholar]
- [34].Wang Z-M, Messi ML, Delbono O. Sustained overexpression of IGF-1 prevents age-dependent decrease in charge movement and intracellular calcium in mouse skeletal muscle. Biophys J 2002; 82: 1338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gonzalez A, Kirsch WG, Shirokova N, Pizarro G, Brum G, Pessah IN, et al. Involvement of multiple intracellular release channels in calcium sparks of skeletal muscle. Proc Natl Acad Sci USA 2000; 97(8): 4380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol Ser A, Biol Sci Med Sci 1995; 50(Spec No): 11–6. [DOI] [PubMed] [Google Scholar]
- [37].Dutta C. Significance of sarcopenia in the elderly. J Nutr 1997; 127(5 Suppl): 992S–3S. [DOI] [PubMed] [Google Scholar]
- [38].Larsson L, Ansved T, Edstrom L, Gorza L, Schiaffino S. Effects of age on physiological, immunohistochemical and biochemical properties of fast-twitch single motor units in the rat. J Physiol (Lond) 1991; 443: 257–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Frontera WR, Suh D, Krivickas LS, Hughes VA, Goldstein R, Roubenoff R. Skeletal muscle fiber quality in older men and women. Am J Physiol Cell Physiol 2000; 279(3): C611–8. [DOI] [PubMed] [Google Scholar]
- [40].Lauretani F, Bandinelli S, Bartali B, Di Iorio A, Giacomini V, Corsi AM, et al. Axonal degeneration affects muscle density in older men and women. Neurobiol Aging 2006; 27(8): 1145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brooks SV, Faulkner JA. Skeletal muscle weakness in old age: underlying mechanisms. Med Sci Sports Exer 1994; 26(4): 432–9. [PubMed] [Google Scholar]
- [42].Hook P, Li X, Sleep J, Hughes S, Larsson L. In vitro motility speed of slow myosin extracted from single soleus fibres from young and old rats. J Physiol (Lond) 1999; 520(Pt 2): 463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Faulkner JA, Brooks SV, Opiteck JA. Injury to skeletal muscle fibers during contractions: conditions of occurrence and prevention. Phys Ther 1993; 73: 911–21. [DOI] [PubMed] [Google Scholar]
- [44].Rader EP, Faulkner JA. Effect of aging on the recovery following contraction-induced injury in muscles of female mice. J Appl Physiol 2006; 101(3): 887–92. [DOI] [PubMed] [Google Scholar]
- [45].Tseng BS, Marsh DR, Hamilton MT, Booth FW. Strength and aerobic training attenuate muscle wasting and improve resistance to the development of disability with aging. J Gerontol A Biol Sci Med Sci 1995; 50 (Spec No): 113–9. [DOI] [PubMed] [Google Scholar]
- [46].Weindruch R. Interventions based on the possibility that oxidative stress contributes to sarcopenia. J Gerontol A Biol Sci Med Sci 1995; 50(Spec No): 157–61. [DOI] [PubMed] [Google Scholar]
- [47].Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 2008; 88(4): 1243–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Renganathan M, Sonntag WE, Delbono O. L-type Ca2+ channel-insulin-like growth factor-1 receptor signaling impairment in aging rat skeletal muscle. Biochem Biophys Res Commun 1997; 235(3): 784–9. [DOI] [PubMed] [Google Scholar]
- [49].Owino W, Yang SY, Goldspink G. Age-related loss of skeletal muscle function and the inability to express the autocrine form of insulin-like growth factor-1 (MGF) in response to mechanical overload. FEBS Lett 2001; 505: 259–63. [DOI] [PubMed] [Google Scholar]
- [50].Shavlakadze T, Winn N, Rosenthal N, Grounds MD. Reconciling data from transgenic mice that overexpress IGF-I specifically in skeletal muscle. Growth Horm IGF Res 2005; 15(1): 4–18. [DOI] [PubMed] [Google Scholar]
- [51].Decary S, Mouly V, Hamida CB, Sautet A, Barbet JP, Butler-Browne GS. Replicative potential and telomere length in human skeletal muscle: implications for satellite cell-mediated gene therapy. Hum Gene Ther 1997; 8(12): 1429–38. [DOI] [PubMed] [Google Scholar]
- [52].Edstrom E, Altun M, Bergman E, Johnson H, Kullberg S, Ramirez-Leon V, et al. Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol Behav 2007; 92(1-2): 129–35. [DOI] [PubMed] [Google Scholar]
- [53].Ceballos D, Cuadras J, Verdu E, Navarro X. Morphometric and ultrastructural changes with ageing in mouse peripheral nerve. J Anat 1999; 195 (Pt 4): 563–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Verdu E, Ceballos D, Vilches JJ, Navarro X. Influence of aging on peripheral nerve function and regeneration. J Peripher Nerv Syst 2000; 5(4): 191–208. [DOI] [PubMed] [Google Scholar]
- [55].Jacob JM. Lumbar motor neuron size and number is affected by age in male F344 rats. Mech Aging Dev 1998; 106: 205–16. [DOI] [PubMed] [Google Scholar]
- [56].Liu R-H, Bertolotto C, Engelhardt JK, Chase MH. Age-related changes in soma size of neurons in the spinal cord motor column of the cat. Neurosci Lett 1996; 211: 163–6. [DOI] [PubMed] [Google Scholar]
- [57].Hashizume K, Kanda K, Burke R. Medial gastrocnemius motor nucleus in the rat: Age-related changes in the number and size of motoneurons. J Comp Neurol 1988; 269: 425–30. [DOI] [PubMed] [Google Scholar]
- [58].Larsson L. Motor units: remodeling in aged animals. J Gerontol A Biol Sci Med Sci 1995; 50(Spec No): 91–5. [DOI] [PubMed] [Google Scholar]
- [59].Urbancheck MG, Picken EB, Kalliainen LK, Kuzon WM. Specific force deficit in skeletal muscles of old rats is partially explained by the existence of denervated muscle fibers. J Gerontol: Biol Sci 2001; 56A: B191–B8. [DOI] [PubMed] [Google Scholar]
- [60].Kanda K, Hashizume K. Changes in properties of the medial gastrocnemius motor units in aging rats. J Neurophysiol 1989; 4: 737–46. [DOI] [PubMed] [Google Scholar]
- [61].Einsiedel LJ, Luff AR. Effect of partial denervation on motor units in the ageing rat medial gastrocnemius. J Neurol Sci 1992; 112: 178–84. [DOI] [PubMed] [Google Scholar]
- [62].Kanda K, Hashizume K. Factors causing difference in force output among motor units in the rat medial gastrocnemius muscle. J Physiol (Lond) 1992; 448: 677–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Doherty TJ, Vandervoort AA, Taylor AW, Brown WF. Effects of motor unit losses on strength in older men and women. J Appl Physiol 1993; 74: 868–74. [DOI] [PubMed] [Google Scholar]
- [64].Johnson H, Mossberg K, Arvidsson U, Piehl F, Hokfelt T, Ulfhake B. Increase in alpha-CGRP and GAP-43 in aged motoneurons: A study of peptides, growth factors, and ChAT mRNA in the lumbar spinal cord of senescent rats with symptoms of hindlimb incapacities. J Comp Neurol 1995; 359: 69–89. [DOI] [PubMed] [Google Scholar]
- [65].Zhang C, Goto N, Suzuki M, Ke M. Age-related reductions in number and size of anterior horn cells at C6 level of the human spinal cord. Okajimas Folia Anat Jpn 1996; 73(4): 171–7. [DOI] [PubMed] [Google Scholar]
- [66].Pette D, Staron RS. Transitions of muscle fiber phenotypic profiles. Histochem Cell Biol 2001; 115(5): 359–72. [DOI] [PubMed] [Google Scholar]
- [67].Buller AJ, Eccles JC, Eccles RM. Differentiation of fast and slow muscles in the cat hind limb. J Physiol (Lond) 1960; 150: 399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Buller AJ, Eccles JC, Eccles RM. Interactions between motoneurones and muscles in respect of the characteristic speeds of their responses. J Physiol (Lond) 1960; 150: 417–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Greensmith L, Vrbova G. Motoneurone survival: a functional approach. Trends Neurosci 1996; 19(11): 450–5. [DOI] [PubMed] [Google Scholar]
- [70].Kadhiresan VA, Hassett CA, Faulkner JA. Properties of single motor units in medial gastrocnemius muscles of adult and old rats. J Physiol (Lond) 1996; 493(Pt 2): 543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 2000; 20(7): 2534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Andersson AM, Olsen M, Zhernosekov D, Gaardsvoll H, Krog L, Linnemann D, et al. Age-related changes in expression of the neural cell adhesion molecule in skeletal muscle: a comparative study of newborn, adult and aged rats. Biochem J 1993; 290 (Pt 3): 641–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gosztonyi G, Naschold U, Grozdanovic Z, Stoltenburg-Didinger G, Gossrau R. Expression of Leu-19 (CD56, N-CAM) and nitric oxide synthase (NOS) I in denervated and reinnervated human skeletal muscle. Microsc Res Tech 2001; 55(3): 187–97. [DOI] [PubMed] [Google Scholar]
- [74].González E, Messi ML, Zheng Z, Delbono O. Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J Physiol 2003; 552(Pt 3): 833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wang ZM, Zheng Z, Messi ML, Delbono O. Extension and magnitude of denervation in skeletal muscle from ageing mice. J Physiol 2005; 565(Pt 3): 757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Trimmer JS, Cooperman SS, Tomiko SA, Zhou JY, Crean SM, Boyle MB, et al. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron 1989; 3(1): 33–49. [DOI] [PubMed] [Google Scholar]
- [77].Kallen RG, Sheng ZH, Yang J, Chen LQ, Rogart RB, Barchi RL. Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle. Neuron 1990; 4(2): 233–42. [DOI] [PubMed] [Google Scholar]
- [78].Yang JS, Sladky JT, Kallen RG, Barchi RL. TTX-sensitive and TTX-insensitive sodium channel mRNA transcripts are independently regulated in adult skeletal muscle after denervation. Neuron 1991; 7(3): 421–7. [DOI] [PubMed] [Google Scholar]
- [79].Redfern P, Lundh H, Thesleff S. Tetrodotoxin resistant action potentials in denervated rat skeletal muscle. Eur J Pharmacol 1970; 11(2): 263–5. [DOI] [PubMed] [Google Scholar]
- [80].Pappone PA. Voltage-clamp experiments in normal and denervated mammalian skeletal muscle fibres. J Physiol 1980; 306: 377–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].White MM, Chen LQ, Kleinfield R, Kallen RG, Barchi RL. SkM2, a Na+ channel cDNA clone from denervated skeletal muscle, encodes a tetrodotoxin-insensitive Na+ channel. Mol Pharmacol 1991; 39(5): 604–8. [PubMed] [Google Scholar]
- [82].Kawabuchi M, Zhou CJ, Wang S, Nakamura K, Liu WT, Hirata K. The spatiotemporal relationship among Schwann cells, axons and postsynaptic acetylcholine receptor regions during muscle reinnervation in aged rats. Anat Rec 2001; 264(2): 183–202. [DOI] [PubMed] [Google Scholar]
- [83].Carlson BM, Dedkov EI, Borisov AB, Faulkner JA. Skeletal muscle regeneration in very old rats. J Gerontol A Biol Sci Med Sci 2001; 56(5): B224–33. [DOI] [PubMed] [Google Scholar]
- [84].Cederna PS, Asato H, Gu X, van der Meulen J, Kuzon WM Jr., Carlson BM, et al. Motor unit properties of nerve-intact extensor digitorum longus muscle grafts in young and old rats. J Gerontol A Biol Sci Med Sci 2001; 56(6): B254–8. [DOI] [PubMed] [Google Scholar]
- [85].Kanda K, Hashizume K. Effects of long-term physical exercise on age-related changes of spinal motoneurons and peripheral nerves in rats. Neurosci Res 1998; 31(1): 69–75. [DOI] [PubMed] [Google Scholar]
- [86].Tabata H, Ikegami H, Kariya K. A parallel comparison of age-related peripheral nerve changes in three different strains of mice. Exp Anim 2000; 49(4): 295–9. [DOI] [PubMed] [Google Scholar]
- [87].Kerezoudi E, Thomas PK. Influence of age on regeneration in the peripheral nervous system. Gerontology 1999; 45(6): 301–6. [DOI] [PubMed] [Google Scholar]
- [88].McQuarrie IG, Brady ST, Lasek RJ. Retardation in the slow axonal transport of cytoskeletal elements during maturation and aging. Neurobiol Aging 1989; 10(4): 359–65. [DOI] [PubMed] [Google Scholar]
- [89].Cross DJ, Flexman JA, Anzai Y, Maravilla KR, Minoshima S. Age-related decrease in axonal transport measured by MR imaging in vivo. Neuroimage 2008; 39(3): 915. [DOI] [PubMed] [Google Scholar]
- [90].Jacob JM, Robbins N. Age differences in morphology of reinnervation of partially denervated mouse muscle. J Neurosci 1990; 10(5): 1530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Balice-Gordon RJ. Age-related changes in neuromuscular innervation. Muscle Nerve 1997; S5: S83–S7. [DOI] [PubMed] [Google Scholar]
- [92].Personius KE, Balice-Gordon RJ. Activity-dependent editing of neuromuscular synaptic connections. Brain Res Bull 2000; 53(5): 513–22. [DOI] [PubMed] [Google Scholar]
- [93].Fahim MA. Endurance exercise modulates neuromuscular junction of C57BL/6NNia aging mice. J Appl Physiol 1997; 83(1): 59–66. [DOI] [PubMed] [Google Scholar]
- [94].Costanzo EM, Barry JA, Ribchester RR. Competition at silent synapses in reinnervated skeletal muscle. Nat Neurosci 2000; 3(7): 694–700. [DOI] [PubMed] [Google Scholar]
- [95].Bezakova G, Lomo T. Muscle activity and muscle agrin regulate the organization of cytoskeletal proteins and attached acetylcholine receptor (AchR) aggregates in skeletal muscle fibers. J Cell Biol 2001; 153(7): 1453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ervasti JM. Costameres: the Achilles' Heel of Herculean Muscle. J Biol Chem 2003; 278(16): 13591–4. [DOI] [PubMed] [Google Scholar]
- [97].Salvatori S, Damiani E, Zorzato F, Volpe P, Pierobon S, Quaglino D, et al. Denervation-induced proliferative changes of triads in rabbit skeletal muscle. Muscle Nerve 1988; 11: 1246–59. [DOI] [PubMed] [Google Scholar]
- [98].Delbono O, Stefani E. Calcium current inactivation in denervated rat skeletal muscle fibres. J Physiol (Lond) 1993; 460: 173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Delbono O, Chu A. Ca2+ release channels in rat denervated skeletal muscles. Exp Physiol 1995; 80(4): 561–74. [DOI] [PubMed] [Google Scholar]
- [100].Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol 1997; 10(2): 168–75. [DOI] [PubMed] [Google Scholar]
- [101].Davies AM. The neurotrophic hypothesis: where does it stand? Philos Trans R Soc Lond B Biol Sci 1996; 351(1338): 389–94. [DOI] [PubMed] [Google Scholar]
- [102].Gibbons A, Wreford N, Pankhurst J, Bailey K. Continuous supply of the neurotrophins BDNF and NT-3 improve chick motor neuron survival in vivo. Int J Dev Neurosci 2005; 23(4): 389. [DOI] [PubMed] [Google Scholar]
- [103].Cowen T, Gavazzi I. Plasticity in adult and ageing sympathetic neurons. Prog Neurobiol 1998; 54(3): 249–88. [DOI] [PubMed] [Google Scholar]
- [104].Orike N, Thrasivoulou C, Wrigley A, Cowen T. Differential regulation of survival and growth in adult sympathetic neurons: an in vitro study of neurotrophin responsiveness. J Neurobiol 2001; 47(4): 295–305. [DOI] [PubMed] [Google Scholar]
- [105].Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature 1993; 363(6427): 350–3. [DOI] [PubMed] [Google Scholar]
- [106].Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci 2000; 23(12): 639–45. [DOI] [PubMed] [Google Scholar]
- [107].Boulanger L, Poo MM. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci 1999; 2(4): 346–51. [DOI] [PubMed] [Google Scholar]
- [108].Leßmann V, Brigadski T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: An update. Neurosci Res 2009; 65(1): 11–22. [DOI] [PubMed] [Google Scholar]
- [109].ang X, Berninger B, Poo M. Localized synaptic actions of neurotrophin-4. J Neurosci 1998; 18(13): 4985–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Funakoshi H, Belluardo N, Arenas E, Yamamoto Y, Casabona A, Persson H, et al. Muscle-derived neurotrophin-4 as an activity-dependent trophic signal for adult motor neurons. Science 1995; 268(5216): 1495–9. [DOI] [PubMed] [Google Scholar]
- [111].Bergman E, Ulfhake B, Fundin BT. Regulation of NGF-family ligands and receptors in adulthood and senescence: correlation to degenerative and regenerative changes in cutaneous innervation. Eur J Neurosci 2000; 12(8): 2694–706. [DOI] [PubMed] [Google Scholar]
- [112].Mathews LS, Hammer RE, Behringer RR, D'Ercole AJ, Bell GI, Brinster RL, et al. Growth enhancement of transgenic mice expressing human insulin-like growth factor I. Endocrinology 1988; 123: 2827–33. [DOI] [PubMed] [Google Scholar]
- [113].Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, et al. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem 1995; 270(20): 12109–16. [DOI] [PubMed] [Google Scholar]
- [114].Adams GR, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol 1998; 84(5): 1716–22. [DOI] [PubMed] [Google Scholar]
- [115].Musaro A, McCullagh KJ, Paul A, Houghton L, Dobrowolny G, Molinaro M, et al. Localized IGF-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 2001; 27: 195–200. [DOI] [PubMed] [Google Scholar]
- [116].Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muslce function. Proc Natl Acad Sci 1998; 95: 15603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chakravarthy MV, Fiorotto ML, Schwartz RJ, Booth FW. Long-term insulin-like growth factor-1 expression in skeletal muscles attenuates the enhanced in vitro proliferation ability of the resident satellite cells in transgenic mice. Mech Ageing Dev 2001; 122: 1303–20. [DOI] [PubMed] [Google Scholar]
- [118].González E, Delbono O. Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J Physiol 2003; 552(Pt.3): 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Neff NT, Prevette DM, Houenou LJ, Lewis ME, Glicksman MA, Yin Q-W, et al. Insulin-like growth factors: Putative muscle-derived trophic agents that promote motoneuron survival. J Neurobiol 1993; 24(12): 1578–88. [DOI] [PubMed] [Google Scholar]
- [120].Rind HB, von Bartheld CS. Target-derived cardiotrophin-1 and insulin-like growth factor-I promote neurite growth and survival of developing oculomotor neurons. Mol Cell Neurosci 2002; 19(1): 58–71. [DOI] [PubMed] [Google Scholar]
- [121].Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol 2005; 168(2): 193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Messi ML, Clark HM, Prevette DM, Oppenheim RW, Delbono O. The lack of effect of specific overexpression of IGF-1 in the central nervous system or skeletal muscle on pathophysiology in the G93A SOD-1 mouse model of ALS. Exp Neurol 2007; 207(1): 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Hammarberg H, Risling M, Hokfelt T, Cullheim S, Piehl F. Expression of insulin-like growth factors and corresponding binding proteins (IGFBP 1-6) in rat spinal cord and peripheral nerve after axonal injuries. J Comp Neurol 1998; 400(1): 57–72. [PubMed] [Google Scholar]
- [124].Pu SF, Zhuang HX, Marsh DJ, Ishii DN. Time-dependent alteration of insulin-like growth factor gene expression during nerve regeneration in regions of muscle enriched with neuromuscular junctions. Brain Res Mol Brain Res 1999; 63(2): 207–16. [DOI] [PubMed] [Google Scholar]
- [125].Vergani L, Di Giulio AM, Losa M, Rossoni G, Muller EE, Gorio A. Systemic administration of insulin-like growth factor decreases motor neuron cell death and promotes muscle reinnervation. J Neurosci Res 1998; 54(6): 840–7. [DOI] [PubMed] [Google Scholar]
- [126].O'Kusky JR, Ye P, D'Ercole J. Insulin-Like growth factor-1 promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J Neurosci 2000; 15: 8435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Ye P, Xing Y, Dai Z, D'Ercole J. In vivo actions of insulin-like growth factor-I (IGF-1) on cerebellum development in transgenic mice: evidence that IGF-1 increases proliferation of granule cells progenitors. Dev Brain Res 1996; 95: 44–54. [DOI] [PubMed] [Google Scholar]
- [128].Moreno R, Messi M, Zheng Z, Wang Z-M, Ye P, D'Ercole JA, et al. Role of sustained overexpression of central nervous system IGF-1 in the age-dependent decline of mouse excitation-contraction coupling. J Membr Biol 2006; 212(3): 147–61. [DOI] [PubMed] [Google Scholar]
- [129].Bondy C, Werner H, Roberts CT Jr., LeRoith D. Cellular pattern of type-I insulin-like growth factor receptor gene expression during maturation of the rat brain: comparison with insulin-like growth factors I and II. Neuroscience 1992; 46(4): 909–23. [DOI] [PubMed] [Google Scholar]
- [130].Zheng Z, Messi ML, Delbono O. Age-dependent IGF-1 regulation of gene transcription of Ca2+ channels in skeletal muscle. Mech Aging Dev 2001; 122: 373–84. [DOI] [PubMed] [Google Scholar]
- [131].Zheng Z, Wang Z-M, Delbono O. Charge movement and transcription regulation of L-type calcium channel alpha-1S in skeletal muscle cells. J Physiol (Lond) 2002; 540(2): 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Payne AM, Messi ML, Zheng Z, Delbono O. Motor neuron targeting of IGF-1 attenuates age-related external Ca2+-dependent skeletal muscle contraction in senescent mice. J Physiol (Lond) 2006; 570(2): 283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Payne AM, Zheng Z, Gonzalez E, Wang ZM, Messi ML, Delbono O. External Ca2+-dependent excitation-contraction coupling in a population of ageing mouse skeletal muscle fibres. J Physiol (Lond) 2004; 560.1: 137–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Payne AM, Messi ML, Zheng Z, Delbono O. Motor neuron targeting of IGF-1 attenuates age-related external Ca(2+)-dependent skeletal muscle contraction in senescent mice. Exp Gerontol 2007; 42(4): 309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Payne AM, Jimenez-Moreno R, Wang ZM, Messi ML, Delbono O. Role of Ca2+, membrane excitability, and Ca2+ stores in failing muscle contraction with aging. Exp Gerontol 2009; 44(4): 261–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Anderson AA, Treves S, Biral D, Betto R, Sandona D, Ronjat M, et al. The novel skeletal muscle sarcoplasmic reticulum JP-45 protein. Molecular cloning, tissue distribution, developmental expression, and interaction with alpha 1.1 subunit of the voltage-gated calcium channel. J Biol Chem 2003; 278(41): 39987–92. [DOI] [PubMed] [Google Scholar]
- [137].Anderson AA, Altafaj X, Zheng Z, Wang ZM, Delbono O, Ronjat M, et al. The junctional SR protein JP-45 affects the functional expression of the voltage-dependent Ca2+ channel Cav1.1. J Cell Sci 2006; 119(Pt 10): 2145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Delbono O, Xia J, Treves S, Wang ZM, Jimenez-Moreno R, Payne AM, et al. Loss of skeletal muscle strength by ablation of the sarcoplasmic reticulum protein JP45. Proc Natl Acad Sci USA 2007; 104(50): 20108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Shimuta M, Komazaki S, Nishi M, Iino M, Nakagawara K, Takeshima H. Structure and expression of mitsugumin29 gene. FEBS Lett 1998; 431(2): 263–7. [DOI] [PubMed] [Google Scholar]
- [140].Takeshima H, Shimuta M, Komazaki S, Ohmi K, Nishi M, Iino M, et al. Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem J 1998; 331 (Pt 1): 317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 2000; 6(1): 11–22. [DOI] [PubMed] [Google Scholar]
- [142].Yazawa M, Ferrante C, Feng J, Mio K, Ogura T, Zhang M, et al. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 2007; 448(7149): 78–82. [DOI] [PubMed] [Google Scholar]
- [143].Bleunven C, Treves S, Jinyu X, Leo E, Ronjat M, De Waard M, et al. SRP-27 is a novel component of the supramolecular signalling complex involved in skeletal muscle excitation-contraction coupling. Biochem J 2008; 411(2): 343–9. [DOI] [PubMed] [Google Scholar]
- [144].Zhao X, Weisleder N, Thornton A, Oppong Y, Campbell R, Ma J, et al. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell 2008; 7(4): 561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Treves S, Feriotto G, Moccagatta L, Gambari R, Zorzato F. Molecular cloning, expression, functional characterization, chromosomal localization, and gene structure of junctate, a novel integral calcium binding protein of sarco(endo)plasmic reticulum membrane. J Biol Chem 2000; 275(50): 39555–68. [DOI] [PubMed] [Google Scholar]
- [146].Treves S, Vukcevic M, Maj M, Thurnheer R, Mosca B, Zorzato F. Minor sarcoplasmic reticulum membrane components that modulate excitation-contraction coupling in striated muscles. J Physiol 2009; 587(Pt 13): 3071–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol (Lond) 1988; 404: 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Jimenez-Moreno R, Wang ZM, Gerring R, Delbono O. Sarcoplasmic reticulum Ca2+ release declines in muscle fibers from aging mice. Biophys J 2008; 94: 3178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 2005; 57(4): 397–409. [DOI] [PubMed] [Google Scholar]
- [150].Flucher BE, Obermair GJ, Tuluc P, Schredelseker J, Kern G, Grabner M. The role of auxiliary dihydropyridine receptor subunits in muscle. J Muscle Res Cell Motil 2005; 26(1): 1–6. [DOI] [PubMed] [Google Scholar]
- [151].Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, et al. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature 2004; 429(6992): 675–80. [DOI] [PubMed] [Google Scholar]
- [152].Gregg RG, Messing A, Strube C, Beurg M, Moss R, Behan M, et al. Absence of the beta subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the alpha 1 subunit and eliminates excitation-contraction coupling. Proc Natl Acad Sci USA 1996; 93(24): 13961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Strube C, Beurg M, Powers PA, Gregg RG, Coronado R. Reduced Ca2+ current, charge movement, and absence of Ca2+ transients in skeletal muscle deficient in dihydropyridine receptor beta 1 subunit. Biophys J 1996; 71(5): 2531–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Beurg M, Sukhareva M, Strube C, Powers PA, Gregg RG, Coronado R. Recovery of Ca2+ current, charge movements, and Ca2+ transients in myotubes deficient in dihydropyridine receptor beta 1 subunit transfected with beta 1 cDNA. Biophys J 1997; 73(2): 807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Neuhuber B, Gerster U, Doring F, Glossmann H, Tanabe T, Flucher BE. Association of calcium channel alpha1S and beta1a subunits is required for the targeting of beta1a but not of alpha1S into skeletal muscle triads. Proc Natl Acad Sci USA 1998; 95(9): 5015–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Cheng W, Altafaj X, Ronjat M, Coronado R. Interaction between the dihydropyridine receptor Ca2+ channel beta-subunit and ryanodine receptor type 1 strengthens excitation-contraction coupling. Proc Natl Acad Sci USA 2005; 102(52): 19225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Schredelseker J, Di Biase V, Obermair GJ, Felder ET, Flucher BE, Franzini-Armstrong C, et al. The beta 1a subunit is essential for the assembly of dihydropyridine-receptor arrays in skeletal muscle. Proc Natl Acad Sci USA 2005; 102(47): 17219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Beguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, et al. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 2001; 411(6838): 701–6. [DOI] [PubMed] [Google Scholar]
- [159].Gonzalez-Gutierrez G, Miranda-Laferte E, Neely A, Hidalgo P. The Src homology 3 domain of the beta-subunit of voltage-gated calcium channels promotes endocytosis via dynamin interaction. J Biol Chem 2007; 282(4): 2156–62. [DOI] [PubMed] [Google Scholar]
- [160].Renganathan M, Messi ML, Delbono O. Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J Membr Biol 1997; 157(3): 247–53. [DOI] [PubMed] [Google Scholar]
- [161].O'Connell K, Gannon J, Doran P, Ohlendieck K. Reduced expression of sarcalumenin and related Ca2+ -regulatory proteins in aged rat skeletal muscle. Exp Gerontol 2008; 43(10): 958–61. [DOI] [PubMed] [Google Scholar]
- [162].Taylor JR, Zheng Z, Wang ZM, Payne AM, Messi ML, Delbono O. Increased CaVbeta1A expression with aging contributes to skeletal muscle weakness. Aging Cell 2009; 8(5): 584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Winograd CH, Gerety MB, Chung M, Goldstein MK, Dominguez FJ, Vallone R. Screening for frailty: criteria and prefictors of outcomes. J Am Geriatr Soc 1991; 39: 778–84. [DOI] [PubMed] [Google Scholar]
- [164].Baumgartner RN, Koehler KM, Gallagher D. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998; 147: 755–63. [DOI] [PubMed] [Google Scholar]
- [165].Ryall J, Schertzer J, Lynch G. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 2008; 9(4): 213. [DOI] [PubMed] [Google Scholar]
- [166].Buonanno A, Fields RD. Gene regulation by patterned electrical activity during neural and skeletal muscle development. Curr Opin Neurobiol 1999; 9(1): 110–20. [DOI] [PubMed] [Google Scholar]
- [167].Carrasco M, Jaimovich E, Kemmerling U, Hidalgo C. Signal transduction and gene expression regulated by calcium release from internal stores in excitable cells. Biol Res 2004; 37(4): 701–12. [DOI] [PubMed] [Google Scholar]
- [168].Meissner G, Lu X. Dihydropyridine receptor-ryanodine receptor interactions in skeletal muscle excitation-contraction coupling. Biosci Rep 1995; 15: 399–408. [DOI] [PubMed] [Google Scholar]
- [169].Beard NA, Casarotto MG, Wei L, Varsanyi M, Laver DR, Dulhunty AF. Regulation of ryanodine receptors by calsequestrin: effect of high luminal Ca2+ and phosphorylation. Biophys J 2005; 88(5): 3444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]