

Abstract

Substitution of exocyclic oxygen with sulfur was shown to substantially influence the properties of RNA/DNA bases, which are crucial for prebiotic chemistry and photodynamic therapies. Upon UV irradiation, thionucleobases were shown to efficiently populate triplet excited states and can be involved in characteristic photochemistry or generation of singlet oxygen. Here, we show that the photochemistry of a thionucleobase can be considerably modified in a nucleoside, that is, by the presence of ribose. Our transient absorption spectroscopy experiments demonstrate that thiocytosine exhibits 5 times longer excited-state lifetime and different excited-state absorption features than thiocytidine. On the basis of accurate quantum chemical simulations, we assign these differences to the dominant population of a shorter-lived triplet nπ* state in the nucleoside and longer-lived triplet ππ* states in the nucleobase. This explains the distinctive photoanomerziation of thiocytidine and indicates that the nucleoside will be a less efficient phototherapeutic agent with regard to singlet oxygen generation.
Structural and functional aspects of the chemistry of DNA and RNA can be greatly altered by the introduction of chemically modified nucleobases. Consequently, sulfur- and bromine-substituted nucleobases have been considered in the development of photodynamic therapies,1−4 isoguanine and isocytosine have been employed for encoding a nonstandard amino acid,5 and 2-aminopurine and thienonucleobases have been applied in structural studies as fluorescent probes.6−8 Nucleosides containing nonbiological hypoxanthine and 2-thiopyrimidine bases were also suggested as important side products or intermediates in prebiotically plausible syntheses of canonical RNA and DNA building blocks.9−11 For instance, α-2-thiocytidine is considered as the key intermediate, which upon UV irradiation and hydrolysis yields biologically relevant pyrimidine ribonucleotides.10 2-Thiouridine and inosine were shown to enhance the rate and fidelity of nonenzymatic RNA template copying.12,13 Finally, other modifications of the purine ring, such as 2,6-diaminopurine and 8-oxoguanine, could promote an abiotic variant of photoinduced repair of the most frequent DNA photodamages, that is, cyclobutane pyrimidine dimers.14−16
Among possible structural modifications, nucleobases containing a thiocarbonyl group have attracted particular interest because of their distinctive photochemical properties.17−28 First, substitution of carbonyl with thiocarbonyl groups results in a significant red-shift of the UV–vis spectrum of the nucleobase.18,23 Furthermore, excited singlet states of thiocarbonyl compounds exhibit very high spin–orbit couplings with the triplet manifold of electronic states, leading to near unity triplet quantum yields.29−31 This has been demonstrated for thiocytosine, thioguanine, and different thionated uracil bases and was suggested as the key source of their rich photoreactivity.26,32,33
Thionation in different positions of the heteroaromatic ring may stabilize different types of low-lying triplet electronic states, resulting in different photochemical properties. In particular, if the lowest energy T1 minimum is dominated by the 3ππring* molecular orbital character, the thionucleobase undergoes aromatic ring puckering upon intersystem crossing and could be involved in destructive photochemistry or generation of singlet oxygen, that is, the key active agent in photodynamic therapies.21,34 This is caused by long excited-state lifetimes of 3ππring, which enable the excited thionucleobase to form a complex with an oxygen molecule and transfer its excitation energy. In contrast, stabilization of the 3ππCS* character on the T1 hypersurface results in C=S bond tilting and stretching, followed by more efficient photodeactivation to the closed shell ground state or sulfur loss.11,21 Therefore, previous works indicated that the relative stability of these two different T1 minima of UV-excited thionucleobases affects the overall photoreactivity and photoproduct ratios, including singlet oxygen yields.11,21 However, a recent UV-assisted prebiotic synthesis of pyrimidine nucleosides demonstrated that 2-thiocyditine undergoes photoinduced flipping of the nucleobase between α and β orientations (photoanomerization), which cannot be promoted by any of these 3ππ* states.10,35 Intriguingly, the triplet 3ππring state was determined to be the key contributor to the photorelaxation of the 2-thiocytosine (nucleobase) by nonadiabatic dynamics simulations in the gas phase, aqueous phase transient absorption spectroscopy in the UV region (UV-TAS), and static excited-state QM calculations with explicit water molecules.26,32
In recent years, numerous efforts aimed to scrutinize the excited-state dynamics of different thionated nucleobases in both theoretical and experimental terms.17−27 Apart from providing the mechanistic rationale for the photoreactivity, these studies have been intended to support modification of these chromophores and optimization of their properties. Here, we demonstrate that consideration of the nearest molecular environment of thionucleobases is essential to obtain a full picture of their photochemical properties. We focus on 2-thiocytosine (thioC) and the α- and β-anomers of its nucleoside (thioCyd) to prove that the presence of sugar may support population of different low-lying triplet states than in the nucleobase. In particular, partial population of the 3nSπ* leads to unique photochemical properties and photoproducts. We track the characteristic spectral features of these states by means of time-resolved transient absorption spectroscopy (TAS) experiments performed in aqueous solution and quantum chemistry calculations.
We optimized the ground-state geometries of thioC and β-thioCyd at the MP2/cc-pVTZ,36,37 and the corresponding structures are presented in Figure 1. Here, we focused on the C2′-endo conformer of β-thioCyd which together with the C3′-endo form were experimentally found to be the most stable conformers of the canonical nucleoside cytidine.38 In particular, our ground-state geometry optimizations suggest that the C2′-endo conformer is the most stable structural arrangement of ribose in β-thioCyd. It is worth noting the near proximity of the C1′–H atom in the sugar to the sulfur atom of the nucleobase, as marked in Figure 1. The distance between these two atoms was suggested as the key contributor to the reaction coordinate of the photoanomerization of thioCyd and amounts to merely 2.36 Å.10
Figure 1.

Ground-state geometries of thioC and β-thioCyd.
We next calculated vertical excitation energies of thioC and thioCyd using the ADC(2) method,39,40 which may be regarded as the MP2 equivalent for excited states and was shown to accurately reproduce excited-state energies and potential energy surfaces for nucleobases.41,42 According to our ADC(2)/cc-pVTZ calculations, both thioC and β-thioCyd are characterized by very similar vertical excitation energies of the four lowest singlet excited states, with the lowest three states being red-shifted by ∼0.1 eV in the nucleoside when compared to the nucleobase (see Table 1). The vertical excitation energy of the optically bright S4(ππCS*) amounts to ∼4.40 eV and is virtually identical in thioC and both anomeric forms of thioCyd.
Table 1. Vertical Excitation Energies of ThioC and β-ThioCyd Calculated by Using the ADC(2)/cc-pVTZ Method Based on S0 Minimum-Energy Geometries Optimized with the ADC(2)/cc-pVTZ Methoda.
| state | transition | Eexc [eV] | fosc | λ [nm] |
|---|---|---|---|---|
| thioC | ||||
| S1 | nSπ* | 3.43 (3.93) | 2.43 × 10–5 | 361.5 (315.5) |
| S2 | ππring* | 3.72 (4.25) | 3.57 × 10–2 | 324.6 (291.7) |
| S3 | nSπ* | 3.83 (4.59) | 3.66 × 10–5 | 323.7 (270.1) |
| S4 | ππCS* | 4.41 (4.57) | 4.36 × 10–1 | 281.1 (271.3) |
| β-thioCyd | ||||
| S1 | nSπ* | 3.27 (3.65) | 8.39 × 10–4 | 379.2 (339.7) |
| S2 | ππring* | 3.62 (4.04) | 3.22 × 10–2 | 342.5 (306.9) |
| S3 | nSπ* | 3.72 (4.33) | 1.66 × 10–2 | 333.3 (286.3) |
| S4 | ππCS* | 4.38 (4.47) | 3.21 × 10–1 | 283.1 (277.4) |
Values in parentheses were calculated assuming the COSMO solvation model of bulk water in the nonequilibrium limit.
In addition to calculations for isolated thioC and thioCyd, we also estimated the solvatochromic shifts of the vertical excitation energies assuming the COSMO implicit solvent model43,44 of bulk water. These results are presented in Table 1 in parentheses and show that the three lowest excited singlet states of thioC are blue-shifted with respect to the corresponding gas phase values by more than 0.5 eV.23 This is the result of very different directions of the electric dipole moment vectors (μ) of the S1–S3 states when compared to the electronic ground state. Weaker solvatochromic shifts of the vertical excitations of β-thioCyd likely originate from the fact that part of the chromophore is shielded by the sugar moiety. The optically bright S4(ππCS*) state is only mildly affected by polar solvent with a hypsochromic shift below 0.2 eV. The μ vector of the S4 state has a similar direction to the ground-state μ vector, albeit with lower magnitude, which results in better energetic stabilization of the electronic ground state and mild blue-shift of the S4 excitation energy. In the following paragraphs, we will describe our time-resolved transient absorption spectroscopy experiments performed with the pump wavelength of 308 nm, which was selected to match similar experiments performed by Mai et al. for thioC.26 Therefore, we expect that both the bright S4 and S2 states could be responsible for the absorption of UV light by the studied compounds in our experimental setup.
It is important to emphasize that the vertical excitation energy of the lowest energy triplet ππCS* state of the nucleobase is practically unaffected by sugar substitution (see Table S1). In contrast, the excitation energy of the lowest energy triplet nSπ* state is red-shifted by ∼0.1 eV in the case of the nucleosides, which indicates that this electronic state might play an important role in the excited-state dynamics of α- and β-thioCyd.
The stabilization of triplet nSπ* state in thioCyd is also evident from the optimized T1 minimum-energy geometries. Our ADC(2)/cc-pVTZ geometry optimizations indicate that the T1 minimum of thioC can be described as having a mixed nSπ* + ππring* excitation character with nearly equal contributions of both electronic configurations. A similar interpretation was reported by Mai et al. based on MRCIS calculations.26 To validate this observation, we also optimized the T1 minimum of thioC at the XMS-CASPT2 level,45,46 which yielded a ring-puckered structure nearly completely defined by the 3ππring state. Therefore, all aforementioned quantum chemical approaches consistently show that the T1 minimum of thioC is characterized by substantial contribution from the 3ππring* configuration. In contrast, the molecular orbital character of the lowest energy T1 minima of α- and β-thioCyd is dominated by the 3nSπ* configuration, which is confirmed by both ADC(2) and XMS-CASPT2 methods. Both approaches also consistently showed that the corresponding T1 minimum-energy geometries of thioCyd are characterized by elongation of the C=S bond from 1.65 to ∼1.75 Å and slight pyramidalization of the C6 atom in the chromophore moiety (see Figures 1 and 2 for comparison). This indicates that the diradical structure of the 3nSπ* state is largely located on the S and C6 atoms. Considering slight shortening of the C1′–H··· S distance in β-thioCyd by 0.06 Å, these structural changes occurring in the 3nSπ* state are strongly indicative of the possibility of C1′–H atom abstraction by the thiocarbonyl group. This photochemical process was previously suggested as the initial step in the photoanomerization of thioCyd.10
Figure 2.

T1 minimum-energy geometries of thioC (panels a and b) and β-thioCyd (panels c and d) optimized in the gas phase and assuming microsolvation using the ADC(2)/cc-pVTZ method (see the section Microhydrated structures in the Supporting Information for more details).
In addition to ground- and excited-state geometry optimizations in the gas phase, we also considered thioC and thioCyd microsolvated with four and three water molecules, respectively. This approach allowed us to reproduce the interaction of the chromophores with the nearest surroundings treated at the same level of theory. As can be seen in Figure 2 (panels a and b), microsolvation stabilizes the 3ππring* molecular orbital character in the T1 minimum of thioC, which is also reflected by somewhat shorter C=S bond than in the case of the mixed 3ππring + 3nSπ* configuration. Nevertheless, β-thioCyd retained the nearly pure 3nSπ* molecular orbital character of its T1 minimum upon microsolvation, which underscores the importance of this excitation for the photochemistry of the nucleoside.
We performed time-resolved transient absorption spectroscopy experiments in the visible probe range at an excitation wavelength of 308 nm to validate the computational prediction of 3nSπ* being the dominant configuration of the T1 state of thioCyd. The spectra collected during the first 20 ps after the photoexcitation of thioC and β-thioCyd at 308 nm are presented in Figures 3a and 3b, respectively. It becomes immediately apparent that both studied molecules exhibit long-lived spectral features that could be attributed to the population of triplet states. In particular, both thioC and β-thioCyd possess a strong excited-state absorption (ESA) band with maxima around ∼511 and ∼516 nm, respectively, which were previously assigned to the absorption from the 3ππring* state in the UV-excited nucleobase. For both molecules, the lifetimes of the excited-state absorption band are on the order of several nanoseconds. More importantly, β-thioCyd exhibits a comparably strong absorption feature between 350 and 400 nm, which is indicative of two partially overlapping bands present at time delays below 10 ps. The maximum of the major band is at 372 nm with a minor shoulder band at 392 nm. The minor band at 392 nm vanishes after 20 ps, whereas the major band exhibits a lifetime of around 1.6 ± 0.1 ns. In comparison, thioC is characterized by a single excited-state absorption band at 345 nm with a longer lifetime of 7.4 ± 0.6 ns. The time-resolved transient absorption spectroscopy data for α-thioCyd show analogous spectral features to β-thioCyd and can be viewed in the Supporting Information alongside a detailed description of experimental procedures.
Figure 3.
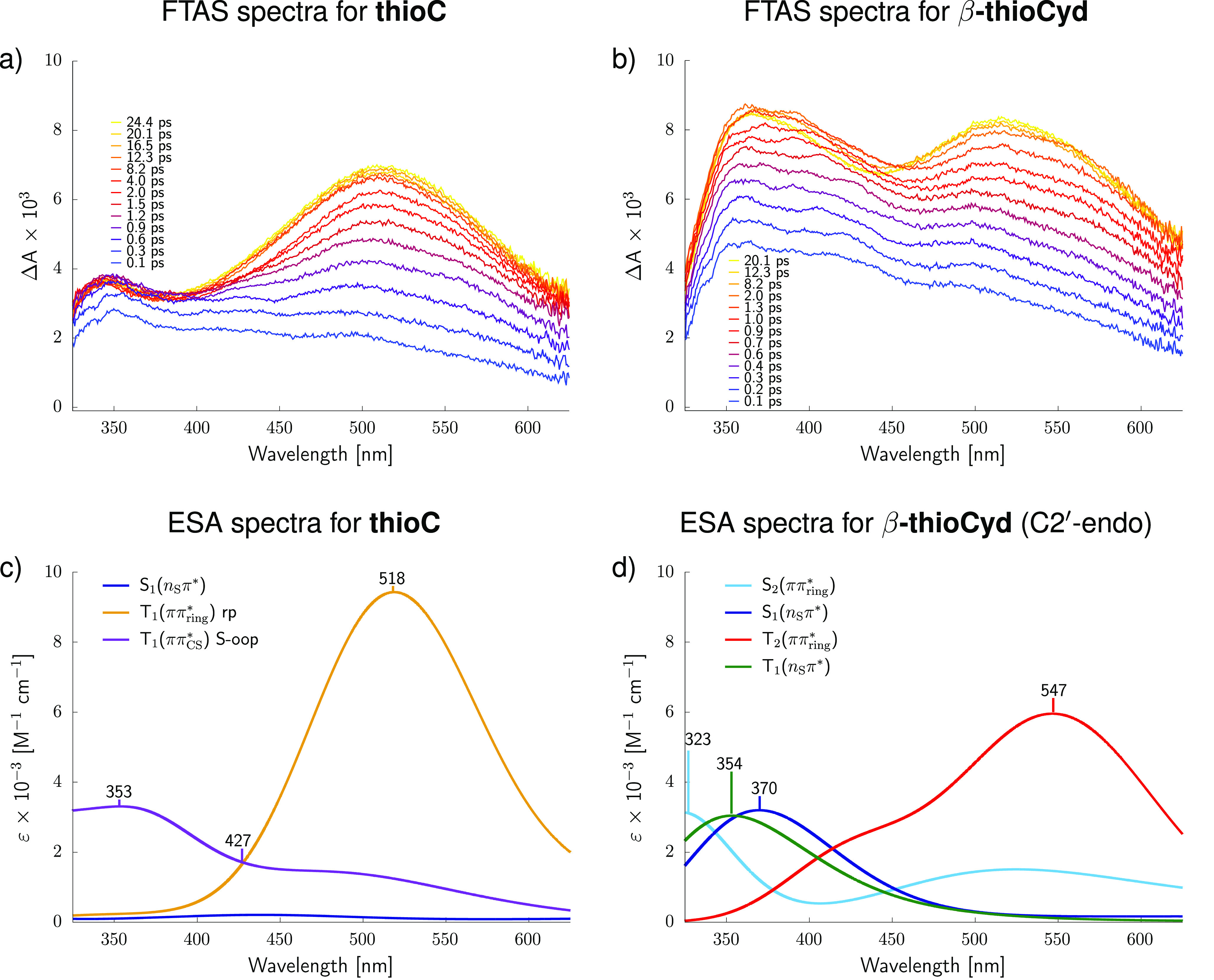
Transient VIS absorption difference spectra of (a) thioC and (b) β-thioCyd following excitation at 308 nm. (c, d) Simulated excited-state absorption (ESA) spectra from specific excited-state (singlet and triplet) minima.
To assign the absorption bands from the time-resolved transient absorption spectroscopy experiments, we simulated excited-state absorption spectra for the key excited-state minima using the ADC(2)/cc-pVTZ method and the independent mode displaced harmonic oscillator (IMDHO)47,48 to explicitly account for the effect of vibrational motion on the band shapes (see Figure 3c,d). Consideration of microsolvated structures and the COSMO implicit solvent model allowed us accurately reproduce specific solvation effects on the width and position of the bands.49 According to these results, the broad and long-lived excited-state absorption bands recorded for thioC and β-thioCyd at around 520 nm correspond to the population of the 3ππring* state. This diabatic state dominates the molecular orbital character of the T1 minimum of thioC, which explains why this spectral feature dominates the transient absorption spectrum of thioC at longer time delays. The assignment of the 3ππring state is also consistent with excited-state geometry optimizations of thioC. Owing to the inversion of energy levels in thioCyd, the 3ππring* character can be assigned to the T2 minimum of the nucleoside (see the red spectrum in Figure 3d). Consequently, our time-resolved transient absorption spectroscopy experiments for α- and β-thioCyd recorded excited-state absorption features of both the T2 and T1 states, the latter being dominated by the 3nSπ* configuration. The simulated excited-state absorption maximum of the T1(3nSπ*) is located at 354 nm and is in good agreement with the long-lived band recorded in our time-resolved transient absorption spectroscopy experiments at 372 nm. It is worth noting that the S1(nSπ*) state of β-thioCyd is characterized by virtually identical excited-state absorption spectrum, with the absorption maximum at slightly longer wavelength, i.e., 370 nm, as according to our simulations. We assign this band to the aforementioned maximum recorded in our time-resolved transient absorption spectroscopy experiments at 392 nm. Therefore, the broader bimodal transient absorption feature visible between 350 and 400 nm during the initial 10 ps is likely the result of the simultaneous population of the S1 and T1 states in β-thioCyd. The assignment of the absorption maximum at 392 nm to the S1(nSπ*) excitation is also supported by the fact that this feature vanishes in β-thioCyd within the initial 20 ps of the excited-state dynamics. This suggests that at time delays beyond 20 ps we predominantly observe population of the triplet 3nSπ* and 3πSπ* states for the nucleoside.
Because we were unable to locate the minimum associated with the 3nSπ* state in thioC, we anticipate that this state is unlikely to be populated in the nucleobase. While our simulations of excited-state absorption spectra do not indicate any clear absorption features for the S1(nSπ*) of thioC, the minor transient absorption band at 345 nm might originate from partial population of another T1(3ππCS*) minimum with the C=S bond elongated and tilted out of the plane of the aromatic ring (S-oop in Figure 3c). Therefore, in contrast to the nucleoside, thioC base exhibits typical equilibrium between the ring-puckered and S out-of-plane (S-oop) T1 minima, which was also reported for thionated uracil bases.21
To evaluate the excited-state lifetimes of the nucleobase and nucleoside in a more accurate manner, we performed global fitting of excited-state decays in our time-resolved transient absorption spectroscopy experiments within the entire range of our visible probe pulse (330–600 nm). This fitting procedure returned global excited-state lifetimes of 7.1 ± 1.4 ns for thioC and 1.5 ± 0.2 ns for β-thioCyd, thus confirming the observations for specific wavelengths discussed above. Most importantly, thioC exhibits a nearly 5 times longer excited-state lifetime than its nucleoside, thioCyd. It is noteworthy that the opposite trend was observed in time-resolved studies of canonical RNA building blocks, for which pyrimidine ribonucleosides exhibited several times longer excited-state lifetimes than their respective nucleobases.50 Here, we assign this surprising trend to the dominant population of longer-lived 3ππring* state in thioC. Significant contribution from the 3nSπ* excitation to the lowest-energy triplet state of thioCyd results in much shorter lifetimes and, consequently, different photoreactivity. More efficient 3nSπ* ⇝ S0 intersystem crossing is explained below based on excited-state potential energy surfaces and spin–orbit couplings calculated for thioC and thioCyd (see Figure 4).
Figure 4.

Excited-state potential energy surfaces calculated for thioC and β-thioCyd at the ADC(2)/cc-pVTZ level of theory.
Possible photorelaxation pathways of thioC and β-thioCyd involving all of the key stationary points and state crossings computed at the ADC(2)/cc-pVTZ level are presented in Figure 4. In the case of thioC, the ring-puckered (rp) T1(3ππring*) minimum is lower in energy by merely 0.23 eV than the S out-of-plane (S-oop) T1(3ππCS) minimum, which demonstrates that both these minima could be populated after photoexcitation and explains why the excited-state absorption feature of the T1(rp) state is dominant. It is worth emphasizing that the T1 state of thioC can be reached in a practically barrierless manner from the S1 state via the T2 state. The calculated spin–orbit coupling (SOC) between the S1 and T2 states amounts to 163 cm–1 in the associated minimum-energy crossing point (MECP), which collectively indicates that this intersystem crossing should be relatively fast. Even though the energy barrier from the S1 minimum of thioCyd to the T2 state amounts to 0.34 eV, the population of triplet states in the nucleoside should still be very efficient, which is also reflected by high SOC value for the S1 and T2 states of 156 cm–1. In fact, this modest barrier explains why part of the excited-state population of β-thioCyd is trapped for the initial ∼10 ps in the S1(nSπ*) minimum. Furthermore, the S1, T2, and T1 minima of β-thioCyd are separated from one another by <0.3 eV (<0.1 eV at XMS-CASPT2 level; see the Supporting Information), which is also consistent with the presence of transient absorption bands for all of these states in our experiments (Figure 3b,d). Finally, we expect relatively efficient depopulation of triplet states to the electronic ground state, considering sizable SOC between the T1 and S0 states in the associated minimum-energy crossing points of thioC and β-thioCyd. In fact, the calculated SOC value is 17 cm–1 higher for the nucleoside. It is worth adding that the ΔE between the T1/S0 state crossings and the corresponding T1 minima may exceed 0.9 eV for thioC and 0.6 eV for β-thioCyd (see the XMS-CASPT2 results in the Supporting Information which more accurately describe state crossings with the S0 state). A lower energy barrier and a higher SOC value determined for the photorelaxation from the T1 state of thioCyd further corroborate our observation of much shorter excited-state lifetime in the nucleoside resulting from the population of the 3nSπ* state.
Similar enhancement of the photorelaxation rate of the T1 state was reported for 2-thiothymidine, a nucleoside of a related thiopyrimidine base.24 Bai and Barbatti22 assigned this observation to the repulsion interaction between the sulfur atom and the sugar moiety, leading to lower energy barriers associated with the photorelaxation from the T1(3ππCS*) state. Therefore, our computational and experimental results obtained for thioC and β-thioCyd allowed us to characterize the additional electronic factor that could affect the excited-state dynamics and lifetimes of specific thionucleosides.
In conclusion, we have demonstrated that ribose may have a far-reaching effect on the photoreactivity of the nucleobase chromophore in thionated nucleosides. Our quantum chemical calculations and time-resolved transient absorption spectroscopy (TAS) measurements consistently show that the thioC nucleobase and its nucleoside thioCyd both undergo efficient intersystem crossing to triplet states upon photoexcitation, albeit of very different excitation character. After initial excited-state relaxation, thioC predominantly populates the T1(3ππring*) minimum of lightly ring-puckered geometry, which was previously shown to facilitate the generation of singlet oxygen, which is the key reactive species in photodynamic therapies. In contrast, both α- and β-anomers of thioCyd exhibit strong transient absorption features characteristic of singlet and triplet nSπ* states. Both singlet and triplet nSπ* states are responsible for the photoanomerization of thioCyd, that is, photochemical interconversion between the α- and β-forms.10 The 5 times shorter excited-state lifetime recorded for β-thioCyd when compared to thioC is another intriguing consequence of the population of nSπ* states. First of all, it will suppress singlet-oxygen generation by the nucleoside, since this process requires triplet photorelexation to occur on comparable or longer time scales to diffusion. This result is also unintuitive, considering that the opposite trend was observed for canonical RNA building blocks; specifically, nucleobases exhibit shorter excited-state lifetimes than nucleosides.50 Most importantly, this work underscores that the consideration of the closest molecular environment of the chromophore is crucial for understanding fundamental photochemical properties, which can be harnessed in applications such as photodynamic therapies or photochemical organic synthesis.
Acknowledgments
The authors thank Prof. Carlos E. Crespo-Hernández for helpful discussions. M.J.J. acknowledges the support of the “Diamond Grant” (0144/DIA/2017/46) from the Polish Ministry of Science and Higher Education and a doctoral scholarship from the National Science Center Poland (2020/36/T/ST4/00564). Some of the quantum-chemical computations were performed with the resources granted by the Wrocław Centre of Networking and Supercomputing (WCSS). R.S. acknowledges the Foundation for Polish Science for support from the START Fellowship. J.S. acknowledges support by the Czech Science Foundation (21-23718S). This work was supported in part by grants from the Simons Foundation (SCOL 290360 to D.D.S. and 290363 to J.W.S.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.1c01384.
Author Present Address
△ Department of Chemistry, University of Guelph, 50 Stone Rd. E., Guelph, Ontario N1G 2W1, Canada
Author Present Address
▲ Department of Earth & Space Sciences, University of Washington, Seattle, Washington 98195
Author Contributions
M.J.J., C.L.K., and Z.R.T. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Reelfs O.; Karran P.; Young A. R. 4-thiothymidine sensitization of DNA to UVA offers potential for a novel photochemotherapy. Photochem. Photobiol. Sci. 2012, 11, 148–154. 10.1039/C1PP05188A. [DOI] [PubMed] [Google Scholar]
- Zdrowowicz M.; Wityk P.; Michalska B.; Rak J. 5-Bromo-2’-deoxycytidine—a potential DNA photosensitizer. Org. Biomol. Chem. 2016, 14, 9312–9321. 10.1039/C6OB01446A. [DOI] [PubMed] [Google Scholar]
- Zdrowowicz M.; Michalska B.; Zylicz-Stachula A.; Rak J. Photoinduced Single Strand Breaks and Intrastrand Cross-Links in an Oligonucleotide Labeled with 5-Bromouracil. J. Phys. Chem. B 2014, 118, 5009–5016. 10.1021/jp500192z. [DOI] [PubMed] [Google Scholar]
- Wityk P.; Wieczór M.; Makurat S.; Chomicz-Mańka L.; Czub J.; Rak J. Dominant Pathways of Adenosyl Radical-Induced DNA Damage Revealed by QM/MM Metadynamics. J. Chem. Theory Comput. 2017, 13, 6415–6423. 10.1021/acs.jctc.7b00978. [DOI] [PubMed] [Google Scholar]
- Piccirilli J. A.; Benner S. A.; Krauch T.; Moroney S. E.; Benner S. A. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature 1990, 343, 33–37. 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- Gedik M.; Brown A. Computational study of the excited state properties of modified RNA nucleobases. J. Photochem. Photobiol., A 2013, 259, 25–32. 10.1016/j.jphotochem.2013.02.023. [DOI] [Google Scholar]
- Jones A. C.; Neely R. K. 2-aminopurine as a fluorescent probe of DNA conformation and the DNA–enzyme interface. Q. Rev. Biophys. 2015, 48, 244–279. 10.1017/S0033583514000158. [DOI] [PubMed] [Google Scholar]
- Xu W.; Chan K. M.; Kool E. T. Fluorescent nucleobases as tools for studying DNA and RNA. Nat. Chem. 2017, 9, 1043–1055. 10.1038/nchem.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Chmela V.; Green N. J.; Russell D. A.; Janicki M. J.; Góra R. W.; Szabla R.; Bond A. D.; Sutherland J. D. Selective prebiotic formation of RNA pyrimidine and DNA purine nucleosides. Nature 2020, 582, 60–66. 10.1038/s41586-020-2330-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Tsanakopoulou M.; Magnani C. J.; Szabla R.; Šponer J. E.; Šponer J.; Góra R. W.; Sutherland J. D. A prebiotically plausible synthesis of pyrimidine β-ribonucleosides and their phosphate derivatives involving photoanomerization. Nat. Chem. 2017, 9, 303–309. 10.1038/nchem.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. J.; Szabla R.; Todd Z. R.; Stairs S.; Bučar D.-K.; Šponer J.; Sasselov D. D.; Powner M. W. Selective prebiotic conversion of pyrimidine and purine anhydronucleosides into Watson-Crick base-pairing arabino -furanosyl nucleosides in water. Nat. Commun. 2018, 9, 4073. 10.1038/s41467-018-06374-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuberger B. D.; Pal A.; Del Frate F.; Topkar V. V.; Szostak J. W. Replacing Uridine with 2-Thiouridine Enhances the Rate and Fidelity of Nonenzymatic RNA Primer Extension. J. Am. Chem. Soc. 2015, 137, 2769–2775. 10.1021/jacs.5b00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. C.; O’Flaherty D. K.; Zhou L.; Lelyveld V. S.; Szostak J. W. Inosine, but none of the 8-oxo-purines, is a plausible component of a primordial version of RNA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 13318–13323. 10.1073/pnas.1814367115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K. V.; Burrows C. J. A Prebiotic Role for 8-Oxoguanosine as a Flavin Mimic in Pyrimidine Dimer Photorepair. J. Am. Chem. Soc. 2011, 133, 14586–14589. 10.1021/ja2072252. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Dood J.; Beckstead A. A.; Li X.-B.; Nguyen K. V.; Burrows C. J.; Improta R.; Kohler B. Photoinduced Electron Transfer in DNA: Charge Shift Dynamics Between 8-Oxo-Guanine Anion and Adenine. J. Phys. Chem. B 2015, 119, 7491–7502. 10.1021/jp511220x. [DOI] [PubMed] [Google Scholar]
- Szabla R.; Zdrowowicz M.; Spisz P.; Green N. J.; Stadlbauer P.; Kruse H.; Šponer J.; Rak J. 2,6-diaminopurine promotes repair of DNA lesions under prebiotic conditions. Nat. Commun. 2021, 12, 1–11. 10.1038/s41467-021-23300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Fernández L.; González L.; Corral I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: the case of 6-thioguanine. Chem. Commun. 2012, 48, 2134–2136. 10.1039/c2cc15775f. [DOI] [PubMed] [Google Scholar]
- Bai S.; Barbatti M. Why Replacing Different Oxygens of Thymine with Sulfur Causes Distinct Absorption and Intersystem Crossing. J. Phys. Chem. A 2016, 120, 6342–6350. 10.1021/acs.jpca.6b05110. [DOI] [PubMed] [Google Scholar]
- Mai S.; Marquetand P.; González L. Intersystem Crossing Pathways in the Noncanonical Nucleobase 2-Thiouracil: A Time-Dependent Picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. 10.1021/acs.jpclett.6b00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai S.; Marquetand P.; González L. A Static Picture of the Relaxation and Intersystem Crossing Mechanisms of Photoexcited 2-Thiouracil. J. Phys. Chem. A 2015, 119, 9524–9533. 10.1021/acs.jpca.5b06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S.; Barbatti M. On the decay of the triplet state of thionucleobases. Phys. Chem. Chem. Phys. 2017, 19, 12674–12682. 10.1039/C7CP02050C. [DOI] [PubMed] [Google Scholar]
- Bai S.; Barbatti M. Mechanism of enhanced triplet decay of thionucleobase by glycosylation and rate-modulating strategies. Phys. Chem. Chem. Phys. 2018, 20, 16428–16436. 10.1039/C8CP02306A. [DOI] [PubMed] [Google Scholar]
- Mai S.; Ashwood B.; Marquetand P.; Crespo-Hernández C. E.; González L. Solvatochromic Effects on the Absorption Spectrum of 2-Thiocytosine. J. Phys. Chem. B 2017, 121, 5187–5196. 10.1021/acs.jpcb.7b02715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood B.; Pollum M.; Crespo-Hernández C. E. Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases. Photochem. Photobiol. 2019, 95, 33–58. 10.1111/php.12975. [DOI] [PubMed] [Google Scholar]
- Brister M. M.; Gustavsson T.; Crespo-Hernández C. E. Excited State Lifetimes of Sulfur-Substituted DNA and RNA Monomers Probed Using the Femtosecond Fluorescence Up-Conversion Technique. Molecules 2020, 25, 584. 10.3390/molecules25030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai S.; Pollum M.; Martínez-Fernández L.; Dunn N.; Marquetand P.; Corral I.; Crespo-Hernández C. E.; González L. The origin of efficient triplet state population in sulfur-substituted nucleobases. Nat. Commun. 2016, 7, 13077. 10.1038/ncomms13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslancan S.; Martínez-Fernández L.; Corral I. Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments. Molecules 2017, 22, 998. 10.3390/molecules22060998. [DOI] [Google Scholar]
- Janicki M. J.; Roberts S. J.; Šponer J.; Powner M. W.; Góra R. W.; Szabla R. Photostability of oxazoline RNA-precursors in UV-rich prebiotic environments. Chem. Commun. 2018, 54, 13407–13410. 10.1039/C8CC07343K. [DOI] [PubMed] [Google Scholar]
- Taras-Goślińska K.; Burdziński G.; Wenska G. Relaxation of the T1 excited state of 2-thiothymine, its riboside and deoxyriboside-enhanced nonradiative decay rate induced by sugar substituent. J. Photochem. Photobiol., A 2014, 275, 89–95. 10.1016/j.jphotochem.2013.11.003. [DOI] [Google Scholar]
- Pollum M.; Jockusch S.; Crespo-Hernández C. E. 2,4-Dithiothymine as a Potent UVA Chemotherapeutic Agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. 10.1021/ja510611j. [DOI] [PubMed] [Google Scholar]
- Sánchez-Rodríguez J. A.; Mohamadzade A.; Mai S.; Ashwood B.; Pollum M.; Marquetand P.; González L.; Crespo-Hernández C. E.; Ullrich S. 2-Thiouracil intersystem crossing photodynamics studied by wavelength-dependent photoelectron and transient absorption spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 19756–19766. 10.1039/C7CP02258A. [DOI] [PubMed] [Google Scholar]
- Janicki M. J.; Szabla R.; Šponer J.; Góra R. W. Solvation effects alter the photochemistry of 2-thiocytosine. Chem. Phys. 2018, 515, 502–508. 10.1016/j.chemphys.2018.06.016. [DOI] [Google Scholar]
- Martínez-Fernández L.; Corral I.; Granucci G.; Persico M. Competing ultrafast intersystem crossing and internal conversion: a time resolved picture for the deactivation of 6-thioguanine. Chem. Sci. 2014, 5, 1336–1347. 10.1039/c3sc52856a. [DOI] [Google Scholar]
- Bai S.; Barbatti M. Divide-to-Conquer: A Kinetic Model for Singlet Oxygen Photosensitization. J. Chem. Theory Comput. 2017, 13, 5528–5538. 10.1021/acs.jctc.7b00619. [DOI] [PubMed] [Google Scholar]
- Colville B. W. F.; Powner M. W. Selective Prebiotic Synthesis of α-Threofuranosyl Cytidine by Photochemical Anomerization. Angew. Chem., Int. Ed. 2021, 60, 10526–10530. 10.1002/anie.202101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigend F.; Häser M. RI-MP2: first derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. 10.1007/s002140050269. [DOI] [Google Scholar]
- Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Plavec J.; Thibaudeau C.; Chattopadhyaya J. How do the energetics of the stereoelectronic gauche and anomeric effects modulate the conformation of nucleos(t)ides?. Pure Appl. Chem. 1996, 68, 2137–2144. 10.1351/pac199668112137. [DOI] [Google Scholar]
- Hättig C. Structure Optimizations for Excited States with Correlated Second-Order Methods: CC2 and ADC(2). Adv. Quantum Chem. 2005, 50, 37–60. 10.1016/S0065-3276(05)50003-0. [DOI] [Google Scholar]
- Dreuw A.; Wormit M. The algebraic diagrammatic construction scheme for the polarization propagator for the calculation of excited states. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 82–95. 10.1002/wcms.1206. [DOI] [Google Scholar]
- Plasser F.; Crespo-Otero R.; Pederzoli M.; Pittner J.; Lischka H.; Barbatti M. Surface Hopping Dynamics with Correlated Single-Reference Methods: 9H-Adenine as a Case Study. J. Chem. Theory Comput. 2014, 10, 1395–1405. 10.1021/ct4011079. [DOI] [PubMed] [Google Scholar]
- Wiebeler C.; Borin V.; Sanchez de Araujo A. V.; Schapiro I.; Borin A. C. Excitation Energies of Canonical Nucleobases Computed by Multiconfigurational Perturbation Theories. Photochem. Photobiol. 2017, 93, 888–902. 10.1111/php.12765. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, 2, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Khani S. K.; Khah A. M.; Hättig C. COSMO-RI-ADC(2) excitation energies and excited state gradients. Phys. Chem. Chem. Phys. 2018, 20, 16354–16363. 10.1039/C8CP00643A. [DOI] [PubMed] [Google Scholar]
- Andersson K.; Malmqvist P.; Roos B. O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. 10.1063/1.462209. [DOI] [Google Scholar]
- Shiozaki T.; Győrffy W.; Celani P.; Werner H.-J. Communication: Extended multi-state complete active space second-order perturbation theory: Energy and nuclear gradients. J. Chem. Phys. 2011, 135, 081106. 10.1063/1.3633329. [DOI] [PubMed] [Google Scholar]
- Petrenko T.; Neese F. Analysis and prediction of absorption band shapes, fluorescence band shapes, resonance Raman intensities, and excitation profiles using the time-dependent theory of electronic spectroscopy. J. Chem. Phys. 2007, 127, 164319. 10.1063/1.2770706. [DOI] [PubMed] [Google Scholar]
- Petrenko T.; Neese F. Efficient and automatic calculation of optical band shapes and resonance Raman spectra for larger molecules within the independent mode displaced harmonic oscillator model. J. Chem. Phys. 2012, 137, 234107. 10.1063/1.4771959. [DOI] [PubMed] [Google Scholar]
- Ferrer F. J. A.; Improta R.; Santoro F.; Barone V. Computing the inhomogeneous broadening of electronic transitions in solution: a first-principle quantum mechanical approach. Phys. Chem. Chem. Phys. 2011, 13, 17007. 10.1039/c1cp22115a. [DOI] [PubMed] [Google Scholar]
- Hare P. M.; Crespo-Hernández C. E.; Kohler B. Internal conversion to the electronic ground state occurs via two distinct pathways for pyrimidine bases in aqueous solution. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 435–440. 10.1073/pnas.0608055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.