

Abstract
Background
Pancreatic ductal adenocarcinoma (PDAC) is one of the leading cancers worldwide and has a poor survival, with a 5-year survival rate of only 8.5%. In this study we investigated altered DNA methylation associated with PDAC severity and prognosis.
Methods
Methylome data, generated using 450 K bead array, was compared between paired PDAC and normal samples in the TCGA cohort (n = 9) and our Indian cohort (n = 7). The total Indian Cohort (n = 75) was split into cohort 1 (n = 7), cohort 2 (n = 22), cohort 3 (n = 26) and cohort 4 (n = 20).Validation of differential methylation (6 selected CpG loci) and associated gene expression for differentially methylated genes (10 selected gDMs) were carried out in separate validation cohorts, using MSP, RT-PCR and IHC correlations between methylation and gene expression were observed in TCGA, GTEx cohorts and in validation cohorts. Kaplan–Meier survival analysis was done to study differential prognosis, during 2–5 years of follow-up.
Results
We identified 156 DMPs, mapped to 91 genes (gDMs), in PDAC; 68 (43.5%) DMPs were found to be differentially methylated both in TCGA cohort and our cohort, with significant concordance at hypo- and hyper-methylated loci. Enrichments of “regulation of ion transport”, “Interferon alpha/beta signalling”, “morphogenesis and development” and “transcriptional dysregulation” pathways were observed among 91 gDMs. Hyper-methylation of NPY and FAIM2 genes with down-regulated expression in PDAC, were significantly associated with poor prognosis in the Indian patient cohort.
Conclusions
Ethnic variations among populations may determine the altered epigenetic landscape in the PDAC patients of the Indian cohort. Our study identified novel differentially methylated genes (mainly NPY and FAIM2) and also validated the previously identified differentially methylated CpG sites associated with PDAC cancer patient’s survival. Comparative analysis of our data with TCGA and CPTAC cohorts showed that both NPY and FAIM2 hyper-methylation and down-regulations can be novel epigenetically regulated genes in the Indian patient population, statistically significantly associated with poor survival and advanced tumour stages.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12935-022-02737-1.
Keywords: Pancreatic-ductal adenocarcinoma, 450K DNA methylation, NPY and FAIM2hyper-methylation, Poor survival, Epigenetically Dysregulated Signalling pathways, Prognostic epigenetic marker
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the leading aggressive cancers and is the third most common cause for cancer deaths worldwide [1]. According to the GLOBOCAN 2020, PDAC caused 4,66,003 deaths with an incidence of 4,95,773 new cases per year [2]. PDAC has a poor prognosis with a 5-year survival rate of less than 8.5%, despite extensive research on the cancer type [3]. Intrinsic heterogeneity across PDAC patients may contribute to poor survival [4].
Epigenetic changes, such as aberrant methylation of DNA, histone deactelyation, chromatin remodelling affect gene expression and are important drivers of carcinogenesis. DNA methylation plays a crucial role in disease outcome [5]. Altered methylation status at the CpG islands at the gene promoters, limits the access to transcription factors, significantly affect gene expression [6]. Hypermethylation at CpG islands with down-regulated expressions of tumour suppressor genes (TSG) and hypomethylation with increased expression of oncogenes, have been documented for many cancers. Epigenomics has become a promising paradigm for PDAC diagnosis and has identified pathways that can be targeted for therapy [7]. Past researches have tried to identify altered methylation of cell-free DNA in PDAC as the potential blood-based diagnostic and prognostic biomarkers. However, limited validation studies across patient population and applicability of such biomarkers was seen till date [8, 9]. Thus, it is important to study the differential methylation marks associated with PDAC development.
Past studies had shown that both genetic and epigenetic alterations contributed to PDAC initiation and progression [7, 10]. PDAC diagnosis based on gene-mutation has only brought partial success in early diagnosis and overall patients’ survival improvement [7]. Distinct epigenetic markers have been observed for specific subtypes of pancreatic cancers and for specific stages of PDAC [11, 12]. Also, studies including pancreatic cancer (PanCa) samples from different populations have identified partially similar methylation marks. The inter-population epigenetic variation has been reported and this may be explained by varied ethnicity, demographic, and occupational factors [13, 14]. Globally few studies have been done on investigating the changing epigenetic landscapes through progressive stages of PDAC [15] and on different ethnicity. In this study, we have reported the altered genome-wide DNA methylation profile of PDAC across progressive stages, compared our results with the TCGA cohort and validated these results in additional 4 independent cohorts.
Material and methods
Study design
In this study, we aimed to identify differential methylation marks associated with PanCa in Indian population, using global methylome analysis. The detailed study design is shown in Additional file 2: Fig S1, which were included Discovery cohort (TCGA pancreatic cancer, n = 185) and Validation cohorts (1, 2, 3, and 4). The description of the study cohorts was mentioned in Additional file 2: Method 1 (Additional file 2: Fig S1).
Sample collection and ethical clearance
The study was approved by the Institutional Review Board (IRB) of Indian Statistical Institute, Kolkata and all involved hospitals. All clinical samples were collected only after taking consent from patient and/or family post debriefing about the study. Our study included samples of PDAC patients, collected from Government and private Hospitals, in the city of Kolkata during the time frame of 2013–2018. A systemic 2–5 year follow up was done for overall survival (OS).Tumour and adjacent normal tissue samples were collected during Whipple’s surgery and stored in RNA later solution (Sigma-Aldrich Co. LLC). Histopathological examination and TNM staging were done according to the 8th edition guidelines by American Joint Committee on Cancer (AJCC), by all the associated clinician. We had investigated the tumour purity through histopathological evaluation, and it was found to be > 80% in all samples (10× imagery by using Leica DM 1000, camera: Leica EC3). We have generated a score for each sample regarding tumour cell percentage by randomly taking 8 images for each slide (Additional file 2: Fig S2).
The inclusion and exclusion criteria of the patients were considered prior to specimen collection and consenting. The inclusion criteria started with collecting samples that were only clinically diagnosed PDAC samples within the age-limit 30–75 years and should positively be sero-negative and also capable of undergoing whipple’s procedure. The exclusion criteria were designed to target PDAC subtype, which is a specific subtype of PanCa. Hence periampullary adenocarcinomas were excluded and also PDAC samples with sero -positivity were excluded. Some other exclusion criteria’s included were age greater than 75 years and if the patient were physically and mentally handicapped.
DNA methylation data generation and analysis
Whole genome DNA methylation profiling was done from the DNA isolated from the tumour and adjacent normal tissues of validation cohort 1 (n = 7) (Additional file 2: Method 2) using Infinium Human Methylation 450 K Bead Chip (Illumina). The bead chip included 485,577 CpG sites and targets 96% of CpG islands in human genome (hg19), using 4 μl of bisulfite-treated DNA, according to the manufacturer’s protocol [16]. The raw signal intensities on bead chips were read and converted to raw data (IDAT files) using the iScan platform (Illumina). The raw data including the IDAT files is available in the GEO database with the entry “GSE181740”.
In this current study, we have followed the analysis done by Aryee et al.,2014 [16] for processing of DNA methylome data. For each CpG site in the genome, methylation level was assessed using the established β value. The β value was calculated as the ratio of fluorescent signal intensity of the methylated (M) and total of signal intensities from the methylated (M) and unmethylated (U) alleles:
We have used the minfi R package R version 4.0.4 to convert raw array data to β value and for data normalisation. To reduce false positive inferences from our data, we have used data quality control as described by Aryee et al.,2014 [16], Probes were excluded from further analysis if: (1) their detection p-value > 0.05, (2) they contain SNPs in their sequences, (3) they are positioned on X and Y chromosomes. With the filtered probe set, we performed both intra-array (Infinium I and Infinium II) normalisation and inter-array normalisation using quantile normalisation approach (using β values).
Prior to differential methylation calculation, purity of tumour samples was estimated using “Infinium Purify R” package using R version 4.0.4 [17]. Differentially methylated positions (DMPs) between the tumour and the normal tissue samples were identified by comparing the average beta values between the study groups. The probes with average |Δβ| (difference between the average β-value among the tumour samples and the average β-value among the normal samples) ≥ 0.2 and Benjamini–Hochberg corrected p < 0.05 were considered as significantly differentially methylated positions (DMPs). Hyper-methylated positions (CpG sites) were DMPs with Δβ ≥ 0.2 and hypo-methylated sites were DMPs with Δβ ≤ − 0.2. The DMPs were annotated to genes, referred to as “gDMs”.
Association of differential methylation at key genes with PanCa, was assessed using MSP in validation cohort 2 (n = 22). The band intensities after electrophoresis were noted for each MSP and the data was represented as percentage (%) methylation among all the samples for a gene. The detailed MSP procedure is mentioned in Additional file 2: Method 3.
Study of relationship between differential methylation and gene expression
Differential methylation at the gene promoters is often inversely associated with its expression. Gene expression data (FPKM) was downloaded for the same set of TCGA samples from GDC data portal (n = 179). Correlations between the expressions of a gene with differential methylation at CpG site (using β values) annotated to that gene, was estimated using Pearson’s correlation. To compare the expressions of selected genes between PanCa and normal pancreas tissues, we additionally investigated the gene expression data (v8) on normal pancreas tissue from GTEx database (https://gtexportal.org/home/datasets) [19] (The Genotype-Tissue Expression [GTEx] project, 2013). Comparison of expression of a gene between PanCa samples (from TCGA cohort) (n = 179) and normal pancreatic tissues (from GTEx database V8) (n = 171) was performed using Gepia V2 (http://gepia2.cancer-pku.cn/#index) [19].
Expression of selected gDMs (with promoter methylation) was studied in the tumour and normal tissue samples collected from the validation cohort 3, using RT-PCR (n = 26) (Additional file 2: Method 4). Due to limitation of tissue availability, simultaneous validation of methylation and expression could not be done on tumour and normal samples from same samples in the validation cohort 2. The relative expressions of selected genes (KCNA6, RASSF1, SIGIRR, NPY, FAIM2, FOXE1, SLITRK3, IRF4, MX2, and GALR1) were studied in PDAC tumour and adjacent counterpart of normal tissue samples compared to two different reference genes (GAPDH and ACTB). All necessary detail procedures were mentioned in and Additional file 2: Method 4.
Functional characterisation of the differentially methylated genes
The DMPs were annotated to their nearest genes with reference genome hg19. The list of mapped genes was designated as genes with differential methylation (gDM). Enrichment of Gene Ontology (GO) terms on biological processes among the gDMs was done using Metascape [20]. Metascape was used to study the enrichment of pathways in the KEGG database. The detailed analysis on the functional enrichment of the gDMs was mentioned in Additional file 2: Method 5.
Validation of the Neurotransmitter Neuropeptide Y (NPY) and Fas Apoptotic Inhibitory Molecule 2 (FAIM2) from publicly available data source
To compare altered methylation at the gDMs in PanCa samples, we further analysed pancreatic cancer methylation database (PCMdb, http://crdd.osdd.net/raghava/pcmdb/), which is a comprehensive collection of differentially methylated genes (n = 4342) in both PanCa cell lines (n = 88) and tissue samples (n = 3078), previously reported to have altered methylation in PanCa [21].
The NPY and FAIM2 methylation pattern across the various CpG sites on their respective chromosomes were investigated on 35 PDAC cell lines using Cancer Cell Line Encyclopedia (CCLE) (https://depmap.org/portal/) [22]. DeMap portal was used to generate map representing the degree of methylation across the cell lines at various CpG locations. Correlations between gene expression and methylation data from TCGA-PAAD dataset of NPY and FAIM2 genes were observed using SMART APP (http://www.bioinfo-zs.com/smartapp) [23]. Protein levels of NPY and FAIM2 genes in normal tissue versus tumour tissue were analysed using UALCAN (http://ualcan.path.uab.edu/) using Clinical ProteomicTumour Analysis Consortium (CPTAC) Confirmatory/Discovery dataset. Finally, Pan-cancer expression analysis of NPY and FAIM2 genes were observed using UALCAN [24].
Validation of differential protein expression using immunohistochemistry (IHC)
We have further validated the differential expressions of Npy and Faim2 proteins in the fourth independent validation cohort (validation cohort 4, n = 20). Protein expression levels of both Npy and Faim2 were compared between the section of cancer and the adjacent normal tissue samples. The detailed process of IHC was described in Additional file 2: Method 6.
Statistical analysis
Differentially methylated positions (DMPs) were identified using F-test to compare the beta values between cancer and normal samples [16]. Linear regression was done to identify the association of methylation values with PanCa, after adjusting for the effects of age, gender, smoking status and tumour purity. Hierarchical clustering was done on Euclidean distance matrix to identify the subgroups of samples differing in the levels of methylation at the DMPs. The R functions “hclust” and “heatmap3” were used for heatmap preparation. Differences in expression of a gene between subgroups were evaluated using Wilcoxon Signed Rank test using GEPIA. Patients in the TCGA cohort was subdivided into high (> 90th percentile) and low (< 10th percentile) subgroups based on the expression levels of target genes. Overall survival (OS) of the subgroups of patients was compared using Kaplan–Meier method of survival analysis. The correlation between differential methylation and gene expression was estimated using Pearson’s correlation coefficient.
The ΔCt values of gene expression data were analysed by using Wilcoxon Rank sum test (Mann Whitney U test) by “nortest” and “ggpubr” (ggplot2) libraries in R [25]. Overall survival analysis was done using IBM®SPSS software (IBM SPSS Statistics 27, Armonk, NY: IBM Corp) by Kaplan–Meier survival analysis. Correlations between gene expressions and clinico-pathological features were observed using two different methods—“Kendall tou” correlation and Pearson’s correlation test using SPSS (https://www.ibm.com/in-en/analytics/spss-statistics-software).
Results
Characteristic of patients recruited in the study
Among the 185 PanCa patients 7 were African-Americans, 11 were Asian, 160 were Caucasian population origin and 7 were from unknown source. Among 9 samples in discovery cohort 6 patients were Caucasian, 1 from Asian and 1 from African-American population origin and 1 was from unknown source. Amongst 185 PanCa patients 3 samples were excluded due to unavailability of histopathological data. Among the 182 PanCa patients in the TCGA cohort, 5 patients had stage IV PanCa (2.7%), 4 patients had stage III PanCa (2.2%), and the remaining patients had stage I + II (95.1%) (Additional file 1: Tables S1, S2). The demographic and clinical features of the patients included in the validation cohorts were summarised in Additional file 1: Table S2. Among the 7 patients in validation cohort1, 5 were males and 2 were females. Average age of the patients was 53.14 years. Among the males, 3 had a history of tobacco intake, either through smoking or chewing habits. None of the females had any history of tobacco intake. Histopathological analysis showed different grades of tumour among the patients with increasing severity: well-differentiated adenocarcinoma (WDA: n = 2), moderately differentiated adenocarcinoma (MDA: n = 2) and poorly differentiated adenocarcinoma (PDA: n = 3). The validation of methylation was done among validation cohort 2 with 22 PDAC patients. Among them, 18.18% (n = 4) of the patients had PDA PDAC, 27.27% (n = 6) had MDA PDAC and 54.54% (n = 12) had WDA PDAC. The validation of gene expression was done among validation cohort 3 with 26 PDAC patients. Among them, 30.76% (n = 8) of the patients had PDA PDAC, 23.07% (n = 6) had MDA PDAC and 46.15% (n = 12) had WDA PDAC. The validation of target genes associated protein level expression by IHC experiment was done among validation cohort 4 with 20 PDAC patients. Among them, 25% (n = 5) of the patients had PDA PDAC, 55% (n = 11) had MDA PDAC and 20% (n = 4) had WDA PDAC (Additional file 1: Table S3).
Differential methylation in pancreatic cancer
The TCGA-PAAD cohort dataset included DNA methylome data on tumour and normal tissue samples from 9 patients. Global differential methylation marks were identified by comparing the 9 PanCa samples with their solid tissue normal (Additional file 1: Table S4). A total of 7832 DMPs (|Δβ|> 0.2, p-value < 0.05) were identified between these tumour and normal samples in the TCGA cohort (Additional file 1: Table S4, Additional file 2: Fig S4A). Among the DMPs, 54% were hyper-methylated and 46% were hypo-methylated (Additional file 2: Fig S4B). The 7832 DMPs could correctly classify cancer and normal samples in separate clusters (Additional file 2: Fig S4A). To compare the differential methylation in the TCGA PanCa samples, we selected the study by Mishra and Guda et al.,2017 [12], where the authors submitted a list of DMPs in all PanCa samples in the TCGA cohort. All the 7832 DMPs were also significantly differentially methylated in PDAC in their study. The delta beta values (i.e. differences in the beta (β) values between cancer and normal samples), estimated in our study were highly correlated with their study results (Additional file 2: Fig S4C).
We next performed the differential methylation analysis in the validation cohort 1 (n = 7), with two aims: (1) the DMPs in PanCa identified from the TCGA cohort and the Indian cohort should be concordant and (2) novel differential methylation marks could be identified specific to the Indian patient cohort, due to ethnicity and cultural distinctions. We identified a total of 156 DMPs (|Δβ|> 0.2), mapped to 91 genes in PDAC tumour compared to adjacent normal counter parts from 7 PDAC patients (Additional file 1: Table S5) after Benjamini Hochberg multiple testing correction (q-value < 0.05). Hierarchical clustering of the samples with these 156 DMPs, showed separate clustering of cancer and the normal samples (Fig. 1A). Among the 156 DMPs, 37.2% (n = 58) were hyper-methylated and 62.8% (n = 98) were hypo-methylated among cancer samples (Fig. 1B). Among the previously identified 7832 DMPs in the TCGA cohort, 68 DMPs overlap with DMPs, identified from our data (Fig. 1C, Additional file 1: Table S6). The differences in beta values between the paired tumour and normal samples at these 68 DMPs in the TCGA data (for 9 PAAD patients) showed a positive correlation with our study (correlation coefficient = 0.98) (Fig. 1D, Additional file 1: Table S6). Excluding the 68 DMPs, methylation at the remaining 88 DMPs (out of 156 DMPs) was significantly associated with PDAC in validation cohort 1 (Additional file 1: Table S7). We have looked into the hemi-methylation at the 156 DMPs in the cancer samples of our cohort (n = 7) and also in the TCGA cohort. Our results showed that the average beta values at the DMPs between 0.2 and 0.8 were 100% in our cohort and 91.6% in the TCGA cohort (Additional file 2: Fig S5).
Fig. 1.

A Hierarchical clustering of the samples based on the top-ranked DMPs (n = 156): Illustrative heat map denoting the Hierarchical Clustering of the PDAC samples (n = 7) based on the top-ranked DMPs (n = 156). Blue to red denoted increase in beta value (hyper-methylation). The cancer samples were marked as Blue and the normal samples were marked as Black (B) Genomic annotations of DMPs-CpG islands (Islands, Shores (± 2 KB from the boundaries of the islands), Shelves (± 2 KB from the boundaries of the shores) and Open Sea) or the transcription start site (5ʹ UTR, Exon 1, Promoter, Body, Non-genic). Distribution of the hypo-methylated and hyper-methylated DMPs across different segments of the genome. C Venn diagram showing common and uncommon DMPs in the TCGA cohort and Indian cohort. Among the 7832 DMPs, 68 DMPs were common to our study finding. D Correlation plot between the delta beta values at these 68 DMPs in TCGA data (for 9 PAAD patients) and in Indian cohort (n = 7)
Change of methylation status at the DMPs with PDAC cancer prognosis
Methylation marks in the DNA can be associated with the severity of cancer stages [7]. We hypothesised that differential methylation signatures at the DMPs were related to poor prognosis in PDAC. We have estimated the average beta value for each cancer stage in the TCGA cohort (stages IV, III, II and I) (Fig. 2). For the hypo-methylated DMPs, the cancer samples with stage I showed the highest average beta values and stage IV samples showed the lowest average beta values (Fig. 2A). Reversely, for the hyper-methylated DMPs, the highest levels of methylation were observed in cancer samples of stages IV and III compared to stage I and stage II (Fig. 2B).
Fig. 2.

Changes of methylation with increasing severity of cancer phenotypes in the TCGA cohort. A Changes in methylation at the hypo-methylated DMPs from Stage I to Stage IV of cancer in the TCGA cohort. B Changes in methylation at the hyper-methylated DMPs from mild to severe stages of cancer in the TCGA cohort. C Higher expression of hypo-methylated gDMs observed from Stage I to Stage IV cancer samples. D Changes in expression of hyper-methylated gDMs across stages of TCGA cohort. E Survival plots showing differences in disease prognosis among the TCGA cohort patients with high and low expressions of the gDMs
Gene methylation was often related inversely to gene expression [12]. We further compared the expressions of the gDMs between the TCGA cohort of PanCa samples and the GTEx cohort of normal pancreas tissues. Out of 91 gDMs, 41 gDMs showed significant differences in expression in the TCGA PanCa samples compared to the pure healthy pancreas tissues in the GTEx cohort (Additional file 1: Table S8). For the hypo-methylated gDMs, higher expressions were observed in the TCGA cancer samples compared to the GTEx cohort samples (Additional file 1: Table S8). The reverse scenario was observed for hyper-methylated gDMs, where PanCa samples in TCGA had lower expression than the normal pancreatic tissues in the GTEx cohort (Additional file 1: Table S8).
We have used GEPIA2 (http://gepia2.cancer-pku.cn/) to identify the significant changes in expression of the 41 gDMs in the TCGA-PDAC cohort. A total 17 genes among the 41 gDMs showed significant differences in expression across cancer stages in the TCGA cohort (Additional file 1: Table S5). Hypo-methylated gDMs like MX2, OAS2, SPRED2, ADAP1, and SLC17A9 genes showed significant increased expression in cancer stage IV compared to stage I (Fig. 2C). Although expressions of 9 hyper-methylated genes were significantly different across cancer stages in the TCGA cohort, reduced expressions with increased cancer severity were observed for 3 hyper-methylated genes- SLITRK3, FOXE1, and TWIST1 (Fig. 2D).
We have further investigated the survival of the patients with different levels of expressions of gDMs in the TCGA cohort, using GEPIA2 [19]. The survival of patients with higher expression of hypo-methylated gDMs was worse than those with low expression (APOL3, RASSF1, OAS2, and MX2). A reverse scenario was observed for hyper-methylated gDMs (CPEB1, KCNA3, FAIM2, and CACNB2) (Fig. 2E).
To further validate our findings of the association of 91 gDMs with PDAC, we searched PCMdb database [21]. Among the 91 gDMs, methylation data of 59 genes were retrieved from the database. For most of the hyper-methylated gDMs, we obtained concordant results from both the tissue samples and cell lines (Additional file 1: Tables S9, S14).
Relation between promoter methylation at DMPs with gene expression
Hyper-methylation at the promoter can cause DNA compaction, giving limited access to the binding sites of the transcription factors and vice versa. Consequently, promoter hyper-methylation can cause reduced gene expression and vice versa. In our study results, among the hypo-methylated DMPs and the hyper-methylated DMPs, 37% and 50% reside on the gene promoters (1500 bp upstream of transcription start site [TSS]), 5ʹUTR, respectively (Data previously shown). Aberrant methylation at the CpG islands and surrounding shores (± 2 kb from CpG islands) was found to be associated with tissue-specific and cancer-specific methylation signatures [15]. Among the hyper-methylated DMPs, 88% resided on the annotated CpG islands and 7% on the CpG shores. Comparatively, hypo-methylated DMPs were less common in the CpG islands (16%) and shores (21%) (Data previously shown).
When compared with the normal pancreas tissue (n = 171) from GTEx database (https://gtexportal.org/home/datasets) [16], we observed genes with promoter methylation to be differentially expressed in the PanCa samples of the TCGA cohort (Additional file 1: Table S8). Up-regulated expressions of the hypo-methylated gDMs in cancer were observed for OAS2, MX2, APOL3, ABHD8, RASSF1, SIGIRR, and SPRED2 (Additional file 2: Fig S6). For the hyper-methylated gDMs, down-regulated expression in cancer samples was observed for CACNB2 and ZIC1 (Additional file 2: Fig S6). To validate promoter associated methylation, MSP was done for 3 promoter CpG sites at 3 gDMs- RASSF1, SIGIRR and KCNA6 in validation cohort 2 (n = 22). Our results showed, hyper-methylation at KCNA6 promoter and hypo-methylation at RASSF1 and SIGIRR promoter in PDAC samples compared to the normal samples, in the validation cohort 2 (Fig. 3A). We further validated differential promoter methylation at FAIM2, NPY, and FOXE1in validation cohort 2 using MSP (Additional file 2: Method 3). Interestingly, data on MSP from the validation cohort showed an increased percentage of promoter methylation among the cancer samples compared to the normal samples (Fig. 3A, B). All detailed primer sequences were mentioned in section of Additional file 1: Table S9 and PCR methods were described in detail in Additional file 2: Method sections 3 and 4.
Fig. 3.

Agarose gel electrophoresis of methylation specific PCR products: In all subfigures first lane represents 100 bp ladder, next two lane methylated tumour and normal PCR products and proceeding two lanes are unmethylated tumour and normal PCR products. A Representative agarose gel images for the methylation status of pathway enriched loci namely KCNA6 (M:145 bp,U:147 bp), RASSF1 (M:193 bp,U:197 bp)and SIGIRR (M:192 bp,U:192 bp). B Representative agarose gel images for the differential promoter methylation status of FAIM2(M:278 bp,U:286 bp), NPY(M:264 bp,U:266 bp) and FOXE1(M:275 bp,U:277 bp). For each subset the same patient’s tumour and normal has been used. C Percentage of methylation in cancer and normal samples was shown in bar graph. Triple independent validation of the respective methylation status of above genes across all 22 (Adjacent tumour and normal) paired samples were done by Agarose gel electrophoresis of methylation specific PCR products. “M” represents methylated PCR product; “U” represents, unmethylated PCR products; “N” represents Adjacent Normal Sample; “T” represents Tumour sample
Furthermore, we consider the RASSF1, we observed a higher percentage of unmethylation than methylation signals in cancer. For SIGIRR, we observed, a reduced amount of methylation in cancer compared to the controls. Both observations suggested hypo-methylation of RASSF1 and SIGIRR in PDAC (Fig. 3C). For the remaining gDMs (NPY, FOXE1, FAIM2, and KCNA6), we found hyper-methylation in PDAC, as confirmed by the higher extent of methylation in cancer samples (Fig. 3C).
We validated differential expressions of 3 hypo-methylated gDMs (RASSF1, SIGIRR, and MX2) (Fig. 4A) and 3 hyper-methylated gDMs (KCNA6, GALR1, and IRF4) (Fig. 4B) in validation cohort 3 (n = 26). The extended promoter methylation in cancer samples of validation cohort 1 was observed. Using the distribution of beta values at all the CpG sites annotated to the gene promoters containing DMPs, we generated density plots that showed differences in average beta values in cancer compared to controls (difference in median |beta value|> = 0.1) at each CpG site in the promoters of six genes (FAIM2, NPY, GSC2, BHLHE23, SLITRK3 and FOXE1) (Fig. 4C, and Additional file 2: Fig S7). The expression levels of FAIM2, NPY, and FOXE1 were also compared in the cancer samples with normal samples in validation cohort 3 and a negative relationship between methylation and gene expression was observed (Fig. 4C, Additional file 1: Tables S10, S12).
Fig. 4.

Validation of differential expressions of gDMs in validation cohort 3 (n = 26). A Hypo-methylated gDMs. B Hyper-methylated gDMs. C gDMs with broad promoter methylation
Molecular significance of differential methylation in PDAC pathogenesis
The 156 DMPs identified in our study were annotated to 91 genes with differential methylation (gDMs) (Additional file 1: Table S5). Enrichment analysis with GO/KEGG terms, canonical pathways, hall mark gene sets, among the 91 genes identified “regulation of ion transport”, “interferon alfa/beta signalling”, “morphogenesis and development” and “transcriptional misregulation” as the 4 most statistically significant enriched terms (Fig. 5A). The genes included in each enriched term were listed in Additional file 1: Table S11. Voltage-gated ion channels are integral membrane proteins, which selectively transport ions and are activated upon a change of membrane potential. Channel activation enables transportation of potassium ions down their electrochemical gradient [26]. The low expression of Kv1.3 in PDAC can be explained by a hyper-methylation of the KCNA3 gene promoter. The ion channel regulation was seen to have a significant role in regulating cell apoptosis, evasion and survival along with invasion and progression in PDAC (Additional file 1: Tables S11, S13).
Fig. 5.

Functional annotations of gDMs. A Enrichment of biological processes among the 91 gDMs. B and C Network analysis with Metascape, where each enriched biological processwas coloured distinctly.Each term in the network is represented by a circle node, where its size is proportional to the number of input genes fall into that term, and its colour represent its cluster identity (i.e., nodes of the same color belong to the same cluster). Terms with a similarity score > 0.3 are linked by an edge (the thickness of the edge represents the similarity score). D MCODE networks showing interconnection between proteins. E Enrichment of transcription factors, known to regulate subsets of gDMs
Network analysis with selected enriched terms with a similarity score > 0.3 were linked with an edge and showed several interactions (Fig. 5B, C). Enriched terms with higher similarity, thicker were the defined edges (Fig. 5B). Stronger interactions were observed between—(1) “regulation of ion transport”, (2) “Interferon alfa/beta signalling”, (3) morphogenesis and development and (4) transcriptional misregulation in cancer. We next ran the MCODE algorithm in Metascape on this network to identify neighbourhoods, where proteins were found to be densely connected [20]. MCODE algorithm identifies two major clusters (Table 1)—MCODE1 (including HLA-A, MX2, IRF4, PML and OAS2) (Fig. 5D), which were mostly hypo-methylated in PanCa and MCODE2 (including ADCY3, NPY, GRM7, GALR1 and OPRK1) which were mostly hyper-methylated in PanCa (Fig. 5D). MCODE1 cluster proteins function in Interferon signalling whereas the MCODE2 cluster proteins function in cell-signalling (Table 1). Differential survival of genes annotated in MCODE1 (MX2,OAS2, IRF4, PML, and HLA-A) and MCODE2 (ADYC3, GALR1, NPY, and OPRK1), in the TCGA PanCa cohort further showed that patients with lower expressions of genes annotated to MCODE1 and higher expressions of genes annotated to MCODE2 showed better disease prognosis (Additional file 2: Fig S8).
Table 1.
Results of GO enrichment analysis on each MCODE network
| Network | Annotation | Genes |
|---|---|---|
| MCODE_1 | R-HSA-913531|Interferon Signaling|-10.8;R-HSA-909733|Interferon alpha/beta signaling|-9.8;R-HSA-877300|Interferon gamma signaling|-9.3 | IRF4, PML, MX2, OAS2, HLA-A |
| MCODE_2 | R-HSA-418594|G alpha (i) signalling events|-9.7;R-HSA-388396|GPCR downstream signalling|-8.3;R-HSA-372790|Signaling by GPCR|-8.0 | ADCY3, NPY, GALR1, OPRK1 |
Log (p-value) is mentioned with each annotation
Altered DNA methylation can affect the binding of the transcription factors (TFs), thus altering gene expressions [27]. We searched for common TFs that could regulate the 91 gDMs. Enrichment analysis showed enrichment of several TFs including IRF7, OCT1, CREBP, ETS1, ELF1 and NERF (− logP < − 3) (Fig. 5).
Assessment of methylation along with gene and protein expression of NPY and FAIM2 genes in PDAC
In regards to our all previous observations starting from cohort 1 till cohort 3 we speculate and might interpreted that the selection of two novel and significantly hyper-methylated genes in our Indian patient population for downstream further validation and reclaimed our prior observation using cohort 4, our Indian patient survival dataset and other global data bases based approaches. In cohort 1 that underwent 450 K methylation 6 genes were chosen out of which some genes underwent methylation and gene expression validation based on their survival information. These genes are not reported previously in TCGA which mostly contains Western world data and are reportedly novel also in terms of Asian (preferably Indian patient population) scenarios. Amongst these genes NPY and FAIM2 showed the highest alteration in differential gene expression. In terms of their methylation status by using MSP the gel images showed significant observation in case of both NPY and FAIM2. In addition to that, the changes in methylation were notably higher for these two genes in normal adjacent tissues with respect to their tumour counterparts. Furthermore, we validated our observations for both NPY and FAIM2 genes by using global databases based in silico approaches and its outcome was torchbearer of our assessment post which we further evaluated our observations in cohort 4.
The Neurotransmitter Neuropeptide Y (NPY) is involved in cell motion, invasion and cell proliferation and associated with multiple carcinomas. Hyper-methylation of NPY is also observed in certain carcinomas. Methylated NPY in circulating tumour DNA is currently a major focus for cancer biomarker detection in colorectal cancers [28, 29, 30, 31]. Fas Apoptotic Inhibitory Molecule 2 (FAIM2) also plays a significant role in apoptosis inhibition by inhibiting Fas/CD95-mediated apoptosis [32].
We observed significant hyper-methylation of NPYand FAIM2 both in the TCGA cohort (NPY:Δβ/p-value: 0.28/1.54E−05, FAIM2:Δβ/p-value: 0.33/8.23E−07) and validation cohorts (FAIM2: p-value-2.46E−06/ Δβ − 0.209; NPY: p-value-4.07E−07/Δβ − 0.205) (Additional file 1: Tables S14, S15, and data previously shown). We also observed that in validation cohort 3, down-regulated expressions of NPY gene in 86.9% patients whereas, FAIM2 gene in 82.6% of PDAC patient. Further assessment were also reinvestigated, patients showing lower expression had poor disease prognosis (log rank p = 0.01) than patients with higher expressions of NPY and FAIM2 in their tumours, which could be indirectly controlled by differential methylation (Fig. 6A). Negative correlation between the methylation and gene expressions of NPY (p-value: 0.00093, correlation coefficient = − 0.24) but not for FAIM2 (p-value: 0.82, R-value: − 0.017), in TCGA cohort was observed (Fig. 6B). The normal tissue samples in GTEx cohort had higher average expressions of both NPY and FAIM2 than in comparison with cancer samples in the TCGA cohort (Fig. 6C). We further data mined for methylation in NPY gene in the CCLE database, which included methylation data on 35 PanCacell lines. The results showed significant hyper-methylation in NPY gene in 35 PanCa cell lines (Additional file 2: Fig S9A). Significant correlation was observed between NPY and FAIM2 expressions (p-value-0.01, R-value-0.14) (Fig. 6D). The expression of the two above genes also showed association with progressive stages of PDAC (Fig. 6E).
Fig. 6.

Differential methylation and expressions of NPY and FAIM2 in the TCGA cohort. A Differential survival of patients in the TCGA cohort with “high” and “low” expressions of NPY and FAIM2. B Correlations between beta value and gene expression of NPY and FAIM2, among the cancer samples of the TCGA cohort. C Differences in expressions of NPY and FAIM2 genes between cancer and normal samples of the TCGA cohort. D Correlation between the expressions of FAIM2 and NPY genes in the TCGA cohort cancer samples. E Expression levels of FAIM2 and NPY genes across stages of cancer
Finally Pan-cancer analysis also showed down-regulation of the two genes in cancer. The expression level was also estimated by using the TCGA RNA seq PAAD dataset by using UALCAN (Additional file 2: Fig S9B).
Cross-validation of both FAIM2 and NPY hyper-methylation by IHC experiment in another validation set (clinical samples).
Using IHC method, in separate validation cohort 4, we further showed that the protein expressions of both Faim2 and Npy were lower in tumour tissues compared to adjacent normal counterpart sections. After assessments of all sections and stained with anti-Npy antibody it has been shown that 84% down-regulation in tumour tissues and 86% up-regulation in normal (Fig. 7A, B). Furthermore, we also observed that Faim2-expression was down-regulated in tumour tissues by 87% compared to 94% upregulation in adjacent normal part of normal tissues (Fig. 7C, D). The results were closely monitored in both 20× and 40× magnifications. Cell morphology was correlated with pathology slides of clinical samples for both tumour and normal sections (Fig. 7E, F). These results were in concordance with the data obtained from MSP and gene expression experiments previously. Identifying more specific PDAC markers that outperform the currently available markers would be very useful in establishing a diagnosis in patients with challenging histopathology. Both Faim2 and Npy protein expressions across all samples supported this motivation.
Fig. 7.
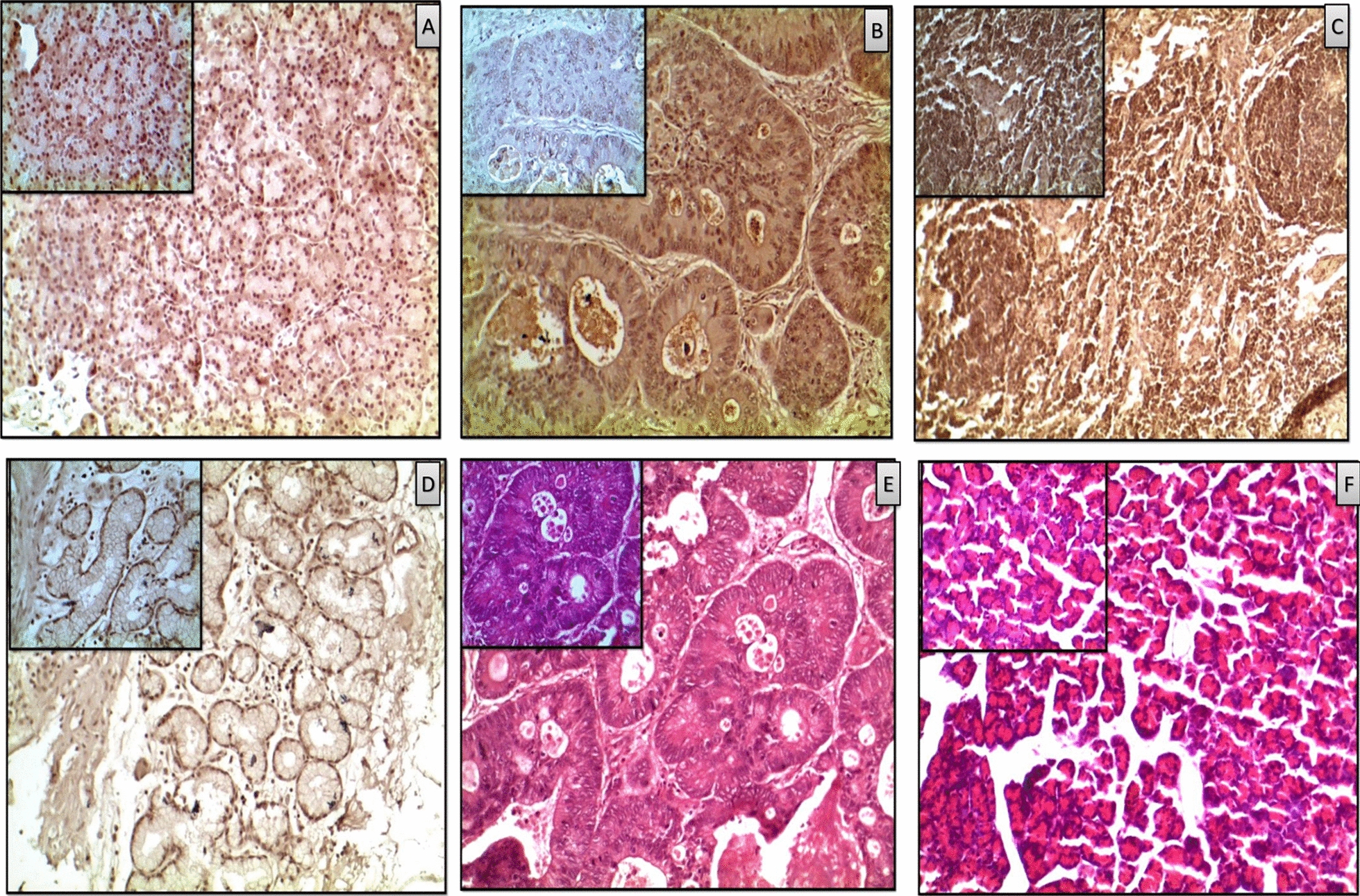
Representative images of immunohistochemistry (IHC) showing expressions of Faim2 and Npy proteins in PDAC and paired control tissues (n = 20): Antibody specific to Npy and Faim2, were used for comparison and images were clicked at both 20× and 40×. Every Slide consisted of 3 independent sections for IHC. The representation is expression of Npy in normal tissues (A) and in cancer (B). Expression of Faim2 in normal (C) and in cancer (D). A and C took more browner spotsin normal cells than (B) and (D) of the proteins in compared to cancer tissues. E and F H&E section of both tumour and normal were taken at similar resolutions. The bigger figures were taken in 20× and the sub-figures in smaller boxes were taken in 40×. Every sample had 3 sections on a slide with similar results to avoid technical biasness
In addition to that, in correlation to mRNA expression levels, protein levels of both Npy and Faim2 in normal pancreatic tissues were significantly higher than in PDAC tumour tissues, in the Clinical Proteomic Tumour Analysis Consortium (CPTAC) Confirmatory/Discovery dataset (Fig. 8A), further validating down-regulation in PDAC (Additional file 2: Fig S9B).
Fig. 8.

A Differential protein expressions of NPY and FAIM2 in the cancer samples compared to control samples in the TCGA cohort. B–D Overall survival analysis showing the difference in disease prognosis among patients in the validation cohort 3, with different levels of expressions of NPY and FAIM2
Association of NPY and FAIM2 expression with clinicopathological risk factors among PDAC patients
Cancer progression and its associated diagnosis and treatment are based on a plethora of parameters taking into consideration clinical, subclinical, and genomic parameters. Overall survival analysis of validation cohort 3 showed that joint down-regulation of NPY and FAIM2 is associated with poor prognosis (p-value: 0.065) (Fig. 8B). Additionally, patients with NPY down-regulation (p-value: 0.041) and high alcohol consumption had poor OS (p-value: 0.039) (Fig. 8C, D). Down-regulations of both FAIM2 and NPY were been strong negatively correlated with diabetes (R/p value: − 0.484/0.026). Dual down-regulation of FAIM2 and NPY has been observed to be correlated with KCNA6 down-regulation (R/p value: 0.592/0.016) and MX2 up-regulation (R/p value: 0.476/0.045). FAIM2 regulation also correlates with SLITRK3 (R/p value: 0.545/0.019). RASSF1A has a very strong correlation with the pancreatic lesion (R/p value: 1.00/0.00). Lastly, GALR1 has also been documented to multiple correlations with tumour stages, lymph node status and the expression levels of SLITRK3 and SIGIRR genes (Additional file 1: Tables S15, S16).
Classical Pearson’s correlation has been done along with “Kendalltau” correlation after considering the non-linearity of the subclinical and clinical data of patients. Starting from an ethnic point of view sex has a significant negative correlation with smoking as the abundance of smokers is higher in the Indian males than females (R/p value: − 0.419/0.037). Negative data correlation between sex and smoking has also been reported after doing “Kendalltau” correlation (p value 0.40). The same has been observed between alcohol and smoking (R/p value: 0.577/0.003). Considering the clinico-pathological parameters significant correlation has been documented between tumour stages with jaundice and lymph node (R/p value: 0.559/0.038). In our sample pool, a significant correlation has been observed between tumour stages (TNM grading) with lymph node metastasis (R/p value: 1.0/0.00).
Discussions
Globally, Pancreatic Cancer, especially ductal adenocarcinoma, is one of the most prevalent cancer types. Late diagnosis, treatment failure, and loco-regional recurrences contribute to its poor prognosis. Along with genetic alterations, studies in the past decade showed that epigenetic alterations contribute to PDAC development and pathogenesis [10, 11, 33, 34, 35]. Recent findings have shown abnormal DNA methylation can mark the spectrum of cancer progression, including the precancerous lesions, thus serving as biomarkers for diagnosis and prognosis [9, 36, 37, 38]. Thus, a specific abnormal methylation profile can serve as a biomarker for that cancer type [39]. Since methylation marks can be reversed to an unmethylated state, epigenetic marks can act as a lucrative therapeutic target.The effects of methylation on gene expression, can vary across different ethnic populations with varied risk factors [33]. In this study, we identified DNA methylation changes in 91 genes (gDMs) associated with PDAC in the Indian population. Among them, differential methylation in 47gDMs (Additional file 1: Table S6) in PDAC concordant with that in the TCGA PAAD cohort. The differential methylation marks were associated with progressive cancer stages and prognosis, both in our cohort and the TCGA cohort. Thus, our results showed both common and unique differential methylation marks in PDAC patients in the Indian cohort.
Promoter hypo-methylation was observed IRF4, PML, MX2, OAS2, HLA-A, and SIGIRR are involved in the interferon signalling pathway (MCODE1, Table 1). In our study, we have observed hypo-methylation of MCODE1, suggesting activation of immune pathways in PDAC. A recent study showed that over-expression of MX2 reduced cell proliferation, migration, and invasion via ERK/P38/NF-κB signalling pathway in glioblastoma cells [34]. Up-regulated expression of OAS2 was previously reported in PanCa [35]. Interferon induced cell killing is a potent anti-tumour immune response in cancer. Previously a study had shown that induction of Type I Interferon signalling made PanCa vulnerable to innate immune systems and showed better response with immune checkpoint therapy [36]. The interferon signalling pathway inhibits cell proliferation and cell migration in PanCa [37]. Up-regulated expression of these genes may thus signify the active anti-tumour immune response in PDAC.
Hyper-methylation and down-regulation of TSG, DNA repair genes are observed in tumours, which aid in cancer progression and metastasis [38]. In current study, we observed hyper-methylation of (1) TSGs including LOC645323, FOXE, and TCERG1L, (2) transcription regulator genes (BHLHE23, GSC2, FOXE1, HOXA10 and TWIST1), (3) Ion transporters—KCNA3, KCNA6, CACNB2 and (4) Immune regulators including HLA-A, IRF4. Previous studies have reported hyper-methylation of these transcription regulators in multiple cancers including PanCa [39, 40, 41] and reduced expression of these genes was associated with a worse prognosis of PAAD cancer in the TCGA cohort. FOXE1 is one of the most frequentlyhyper-methylated TSGs in PanCa [40, 41]. Hyper-methylation of another previously reported TSG TCERG1L was observed in PDAC compared to normal tissues [12, 39]. FOXE1, TWIST1 and other hyper-methylated genes involved in cell cycle regulation were observed in PDAC cancers, thus suggesting that their down-regulation may encourage uncontrolled cell proliferation in PDAC. Deregulations of ion transporters (Calcium, Potassium and Sodium) were observed in many cancers including PanCa’s and often represented as biomarkers [26]. Ion transporters can help in angiogenesis and cancer metastasis [42]. We observed hyper-methylation of potassium (KCNA3, and KCNA6) and calcium (CACNB2) ion channels in PDAC. Studies on animal models showed that epigenetic modification of KCNA3 gene limits T-cell activation [43, 44]. The Ca2+ and K+ ion channels thus can inhibit T-cell activation and proliferation upon antigen recognition by epigenetic modulation, creating an immunosuppressive tumour microenvironment in PDAC, which is again associated with poor prognosis and recurrence. Hyper-methylation of potassium channels may thus alter the immune context in PDAC and limit anti-tumour immunity [52, 57]. One of the recent finding documented epigenetic dysregulation (hyper-methylation) of Ca2+ ion transporters in PDAC [45]. Additionally, hyper-methylation of HLA-A also suggested suppression of antigen presentation and antitumor activity.
In this current study, we observed hyper-methylation of two genes—NPY and FAIM2 in Indian PanCa samples, which were also observed in the TCGA cohort and in the PanCa cell lines in the CCLE database.Hyper-methylation of both NPY and FAIM2 was correlated with reduced expression and was associated with poor survival, both in the TCGA and the Indian patient cohorts. The significant poorer survival also observed when FAIM2 and NPY act jointly. Our data strongly represents that the NPY and FAIM2 cumulatively contributed to significant poorer survival in the Indian Cohort. The survival curve using NPY and FAIM2 status couldn’t be derived from our validation cohorts based on the status that approximately > 80% of samples showed down-regulation of both the genes making the cohort not suitable for Kaplan–Meier survival analysis. In IHC study, validation cohort 4 we also reclaimed down-regulation of protein expression for both genes in PanCa samples with respect to normal counterparts. The result is consistent with the gene expression and promoter methylation. All our patient cohorts including discovery and validation cohorts have been revealed that hyper-methylation and down-regulation properties in mRNA and finally in protein levels of NPY and FAIM2. Considering CPTAC database it has been further proved that both Npy and Faim2 protein levels were also shown down-regulated in PanCa. All our data and from publicly available data sets strongly suggests and support that NPY and FAIM2 are the novel potential frequently hyper-methylated genes and trigger the PanCa progression and development in our patient cohort (Indian PanCa patient population). But surprisingly it has not been proven in other previously reported PanCa methylation studies around the globe. The cumulative down-regulation of NPY and FAIM2 also correlates with poor prognosis in the TCGA cohort data. Cumulatively, under expression of both FAIM2 and NPY have shown strong negative correlation between diabetes and hyper-methylation of both genes which are significant co-morbidity factors in the Indian patient population.
To our knowledge, hyper-methylation of FAIM2 was previously observed in ductal carcinoma of breast cancer [46], where the authors have shown an association of DNA methylation with progressive stages. Fas-apoptotic inhibitory molecule 2 is a member of the transmembrane BAX inhibitor motif-containing (TMBIM) family which comprised of 6 anti-apoptotic proteins. FAIM2 can suppress cell death by regulating Calcium ion homeostasis in the endoplasmic reticulum [32]. Thus hyper-methylation and downregulation of FAIM2 can help the tumour to evade apoptosis. However in current scenario, nothing is known about the effects of FAIM2 in PanCa.
The role of FAIM2 in obesity has already been documented which is a prime risk factor in PDAC development. It has also been reported by Kang et al. 2016, that FAIM2 acts as an novel biomarker in SCLC therefore emphasizing FAIM2's role as cancer biomarkers [47, 48, 49, 50]. Another hyper-methylated gene in PDAC was a NPY, which was included in MCODE2 (Table 1) along with ADCY3, GALR1, OPRK1. NPY stimulates cell proliferation and has been implicated as growth-promoting factor in various malignancies, but little is known about the effects of NPY on PanCa. NPY promoter showed frequently hyper-methylated in colorectal, and rectal cancers, surprisingly, in prostate and cholangiocarcinoma, NPY overexpression was observed but no correlation with hypo-methylation has been found [28, 29, 51, 52]. A physiological function of NPY is to regulate food intake and increase fat storage which is a risk factor for PanCa [28, 29, 51, 52, 53]. In addition, in our study showed that NPY downregulation (p-value: 0.041) and high alcohol consumption were associated with poor OS. All previous findings, including our data strongly support that in PDAC, hyper-methylation of NPY promoter might be correlated with inactivation of gene expression and might promote carcinogenesis.
Considering PDAC aggressive nature and its associated high mortality rate, the urgency of novel therapeutic strategies to combat is of priority. Advances in the methylation landscape are crucial to understand the development of epigenetically targeted biomarker. Our novel findings in this ongoing study can direct that a combination of epigenetic drugs along with existing targeted therapies will be a determinant targeted therapeutic approach that emphasizes the importance of our 450 K methylome study in PDAC patients across our nation (India) [54, 55, 56]. Epigenetic and genetic signatures of PDAC partially vary across the globe based on expression frequency and alternated driver behaviors. According to our previous study, Saha et al.,2020 [57] explained KRAS hotspot mutation was observed in low frequency in the Indian PDAC population whereas other parts of the world, like USA/Canada/European countries and Australia based studies documented KRAS as a high-frequency gene around 90% in the same disease. This partially variable profile may be contributed by the patient pool led by combined factors such as ethnicity, environmental, lifestyle and occupational risk factors [57]. Like the previous findings, it has also been observed in our methylome data. In our current study, correlation between sex, smoking and alcohol consumption has been documented which is specific to our demographic and ethnic beliefs. To gain a clear understanding of the epigenetic landscapes of PanCa, our study strongly suggests a novel architecture of epigenetic landmarks and insight into potential epigenetic outcome on PDAC in the Indian patient population.
Conclusion
The reversible nature of methylation signatures is a key to the development of markers targeting disease diagnosis and prognosis followed by therapeutic aids. Decoding the methylation landscape using 450 K methylome and parallel comparison with global datasets from TCGA makes our understanding even magnified and clinically more suitable for biomarker development. Two novel genes namely NPY and FAIM2 have been observed from our work to be frequently hyper-methylated significantly in all Indian cohorts and the result are in concordance with mRNA and protein expression, and protein databases making them translational importance. A statistically significant correlation with clinical and subclinical parameters also concludes to the fact that demographic and ethnic reasons do play a role in the epigenetic marker development. The methylation status of both NPY and FAIM2 and correlation with the TCGA validates our data against Western world data but the variance in the level of expression and methylation also concludes the demographic and ethnic inputs specific to our patient pool making it a significant study. This helps in gaining a clear understanding of the epigenetic landscapes of PanCa, as our study strongly suggests and support novel architecture of epigenetic landmarks including therapeutic strategies, and insight into potential epigenetic outcome on PDAC in the Indian patient population.
Future perspectives
The reversible role of DNA methylation can be a boon in the field of targeted therapeutic development and a prospective alternative to surgical resection. Considering the fact that PDAC is asymptomatic in nature and diagnosis happens mostly at late stages hence development of biomarkers for detection and targeted drug treatment is of great use. Further work using NPY and FAIM2 on a bigger sample size with uniform stage participants can unveil the role of the methylation based biomarkers in PDAC staging which can be useful for early detection. In regards to the role of both of the genes NPY and FAIM2, it can be assumed that they might have TSG like functional involvement. These hypotheses can be validated if knock-out experiments in PanCa cell lines stating a more functional aspect were to be done. Future studies enabling knockout of NPY and FAIM2 can also reveal crucial information’s. For easy detection the proteome data is of great use. The lack of availability of proteome data is a crucial limitation across the globe. Further proteomic analysis targeting the NPY and FAIM2 methylation status can provide very useful insight.
Limitations of the study
We declare two major limitations of this study. The sample size for 450 K analysis (validation cohort 1) was low (n = 7). Identification of differentially methylated positions in PDAC cancer was done using regression model, after adjusting for the effects of age and gender. Although we have validated our findings using publicly available datasets, literature searches, and through a respectively small validation cohorts, our findings need to be validated in a larger cohort. Secondly, due to insufficient sample availability, we could not validate the methylation marks and gene expression on the same samples.
Supplementary Information
Additional file 1.Details of the PAAD samples in the TCGA cohort (n=185). The red marked samples were excluded from the analysis. Highlighted samples wre used in discovery cohort
Additional file 2. DNA methylome in pancreatic cancer identified novel promoter hypermethylation in NPY and FAIM2 genes associated with poor prognosis in Indian patient cohort
Acknowledgements
Authors also acknowledged the National Institute of Biomedical Genomics (NIBMG) for instrumental support for performing experiments on an outsourcing basis under the Core Technologies Research Initiative (Co-TeRi) Initiative, National Institute of Biomedical Genomics (NIBMG), Kalyani, West Bengal carried out the Illumina 450 K bead chip experiment, directed by Prof. Arindam Maitra. Authors are also thankful to Dr. Dheeraj Anchlia, Dr. Sahid Khondaker, Dr. Abhisekh Mohata, Dr. Jitesh Midya, Dr. Vinu Shankar, Dr. Shuchismita Chakraborty, Dr. Debtanu Halder and Dr. Nilanjan Ghosh for collecting samples from respective hospitals. Authors specially thank Mr. Gourab Saha, Miss. Suchandra Pal and Mr. Sanjoy Kr. Dey from NS laboratory for collecting clinical specimens and DNA and RNA isolation. Authors are also thankful to Dr. Sudipto Saha, from Bose Institute (BI), Kolkata for support in IPA pathway analysis. Mr. Sandip Ghosh, and Dr. Biswarup Basu, from Chittaranjan National Cancer Research Institute (CNCRI), helped in IHC staining and scoring. The authors thank Dr. Meenakshi Munshi, from Department of Biotechnology, GOI for supporting and processing the fund. Associate Prof. Aniruddha Chatterjee would like to thank the Rutherford Discovery Fellowship (Royal Society of NZ) for supporting his current position. Authors are also thankful to Prof. Bidyut Roy for strategic input and proofreading of the manuscript. N.S. also thanks to Master. Saraswan Sikdar.
Abbreviations
- PDAC
Pancreatic ductal adenocarcinoma
- PanCa
Pancreatic cancer
- TSG
Tumour suppression gene
- DMP
Differentially methylated position
- IRB
Institutional Review Board
- PDA
Poorly differentiated adenocarcinoma
- MDA
Moderately differentiated adenocarcinoma
- WDA
Well differentiated adenocarcinoma
- AJCC
American Joint Committee on Cancer
- MSP
Methylation specific PCR
- MS-HRM
Methylation specific high resolution melting curve
- WGBS
Whole genome bisulfate sequencing
- TSG
Tumour suppressor gene
- TSS
Transcription start site
- DMR
Differentially methylated region
- TFs
Transcription factors
- gDMs
Genes with differential methylation
- PCMD
Pancreatic cancer methylation database
- CCLE
Cancer Cell Line Encyclopedia
- CPTAC
Clinical Proteomic Tumour Analysis Consortium
- NPY
Neurotransmitter Neuropeptide Y
- FAIM2
Fas apoptotic inhibitory molecule 2
- RASSF1
Ras Association Domain Family Member 1
- FOXE1
Forkhead Box E1
- SLITRK3
SLIT and NTRK Like Family Member 3
- SIGIRR
Single Ig and TIR Domain Containing
- KCNA6
Potassium Voltage-Gated Channel Subfamily A Member 6
- IRF4
Interferon Regulatory Factor 4
- MX2
MX Dynamin Like GTPase 2
- GALR1
Galanin Receptor 1
- OS
Overall survival
Author contributions
AnkC and AB both authors contributed equally in this manuscript work and preparation (*). AnkC, AB and DG carried out the gene expression profiling, methylation Specific PCR, and IHC validation experiments. The methylation data analysis and other data analysis were done by AnkC, AB, and AniC. All figures and tables were made by AnkC and AB. PKM checked and helped in statistical analysis part (experiments and written part). Clinically confirmed surgical/resected samples along with patient clinico-pathological and demographic data and histology reports for the study from different hospitals were provided by SumG, ShG, SupG, and PR gave input in histopathological findings and TNM staging for clinical samples. BKC and SB are clinical collaborators. P.B. helped in resources and for laboratory experiments. AnkC, AB, AniC and NS prepared the manuscript and did the interpretation of the findings. NS conceived, designed, edited, prepared and correspond the manuscript. All authors read and approved the final manuscript.
Funding
The study was supported by the Department of Biotechnology (DBT), Government of India (GOI) Grant Sanction Number: Ramalingaswami Re-entry fellowship (RLS/BT/Re-entry/05/2012).
Data availability statement
The original raw and cured datasets used and/or analysed during the current study are publicly available from Gene Expression Omnibus (GEO) database under the accession number GSE181740 (URL for data: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181740).
Declarations
Ethics approval and consent to participate
The authors sincerely thank the participants for their help and willingness to take part in this study. Patient samples were collected from two State Government Hospitals (Medical College & Hospital, Kolkata, SSKM & I.P.G.M.E.&R Hospital, Kolkata), Government of West Bengal, and two private hospitals Calcutta Medical Research Institute (CMRI), a C. K. Birla Hospital and Tata Medical Center (TMC), Rajarhat, Kolkata. The study was approved by the Institutional Review Board (IRB) of Indian Statistical Institute, Kolkata and also from all above mentioned Hospitals. The consents were taken from each patient and their families for this study.
Consent for publication
Not applicable.
Competing interests
The authors don’t have any competing interest to disclose.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
AnkC and AB both authors contributed equally in this manuscript work and preparation.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 3.Lucas AL, Malvezzi M, Carioli G, Negri E, La Vecchia C, Boffetta P, et al. Global trends in pancreatic cancer mortality from 1980 through 2013 and predictions for 2017. Clin Gastroenterol Hepatol. 2016;14:1452–1462.e4. doi: 10.1016/j.cgh.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee A, Rodger EJ, Eccles MR. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin Cancer Biol. 2018;51:149–159. doi: 10.1016/j.semcancer.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee A, Eccles MR. DNA methylation and epigenomics: new technologies and emerging concepts. Genome Biol. 2015;16:103. doi: 10.1186/s13059-015-0674-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lomberk G, Dusetti N, Iovanna J, Urrutia R. Emerging epigenomic landscapes of pancreatic cancer in the era of precision medicine. Nat Commun. 2019 doi: 10.1038/s41467-019-11812-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vrba L, Futscher BW, Oshiro M, Watts GS, Menashi E, Hu C, et al. Liquid biopsy, using a novel DNA methylation signature, distinguishes pancreatic adenocarcinoma from benign pancreatic disease. Clin Epigenet. 2022;14:1–6. doi: 10.1186/s13148-022-01246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henriksen SD, Thorlacius-Ussing O. Cell-free DNA methylation as blood-based biomarkers for pancreatic adenocarcinoma—a literature update. Epigenomes. 2021;5:8. doi: 10.3390/epigenomes5020008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bararia A, Dey S, Gulati S, Ghatak S, Ghosh S, Banerjee S, et al. Differential methylation landscape of pancreatic ductal adenocarcinoma and its precancerous lesions. Hepatobiliary Pancreat Dis Int. 2020;19:205–217. doi: 10.1016/j.hbpd.2020.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mishra NK, Guda C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget. 2017;8:28990–29012. doi: 10.18632/oncotarget.15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ansari D, Tingstedt B, Andersson B, Holmquist F, Sturesson C, Williamsson C, et al. Pancreatic cancer: Yesterday, today and tomorrow. Futur Oncol. 2016;12:1929–1946. doi: 10.2217/fon-2016-0010. [DOI] [PubMed] [Google Scholar]
- 14.Storz P, Crawford HC. Carcinogenesis of pancreatic ductal adenocarcinoma. Gastroenterology. 2020;158:2072–2081. doi: 10.1053/j.gastro.2020.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nones K, Waddell N, Song S, Patch AM, Miller D, Johns A, et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014;135:1110–1118. doi: 10.1002/ijc.28765. [DOI] [PubMed] [Google Scholar]
- 16.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin Y, Feng H, Chen M, Wu H, Zheng X. InfiniumPurify: an R package for estimating and accounting for tumor purity in cancer methylation research. Genes Dis. 2018;5:43–45. doi: 10.1016/j.gendis.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagpal G, Sharma M, Kumar S, Chaudhary K, Gupta S, Gautam A, et al. PCMdb: pancreatic cancer methylation database. Sci Rep. 2014;4:4197. doi: 10.1038/srep04197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Ge D, Lu C. The SMART App: an interactive web application for comprehensive DNA methylation analysis and visualization. Epigenet Chromatin. 2019;12:71. doi: 10.1186/s13072-019-0316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.R Core Team. R: a language and environment for statistical computing. R Found Stat Comput. 2021.
- 26.Anderson KJ, Cormier RT, Scott PM. Role of ion channels in gastrointestinal cancer. World J Gastroenterol. 2019;25:5732–5772. doi: 10.3748/wjg.v25.i38.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MSI, Kawaji H, et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics. 2014;15:119. doi: 10.1186/1471-2164-15-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Overs A, Flammang M, Hervouet E, Bermont L, Pretet JL, Christophe B, et al. The detection of specific hypermethylated WIF1 and NPY genes in circulating DNA by crystal digital PCRTM is a powerful new tool for colorectal cancer diagnosis and screening. BMC Cancer. 2021;21:1092. doi: 10.1186/s12885-021-08816-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen LH, Olesen R, Petersen LN, Boysen AK, Andersen RF, Lindebjerg J, et al. NPY gene methylation as a universal, longitudinal plasma marker for evaluating the clinical benefit from last-line treatment with regorafenib in metastatic colorectal cancer. Cancers (Basel) 2019;11:1649. doi: 10.3390/cancers11111649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai Y, Wei C, Zhong Y, Zhang Y, Long J, Huang S, et al. Development and validation of a prognostic nomogram for gastric cancer based on DNA methylation-driven differentially expressed genes. Int J Biol Sci. 2020;16:1153–1165. doi: 10.7150/ijbs.41587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Tian Y, Wu A. Neuropeptide Y receptors: a promising target for cancer imaging and therapy. Regen Biomater. 2015;2:215–219. doi: 10.1093/rb/rbv013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong CJ, Yeon J, Yeo BK, Woo H, An HK, Heo W, et al. Fas-apoptotic inhibitory molecule 2 localizes to the lysosome and facilitates autophagosome-lysosome fusion through the LC3 interaction region motif-dependent interaction with LC3. FASEB J. 2020;34:161–179. doi: 10.1096/fj.201901626R. [DOI] [PubMed] [Google Scholar]
- 33.Kader F, Ghai M. DNA methylation-based variation between human populations. Mol Genet Genomics. 2017;292:5–35. doi: 10.1007/s00438-016-1264-2. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Guan Q, Nan Y, Ma Q, Zhong Y. Overexpression of human MX2 gene suppresses cell proliferation, migration, and invasion via ERK/P38/NF-κB pathway in glioblastoma cells. J Cell Biochem. 2019;120:18762–18770. doi: 10.1002/jcb.29189. [DOI] [PubMed] [Google Scholar]
- 35.Kim JC, Ha YJ, Tak KH, Roh SA, Kwon YH, Kim CW, et al. Opposite functions of GSN and OAS2 on colorectal cancer metastasis, mediating perineural and lymphovascular invasion, respectively. PLoS ONE. 2018;13:e0202856. doi: 10.1371/journal.pone.0202856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (80−) 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujisawa M, Kanda T, Shibata T, Sasaki R, Masuzaki R, Matsumoto N, et al. Involvement of the interferon signaling pathways in pancreatic cancer cells. Anticancer Res. 2020;40:4445–4455. doi: 10.21873/anticanres.14449. [DOI] [PubMed] [Google Scholar]
- 38.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vincent A, Omura N, Hong SM, Jaffe A, Eshleman J, Goggins M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin Cancer Res. 2011;17:4341–4354. doi: 10.1158/1078-0432.CCR-10-3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinugawa Y, Uehara T, Sano K, Matsuda K, Maruyama Y, Kobayashi Y, et al. Methylation of tumor suppressor genes in autoimmune pancreatitis. Pancreas. 2017;46:614–618. doi: 10.1097/MPA.0000000000000804. [DOI] [PubMed] [Google Scholar]
- 41.Natale F, Vivo M, Falco G, Angrisano T. Deciphering DNA methylation signatures of pancreatic cancer and pancreatitis. Clin Epigenet. 2019 doi: 10.1186/s13148-019-0728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munaron L. Systems biology of ion channels and transporters in tumor angiogenesis: an omics view. Biochim Biophys Acta Biomembr. 2015;1848:2647–2656. doi: 10.1016/j.bbamem.2014.10.031. [DOI] [PubMed] [Google Scholar]
- 43.Kang JA, Park SH, Jeong SP, Han MH, Lee CR, Lee KM, et al. Epigenetic regulation of Kcna3-encoding Kv1.3 potassium channel by cereblon contributes to regulation of CD4+ T-cell activation. Proc Natl Acad Sci USA. 2016;113:8771–8776. doi: 10.1073/pnas.1502166113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel SH, Edwards MJ, Ahmad SA. Intracellular ion channels in pancreas cancer. Cell Physiol Biochem. 2019;53:44–51. doi: 10.33594/000000193. [DOI] [PubMed] [Google Scholar]
- 45.Gregório C, Soares-Lima SC, Alemar B, Recamonde-Mendoza M, Camuzi D, de Souza-Santos PT, et al. Calcium signaling alterations caused by epigenetic mechanisms in pancreatic cancer: From early markers to prognostic impact. Cancers (Basel) 2020;12:1–22. doi: 10.3390/cancers12071735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson KC, Koestler DC, Fleischer T, Chen P, Jenson EG, Marotti JD, et al. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. Clin Epigenet. 2015;7:75. doi: 10.1186/s13148-015-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu L, Zhao X, Shen Y, Zhang MX, Yan Y, Hou D, et al. Promoter methylation of fas apoptotic inhibitory molecule 2 gene is associated with obesity and dyslipidaemia in Chinese children. Diabetes Vasc Dis Res. 2015;12:217–220. doi: 10.1177/1479164114565630. [DOI] [PubMed] [Google Scholar]
- 48.Kang HC, Kim JI, Chang HK, Woodard G, Choi YS, Ku JL, et al. FAIM2, as a novel diagnostic maker and a potential therapeutic target for small-cell lung cancer and atypical carcinoid. Sci Rep. 2016;6:34022. doi: 10.1038/srep34022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Planells-Ferrer L, Urresti J, Coccia E, Galenkamp KMO, Calleja-Yagüe I, López-Soriano J, et al. Fas apoptosis inhibitory molecules: more than death-receptor antagonists in the nervous system. J Neurochem. 2016;139:11–21. doi: 10.1111/jnc.13729. [DOI] [PubMed] [Google Scholar]
- 50.She K, Yang W, Li M, Xiong W, Zhou M. FAIM2 promotes non-small cell lung cancer cell growth and bone metastasis by activating the Wnt/β-catenin pathway. Front Oncol. 2021;11:3373. doi: 10.3389/fonc.2021.690142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeMorrow S, Onori P, Venter J, Invernizzi P, Frampton G, White M, et al. Neuropeptide Y inhibits cholangiocarcinoma cell growth and invasion. Am J Physiol Cell Physiol. 2011;300:C1078–C1089. doi: 10.1152/ajpcell.00358.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alshalalfa M, Nguyen PL, Beltran H, Chen WS, Davicioni E, Zhao SG, et al. Transcriptomic and clinical characterization of neuropeptide Y expression in localized and metastatic prostate cancer: identification of novel prostate cancer subtype with clinical implications. Eur Urol Oncol. 2019;2:405–412. doi: 10.1016/j.euo.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Bijker MS, Herzog H. The neuropeptide Y system: pathophysiological and therapeutic implications in obesity and cancer. Pharmacol Ther. 2011;131:91–113. doi: 10.1016/j.pharmthera.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 54.Sikdar N, Saha G, Dutta A, Ghosh S, Shrikhande SV, Banerjee S. Genetic alterations of periampullary and pancreatic ductal adenocarcinoma: an overview. Curr Genomics. 2018;19:444–463. doi: 10.2174/1389202919666180221160753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 56.Lomberk GA, Iovanna J, Urrutia R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics. 2016;8:831–842. doi: 10.2217/epi-2015-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saha G, Singh R, Mandal A, Das S, Chattopadhyay E, Panja P, et al. A novel hotspot and rare somatic mutation p.A138V, at TP53 is associated with poor survival of pancreatic ductal and periampullary adenocarcinoma patients. Mol Med. 2020;26:59. doi: 10.1186/s10020-020-00183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1.Details of the PAAD samples in the TCGA cohort (n=185). The red marked samples were excluded from the analysis. Highlighted samples wre used in discovery cohort
Additional file 2. DNA methylome in pancreatic cancer identified novel promoter hypermethylation in NPY and FAIM2 genes associated with poor prognosis in Indian patient cohort
Data Availability Statement
The original raw and cured datasets used and/or analysed during the current study are publicly available from Gene Expression Omnibus (GEO) database under the accession number GSE181740 (URL for data: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181740).