

Abstract
Amide synthesis is one of the most widely practiced chemical reactions, owing to its use in drug development and peptide synthesis. Despite the importance of these applications, the attendant effort to eliminate waste associated with these protocols has met with limited success, and pernicious α-epimerization is most often minimized but not eliminated when targeting challenging amides (e.g. N-aryl amides). This effort has focused on what is essentially a single paradigm in amide formation wherein an electrophilic acyl donor reacts with a nucleophilic amine. Umpolung Amide Synthesis (UmAS) emerged from α-halo nitroalkane reactions with amines and has since been developed into a method for the synthesis of enantiopure amides using entirely catalytic, enantioselective synthesis. However, its inability to forge N-aryl amides has been a longstanding problem, one limiting its application more broadly in drug development where α-chiral N-aryl amides are increasingly common. We report here the reaction of α-fluoronitroalkanes and N-aryl hydroxyl amines for the direct synthesis of N-aryl amides using a simple Brønsted base as the promoter. No other activating agents are required, and experiments guided by mechanistic hypotheses outline a mechanism based on the UmAS paradigm and confirm that the N-aryl amide, not the N-aryl hydroxamic acid, is the direct product. Ultimately, select chiral α-amino-N-aryl amides were prepared with complete conservation of enantioenrichment, in contrast to a parallel demonstration of their ability to epimerize using the conventional amide synthesis alternative.
Graphical Abstract
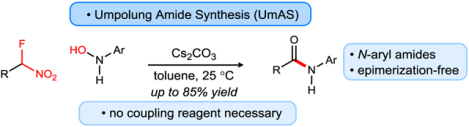
Amide synthesis is arguably one of the most broadly- and frequently-used organic reactions, stimulating a massive infrastructure development that includes technology (e.g. solid-phase peptide synthesis)1 and a broad array of raw materials (e.g. commercially-available carboxylic acids and amines).2 The protocols practiced for amide synthesis are economically unsustainable,3,4,5,6 and they are increasingly associated with health hazards that include human allergic response to the coupling reagents.7 At research scale where external safety measures may derisk the use of coupling agents and nucleophilic promoters, the overall strategy to prepare α-chiral amides is not only step-intensive, but also susceptible to epimerization.8,9,10 Armed with the belief that improvements within this paradigm may be only marginal, we have developed Umpolung Amide Synthesis (UmAS) into a method that can alleviate many of these issues, providing a means to prepare complex amides using entirely catalytic, enantioselective tactics.11,12,13,14,15 A key limitation, however, is the fact that aryl amines (anilines) have been uniformly unsuccessful in UmAS. We now address this shortcoming by the discovery that N-aryl hydroxyl amines are suitable acceptors in UmAS, unexpectedly leading directly to N-aryl amides instead of hydroxamic acids.16,17
Recent reports of the use of activated hydroxyl amines as electrophilic nitrogen sources led us to investigate the reactivity of α-halo nitronate nucleophiles with N-aryl hydroxyl amines and their activated derivatives (Figure 1).18,19 These efforts were largely unsuccessful with vanishingly small amounts of the desired N-aryl amide forming in the best cases. Within this dataset, use of an α-bromo nitroalkane (1a), the most common type of UmAS donor, with N-aryl hydroxyl amine 2 gave only 1% and 16% yield of the desired N-aryl amide (Table 1, entries 1–2). Although base selection can dramatically improve the yield of UmAS, the investigation of alternatives was met with limited improvement (Table 1, entries 3–4), but nitronate formation was clearly necessary (Table 1, entry 5). Cesium carbonate consistently outperformed alternative bases, so it was selected for subsequent studies.20 A lower concentration aided stirring since the carbonate base was minimally soluble, and this had little effect otherwise on the reaction outcome (Table 1, entry 6 vs. 7). Reasoning that the reactivity of the α-halonitronate might be better matched to the unusual N-aryl electrophile, we compared the reactivity of α-bromo nitroalkane 1a to α-fluoro nitroalkane 1b (Table 1, entries 7–8). This experiment could be performed at room temperature in both cases but the yield of the desired N-aryl amide was doubled when using the α-fluoronitroalkane. Further optimization of the protocol led to a 76% yield from 1b using three equivalents of cesium carbonate at room temperature (Table 1, entry 9). Throughout these experiments, formation of azoxy 4 from the hydroxyl amine substrate was noted.21
Figure 1.

Comparison of conventional amide synthesis (top) and initial UmAS approach to N-aryl amide synthesis (bottom).
Table 1.
Reaction optimization for α-halo nitroalkane conversion to N-aryl amide.a

| |||||
|---|---|---|---|---|---|
| entry | X | 1 | base | temp (°C) |
yield (%) |
| 1 | Br | a | TEA | 60 | 1 |
| 2 | Br | a | K2CO3 | 60 | 16 |
| 3 | Br | a | KOH | 60 | 13 |
| 4 | Br | a | Cs2CO3 | 60 | 30 |
| 5 | Br | a | none | 60 | 0 |
| 6 | Br | a | Cs2CO3 | rt | 33 |
| 7b | Br | a | Cs2CO3 | rt | 30 |
| 8b | F | b | Cs2CO3 | rt | 64 |
| 9b,c | F | b | Cs2CO3 | rt | 76 |
General procedure: nitroalkane (1 equiv.), hydroxylamine (1.5 equiv.), and base (2 equiv) in toluene (0.1 M) were sparged with Ar for ~2 min. The reaction was stirred for 16–26 hours.
Yields measured by 1H NMR relative to CH2Br2.
0.05 M.
3 equiv. base used.

The initial limits of N-aryl amide synthesis from a common α-fluoro nitroalkane (1b) and a variety of N-aryl hydroxyl amines were explored as summarized in Table 2. Electron-rich N-aryl amides were of particular interest, and the most common substituents (-NH2 (7b), -OMe (7e)) were tolerated. Although double acylation was not expected based on our mechanistic hypothesis, we confirmed that no double acylation product could be detected for 7b. Common electron-deficient N-aryl amides (7f, 7h, 7i) were also prepared in moderate to good yield (Table 2). A variety of α-fluoro nitroalkane donors were also prepared and compared using a common N-aryl hydroxyl amine (2). These included aryl (7k-l, q-s) and alkyl amides (7m-p, t-u).
Table 2.
N-Aryl amide synthesis from α-fluoronitroalkane and N-aryl hydroxyl amine.a

|
General procedure: The aryl hydroxylamine (1.5 equiv), Cs2CO3 (3 equiv), and fluoronitroalkane were stirred in solvent (0.05 M) in a vial for 24 h.
Yields were measured by 1H NMR (relative to CH2Br2) after filtration through a pad of silica gel. Isolated yields (after chromatography for most compounds) are reported in the SI.
Chemoselective amidations are particularly useful, and this method provides the amide derived exclusively from N-hydroxyl amines (Scheme 1, 8→9, 10→11). Finally, some reactions of cyclopropyl-substituted bromonitromethane are not stereospecific,22 but use of α-fluoro nitronate 5a in these studies led to N-aryl amide 7o as a single diastereomer. Intramolecular cyclization by Michael addition followed N-aryl amide formation in the case leading to 7u due to the electrophilic enoate functionality.
Scheme 1.
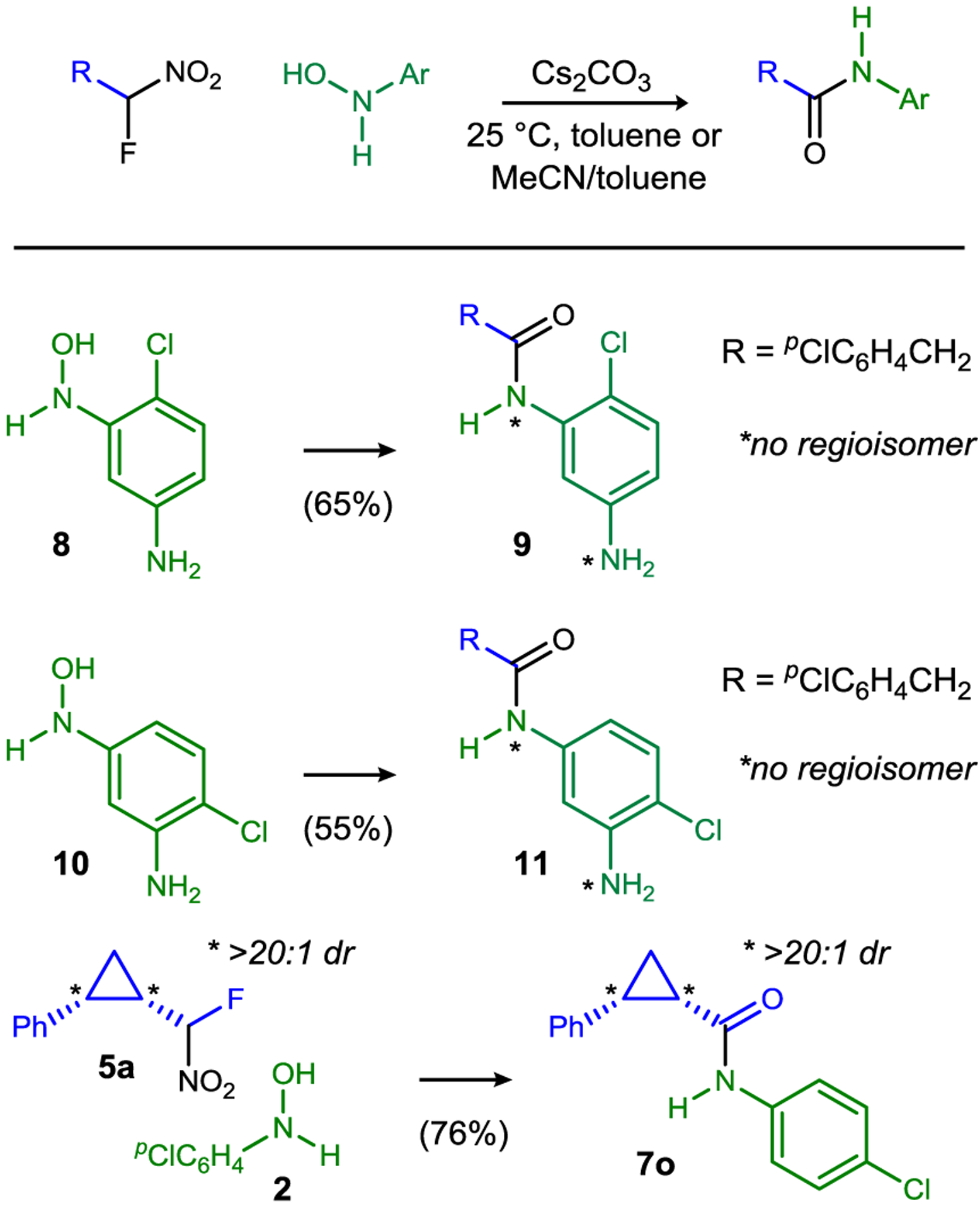
Umpolung N-aryl amide synthesis: regio- and diastereospecific reactions.a
a Reaction conditions mirrored those for Table 2. See SI for complete details. Isolated yield shown for 7o.
Despite the welcome simplicity of the reaction, using only a common base to promote the coupling, we were mystified by the observation that attempts to further activate the hydroxyl amine by tosylation, or acylation at oxygen (e.g. Figure 1), led only to recovery of starting materials. A formal substitution at nitrogen would lead directly to the amide products observed. Ultimately, our mechanistic hypotheses for the experiments in Table 1 were based on the oxidation of the hydroxyl amine to an aryl nitroso, but this redox pathway should lead to an N-aryl hydroxamic acid product instead of N-aryl amide.16
Initial mechanistic investigations therefore first reconsidered the non-umpolung paradigm for amide synthesis. Namely, if the N-aryl hydroxylamine is functioning as a nucleophile, as either a hydroxylamine N-nucleophile, or precursor to its aniline derivative, then these conditions should apply successfully to an aniline (Scheme 2). The competition between N-aryl hyroxylamine 2 and para-fluoro aniline (12) with α-fluoronitroalkane 1b (1:1:1 ratio) was found to produce solely the hydroxylamine-derived N-aryl amide (3) in 71% yield (Scheme 2, eq 2), consistent with the experiments in Scheme 1.23 A similar competition experiment with a more nucleophilic amine was also examined, leading to a similar result and a trace amount of benzyl amine-derived amide 15 (3%, Scheme 2, eq 3). A series of experiments using possible hydroxylamine-derived products as reactants led to the knowledge that azoxy 4 and nitroarene (para-chloro nitrobenzene)24 are not competent precursors to amide. Collectively, these experiments directed our hypothesis-driven mechanistic experiments back to an umpolung mechanism.
Scheme 2.
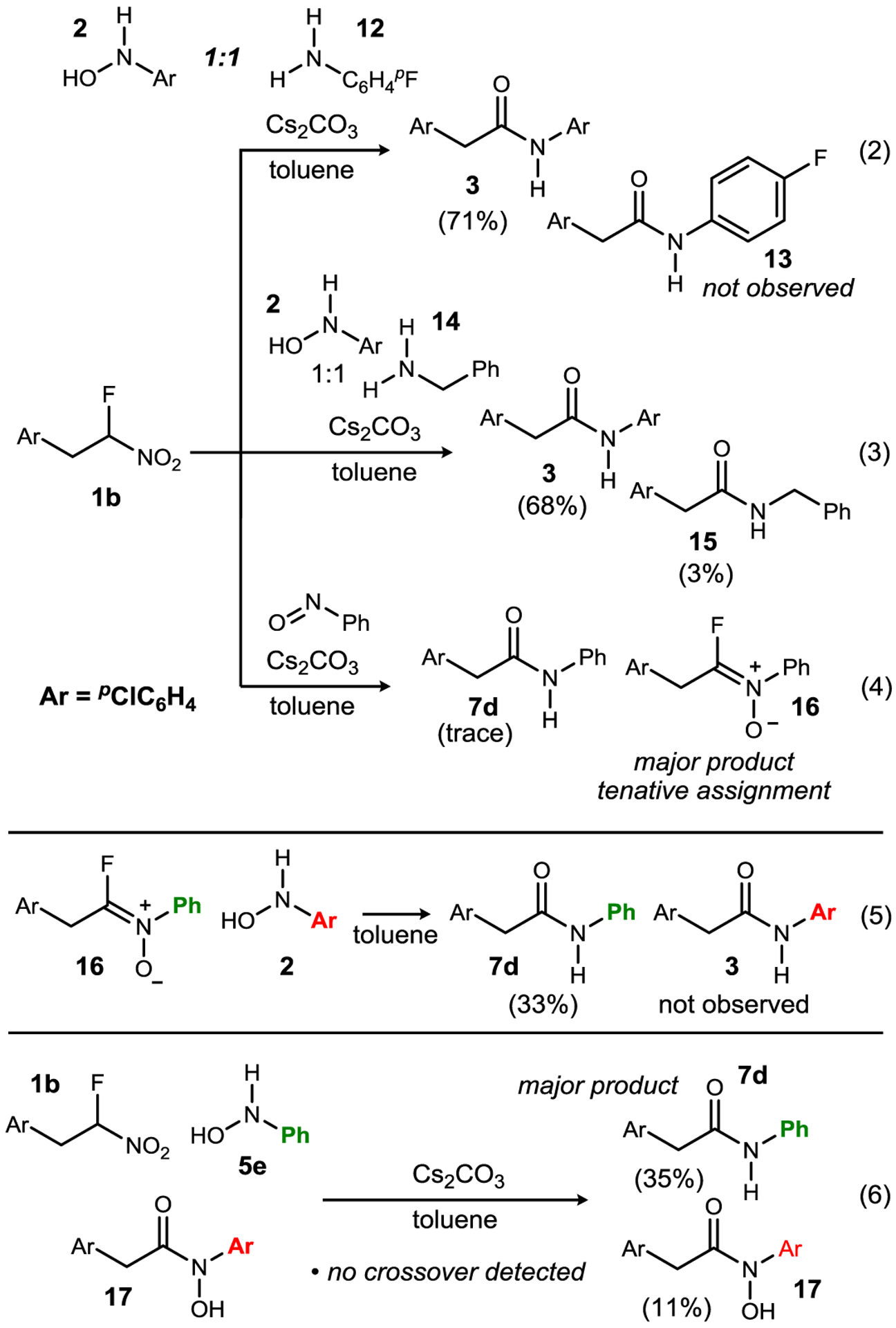
Experiments probing the initial mechanistic hypothesis.a
a Reaction conditions generally mirrored those for Table 2. Yields were measured by 1H NMR (relative to CH2Br2) after filtration through a pad of silica gel.
A working hypothesis consistent with these observations is summarized in Scheme 3. The N-aryl hydroxylamine is first oxidized to an aryl nitroso (20). This electrophile reacts with the α-fluoronitronate (21) to form haloamino nitroalkane (HANA)25 22 which then collapses to fluoronitrone 23. A new hydroxyl amine (19) then converts 23 to amide product, and consequently, forms a new aryl nitroso (20). An interesting feature of the catalytic cycle in Scheme 3 is the requirement for an aryl nitroso intermediate (20), but a parallel need for N-aryl hydroxyl amine (19) to effect the final step from 23 to amide 24. Hence, substitution of the hydroxyl amine by an aryl nitroso should arrest the sequence at 23, owing to the removal of the N-aryl hydroxyl amine and its putative role as a key reductant. In the event (Scheme 2, eq 4), use of nitrosobenzene provided traces of amide 7d alongside a major product assigned the structure of fluoronitrone 16 as a 77:23 mixture of geometric isomers. This species was unstable, but immediate analysis was revealing by 1H NMR (pair of doublets at 3.78, 3.61 ppm) and 19F NMR (singlets at −26.4, −27.4 ppm), leading to our assignment (16). Additional experiments established the coupling of fluorine with proton. Although no direct analogies for 16 could be found in the literature, it has been postulated previously26,27 and other α-heteroatom nitrones are known.28,29 HNO2 elimination from a HANA is analogous to formation of nitrones from nitroalkane addition to nitrosoarenes30 and azomethine ylide formation from α-fluoro nitroalkane addition to azodicarboxylate.25
Scheme 3.
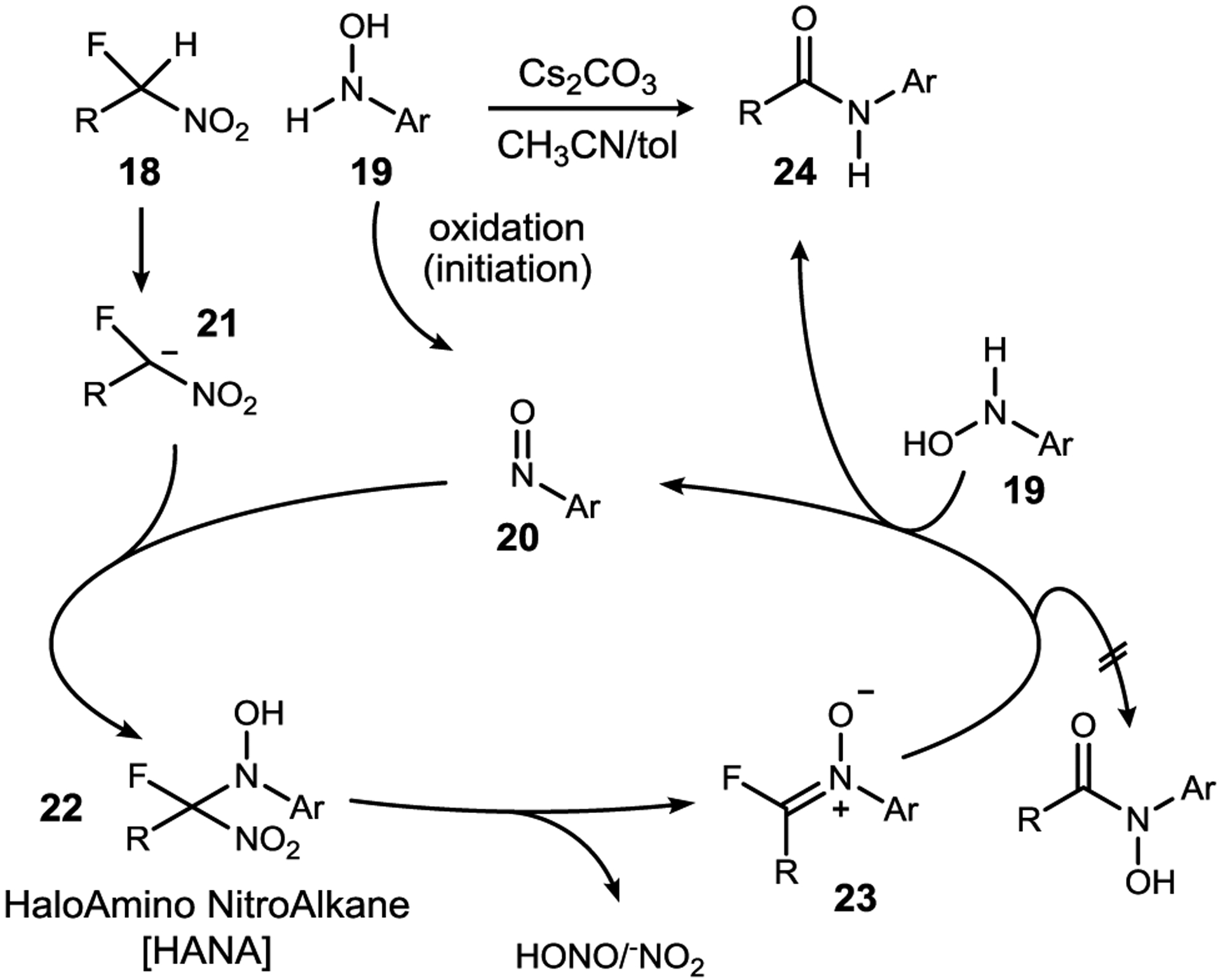
Umpolung N-aryl amide synthesis: mechanistic hypothesis.
The mechanistic hypothesis in Scheme 3 suggests that when the putative fluoro nitrone (23) is formed stoichiometrically from an aryl nitroso, a separate N-aryl hydroxyl amine should facilitate its conversion to amide product by acting as the reductant. Indeed, this was the outcome when using reactants with distinct aryl groups (Scheme 2, eq 5). A similar crossover experiment was performed to establish that a discrete N-aryl hydroxamic acid is not formed during the reaction (Scheme 2, eq 6). The interdependency of nitrosoarene formation and fluoronitrone conversion to amide (23+19→24) is an essential component of this ‘direct’ umpolung N-aryl amide synthesis.
An umpolung mechanism for N-aryl amide synthesis from N-aryl hydroxyl amines provides a unique opportunity to target α-chiral, highly acidic N-aryl amides. Aryl glycines are more susceptible to epimerization relative to other α-amino acids,31,32 with the rate of phenyl glycine epimerization measured at 9-fold higher than that of alanine. We recapitulated this behavior with an N-aryl amide of an N-aryl glycine and then designed experiments to compare the degree of conservation of configurational integrity during its synthesis. Scheme 4 describes the results when preparing 26 from 25 (90% ee) using EDC/HOAt under basic conditions in DMF. The desired N-aryl amide is obtained in 57% yield, but only 65% ee (Scheme 4, eq 7). Similarly, the product was prepared in an improved 82% ee and 83% yield when using DEPBT (eq 8), a coupling reagent known to be effective with epimerization-prone amides.33 By comparison, α-fluoronitroalkane 27 was deployed as a mixture of diastereomers (1:1) homochiral at the benzylic amine carbon (98% ee for each diastereomer),34,35 and when stirred with N-para-chlorophenyl hydroxylamine, the desired amide 26 was formed with complete conservation of ee (eq 9). Although the yield (41%) requires further optimization, the outcome is consistent with the mechanism of UmAS wherein no electrophilic intermediate is formed (Scheme 3). We noted, however, that extended exposure of the product to the basic conditions can result in racemization of 26 (t1/2 ~ 220 h, toluene, 0 °C, 98→75% ee at 105 h). This further illustrates the heightened acidity of N-aryl amides, and the challenge they present for synthesis.
Scheme 4.
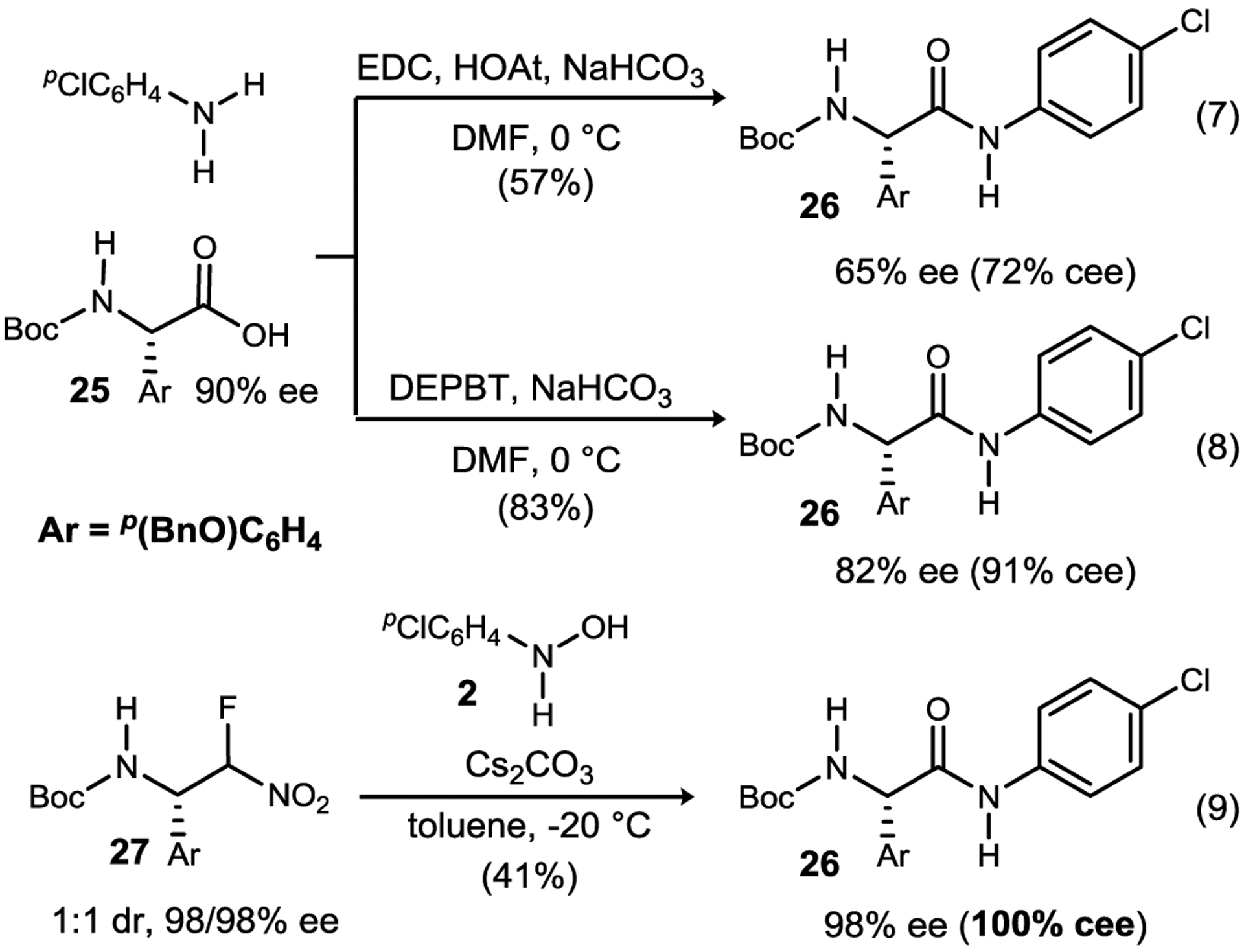
Comparison between conventional amide synthesis (EDC, DEPBT) and UmAS of an N-Aryl-α-Aryl glycinamide target to assess conservation of enantiomeric excess (% cee).
a See SI for complete details. Reaction conditions generally mirrored those for Table 2. Yields were measured by 1H NMR (relative to CH2Br2) after filtration through a pad of silica gel.
Incorporation of this umpolung N-aryl amide synthesis into synthesis planning was targeted next. N-Aryl amide 2836 is a member of the Globalagliatin-class of glucokinase activators for the treatment of diabetes.37 We envisioned an approach to 28 that involved the two key disconnections illustrated in Scheme 5, requiring chemistry that allows an inverted ketene synthon.38,39 Friedel-Crafts acylation of 29 provided aryl ketone 31, although the reaction was somewhat dependent on the age of the aluminum chloride used. Oxidation to the sulfone, and Wittig methylenation delivered terminal alkene 32. Nitration prepared the substrate for enantioselective reduction using 34. The product was obtained in good yield and encouraging enantioselection (86% ee) using a new enantioselective conjugate reduction.40 Fluorination readied 36 for N-aryl amidation using 2-pyridyl hydroxyl amine. Complete conservation of enantiomeric enrichment was observed. Forging the key aryl amide bond was met first with some disappointment, as 28 was formed in only 26% yield. However, experimentation revealed that copper(II) acetate enhanced the amidation, providing 28 in 69% yield. This discovery was based on the hypothesis that a Lewis acidic and potentially redox-active metal might affect the rate of HANA formation and/or turnover.16d Overall, the combination of nitroalkene reduction and UmAS with arylated hydroxyl amine realized the inverted ketene character for the first time in an N-aryl amide synthesis.
Scheme 5.
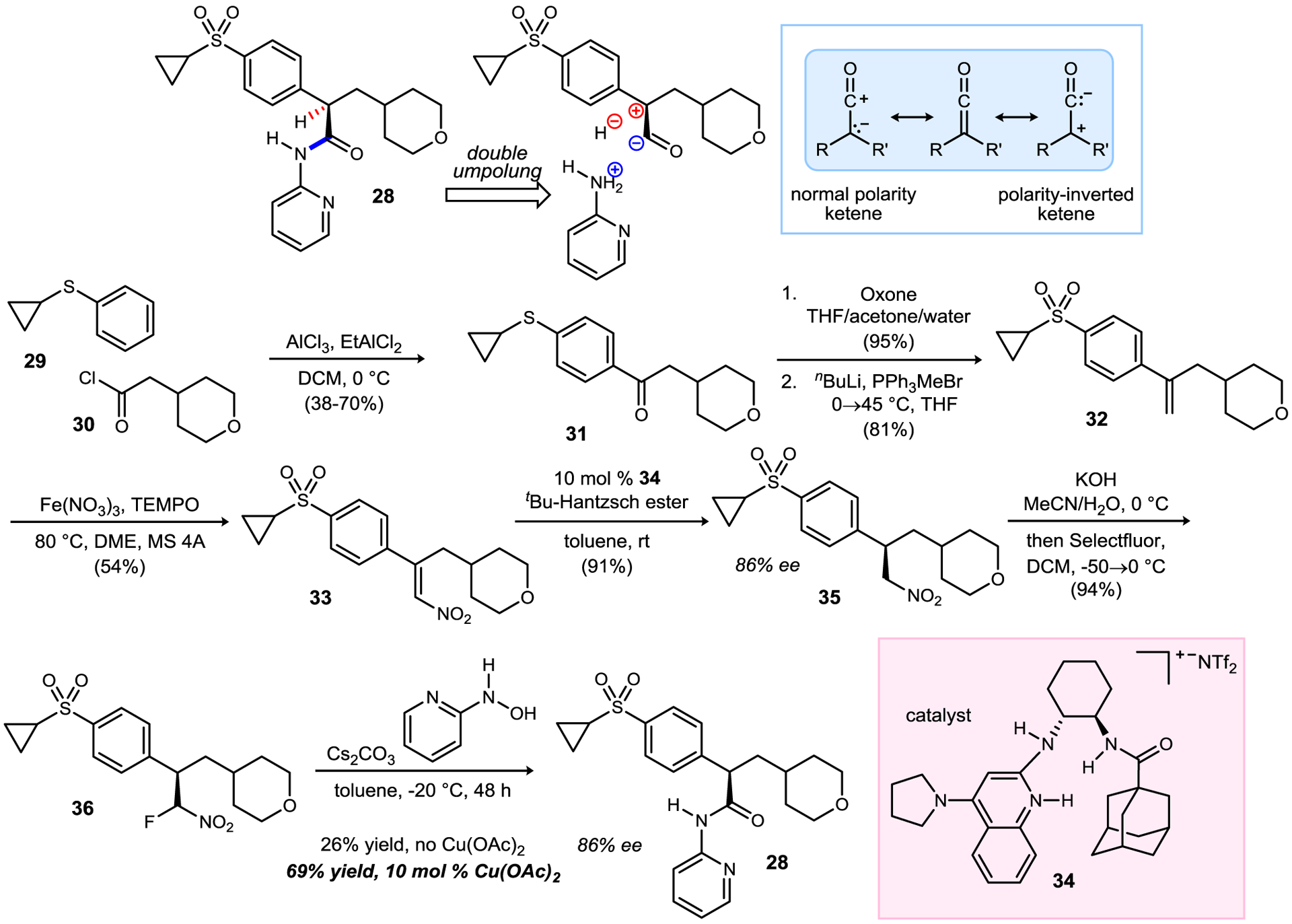
Development of a double umpolung approach using N-Aryl UmAS to a glucokinase activator.
In conclusion, we have developed an umpolung amide synthesis that forms N-aryl amides directly. Anilines have been uniformly unsuccessful amines in UmAS since its introduction, but now N-aryl hydroxyl amines enable a remarkably simple protocol, requiring only base to access N-aryl amides. The overall simplicity, however, belies a more complex hydroxylamine-nitroso redox process. The redox step is coupled to ‘turnover’ of a putative α-fluoro nitrone intermediate, a mechanistic hypothesis supported by a study that outlines the involvement of key intermediates while establishing that the expected hydroxamic acid product is not formed or otherwise involved. Central to the UmAS approach is the avoidance of an epimerization-prone electrophilic acyl donor. UmAS with an N-aryl hydroxyl amine provided the N-aryl amide product directly, and without epimerization, unlike two leading coupling reagents used in conventional amide synthesis where the lower nucleophilicity of anilines is known to be problematic for yield and epimerization. Future studies will target other unsolved challenges in conventional amide synthesis while further probing the mechanistic details of HANA intermediates formed from nitroso electrophiles.
Supplementary Material
ACKNOWLEDGMENT
M.S.C. was supported in part by an NSF Graduate Research Fellowship. We are grateful to the National Institute of General Medical Sciences (NIH GM 063557) for financial support. The Indiana University Mass Spectrometry Facility acknowledges support from the NSF (CHE1726633).
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org: Complete experimental details (PDF); NMR and HPLC trace data (PDF)
REFERENCES
- 1.Merrifield RB Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide, J. Am. Chem. Soc 1963, 85, 2149. [Google Scholar]
- 2.a) Bode JW Emerging methods in amide- and peptide-bond formation, Curr. Opin. Drug Discovery Dev 2006, 9, 765. [PubMed] [Google Scholar]; b) Pattabiraman VR; Bode JW Rethinking amide bond synthesis, Nature 2011, 480, 471. [DOI] [PubMed] [Google Scholar]; c) Lanigan RM; Sheppard TD Recent Developments in Amide Synthesis: Direct Amidation of Carboxylic Acids and Transamidation Reactions, Eur. J. Org. Chem 2013, 2013, 7453. [Google Scholar]; d) de Figueiredo RM; Suppo J-S; Campagne J-M Nonclassical Routes for Amide Bond Formation, Chem. Rev 2016, 116, 12029. [DOI] [PubMed] [Google Scholar]; e) Hollanders K; Maes BUW; Ballet SA New Wave of Amide Bond Formations for Peptide Synthesis, Synthesis 2019, 51, 2261. [Google Scholar]; f) Narendar Reddy T; Beatriz A; Jayathirtha Rao V; de Lima DP Carbonyl Compounds′ Journey to Amide Bond Formation, Chem. - Asian J 2019, 14, 344. [DOI] [PubMed] [Google Scholar]
- 3.a) Bryan MC; Dunn PJ; Entwistle D; Gallou F; Koenig SG; Hayler JD; Hickey MR; Hughes S; Kopach ME; Moine G; Richardson P; Roschangar F; Steven A; Weiberth FJ Key Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited, Green Chem. 2018, 20, 5082. [Google Scholar]; b) Constable DJC; Dunn PJ; Hayler JD; Humphrey GR; Leazer JJL; Linderman RJ; Lorenz K; Manley J; Pearlman BA; Wells A; Zaks A; Zhang TY Key green chemistry research areas-a perspective from pharmaceutical manufacturers, Green Chem. 2007, 9, 411. [Google Scholar]
- 4.Sabatini MT; Boulton LT; Sneddon HF; Sheppard TD A green chemistry perspective on catalytic amide bond formation, Nature Catalysis 2019, 2, 10. [Google Scholar]
- 5.Bryan MC; Dillon B; Hamann LG; Hughes GJ; Kopach ME; Peterson EA; Pourashraf M; Raheem I; Richardson P; Richter D; Sneddon HF Sustainable Practices in Medicinal Chemistry: Current State and Future Directions, J. Med. Chem 2013, 56, 6007. [DOI] [PubMed] [Google Scholar]
- 6.Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates, J. Med. Chem 2011, 54, 3451. [DOI] [PubMed] [Google Scholar]
- 7.McKnelly KJ; Sokol W; Nowick JS Anaphylaxis Induced by Peptide Coupling Agents: Lessons Learned from Repeated Exposure to HATU, HBTU, and HCTU, J. Org. Chem 2020, 85, 1764. [DOI] [PubMed] [Google Scholar]
- 8.Valeur E; Bradley M Amide bond formation: beyond the myth of coupling reagents, Chem. Soc. Rev 2009, 38, 606. [DOI] [PubMed] [Google Scholar]
- 9.Review:; Tailhades J Arylglycine: A Focus on Amino Acid Preparation and Peptide Synthesis, Int. J. Pept. Res. Ther 2021, 28, 10. [Google Scholar]
- 10.Schwieter KE; Johnston JN On-Demand Complex Peptide Synthesis: An Aspirational (and Elusive?) Goal for Peptide Synthesis, J. Am. Chem. Soc 2016, 138, 14160. [DOI] [PubMed] [Google Scholar]
- 11.a) Schwieter KE; Johnston JN A one-pot amidation of primary nitroalkanes, Chem. Commun 2016, 52, 152. [DOI] [PubMed] [Google Scholar]; b) Schwieter KE; Shen B; Shackleford JP; Leighty MW; Johnston JN Umpolung Amide Synthesis Using Substoichiometric N-Iodosuccinimide (NIS) and Oxygen as a Terminal Oxidant, Org. Lett 2014, 16, 4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Schwieter KE; Johnston JN A one-pot amidation of primary nitroalkanes, Chem. Commun 2016, 52, 152. [DOI] [PubMed] [Google Scholar]; b) Schwieter KE; Johnston JN Enantioselective Addition of Bromonitromethane to Aliphatic N-Boc Aldimines Using a Homogeneous Bifunctional Chiral Organocatalyst, ACS Catal. 2015, 5, 6559. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schwieter KE; Johnston JN Enantioselective synthesis of D-α-amino amides from aliphatic aldehydes, Chem. Sci 2015, 6, 2590. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Makley DM; Johnston JN Silyl Imine Electrophiles in Enantioselective Catalysis: A Rosetta Stone for Peptide Homologation, Enabling Diverse N-Protected Aryl Glycines from Aldehydes in Three Steps, Org. Lett 2014, 16, 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shen B; Makley DM; Johnston JN Umpolung reactivity in amide and peptide synthesis, Nature 2010, 465, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Batiste SM; Johnston JN Evidence for Ion-Templation During Macrocyclooligomerization of Depsipeptides, J. Am. Chem. Soc 2018, 140, 4560. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Batiste SM; Johnston JN Rapid synthesis of cyclic oligomeric depsipeptides with positional, stereochemical, and macrocycle size distribution control, Proc. Natl. Acad. Sci. U. S. A 2016, 113, 14893. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Leighty MW; Shen B; Johnston JN Enantioselective Synthesis of α-Oxy Amides via Umpolung Amide Synthesis, J. Am. Chem. Soc 2012, 134, 15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Tokumaru K; Bera K; Johnston JN 1,3,4-Oxadiazole and Heteroaromatic-Fused 1,2,4-Triazole Synthesis Using Diverted Umpolung Amide Synthesis, Synthesis 2017, 49, 4670. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tokumaru K; Johnston JN A convergent synthesis of 1,3,4-oxadiazoles from acyl hydrazides under semiaqueous conditions, Chem. Sci 2017, 8, 3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White AM; Palombi IR; Malins LR Umpolung strategies for the functionalization of peptides and proteins, Chem. Sci 2022, 13, 2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For hydroxamic acid synthesis from nitrosoarene electrophiles, see:; a) Wong FT; Patra PK; Seayad J; Zhang Y; Ying JY N-Heterocyclic Carbene (NHC)-Catalyzed Direct Amidation of Aldehydes with Nitroso Compounds, Org. Lett 2008, 10, 2333. [DOI] [PubMed] [Google Scholar]; b) Ning Y; Wang S; Li M; Han J; Zhu C; Xie J Site-specific Umpolung amidation of carboxylic acids via triplet synergistic catalysis, Nat. Commun 2021, 12, 4637. [DOI] [PMC free article] [PubMed] [Google Scholar]; The electrophilicity of acyl radicals in their additions to nitrogen π radicalophiles has been shown to dominate the transition state interactions, best characterized by the traditional N-nucleophile/C=O electrophile C-N bond formation:; c) Schiesser CH; Wille U; Matsubara H; Ryu I Radicals Masquerading as Electrophiles: Dual Orbital Effects in Nitrogen-Philic Acyl Radical Cyclization and Related Addition Reactions, Acc. Chem. Res 2007, 40, 303. [DOI] [PubMed] [Google Scholar]; See also:; d) Fisher DJ; Burnett GL; Velasco R; Read de Alaniz J Synthesis of Hindered α-Amino Carbonyls: Copper-Catalyzed Radical Addition with Nitroso Compounds, J. Am. Chem. Soc 2015, 137, 11614. [DOI] [PubMed] [Google Scholar]; e) Fisher DJ; Shaum JB; Mills CL; Read de Alaniz J Synthesis of Hindered Anilines: Three-Component Coupling of Arylboronic Acids, tert-Butyl Nitrite, and Alkyl Bromides, Org. Lett 2016, 18, 5074. [DOI] [PubMed] [Google Scholar]
- 17.Zuman P; Shah B Addition, Reduction, and Oxidation Reactions of Nitrosobenzene, Chem. Rev 1994, 94, 1621. [Google Scholar]
- 18.a) Sheradsky T; Nir Z The animation of carbanions, Tetrahedron Lett. 1969, 10, 77. [Google Scholar]; b) Berman AM; Johnson JS Copper-Catalyzed Electrophilic Amination of Organozinc Nucleophiles: Documentation of O-Benzoyl Hydroxylamines as Broadly Useful R2N(+) and RHN(+) Synthons, J. Org. Chem 2006, 71, 219. [DOI] [PubMed] [Google Scholar]; c) Erdik E; Ay M Electrophilic amination of carbanions, Chem. Rev 1989, 89, 1947. [Google Scholar]
- 19.Ohmatsu K; Ando Y; Nakashima T; Ooi T A Modular Strategy for the Direct Catalytic Asymmetric α-Amination of Carbonyl Compounds, Chem 2016, 1, 802. [Google Scholar]
- 20.Dijkstra G; Kruizinga WH; Kellogg RM An assessment of the causes of the “cesium effect”, J. Org. Chem 1987, 52, 4230. [Google Scholar]
- 21.Chen Y-F; Chen J; Lin L-J; Chuang GJ Synthesis of Azoxybenzenes by Reductive Dimerization of Nitrosobenzene, J. Org. Chem 2017, 82, 11626. [DOI] [PubMed] [Google Scholar]
- 22.Li J; Lear MJ; Kawamoto Y; Umemiya S; Wong AR; Kwon E; Sato I; Hayashi Y Oxidative Amidation of Nitroalkanes with Amine Nucleophiles using Molecular Oxygen and Iodine, Angew. Chem. Int. Ed 2015, 54, 12986. [DOI] [PubMed] [Google Scholar]
- 23.In a competition experiment where aryl hydroxyl amine, benzyl amine, and para-chlorophenacyl chloride (1:1:1) were treated with Hunig’s base in toluene, the hydroxamic acid and N-benzyl amide were formed in 41% and 56% yield, respectively, based on the acid chloride. This reflects a slightly slower reaction rate of the aryl hydroxyl amine with the acid chloride.
- 24.For an example of a nitroarene precursor to N-aryl amide or hydroxamic acid, see:; Jain SK; Aravinda Kumar KA; Bharate SB; Vishwakarma RA Facile access to amides and hydroxamic acids directly from nitroarenes, Org. Biomol. Chem 2014, 12, 6465. [DOI] [PubMed] [Google Scholar]
- 25.Crocker MS; Foy H; Tokumaru K; Dudding T; Pink M; Johnston JN Direct Observation and Analysis of the Halo-Amino-Nitro Alkane Functional Group, Chem 2019, 5, 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarantakis D; Sutherland JK; Tortorella C; Tortorella V 2-Fluoropyridine N-oxide in peptide chemistry, Chem. Commun 1966, 105. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X Fluorine radical interception with radical traps: An experimental and DFT study, Journal of Molecular Structure 2011, 1002, 121. [Google Scholar]
- 28.Voinov MA; Shevelev TG; Rybalova TV; Gatilov YV; Pervukhina NV; Burdukov AB; Grigor’ev IA α-Organoelement Nitrones: Synthesis, Properties, and IR and 13C NMR Spectral and X-ray Structural Characterization, Organometallics 2007, 26, 1607. [Google Scholar]
- 29.Voinov MA; Grigor’ev IA A route to the synthesis of previously unknown α-heteroatom substituted nitrones, Tetrahedron Letters 2002, 43, 2445. [Google Scholar]
- 30.Lyapkalo IM; Ioffe SL; Strelenko YA; Tartakovsky VA A novel general method for the synthesis of nitrones by reaction of nitroso compounds with anions of aliphatic nitro compounds, Russian Chemical Bulletin 1996, 45, 856. [Google Scholar]
- 31.Nicolaou KC; Boddy CNC; Bräse S; Winssinger N Chemistry, Biology, and Medicine of the Glycopeptide Antibiotics, Angew. Chem. Int. Ed 1999, 38, 2096. [DOI] [PubMed] [Google Scholar]
- 32.Matsuo H; Kawazoe Y; Sato M; Ohnishi M; Tatsuno T Studies on Racemization of Amino Acids and Their Derivatives .2. On Deuterium-Hydrogen Exchange Reaction of Amino Acid Derivatives in a Medium of Deuterated Acetic Acid, Chem. Pharm. Bull 1970, 18, 1788. [DOI] [PubMed] [Google Scholar]
- 33.a) Liang C; Behnam MAM; Sundermann TR; Klein CD Phenylglycine racemization in Fmoc-based solid-phase peptide synthesis: Stereochemical stability is achieved by choice of reaction conditions, Tetrahedron Lett. 2017, 58, 2325. [Google Scholar]; b) Li H; Jiang X; Ye Y.-h.; Fan C; Romoff T; Goodman M 3-(Diethoxyphosphoryloxy)-1,2,3- benzotriazin-4(3H)-one (DEPBT): A New Coupling Reagent with Remarkable Resistance to Racemization, Org. Lett 1999, 1, 91. [DOI] [PubMed] [Google Scholar]
- 34.a) Knowe MT; Tsukanov SV; Johnston JN Preparation of Benzyl((R)-2-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)acetyl)-D-phenylalaninate using Umpolung Amide Synthesis Org. Synth 2017, 94, 388. [Google Scholar]; b) Lim VT; Tsukanov SV; Stephens AB; Johnston JN Enantioselective Synthesis of α-Bromonitroalkanes for Umpolung Amide Synthesis: Preparation of tert-Butyl ((1R)-1-(4-(benzyloxy)phenyl)-2-bromo-2-nitroethyl)carbamate, Org. Synth 2016, 93, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Davis TA; Dobish MC; Schwieter KE; Chun AC; Johnston JN Preparation of H,4PyrrolidineQuin-BAM (PBAM), Org. Synth 2012, 89, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis TA; Wilt JC; Johnston JN Bifunctional Asymmetric Catalysis: Amplification of Bronsted Basicity Can Orthogonally Increase the Reactivity of a Chiral Bronsted Acid, J. Am. Chem. Soc 2010, 132, 2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fyfe MCT; Proctor MJ Tricyclo substituted amides, processes for preparing them, pharmaceutical compositions containing them, and their use as glucokinase modulators. WO2007051846A1, 2007.
- 37.a) Efanov AM; Barrett DG; Brenner MB; Briggs SL; Delaunois A; Durbin JD; Giese U; Guo H; Radloff M; Gil GS; Sewing S; Wang Y; Weichert A; Zaliani A; Gromada J A Novel Glucokinase Activator Modulates Pancreatic Islet and Hepatocyte Function, Endocrinology 2005, 146, 3696. [DOI] [PubMed] [Google Scholar]; b) Bueno Melendo AB; Agejas-Chicharro FJ N-Thiazolyl-2-cyclohexyl-1-phenylcyclopropanecarboxamides as glucokinase activator and their preparation and use for the treatment of diabetes. WO2010080333A1, 2010.
- 38.Seebach D; Henning R; Gonnermann J Zweifache Deprotonierung von 2-Aryl-1-nitroethanen Umpolung der Nitroolefin-Reaktivität, Chem. Ber 1979, 112, 234. [Google Scholar]
- 39.Vishe M; Johnston JN The inverted ketene synthon: a double umpolung approach to enantioselective β2,3-amino amide synthesis, Chem. Sci 2019, 10, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Details of this reaction will be disclosed in due course. Existing organocatalyst alternatives were substantially less selective.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.