

Abstract

We report a general synthetic entry to dihydrooxepine-spiroisoxazoline (DOSI) natural products that culminated in the first racemic total synthesis of psammaplysin A. For the synthesis of the unique spirocyclic fragment we employed a strategy that features two key transformations: (1) a diastereoselective Henry reaction/cyclization sequence to access the C7 hydroxylated isoxazoline scaffold in one step and (2) a regioselective Baeyer–Villiger ring expansion to install the fully substituted dihydrooxepine and avoid the risk of a previously observed oxepine-arene oxide rearrangement. The overall synthesis proceeds in 13 steps from an inexpensive starting material.
Dihydrooxepine-spiroisoxazoline (DOSI) natural products are a structurally unique family of marine alkaloids that comprises the psammaplysins, ceratinamides, ceratinadins, and frondoplysins.1 Among them, the psammaplysins stand out as the largest class (>35 members), with many displaying anticancer, antimalarial, anti-HIV, or antibiotic activities.2 In 1982, psammaplysin A (1) was isolated as the first member from the marine sponge Psammaplysilla purpurea by Kashman (Scheme 1A).3 The structure of 1 was initially proposed to feature a C6 cyclohexadiene-spiroisoxazoline motif as present in aerothionin (2). While this assignment was revised in 1985 by Clardy based on detailed NMR studies and single-crystal X-ray crystallographic analysis,4 it took another 30 years to validate the proposed absolute configuration.5 The structurally unique dihydrooxepine motif is thought to be biosynthetically derived from the oxidation of dibromotyrosine (3) followed by ring opening.4 For the formation of aerothionin (2), a direct intramolecular opening of the putative epoxide intermediate at C6 was postulated and a 6π-electrocyclic ring-opening event that initiates the rearrangement of the arene oxide to the oxepine might be involved in the formation of 1 (compare with Scheme 1B).6 This divergent process was investigated in the seminal work by Clardy, which enabled the formal synthesis of 2, but it did not lead to 1.7 The S-adenosylmethionine (SAM, 4) derived three-carbon amide linker of 1 connects the DOSI to a modified dibromotyrosine and distinguishes it from its natural congeners, some of which possess an additional C19 stereocenter.6c,8 The intriguing biological properties and unique functionalization pattern of the molecular framework render 1 a formidable synthetic target.
Scheme 1. Dihydrooxepine-spiroisoxazoline (DOSI) Natural Products and Preliminary Insights.
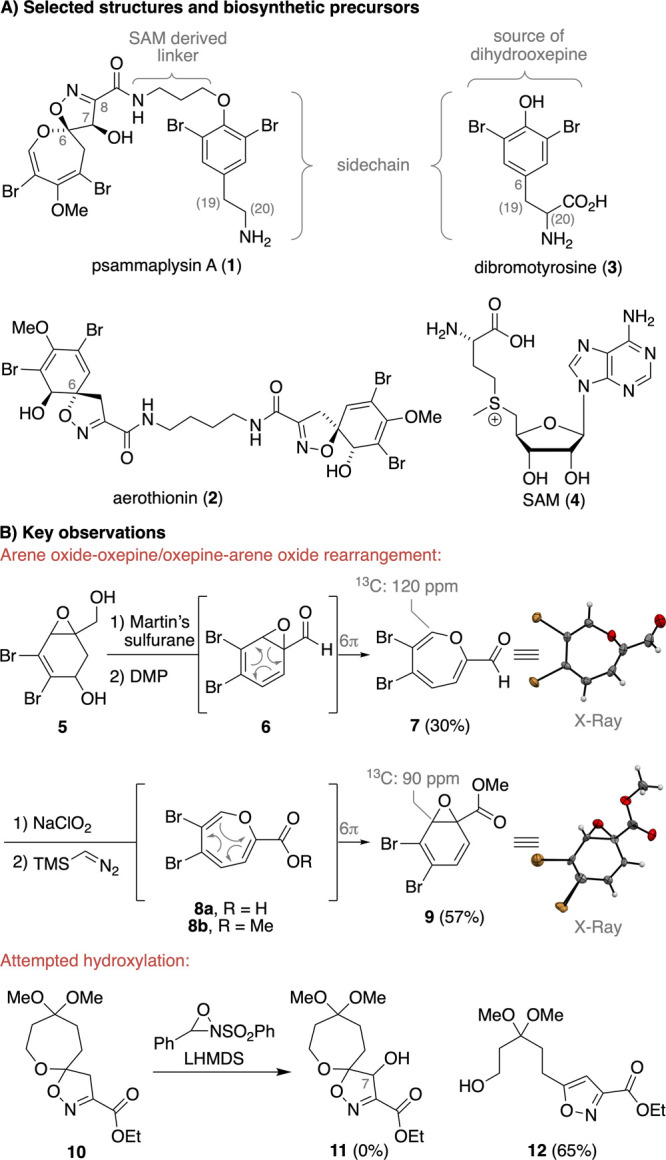
Surprisingly, despite the early studies by Clardy,7 an increasing number of powerful synthetic methods for construction of dihydrooxepines,9 and more recent work by Vanderwal and Yang,10 a synthesis of DOSI natural products has yet to be accomplished. As part of our ring expansion program,11 we set out to develop a general synthetic route to 1 that would permit access to the entire DOSI family.
During preliminary studies in our laboratories, we found that aldehyde 6, obtained from the dehydration with Martin’s sulfurane and oxidation of diol 5 with Dess–Martin periodinane (DMP), undergoes spontaneous arene oxide-oxepine rearrangement12 to oxepine 7 in 30% yield (Scheme 1B). We initially considered oxepine 7 as a valuable precursor for 1, but 7 proved to be unstable upon storage and underwent slow decomposition to unidentified aromatic byproducts. Unexpectedly, attempted conversion of oxepine 7 to ester 8b was even more problematic as arene oxide 9 was obtained as the only product. A second key observation was made when we investigated the hydroxylation of isoxazoline 10, the product of a preceding nitrile oxide [3 + 2]-cycloaddition reaction (see Supporting Information for details).10b,13 In this case, hydroxylation to isoxazoline 11 was outcompeted by rapid ring opening of the aza enolate to give isoxazole 12 as the sole product in 65% yield.14,15 These key insights were crucial for our further synthetic planning and ultimately led us to the development of the revised retrosynthesis outlined in Scheme 2.
Scheme 2. Retrosynthetic Strategy for Psammaplysin A.
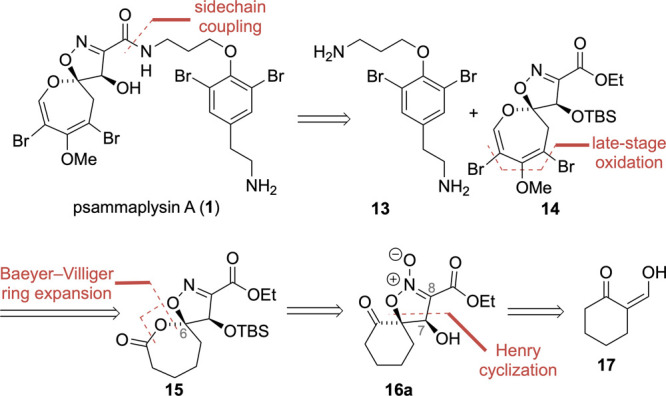
We began with the disconnection of the known linker side chain moloka’iamine (13)16 from 1 and focused our further analysis on the resulting DOSI 14. We anticipated that the lack of a fully decorated oxepine should eliminate the risk of unwanted arene oxide formation. Sequential removal of the enol ether decoration of ester 14 revealed structurally simplified lactone 15. The lactone acetal contains the retron for a Baeyer–Villiger oxidation of C6 producing spirocyclic ketone 16a.17,11a To avoid potential ring opening as observed for isoxazoline 10, we considered simultaneous installation of the requisite C7 hydroxy group and the isoxazoline scaffold in one step employing a powerful diastereoselective Henry reaction/cyclization protocol.18 To this end, nitronate 16a was traced back to commercially available 2-hydroxymethylenecyclohexanone (17).
We began our forward synthesis with the bromination of compound 17 (Scheme 3A), which was obtained on large scale from the formylation of cyclohexanone in 89% yield. The use of N-bromosuccinimide (NBS) afforded the crude α-bromo ketoaldehyde in excellent yields; however, we found it to be air-sensitive. For this reason, we decided to telescope the following step and conduct the overall process as a one-pot reaction. According to the seminal report of Rosini,18 the intermediate bromide was treated with ethyl nitroacetate and triethylamine to give nitronate 16a and its C7 diastereomer 16b as a 3:1 mixture as determined from the crude 1H NMR (see Supporting Information for details). After chromatographic separation of the highly polar 16b, we obtained nitronate 16a in 51% yield. The relative configuration of the desired diastereomer 16a was validated by single crystal X-ray analysis. Performing the reaction in acetonitrile at −25 °C was crucial, as lower diastereoselectivities were observed when performing the reaction at higher temperatures or changing the solvent to methanol in which an equimolar mixture of nitronates 16a and 16b was obtained. We then proceeded with the deoxygenation of nitronate 16a by employing triethyl phosphite in 1,4-dioxane at 100 °C to isolate the spirocyclic isoxazoline 18 in up to 99% yield.
Scheme 3. Synthesis of Psammaplysin A.

See the Supporting Information for detailed procedures and characterization data.
Having installed the 4-hydroxyisoxazoline motif and with multigram quantities of ketone 18 in hand, our attention was directed toward investigation of the Baeyer–Villiger oxidation to enable installation of the DOSI motif. We were pleased to see that exposure of ketone 18 to m-chloroperoxybenzoic acid (m-CPBA) in the presence of disodium hydrogen phosphate afforded the corresponding lactone 19 in nearly quantitative yield. To our surprise, the protection of the hydroxy group in intermediate 19 with tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) and triethylamine failed to give the desired silyl ether.19 Instead, formation of carbonate 20 was observed. This product might be derived from the reaction of lactone 19 with ethyl cyanoformate, which could form via decomposition of lactone 19. As a part of the biosynthesis of ceratinamine A, a similar isoxazoline fragmentation was studied by Ganem.16a
In parallel, we took advantage of the reaction of nitronate 16a with dimethylvinylsilyl chloride in the presence of imidazole.20 Under these conditions, clean silylation of the C7 hydroxy group followed by intramolecular 1,3-dipolar cycloaddition of the vinyl group with the nitronate took place to furnish the tricyclic acetal 21 in 95% yield. This motif serves a dual role as it (1) protects the hydroxy group and (2) enables one-step unmasking of the isoxazoline as described by Rosini.20 The following Baeyer–Villiger oxidation provided lactone 22 as the only regioisomer in 88% yield. However, subsequent attempts to form the ketene acetal phosphate 23 by employing Nicolaou’s conditions (diphenyl chlorophosphate, KHMDS, THF, HMPA, −78 °C),21 were low yielding due to decomposition of the product during isolation.
For this reason, we returned to isoxazoline 18 and protected the hydroxy group (imH, TBSCl) prior to the ring expansion. The subsequent Baeyer–Villiger oxidation proceeded with excellent regioselectivity and furnished the desired lactone 15 in 87% yield over two steps on a 6-g scale. When performing the following ketene acetal phosphate formation at −78 °C, varying yields between 36–67% of 24 were obtained and large amounts of lactone 15 were recovered. After further optimization, we found that slow addition of potassium hexamethyldisilazide (KHMDS) solution in THF at −95 °C was crucial for complete consumption of the starting material and to give ketene acetal phosphate 24 in reproducible 86% yield.
The palladium-catalyzed reduction of the ketene acetal phosphate to enol ether 25 also required some optimization. The use of Pd(PPh3)4 and Et3Al gave enol ether 25 as a complex mixture together with an unstable byproduct resulting from a competing cross-coupling reaction with Et3Al. We then switched to LiBH4 as the reductant;21 however, this led to competing reduction of the ester moiety. The chemoselectivity issue could be avoided by replacing LiBH4 with formic acid/triethylamine buffer. This allowed for clean conversion of ketene acetal phosphate 24 to tetrahydrooxepine-spiroisoxazoline 25 for the first time. Further screening revealed PhMe2SiH as the ideal reagent to give enol ether 25 in 66% yield.
With a scalable route to enol ether 25 in hand, we continued with the investigation of its sequential functionalization (Scheme 3B). We exposed enol ether 25 to a panel of oxidation conditions, most of which employed tert-butyl hydroperoxide (TBHP) in combination with a transition metal catalyst (CuBr; CuI; RuCl3; Mn(OAc)3; Mn(dpm)3; Pd(OH)2/C; see Supporting Information for details).22 The screening revealed that the desired vinylogous lactone 26 was formed under most conditions accompanied only by its regioisomeric α,β-unsaturated lactone 27. The use of Pd(OH)2/C as the catalyst afforded vinylogous lactone 26 and lactone 27 as a 2.5:1 mixture of regioisomers; however, the reaction suffered from low conversion. The best performing conditions in terms of conversion involved Mn(dpm)3 and dropwise addition of TBHP overnight to give 26 in 32% isolated yield. Direct conversion of the undesired regioisomer 27 (19%) to ketene acetal phosphate 24 was unsuccessful in our hands but allowed for the isolation of lactone 15 in 67% yield.
Subsequent α-bromination of vinylogous lactone 26 with CuBr2 and 2,6-di-tert-butylpyridine (DTBP) in a mixture of chloroform and ethyl acetate at 100 °C proceeded cleanly to deliver the C4-brominated product in 78% yield. The second bromination was accomplished upon exposure to tetra-n-butylammonium tribromide (TBATB) and DTBP at 60 °C in chloroform to afford vinylogous lactone 28 in 72% yield.10b We found that the overall yield of vinylogous lactone 28 could be improved by telescoping the three oxidations without chromatographic purification of the intermediates. In this way, we obtained vinylogous lactone 28 in 27% yield over three steps from 25. Finally, sequential treatment of vinylogous lactone 28 with KHMDS and methyl trifluoromethanesulfonate (MeOTf) gave the fully substituted DOSI 14 in 53% yield.
For the late-stage attachment of the protected moloka′iamine unit 29, which was prepared from tyramine in four steps (see Supporting Information), we first attempted conversion of ethyl ester 14 into the corresponding carboxylic acid. Saponification under aqueous conditions (LiOH, THF, H2O, 23 °C) followed by isolation of the acid turned out be impossible in our hands as decomposition, presumably via decarboxylation, was observed even at low temperatures.23 For this reason, we decided to treat ester 14 with potassium trimethylsilanolate (TMSOK) at −15 °C under nonaqueous conditions followed by direct addition of ammonium salt 29, N-methylimidazole (NMI), and chloro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate (TCFH).10b This one-pot procedure bypassed the problematic isolation of the acid and allowed us to obtain the desired amide 30 in 79% yield from ester 14. The following deprotections of the C7 oxygen and C20 nitrogen proceeded smoothly to furnish psammaplysin A (1) in 60% yield. The analytical data (1H NMR, 13C NMR, HRMS) for 1 and its bisacetylated derivative fully matched those reported for the natural compound.4 Overall, psammaplysin A (1) was synthesized in 13 steps from commercially available starting materials.
In summary, we have accomplished the first synthesis of psammaplysin A (1) nearly 40 years after its first isolation. The key to success of the developed strategy was the use of a Henry addition/O-alkylation sequence instead of a nitrile oxide [3 + 2]-cycloaddition. This granted access to the fully substituted C7 hydroxylated heterocyclic scaffold in one step. For the construction of the 7-membered ring-system a high-yielding Baeyer–Villiger oxidation turned out to be the method of choice. This avoided the risk of a previously encountered oxepine-arene oxide rearrangement and paved the way for the selective installation of the fully intact DOSI motif. The late-stage attachment of the side chain ensures the high flexibility and modularity of the synthesis. This approach should allow for the preparation of several structurally related members as well as analogs with deep-seated structural modifications and facilitate future bioactivity studies.
Acknowledgments
T.M. acknowledges the European Research Council under the European Union’s Horizon 2020 research and innovation program HALODRUGSYN (grant agreement No 714049), the Austrian Science Fund FWF (P31023-NBL and P33894-N), and the Center for Molecular Biosciences (CMBI). Furthermore, we thank Dr. Kevin Mellem (Maze Therapeutics) for helpful discussions during the preparation of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10010.
Experimental details, spectroscopic data, and X-ray data (PDF)
Open Access is funded by the Austrian Science Fund (FWF).
The authors declare no competing financial interest.
Supplementary Material
References
- For a complete and chronological overview, see:Sokol K. R.Studies toward the Total Synthesis of Psammaplysin A and Acid-catalyzed Cycloisomerization of Neopentylic Epoxides. Ph.D. Dissertation, Ludwig- Maximilians-University Munich, Munich, Germany, 2020. [Google Scholar]
- For selected screens, see:; a Copp B. R.; Ireland C. M.; Barrows L. R. Psammaplysin C: A new cytotoxic dibromotyrosine-derived metabolite from the marine sponge Druinella (= Psammaplysilla) purpurea. J. Nat. Prod. 1992, 55, 822–823. 10.1021/np50084a021. [DOI] [PubMed] [Google Scholar]; b Mudianta I. W.; Skinner-Adams T.; Andrews K. T.; Davis R. A.; Hadi T. A.; Hayes P. Y.; Garson M. J. Psammaplysin derivatives from the Balinese marine sponge Aplysinella strongylata. J. Nat. Prod. 2012, 75, 2132–2143. 10.1021/np300560b. [DOI] [PubMed] [Google Scholar]; c Ichiba T.; Scheuer P. J.; Kelly-Borges M. Three bromotyrosine derivatives, one terminating in an unprecedented diketocyclopentenylidene enamine. J. Org. Chem. 1993, 58, 4149–4150. 10.1021/jo00067a062. [DOI] [Google Scholar]; d Ramsey D. M.; Islam M. A.; Turnbull L.; Davis R. A.; Whitchurch C. B.; McAlpine S. R. Psammaplysin F: A unique inhibitor of bacterial chromosomal partitioning. Bioorg. Med. Chem. Lett. 2013, 23, 4862–4866. 10.1016/j.bmcl.2013.06.082. [DOI] [PubMed] [Google Scholar]
- a Kashman Y.; Groweiss A.; Carmely S.; Kinamoni Z.; Czarkie D.; Rotem M. Recent research in marine natural products from the Red Sea. Pure Appl. Chem. 1982, 54, 1995–2010. 10.1351/pac198254101995. [DOI] [Google Scholar]; b Rotem M.; Carmely S.; Kashman Y.; Loya Y. Two new antibiotics from the red sea sponge Psammaplysilla purpurea: Total 13C-NMR line assignment of psammaplysins A and B and aerothionin. Tetrahedron 1983, 39, 667–676. 10.1016/S0040-4020(01)91843-5. [DOI] [Google Scholar]
- Roll D. M.; Chang C. W. J.; Scheuer P. J.; Gray G. A.; Shoolery J. N.; Matsumoto G. K.; Van Duyne G. D.; Clardy J. Structure of the psammaplysins. J. Am. Chem. Soc. 1985, 107, 2916–2920. 10.1021/ja00296a014. [DOI] [Google Scholar]
- Mándi A.; Mudianta I. W.; Kurtán T.; Garson M. J. Absolute configuration and conformational study of psammaplysins A and B from the Balinese marine sponge Aplysinella strongylata. J. Nat. Prod. 2015, 78, 2051–2056. 10.1021/acs.jnatprod.5b00369. [DOI] [PubMed] [Google Scholar]
- a Ragini K.; Fromont J.; Piggott A. M.; Karuso P. Enantiodivergence in the Biosynthesis of Bromotyrosine Alkaloids from Sponges?. J. Nat. Prod. 2017, 80, 215–219. 10.1021/acs.jnatprod.6b01038. [DOI] [PubMed] [Google Scholar]; b Hentschel F.; Lindel T. Synthesis of Oximinotyrosine-Derived Marine Natural Products. Synthesis 2010, 2010, 181–204. 10.1055/s-0029-1218615. [DOI] [Google Scholar]; c Rogers E. W.; Molinski T. F. Highly Polar Spiroisoxazolines from the Sponge Aplysina fulva. J. Nat. Prod. 2007, 70, 1191–1194. 10.1021/np070109l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Okamoto K. T.; Clardy J. Studies on a Biomimetic Approach to Aerothionin and Psammaplysin-A. Tetrahedron Lett. 1987, 28, 4969–4972. 10.1016/S0040-4039(00)96672-3. [DOI] [Google Scholar]; b Nishiyama S.; Yamamura S. Total Syntheses of (±)-Aerothionin and (±)-Homoaerothionin. Tetrahedron Lett. 1983, 24, 3351–3352. 10.1016/S0040-4039(00)86267-X. [DOI] [Google Scholar]; c Nishiyama S.; Yamamura S. Total Syntheses of (±)-Aerothionin, (±)-Homoaerothionin, and (±)-Aerophobin-1. Bull. Chem. Soc. Jpn. 1985, 58, 3453–3456. 10.1246/bcsj.58.3453. [DOI] [Google Scholar]; d Ogamino T.; Obata R.; Nishiyama S. Asymmetric synthesis of aerothionin, a marine dimeric spiroisoxazoline natural product, employing optically active spiroisoxazoline derivative. Tetrahedron Lett. 2006, 47, 727–731. 10.1016/j.tetlet.2005.11.097. [DOI] [Google Scholar]
- For selected examples, see:; a Liu S.; Fu X.; Schmitz F. J.; Kelly-Borges M. Psammaplysin F, a New Bromotyrosine Derivative from a Sponge, Aplysinella sp. J. Nat. Prod. 1997, 60, 614–615. 10.1021/np970070s. [DOI] [PubMed] [Google Scholar]; b Wright A. D.; Schupp P. J.; Schrör J.-P.; Engemann A.; Rohde S.; Kelman D.; de Voogd N.; Carroll A.; Motti C. A. Twilight Zone Sponges from Guam Yield Theonellin Isocyanate and Psammaplysins I and J. J. Nat. Prod. 2012, 75, 502–506. 10.1021/np200939d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol K. R.; Magauer T. Total Synthesis of Oxepin and Dihydrooxepin Containing Natural Products. Synthesis 2021, 53, 4187–4202. 10.1055/s-0037-1610776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Carlson J. S.General Approach to the Synthesis of the Chlorosulfolipids Danicalipin A, Mytilipin A, and Malhamensilipin A in Enantioenriched Form and Progress towards the Synthesis of the Psammaplysin Family of Natural Products. Ph.D. Dissertation, UC Irvine, USA, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang L.; Wang R.; Wang C.; Liu B.; Yang J.; Zhang Z.; Huang J.; Yang Z. Concise Synthesis of 7-Deoxypsammaplysins K and O and 7-Deoxyceratinamide A by 1,3-Dipole Cycloaddition. Org. Lett. 2022, 24, 3786–3791. 10.1021/acs.orglett.2c01298. [DOI] [PubMed] [Google Scholar]
- a Schmid M.; Grossmann A.; Wurst K.; Magauer T. Total Synthesis of Salimabromide: A Tetracyclic Polyketide from a Marine Myxobacterium. J. Am. Chem. Soc. 2018, 140, 8444–8447. 10.1021/jacs.8b06228. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zamarija I.; Marsh B.; Magauer T. Ring Expansion of 1-Indanones to 2-Halo-1-naphthols as an Entry Point to Gilvocarcin Natural Products. Org. Lett. 2021, 23, 9221–9226. 10.1021/acs.orglett.1c03530. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Unzner T. A.; Grossmann A.; Magauer T. Rapid Access to Orthogonally Functionalized Naphthalenes: Application to the Total Synthesis of the Anticancer Agent Chartarin. Angew. Chem., Int. Ed. 2016, 55, 9763–9767. 10.1002/anie.201605071. [DOI] [PubMed] [Google Scholar]
- a Vogel E.; Günther H. Benzene Oxide-Oxepin Valence Tautomerism. Angew. Chem., Int. Ed. 1967, 6, 385–401. 10.1002/anie.196703851. [DOI] [Google Scholar]; b Boyd D. R.; Berchtold G. A. Synthesis and aromatization of 2-carboxy- and 2-carbomethoxyoxepin-benzene oxide. J. Am. Chem. Soc. 1978, 100, 3958–3959. 10.1021/ja00480a067. [DOI] [Google Scholar]; c Chao H. S. I.; Berchtold G. A. Aromatization of arene 1,2-oxides. Comparison of the aromatization pathways of 1-carboxy-, 1-carbomethoxy-, 1-formyl-, 1-(hydroxymethyl)-, and 1-(2-hydroxy-2-propyl)benzene oxide. J. Am. Chem. Soc. 1981, 103, 898–902. 10.1021/ja00394a029. [DOI] [Google Scholar]
- Rakesh; Sun D.; Lee R. B.; Tangallapally R. P.; Lee R. E. Synthesis, Optimization and Structure–Activity Relationships of 3,5- Disubstituted Isoxazolines as New Anti-Tuberculosis Agents. Eur. J. Med. Chem. 2009, 44, 460–472. 10.1016/j.ejmech.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis F. A.; Kumar A.; Reddy R. E.; Chen B.-C.; Wade P. A.; Shah S. W. Hydroxylation of dihydroisoxazoles using N-sulfonyloxaziridines. J. Org. Chem. 1993, 58, 7591–7593. 10.1021/jo00078a049. [DOI] [Google Scholar]
- a Efforts to close the 7-membered ring through oxidative bromo-cyclization were not made due to the instability of a substrate closely related to 12; see ref (1); b Das P.; Hamme A. T. II Divergent and Concise Syntheses of Spiroisoxazolines: First Total Synthesis of 11- Deoxyfistularin-3. Eur. J. Org. Chem. 2015, 2015, 5159–5166. 10.1002/ejoc.201500603. [DOI] [Google Scholar]
- a Schoenfeld R. C.; Ganem B. Synthesis of Ceratinamine and Moloka’iamine: Antifouling Agents from the Marine Sponge Pseudoceratina Purpurea. Tetrahedron Lett. 1998, 39, 4147–4150. 10.1016/S0040-4039(98)00771-0. [DOI] [Google Scholar]; b Hamann M. T.; Scheuer P. J.; Kelly-Borges M. Biogenetically diverse, bioactive constituents of a sponge, order Verongida: bromotyramines and sesquiterpene-shikimate derived metabolites. J. Org. Chem. 1993, 58, 6565–6569. 10.1021/jo00076a012. [DOI] [Google Scholar]
- a ten Brink G.-J.; Arends I. W. C. E.; Sheldon R. A. The Baeyer-Villiger Reaction: New Developments toward Greener Procedures. Chem. Rev. 2004, 104, 4105–4124. 10.1021/cr030011l. [DOI] [PubMed] [Google Scholar]; b Krow G. The Baeyer–Villiger Oxidation of Ketones and Aldehydes. Org. React. 1993, 43, 251–798. 10.1002/0471264180.or043.03. [DOI] [Google Scholar]; c Renz M.; Meunier B. 100 Years of Baeyer–Villiger Oxidations. Eur. J. Org. Chem. 1999, 1999, 737–750. . [DOI] [Google Scholar]
- Rosini G.; Marotta E.; Righi P.; Seerden J.-P. StereocontroIIed Synthesis of 3-(Ethoxycarbonyl)-4-hydroxy-2-isoxazoline 2-Oxides. A New Approach to the Synthesis of 4-Hydroxylated 2-Isoxazolines. J. Org. Chem. 1991, 56, 6258–6260. 10.1021/jo00022a005. [DOI] [Google Scholar]
- We later found that 15 can be obtained in 99% yield from 19 when employing TBSCl, imH.
- Righi P.; Marotta E.; Rosini G. Linear Aminopolyhydroxylated Structures Through Rapid Domino Assembly of a Highly Functionalized Heterotricyclic System and Its Selective Cleavage. Chem.—Eur. J. 1998, 4, 2501–2512. . [DOI] [Google Scholar]
- Nicolaou K. C.; Yu R.; Shi L.; Cai Q.; Lu M.; Heretsch P. General Synthetic Approach to Functionalized Dihydrooxepines. Org. Lett. 2013, 15, 1994–1997. 10.1021/ol4006689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A.; Nakada M. Allylic Oxidations in Natural Product Synthesis. Synthesis 2013, 45, 1421–1451. 10.1055/s-0033-1338426. [DOI] [Google Scholar]
- Ogamino T.; Nishiyama S. A new ring-opening access to aeroplysinin-1, a secondary metabolite of Verongia aerophoba. Tetrahedron 2003, 59, 9419–9423. 10.1016/j.tet.2003.09.075. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.