

Abstract
Accumulating evidence indicates that mitochondrial dysfunction is involved in the pathogenesis of neurodegenerative diseases. Both of these conditions are often associated with an increase in protein aggregation. However, still unknown are the specific defects of mitochondrial biology that play a critical role in the development of Alzheimer’s disease, in which Tau protein aggregates are observed in the brains of some patients. Here, we report that long-term mitochondrial stress triggered Tau dimerization, which is the first step of protein aggregation. Mitochondrial dysfunction was induced in HEK293T cells that received prolonged treatment with rotenone and in HEK293T cells with the knockout of NDUFA11 protein. To monitor changes in Tau protein aggregation, we took advantage of the bimolecular fluorescence complementation assay using HEK293T cells that were transfected with plasmids that encoded Tau. Inhibition of the ISR with ISRIB induced Tau dimerization, whereas ISR activation with salubrinal, guanabenz, and sephin1 partially reversed this process. Cells that were treated with ROS scavengers, N-acetyl-l-cysteine or MitoQ, significantly reduced the amount of ROS and Tau dimerization, indicating the involvement of oxidative stress in Tau aggregation. Our results indicate that long-term mitochondrial stress may induce early steps of Tau protein aggregation by affecting oxidative balance and cellular proteostasis.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive brain disorder and the most common form of dementia (Weidling and Swerdlow, 2020). Many reports indicate a decrease in mitochondrial respiration and changes in mitochondrial morphology that are associated with AD (Cabral-Costa and Kowaltowski, 2020). However, still mostly unknown is whether mitochondrial dysfunction can trigger these cellular adaptive responses and affect signaling pathways, which are involved in protein aggregation and other defects of protein homeostasis that are observed in neurodegenerative diseases.
Mitochondria are nowadays recognized as very important signaling organelles. Under stress conditions, they can signal their state to other organelles in the cell (Nunnari and Suomalainen, 2012; Chandel, 2014), also via increased production of reactive oxygen species (ROS) (Reczek and Chandel, 2015; Samluk et al., 2018, 2019; Topf et al., 2018). This allows the adjustment of cellular protein homeostasis according to mitochondrial activity and enables cell survival, especially because almost all mitochondrial proteins are encoded by nuclear genes (Mohanraj et al., 2020). Mitochondria under stress conditions can also inhibit cellular protein synthesis, which was shown to reduce mitochondrial degeneration (Wang et al., 2008). One pathway that is involved in regulating protein synthesis in the cell is the integrated stress response (ISR) (Harding et al., 2003; Pakos-Zebrucka et al., 2016). The ISR can decrease the efficiency of cap-dependent protein synthesis and induce the cap-independent translation of selected mRNAs via the phosphorylation of its core component, eukaryotic translation initiation factor 2α (eIF2α). It was reported that dysfunctional mitochondria may induce the phosphorylation of eIF2α in Caenorhabditis elegans, which was required for extending the lifespan of C. elegans in the presence of mitochondrial stress, suggesting its protective role (Baker et al., 2012). However, in Drosophila melanogaster, eIF2α phosphorylation that was induced by mitochondrial dysfunction caused selective dendritic loss of class IV dendritic arborization neurons (Tsuyama et al., 2017). A long-lasting reduction of protein synthesis that is induced by prolonged mitochondrial stress can be harmful to the cell. Contrary to short-term mitochondrial stress, long-term mitochondrial dysfunction is leading to a decrease in eIF2α phosphorylation in human cells (Samluk et al., 2019). This adaptive response enables sufficient protein synthesis for the cells to survive under stress conditions but may also have pathological consequences, such as protein aggregation.
Tau is a protein that is present almost exclusively in neurons and associated with microtubules. Pathological conditions induce Tau dissociation from microtubules. This is believed to cause microtubule destabilization and the formation of Tau insoluble aggregates called neurofibrillary tangles (NFTs), resulting in neuronal degeneration. Tau aggregation is often associated with an increase in Tau phosphorylation, which is a notable characteristic of several neurodegenerative disorders, including AD (Spillantini and Goedert, 2013). Mitochondrial dysfunction was also shown to induce Tau protein hyperphosphorylation and neurodegeneration (Merkwirth et al., 2012; Kondadi et al., 2014). Interestingly, the treatment of neurons with antioxidants, such as N-acetyl-l-cysteine (NAC) or vitamin E, reduced phosphorylated Tau levels and rescued anterograde transport defects under mitochondrial stress (Kondadi et al., 2014). Strikingly, Tau protein, which also facilitates efficient axonal transport in neurons, was hyperphosphorylated and aggregated in mitochondrial prohibitin-2 (PHB2)-deficient neurons (Merkwirth et al., 2012). This was the first genetic evidence that mitochondrial dysfunction can trigger Tau hyperphosphorylation and aggregation, but the precise mechanism is still unknown.
Accumulating evidence demonstrates the involvement of mitochondrial dysfunction in the pathogenesis of neurodegenerative diseases, including AD (Sorrentino et al., 2017). However, many of these studies focused on mitochondrial defects themselves. The present study focused on the very early steps of the Tau protein aggregation process that involved the dysfunction of mitochondrial respiratory chain complex I. We investigated the contribution of mitochondrial defects (e.g., increase in ROS production) in Tau protein aggregation. Moreover, we tested a new concept of the involvement of adaptive cellular responses that are triggered by long-term mitochondrial stress, such as ISR inhibition, in this process. We induced long-term mitochondrial stress in HEK293T and SH-SY5Y cells that were transiently transfected with plasmids that encoded Tau to test whether Tau protein aggregation increases under these conditions. We took advantage of the bimolecular fluorescence complementation (BiFC) assay to monitor early steps of Tau aggregation, dimerization, and oligomerization. Tau dimerization was significantly increased in SH-SY5Y cells that were treated with rotenone and HEK293T cells under conditions of mitochondrial stress that was induced by NADH:ubiquinone oxidoreductase subunit A11 (NDUFA11) protein knockout or prolonged treatment with rotenone. Surprisingly, the increase in Tau phosphorylation did not appear to be directly involved in the aggregation process. Previously it was shown that ISR was inhibited under conditions of long-term mitochondrial stress (Samluk et al., 2019). To reverse the increase in Tau dimerization in HEK293T cells, we activated the ISR with blockers of eIF2α dephosphorylation (i.e., salubrinal, guanabenz, and sephin1), but the effect was moderate. Under conditions of induced mitochondrial stress, we observed inhibition of Tau dimerization when the cells were pretreated with ROS scavengers, NAC or mitochondria-targeted MitoQ. Overall, we propose that early steps of Tau protein aggregation under conditions of long-term mitochondrial stress are triggered by oxidative imbalance and the disruption of cellular proteostasis.
RESULTS
Long-term mitochondrial stress induces early steps of Tau aggregation
In our previous study, we found that long-term mitochondrial stress induced cytosolic adaptive responses, such as inhibition of the ISR, that ensured cell survival but affected protein homeostasis (Samluk et al., 2019). These stress conditions may increase protein aggregation, which is a hallmark of neurodegeneration. Tau protein aggregates are observed in the brains of some AD patients. To determine whether mitochondrial dysfunction leads to an increase in Tau protein dimerization (i.e., the first step of its aggregation) (Meraz-Rios et al., 2010), we performed the BiFC assay (Figure 1A) (Tak et al., 2013; Lim et al., 2014; Blum et al., 2015). To model long-term mitochondrial stress, we used HEK293T cells that lacked the expression of NDUFA11, an accessory subunit of mitochondrial complex I (NADH:ubiquinone oxidoreductase) (Stroud et al., 2016), and HEK293T cells that were treated for 68 h with rotenone, an inhibitor of mitochondrial respiratory chain complex I. We demonstrated that the NDUFA11 knockout in HEK293T cells caused a significant loss of mitochondrial complex I activity, which reflects this enzyme assembly deficiency (Supplemental Figure 1) (Stroud et al., 2016). HEK293T cells, which do not express Tau endogenously, were transfected for 72 h with a pair of BiFC plasmids that encoded Tau protein that was fused to the N-terminal part of Venus protein (Tau-VN) and Tau protein that was fused to the C-terminal part of Venus protein (Tau-VC). The close proximity of two parts of Venus, meaning the dimerization of Tau, enabled the reconstitution of functional fluorescent Venus protein (Figure 1A). We positively validated the BiFC assay by HEK293T cell treatment with okadaic acid, which was previously shown to increase Tau dimerization (Supplemental Figure 2) (Tak et al., 2013). The results of the BiFC assay showed an increase in the fluorescence of Venus protein in HEK293T NDUFA11 knockout cells and HEK293T cells after 68 h of rotenone treatment (100 and 200 nM) (Figure 1, B and D). We observed the lower expression of Tau in NDUFA11-deficient cells than in HEK293T wild-type cells (Figure 1B), most likely because of a reduction of global protein synthesis in this cell line, which we observed in our previous study (Samluk et al., 2019). To avoid the potential influence of unequal cell transfection or Tau expression, the results of the fluorescence measurements were normalized to levels of Tau expression, which was determined by immunoblotting (Figure 1, B and D). Notably, although cells were transfected with two BiFC plasmids that encoded Tau, both Tau-Venus proteins migrated in the polyacrylamide gel as one band (Figure 1, B and D). The increase in Venus fluorescence in cells that were treated with rotenone was concentration dependent (Figure 1D). The increase in fluorescence was around 1.5-fold after cells were treated with 100 nM rotenone, similar to the increase in fluorescence in HEK293T NDUFA11 knockout cells compared with wild-type cells. HEK293T cells that were treated with 200 nM rotenone exhibited twofold higher fluorescence of Venus compared with control cells, suggesting that not only the type but also the strength of mitochondrial stress affects Tau dimerization. Additionally, SH-SY5Y human neuroblastoma cells that were transfected with BiFC plasmids and treated with 2 µM rotenone for 24 h exhibited an increased Venus fluorescence, suggesting that Tau dimerization may take place also in neuronal-like cells under mitochondrial stress (Supplemental Figure 3). Moreover, confocal imaging revealed an increase in fluorescence after the BiFC plasmid transfection of HEK293T NDUFA11-deficient cells and HEK293T cells that were treated with 100 nM rotenone for 68 h, confirming the increase in Tau dimerization under conditions of mitochondrial stress (Figure 1, C and E). We also noticed a negligible baseline level of fluorescence under control conditions, suggesting that most Tau molecules exist in the cytoplasm as monomers under nonstress conditions (Figure 1, C and E) (Tak et al., 2013; Lim et al., 2014). Furthermore, it is noteworthy that in HEK293T cells that were treated for 68 h with 2.5 µM antimycin A, an inhibitor of mitochondrial complex III, Tau dimerization was only moderately increased (Supplemental Figure 4). All these results demonstrate that mitochondrial respiratory chain complex I dysfunction leads to an increase in Tau protein aggregation.
FIGURE 1:

Knockout of NDUFA11 in HEK293T cells and treatment with rotenone in HEK293T cells induced early steps of Tau aggregation. (A) Schematic illustration of BiFC assay (yellow fluorescent protein [YFP] turn-on sensor). Cells were transfected for 72 h with plasmids that encoded Tau protein that was fused with the N-terminal part of Venus protein (Tau-VN) and Tau protein that was fused with the C-terminal part of Venus protein (Tau-VC). The dimerization and oligomerization of Tau protein enabled the reconstitution of functional Venus protein, resulting in an increase in fluorescence. (B) Fold change in Venus fluorescence normalized to the level of Tau expression in HEK293T wild-type (WT) and HEK293T NDUFA11 knockout cells. The data are expressed as mean ± SEM. n = 12. (C) Increase in Venus fluorescence in HEK293T NDUFA11 knockout cells. Nuclei were stained with DAPI. Scale bar = 50 μm. The graph shows the quantification of fold changes in mean Venus fluorescence per cell in the confocal microscopy experiments. n = 3. (D) Fold change in Venus fluorescence normalized to the level of Tau expression in HEK293T control (Control) cells and HEK293T cells that were treated with rotenone for 68 h as indicated. The data are expressed as mean ± SEM. n = 3. (E) Increase in Venus fluorescence in HEK293T cells that were treated with rotenone for 68 h as indicated. Nuclei were stained with DAPI. Scale bar = 50 μm. The graph shows the quantification of fold changes in mean Venus fluorescence per cell in the confocal microscopy experiments. n = 3. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test or one-way ANOVA followed by Dunnett’s multiple comparisons test in D).
Tau dimerization increases independently of its phosphorylation under conditions of long-term mitochondrial stress
Previous studies demonstrated that mitochondrial dysfunction leads to an increase in Tau protein phosphorylation (Merkwirth et al., 2012; Kondadi et al., 2014). Moreover, an increase in Tau protein phosphorylation was shown to be involved in its increase in aggregation (Despres et al., 2017). Tau protein phosphorylation status depends on the activity of PP2A phosphatase (Sontag and Sontag, 2014), among other factors. We previously found a decrease in mRNA levels of catalytic and regulatory subunits of PP2A in HEK293T NDUFA11-deficient cells (Samluk et al., 2019). Therefore, we evaluated the activity of PP2A and Tau protein phosphorylation in this cell line. As shown in Figure 2, the activity of PP2A decreased, as assessed by its ability to dephosphorylate the phosphopeptide K-R-pT-I-R-R using the malachite green phosphatase assay (Figure 2A). Tau protein phosphorylation (Ser-202, Thr-205) increased in HEK293T NDUFA11 knockout cells, detected by immunoblotting (Figure 2B). The phosphorylation of Tau residues Ser-202/Thr-205 is a very well-characterized feature of AD (Neddens et al., 2018). To determine whether Tau protein phosphorylation impacts its aggregation under conditions of mitochondrial stress, we transfected cells with BiFC plasmids that encoded Tau protein that could not be phosphorylated, in which all serine and threonine phosphorylation sites were mutated to alanine (Figure 2, C and D). We found that these Tau Ser/Thr phosphomutants (Tau-VN-AP and Tau-VC-AP) were not detected by anti–phospho-Tau antibody, which recognizes Tau phosphorylation at Ser-202 and Thr-205, thus confirming the mutagenesis of Tau serine and threonine phosphorylation sites at BiFC vectors (Figure 2D). In contrast, wild-type Tau phosphorylation was detected using anti–phospho-Tau antibody (Ser-202, Thr-205) and enhanced in HEK293T NDUFA11-deficient cells compared with HEK293T wild-type cells (Figure 2D). Interestingly, we observed that Tau-Venus proteins that were encoded by BiFC vectors with mutated phosphorylation sites migrated slightly differently in SDS–PAGE than their unmutated forms (Figure 2, D and E). We noticed a shift in the electrophoretic mobility of both Tau Ser/Thr phosphomutants (Tau-VN-AP and Tau-VC-AP). In contrast to cells that were transfected with a pair of BiFC plasmids that encoded wild-type Tau (Tau-VN and Tau-VC), two separate Tau bands were detected by immunoblotting (Figure 2, D and E). This phenomenon is well known. Many proteins exhibit a phosphorylation-dependent electrophoretic mobility shift that is caused by the distribution of negatively charged amino acids around the phosphorylation site and changes in the binding of SDS to proteins (Lee et al., 2019). We next performed a BiFC assay using pairs of BiFC plasmids that encoded wild-type Tau (Tau-VN and Tau-VC) or mutated Tau (Tau-VN-AP and Tau-VC-AP) and compared relative fluorescence between the experiments (Figure 2, C and E). Surprisingly, the increases in Tau dimerization were comparable in both cases, suggesting that the increase in phosphorylation at sites that were mutated is not directly needed for early steps of Tau aggregation, at least under conditions of long-term mitochondrial stress (Figure 2E). These results indicated that the dysfunction of mitochondrial respiratory chain complex I resulted in a decrease in the activity of Tau protein phosphatase (PP2A) and an increase in Tau protein phosphorylation, but this was not a prerequisite for Tau dimerization under conditions of mitochondrial stress.
FIGURE 2:

Mutation of phosphorylation sites did not reduce Tau dimerization under conditions of long-term mitochondrial stress. (A) PP2A phosphatase activity measured with the malachite green phosphatase assay in HEK293T wild-type cells and HEK293T NDUFA11 knockout cells. The data are expressed as mean ± SEM. n = 3. (B) Phosphorylation of Tau protein determined by immunoblotting with phospho-Tau (Ser-202, Thr-205) antibody in HEK293T wild-type cells and HEK293T NDUFA11 knockout cells. The data are expressed as mean ± SEM. n = 3. (C) Schematic illustration of BiFC assay. Cells were transfected for 72 h with a pair of BiFC vectors that encoded wild-type (WT) Tau (Tau-VN and Tau-VC) or a pair of BiFC vectors that encoded mutated Tau (Tau Ser/Thr phosphomutants; Tau-VN-AP and Tau-VC-AP). Tau was mutated to alanine in all 14 S/P or T/P amino acid residues (T111, T153, T175, T181, S199, S202, T205, T212, T217, T231, S235, S396, S404, and S422; numbering based on the longest 441-amino-acid brain isoform of hTau). BiFC plasmids encoded Tau protein that was fused with the N-terminal part of Venus protein (Tau-VN, Tau-VN-AP) or Tau protein that was fused with the C-terminal part of Venus protein (Tau-VC, Tau-VC-AP). The dimerization and oligomerization of Tau protein enabled the reconstitution of functional Venus protein, resulting in an increase in fluorescence. (D) Phosphorylation of Tau protein determined by immunoblotting with phospho-Tau (Ser-202, Thr-205) antibody in HEK293T wild-type cells and HEK293T NDUFA11 knockout cells. Cells were transfected with BiFC vectors that encoded wild-type (WT) Tau (Tau-VN and Tau-VC) or BiFC vectors that encoded Tau, in which all 14 S/P or T/P amino acid residues (T111, T153, T175, T181, S199, S202, T205, T212, T217, T231, S235, S396, S404, and S422; numbering based on the longest 441-amino-acid brain isoform of hTau) were mutated to alanine (Tau Ser/Thr phosphomutants; Tau-VN-AP and Tau-VC-AP). n = 3. (E) Fold change in Venus fluorescence normalized to the level of Tau expression in HEK293T wild-type (WT) cells and HEK293T NDUFA11 knockout (KO) cells. Cells were transfected with BiFC vectors that encoded wild-type Tau (Tau-VN and Tau-VC) or BiFC vectors that encoded Tau, in which all 14 S/P or T/P amino acid residues (T111, T153, T175, T181, S199, S202, T205, T212, T217, T231, S235, S396, S404, and S422; numbering based on the longest 441-amino-acid brain isoform of hTau) were mutated to alanine (Tau-VN-AP and Tau-VC-AP). The data are expressed as mean ± SEM. n = 4. *p < 0.05, **p < 0.01; ns, not significant (p > 0.05) (Student’s t test or one-way ANOVA followed by Tukey’s multiple comparisons test in E).
Inhibition of ISR induces Tau dimerization
We did not observe a direct impact of an increase in Tau phosphorylation on early steps of Tau aggregation under conditions of long-term mitochondrial stress. Thus, we investigated the involvement of the ISR in this pathological process. We confirmed observations from our previous study, in which long-term mitochondrial stress led to a decrease in the phosphorylation of eIF2α, which is a key player in the ISR (Figures 3, A and B, and 4A). eIF2α phosphorylation was decreased in HEK293T NDUFA11-deficient cells and HEK293T cells that were treated with rotenone for 48 h in a dose-dependent manner (Figure 3, A and B). Higher rotenone concentrations resulted in a higher reduction of eIF2α phosphorylation (Figure 3B), suggesting that the intensity of mitochondrial stress is an important factor for this process. Under stress conditions, an increase in eIF2α phosphorylation led to the global inhibition of translation (Figure 4A), which is beneficial for cells under short-term stress conditions, but protein synthesis inhibition for too long can induce cellular death. We conclude that the reduction of eIF2α phosphorylation under conditions of long-term mitochondrial stress allows cells to maintain protein synthesis at a sufficient level for survival. However, inhibition of the ISR that leads to the stimulation of protein synthesis under conditions of long-term mitochondrial stress in an inefficient protein-folding environment may have pathological consequences for cellular proteostasis, such as an increase in protein aggregation. This possibility was supported by a study in which inhibition of the ISR was suggested to be involved in β-amyloid aggregation in mammalian cells (Sorrentino et al., 2017). To determine whether the decrease in the activity of the ISR leads to an increase in the aggregation of Tau protein, we performed the BiFC assay in HEK293T cells that were transfected for 72 h with Tau-BiFC plasmids and treated for 68 h with ISR inhibitor (ISRIB) (Figure 4A). ISRIB reduced the expression of activating transcription factor 4 (ATF4) (Figure 3C), a central effector of the ISR (Harding et al., 2003; Pakos-Zebrucka et al., 2016). Interestingly, a few studies showed that ATF4 is a major player in the mitochondrial stress response in mammalian cells (Khan et al., 2017; Kuhl et al., 2017; Quiros et al., 2017). As shown in Figure 3, C and D, HEK293T cells that were treated with ISRIB for 68 h exhibited an increase in Venus protein fluorescence, indicating higher Tau protein dimerization. These results confirmed that attenuation of the ISR, which also occurred under conditions of long-term mitochondrial stress, induced Tau aggregation.
FIGURE 3:

HEK293T cells that were treated with ISRIB exhibited Tau dimerization. (A) Phosphorylation of eIF2α (Ser-51) in HEK293T wild-type cells and HEK293T NDUFA11 knockout cells. The data are expressed as mean ± SEM. n = 3. (B) Phosphorylation of eIF2α (Ser-51) in HEK293T cells that were treated for 48 h with rotenone. The data are expressed as mean ± SEM. n = 3. (C) Fold change in Venus fluorescence normalized to the level of Tau expression in HEK293T control cells and HEK293T cells that were treated with ISRIB for 68 h as indicated. The data are expressed as mean ± SEM. n = 5. (D) Increase in Venus fluorescence in HEK293T cells that were treated with ISRIB for 68 h as indicated. Nuclei were stained with DAPI. Scale bar = 50 μm. The graph shows the quantification of fold changes in mean Venus fluorescence per cell in the confocal microscopy experiments. n = 3. Rot., rotenone. *p < 0.05, ***p < 0.001; ns, not significant (p > 0.05) (Student’s t test or one-way ANOVA followed by Dunnett’s multiple comparisons test in B).
FIGURE 4:

Inhibition of PP1 phosphatase subunits increased eIF2α phosphorylation in HEK293T NDUFA11 knockout cells. (A) Schematic diagram of ISR signaling. Various stress conditions can activate kinases, resulting in the phosphorylation of eIF2α. The activation of eIF2α leads to the global attenuation of cap-dependent translation and induction of selected genes (e.g., via ATF4 transcription factor). ISR signaling can be modulated by specific compounds that block ATF4 expression (e.g., ISRIB), inhibitors of PP1 subunits that block GADD34 (e.g., sephin1 and guanabenz), or inhibitors of CReP and GADD34 (e.g., salubrinal). CReP is constitutively expressed, and GADD34 is a stress-inducible PP1 phosphatase subunit that dephosphorylates eIF2α. (B) GADD34 mRNA levels in HEK293T wild-type cells and HEK293T NDUFA11 knockout cells. The data are expressed as mean ± SEM. n = 3. (C–E) Fold change in eIF2α phosphorylation in HEK293T NDUFA11 knockout cells that were treated for 24 h with salubrinal (C), guanabenz (D), or sephin1 (E) as indicated. The data are expressed as mean ± SEM. n = 5 (C); n = 6 (D and E). *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test).
Activation of the ISR in cells under conditions of long-term mitochondrial stress partially reverses Tau dimerization
We then investigated whether activation of the ISR under conditions of long-term mitochondrial stress reduces Tau dimerization. Our previous study found that the ISR activator tunicamycin did not induce the strong induction of eIF2α phosphorylation (Ser-51) upon 72 h of the small interfering RNA–mediated knockdown of mitochondrial intermembrane space import and assembly protein 40 (MIA40) in HeLa and HEK293 cells (Samluk et al., 2019). MIA40 is a mitochondrial oxidoreductase that is responsible for the import of proteins into the intermembrane space of mitochondria (Chacinska et al., 2004). Tunicamycin activates the ISR and protein kinase R (PKR)-like endoplasmic reticulum (ER) kinase (PERK) by inducing ER stress in cells by inhibiting the first step of the biosynthesis of N-linked glycans in proteins, resulting in many misfolded proteins (Harding et al., 2005; Guha et al., 2017). In the present study, we used more-specific ISR activators, namely blockers of eIF2α dephosphorylation (salubrinal, guanabenz, and sephin1). Protein phosphatase 1 (PP1) contains two subunits that dephosphorylate eIF2α, stress-inducible growth arrest and DNA damage–inducible protein 34 (GADD34) (PPP1R15A) and constitutively expressed repressor of eIF2α phosphorylation (CReP) (PPP1R15B). Sephin1 and guanabenz inhibited GADD34, and salubrinal inhibited both CReP and GADD34 (Figure 4A). Interestingly, we observed higher levels of mRNA for GADD34 in NDUFA11-deficient cells (Figure 4B). In our previous study, we found an increase in GADD34 protein levels in HEK293 cells that were treated with rotenone for 48 h (Samluk et al., 2019). HEK293T NDUFA11 knockout cells that were treated for 24 h with maximal nontoxic concentrations of inhibitors of PP1 subunits exhibited a significant increase in eIF2α phosphorylation (Figure 4, C–E), demonstrating that inhibition of the ISR under conditions of long-term mitochondrial stress could be overcome, at least to some extent. Next, to evaluate the influence of PP1 inhibitors on Tau dimerization, we performed the BiFC assay by transfecting HEK293T NDUFA11 knockout cells for 72 h with BiFC plasmids that encoded Tau protein that was fused with different parts of fluorescent Venus protein. Cells were treated for the last 24 h with salubrinal, guanabenz, or sephin1, followed by measurements of Venus fluorescence. Tau dimerization, which was revealed by changes in fluorescence, was partially decreased (Figure 5, A–C), and this reduction correlated with the increase in eIF2α phosphorylation that was induced by salubrinal, guanabenz, and sephin1 (Figure 4, C–E). Although salubrinal, guanabenz, and sephin1 can reduce Tau aggregation only partially, these results suggest the involvement of more-significant mechanisms of Tau dimerization under conditions of long-term mitochondrial stress beyond inhibition of the ISR.
FIGURE 5:

Treatment of HEK293T NDUFA11 knockout cells with salubrinal, guanabenz, or sephin1 partly reversed Tau dimerization. (A–C) Fold change in Venus fluorescence normalized to the level of Tau expression in untreated HEK293T NDUFA11 knockout cells and HEK293T NDUFA11 knockout cells that were treated for 24 h with salubrinal (A), guanabenz (B), or sephin1 (C) as indicated. The data are expressed as mean ± SEM. n = 4 (A and C); n = 3. (B) Sal., salubrinal; Gua., guanabenz; Seph.1, sephin1. *p < 0.05, **p < 0.01; ns, not significant (p > 0.05) (Student’s t test).
Scavenging of ROS reverses Tau dimerization in cells under conditions of long-term mitochondrial stress
ROS are produced in the cell mainly by mitochondria. This process is often enhanced under pathological conditions. The overproduction of mitochondrial ROS (mROS) can induce oxidative stress in the cell, but mROS can also serve as secondary messengers (Reczek and Chandel, 2015). One example of such signaling is the oxidation of cysteine residues in several ribosomal proteins by mitochondrially born ROS to reduce global protein synthesis (Topf et al., 2018). In the present study, we investigated the role of mROS in Tau dimerization under conditions of long-term mitochondrial stress. We observed a significant increase in mitochondrial superoxide production, detected by MitoSOX (Figures 6, A and B, and 7, A and B), and general ROS production, detected by CM-H2DCFDA dye (Figures 6, C and D, and 7, C and D), in HEK293T NDUFA11 knockout cells (Figures 6, A and C, and 7, A and C) and HEK293T cells that were treated with rotenone for 68 h (Figures 6, B and D, and 7, B and D). Cells that were treated for 24 h with ROS scavengers, NAC or mitochondria-targeted MitoQ, moderately decreased the production of superoxide by mitochondria (Figures 6, A and B, and 7, A and B) and significantly decreased general oxidative stress (Figures 6, C and D, and 7, C and D). The decrease in ROS production after cells were treated with NAC or MitoQ was much higher in the case of general ROS that was detected by CM-H2DCFDA dye (Figures 6, C and D, and 7, C and D) than the decrease in mitochondrial superoxide production that was detected by MitoSOX (Figures 6, A and B, and 7, A and B). This was likely caused by the fact that the use of CM-H2DCFDA detected ROS mainly outside mitochondria, whereas MitoSOX detected superoxide inside mitochondria where ROS production under mitochondrial stress is too high to be efficiently reduced by ROS scavengers. Our previous study found that ROS scavenging by NAC restored protein synthesis under conditions of mitochondrial stress, suggesting the recovery of protein homeostasis (Samluk et al., 2019). To analyze the dependence of oxidative stress and Tau dimerization under conditions of mitochondrial stress, we performed the BiFC assay using HEK293T NDUFA11-deficient cells and HEK293T cells that were treated with rotenone for 68 h in the presence of NAC or MitoQ. Consistent with our previous experiments, we observed a significant increase in fluorescence in cells that were transfected with Tau-Venus BiFC plasmids under conditions of long-term mitochondrial stress, suggesting an increase in Tau dimerization (Figure 8, A–D). Strikingly, cells that were treated for 24 h with NAC or MitoQ exhibited a reduction of Tau dimerization in HEK293T NDUFA11 knockout cells and HEK293T cells that were treated with rotenone (Figure 8, A–D). Interestingly, in NDUFA11-deficient cells, the strongest effect was observed for the lowest applied concentration of NAC (1 mM), suggesting that a certain level of ROS was beneficial for these cells (Figures 6C and 8A) and could reflect a mitohormetic response (Messina et al., 2020). In addition to its own scavenging properties, NAC also enhances levels of cellular glutathione, which possesses anti-ROS activity (Halasi et al., 2013). We checked the level of protein glutathionylation, using specific anti-glutathione antibody and flow cytometry, in HEK293T NDUFA11 knockout cells and HEK293T cells that were treated with rotenone for 68 h in the presence of NAC. We observed increased protein glutathionylation under mitochondrial stress, reflecting the presence of oxidative stress in these cells and concentration-dependent reduction of protein glutathionylation in the cells that were additionally treated with NAC (Supplemental Figure 5, A and B). However, there was no direct correlation between general protein glutathionylation and Tau dimerization because cell treatment with 1 mM NAC did not change protein glutathionylation but caused a significant reduction of Tau dimerization (Figure 8, A and C, and Supplemental Figure 5, A and B). Collectively, these results indicated that the increase in mitochondrial-derived ROS levels led to the enhancement of early steps of Tau aggregation.
FIGURE 6:

NAC attenuated the increase in ROS production in cells with prolonged mitochondrial dysfunction. (A) Mitochondrial superoxide production measured with MitoSOX in HEK293T NDUFA11 knockout cells and in the presence of NAC (24 h) as indicated. The data are expressed as mean ± SEM. n = 4. (B) Mitochondrial superoxide production measured with MitoSOX in HEK293T cells that were treated with rotenone for 68 h and in the presence of NAC (24 h) as indicated. The data are expressed as mean ± SEM. n = 4. (C) ROS production measured with CM-H2DCFDA in HEK293T NDUFA11 knockout cells and in the presence of NAC (24 h) as indicated. The data are expressed as mean ± SEM. n = 4. (D) ROS production measured with CM-H2DCFDA in HEK293T cells that were treated with rotenone for 68 h and in the presence of NAC (24 h) as indicated. The data are expressed as mean ± SEM. n = 4. NAC, N-acetyl-l-cysteine; Rot., rotenone; ROS, reactive oxygen species. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant (p > 0.05) (one-way ANOVA followed by Tukey’s multiple comparisons test).
FIGURE 7:
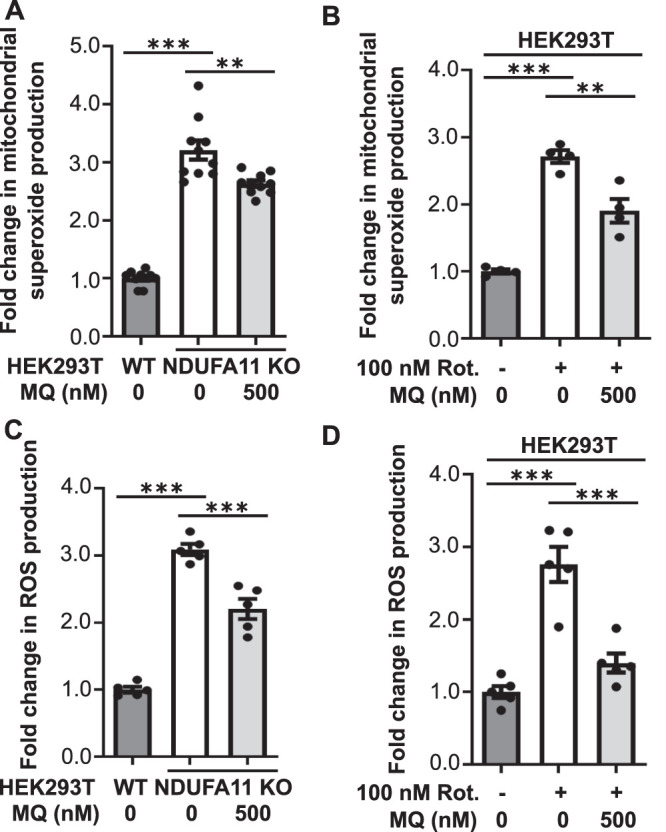
MitoQ attenuated the increase in ROS production in cells with prolonged mitochondrial dysfunction. (A) Mitochondrial superoxide production measured with MitoSOX in HEK293T NDUFA11 knockout cells and in the presence of MitoQ (24 h) as indicated. The data are expressed as mean ± SEM. n = 10. (B) Mitochondrial superoxide production measured with MitoSOX in HEK293T cells that were treated with rotenone for 68 h and in the presence of MitoQ (24 h) as indicated. The data are expressed as mean ± SEM. n = 4. (C) ROS production measured with CM-H2DCFDA in HEK293T NDUFA11 knockout cells and in the presence of MitoQ (24 h) as indicated. The data are expressed as mean ± SEM. n = 5. (D) ROS production measured with CM-H2DCFDA in HEK293T cells that were treated with rotenone for 68 h and in the presence of MitoQ (24 h) as indicated. The data are expressed as mean ± SEM. n = 5. MQ, MitoQ; Rot., rotenone; ROS, reactive oxygen species. **p < 0.01, ***p < 0.001 (one-way ANOVA followed by Tukey’s multiple comparisons test).
FIGURE 8:

NAC and MitoQ reversed Tau dimerization in cells under conditions of long-term mitochondrial stress. (A) Fold change in Venus fluorescence normalized to the level of Tau expression in untreated HEK293T NDUFA11 knockout cells and HEK293T NDUFA11 knockout cells that were treated for 24 h with NAC. The data are expressed as mean ± SEM. n = 6. (B) Fold change in Venus fluorescence normalized to the level of Tau expression in untreated HEK293T NDUFA11 knockout cells and HEK293T NDUFA11 knockout cells that were treated for 24 h with MitoQ. The data are expressed as mean ± SEM. n = 6. (C) Fold change in Venus fluorescence normalized to the level of Tau expression in HEK293T cells that were treated with rotenone for 68 h and in the presence of NAC (24 h) as indicated. The data are expressed as mean ± SEM. n = 3. (D) Fold change in Venus fluorescence normalized to the level of Tau expression in HEK293T cells that were treated with rotenone for 68 h and in the presence of MitoQ (24 h) as indicated. The data are expressed as mean ± SEM. n = 5. (E) Schematic illustration of the influence of long-term mitochondrial stress on Tau protein aggregation. Long-term mitochondrial stress led to oxidative stress (i.e., increase in ROS production) and inhibition of the ISR (i.e., decrease in eIF2α phosphorylation). The increase in ROS production was the main factor that induced early steps of Tau aggregation, but the reduction of eIF2α phosphorylation also partially triggered this process. Tau phosphorylation increased, but this was not a prerequisite of Tau oligomerization under conditions of long-term mitochondrial stress. NAC, N-acetyl-l-cysteine; MQ, MitoQ; Rot., rotenone; ROS, reactive oxygen species. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant (p > 0.05) (one-way ANOVA followed by Tukey’s multiple comparisons test).
DISCUSSION
Accumulating evidence indicates that dysfunctional mitochondria are important effectors of neurodegeneration. For example, the loss of PHB2, which is a scaffold protein of the inner mitochondrial membrane, leads to extensive neurodegeneration that is associated with behavioral and cognitive deficiencies in mice (Merkwirth et al., 2012). Another study linked mitochondrial dysfunction that was induced by the depletion of mitochondrial protease AFG3-like protein 2 (AFG3L2) in murine neurons with Tau hyperphosphorylation and a defect of the anterograde transport of mitochondria (Kondadi et al., 2014). Recently, it was shown that mitochondrial protein import defects and the accumulation of mitochondrial precursor proteins in the cytosol may induce cellular protein aggregation, including α-synuclein and amyloid β (Nowicka et al., 2021a). Moreover, the stimulation of mitochondrial protein import enhanced protein aggregate clearance in the cytosol (Nowicka et al., 2021b; Schlagowski et al., 2021). These observations support the hypothesis of the involvement of dysfunctional mitochondria in the development of neurodegenerative diseases by collapsing cellular proteostasis.
In the present study, we sought to understand the interplay between mitochondrial stress, cellular proteostasis, and Tau protein aggregation. Interestingly, mitochondrial protein import blockade activates such mechanisms as mitochondria-associated ribosome quality control (mitoRQC), mitochondrial compromised protein import response (mitoCPR), and mitochondrial protein translocation–associated degradation (mitoTAD), which remove stalled mitochondrial proteins (Izawa et al., 2017; Weidberg and Amon, 2018; Martensson et al., 2019). These clearance mechanisms are very important for proteostasis because stalled proteins can form aggregates (Choe et al., 2016). Additionally, mitochondria can actively deal with cytosolic protein aggregates by importing them into mitochondria, followed by their degradation or sequestration (Ruan et al., 2017; Bruderek et al., 2018). The multiplicity of discovered mechanisms that restore mitochondrial function and safeguard cytosolic proteostasis have been cumulatively referred to as the mitoprotein-induced stress response (Boos et al., 2020), but the potential involvement of these mechanisms in neurodegenerative diseases, such as tauopathies, has not been clarified.
We found that long-term mitochondrial stress induced Tau dimerization, which is the first step of protein aggregation (Meraz-Rios et al., 2010). Our microscopic analysis revealed that under conditions of applied stress, Tau likely did not aggregate in the form of insoluble NFTs because Tau oligomers were dispersed in cells. However, some studies suggested that soluble Tau aggregates can be even more harmful to neurons than large insoluble filaments, especially if they contain hyperphosphorylated Tau (Alonso et al., 1994). Interestingly, we observed an increase in Tau phosphorylation under conditions of mitochondrial stress, but this was not a prerequisite of Tau dimerization. In fact, the increase in Tau phosphorylation decreased microtubule binding affinity and stimulated its aggregation. Interestingly, however, Tau hyperphosphorylation during fetal brain development did not cause significant toxicity (Hefti et al., 2019).
Similar to our previous study (Samluk et al., 2019), we found that long-term mitochondrial stress inhibited the ISR by reducing the phosphorylation of eIF2α. Sorrentino et al. (2017) proposed that a reduction of ISR activity may cause amyloid β aggregation in mammalian cells, suggesting the involvement of this pathway in AD pathogenesis. Thus, one issue is whether the observed reduction of ISR activity in cells with prolonged mitochondrial dysfunction induces Tau protein aggregation. Our results demonstrated that inhibition of the ISR triggered Tau dimerization and activation of the ISR under conditions of long-term mitochondrial stress at least partially reduced early steps of Tau aggregation. These results suggested the involvement of ISR inhibition in tauopathies. However, the effect of ISR activation on the reduction of Tau dimerization was subtle and its physiological significance must be clarified in the future. Interestingly, the pharmacological activation of ISR signaling with compounds that inhibit eIF2α dephosphorylation had beneficial effects in experimental models of many neurodegenerative disorders (Reijonen et al., 2008; Lee et al., 2010; Jiang et al., 2014; Das et al., 2015; Dash et al., 2015; Way et al., 2015), whereas inhibition of the ISR with ISRIB enhanced cognitive memory (Sidrauski et al., 2013). Thus, no consensus has been reached about whether activation or inhibition of the ISR is more beneficial for reducing symptoms of neurodegenerative disorders. Costa-Mattioli and Walter (2020) proposed that depending on the disease or pathology and the optimal homeostatic set point for a particular phenotype, activation of the ISR or inhibition of the ISR would restore homeostasis to optimal cell fitness. We also believe that ISR activity needs to be fine-tuned to specific stress conditions, and pharmacological manipulations of this signaling pathway would need to be tailored to the particular pathology.
Many studies have reported that mitochondrial disorders are associated with oxidative stress (Yamada et al., 2020). We observed an increase in ROS production by cells with the dysfunction of mitochondrial respiratory chain complex I. We also found that this oxidative stress was mitigated by the ROS scavengers, NAC and mitochondria-targeted MitoQ. Interestingly, cells that were treated with NAC or MitoQ exhibited a reduction of Tau dimerization, suggesting a key role for ROS overproduction in the early steps of Tau aggregation under conditions of long-term mitochondrial stress. These results are generally consistent with observations that antioxidants decreased levels of phospho-Tau and rescued anterograde transport defects in neurons with dysfunctional mitochondria (Kondadi et al., 2014). NAC also increases levels of glutathione, which is an important cellular antioxidant (Halasi et al., 2013). However, we did not observe the direct correlation between cellular protein glutathionylation and Tau dimerization. Oxidative stress is believed to be an early event in the pathogenesis of AD, in which it was detected long before symptoms appeared (Wang et al., 2014). Interestingly, patients with AD had lower levels of antioxidants, such as uric acid, vitamin C, vitamin E, and glutathione, and antioxidant enzymes, such as superoxide dismutase and catalase (Singh et al., 2019). A few hypotheses have been proposed to explain the involvement of mitochondrial dysfunction and oxidative imbalance in the development of AD. The best-documented abnormalities include a decrease in energy metabolism, a decrease in the neuronal expression of genes that encode several key enzymes of oxidative metabolism, calcium dyshomeostasis, oxidative damage in mitochondrial DNA (mtDNA), and abnormal mitochondrial dynamics (Wang et al., 2014; Elfawy and Das, 2019; Messina et al., 2020). The present results support the involvement of oxidative imbalance in early events of AD. We propose that mitochondrially born oxidative stress triggers neurodegeneration by affecting cellular proteostasis and inducing early steps of protein aggregation, including Tau. The data suggest that mitochondrial dysfunction and oxidative stress could activate signaling pathways that play a role in the hyperphosphorylation and aggregation of Tau protein, but the precise mechanisms need further elucidation.
NDUFA11 is an accessory subunit of mitochondrial complex I. Interestingly, the dysfunction of subunits of this complex was shown to lead to neurodegenerative diseases, such as Leigh’s syndrome (Berger et al., 2008; Rodenburg, 2016). Complex I is also a major source of damaging ROS, and its deficiency is the most frequent mitochondrial disorder present in childhood (Stroud et al., 2016; Peralta et al., 2020). We demonstrated that knockout of NDUFA11 greatly reduced mitochondrial complex I activity and the impact of inhibition of mitochondrial complex III on Tau dimerization was not very pronounced, suggesting specific involvement of complex I deficiency in the induction of Tau aggregation. Importantly, rotenone, which was used in the present study to induce long-term mitochondrial stress, is also considered a cause of Parkinson’s disease. Rotenone is a naturally occurring compound that is derived from the roots of certain plant species. It was commonly used as an insecticide and pesticide. Rotenone easily crosses all biological membranes, including the blood-brain barrier because of its high lipophilicity (Betarbet et al., 2000; Sherer et al., 2003; Tanner et al., 2011; Johnson and Bobrovskaya, 2015; Innos and Hickey, 2021). People who used rotenone had a 2.5-fold higher risk of Parkinson’s disease (Tanner et al., 2011). The present data also suggest the involvement of rotenone in the development of AD through an increase in the early steps of Tau aggregation (Figure 8E).
Many intriguing findings shed light on different aspects of mitochondrial biology in neurodegeneration. Altogether, these important discoveries paint a broader picture of the involvement of mitochondrial defects in neurodegenerative diseases. Despite great efforts that have been made, still unknown are whether and how mitochondrial functions may be targets for the treatment of neurodegenerative defects. The present study, showing that the excess of mitochondrial ROS induces early steps of Tau aggregation, provides insights that could improve our understanding of processes that are triggered by mitochondrial stress in neurodegenerative diseases.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Cell culture conditions
HEK293T wild-type and HEK293T NDUFA11 knockout cell lines were cultured in high-glucose (4.5 g/l) 90% DMEM (Sigma; catalogue no. D5671) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 50 μg/ml uridine at 37°C in a 5% CO2 humidified atmosphere. The culture medium was changed every other day. The cells were treated with rotenone (48 or 68 h) (Sigma; catalogue no. R8875), ISRIB (68 h) (Sigma; catalogue no. SML0843), salubrinal (24 h) (Sigma; catalogue no. SML0951), guanabenz (24 h) (Sigma; catalogue no. G110), sephin1 (24 h) (Sigma; catalogue no. SML1356), NAC (24 h) (Sigma; catalogue no. A9165), MitoQ (24 h) (Cayman Chemicals; catalogue no. 89950), antimycin A (68 h) (Sigma; catalogue no. A8674), and okadaic acid (24 h) (Abcam; catalogue no. ab120375) where indicated. HEK293T wild-type and HEK293T NDUFA11 knockout cells were provided by David Stroud and Michael Ryan (Monash University, Melbourne, Australia) (Stroud et al., 2016).
The human neuroblastoma cell line SH-SY5Y was maintained in DMEM with nutrient Mixture F-12 Ham (DMEM/F12; Life Technologies; catalogue no. 11320033) supplemented with 10% FBS and 5 mg/ml penicillin/streptomycin at 37°C in a 5% CO2 humidified atmosphere. The medium was renewed every 3 d. For differentiation, SH-SY5Y cells were plated on 10 μg/ml laminin-coated plates in DMEM/F12 supplemented with 10% FBS. After 24 h, the medium was replaced with DMEM/F12 containing 3% FBS and 10 μM all-trans retinoic acid (RA; Sigma; catalogue no. R2625) in order to induce differentiation. After 72 h the differentiation medium was replaced and cells were transfected for 72 h and treated with rotenone for the last 24 h.
Complex I enzyme activity assay
To analyze the activity of Complex I in HEK293T wild-type and HEK293T NDUFA11 knockout cells, we used the Complex I Enzyme Activity Assay Kit according to the manufacturer’s instructions (Abcam; catalogue no. ab109721). Cells were washed twice with phosphate-buffered saline (PBS) and then lysed in PBS containing 10% detergent provided by the manufacturer for 30 min on ice. Then, total cellular protein extracts were centrifuged at 16,000 × g for 20 min at 4°C, and the protein concentration was measured in collected supernatants. Next, samples containing 200 µg of protein were diluted to 200 µl using Incubation Solution, loaded on a 96-well plate provided in the kit, and incubated for 3 h at room temperature. In the next step, wells were washed three times with 300 µl of 1× Wash Buffer, and directly after the final wash, 200 µl of Assay Solution was added to each well. The absorbance was measured using a Multiskan SkyHigh Microplate Spectrophotometer (Thermo Scientific) at 450 nm for 30 min with 1 min intervals and a shake between readings at room temperature.
Transfection of HEK293T wild-type cells, HEK293T NDUFA11 knockout cells, and SH-SY5Y cells
For transfection, HEK293T wild-type cells, HEK293T NDUFA11 knockout cells, and SH-SY5Y cells were seeded in 60 mm cell culture dishes, HEK293T cells at a density of 1.25 × 105 and grown for 24 h in high-glucose DMEM to reach 20–25% confluence and SH-SY5Y cells at a density of 1 × 106 and grown and differentiated for 72 h in DMEM/F12 to reach 60–70% confluence on the day of transfection. Cells were transfected using GeneJuice Transfection Reagent (Sigma; catalogue no. 70967). For each transfected plate, 6 µl (HEK293T) or 12 µl (SH-SY5Y) of GeneJuice was added to 250 µl of Opti-MEM I Reduced Serum Medium (Life Technologies; catalogue no. 31-985-070) and mixed thoroughly by vortexing. After 5 min of incubation at room temperature, purified plasmid DNA was added at a concentration of 0.5 or 1 µg/plate (HEK293T) or 2 µg/plate (SH-SY5Y) according to the specific experiment. The mixture was then gently mixed by pipetting and incubated for 15 min at room temperature. After incubation, GeneJuice/DNA mixture was added dropwise to each plate that contained 5 ml of complete high-glucose DMEM (HEK293T) or complete DMEM/F12 (SH-SY5Y). The plates were gently rocked to ensure an even distribution of the transfection mixture.
BiFC
To monitor the oligomerization of Tau protein, we performed a BiFC assay (Figure 1A), which was optimized in the previous studies (Tak et al., 2013; Blum et al., 2015). Blum et al. (2015) initially inserted Tau into the Venus BiFC vectors in N- and C-terminal positions and found the combination of constructs that provides an optimal BiFC signal, which we used in our study. Untreated HEK293T and SH-SY5Y cells, HEK293T cells that were treated with rotenone or antimycin A for the last 68 h or okadaic acid for the last 24 h, SH-SY5Y cells that were treated with rotenone for the last 24 h, and HEK293T NDUFA11 knockout cells were transfected for 72 h with plasmids that encoded Tau protein that was fused to the N-terminal part of Venus protein (N-terminal on backbone), Tau-VN (VN-Tau [wt]; Addgene; catalogue no. 87368) and Tau protein that was fused to the C-terminal part of Venus protein (C terminal on backbone), Tau-VC (Tau [wt]-VC; Addgene; catalogue no. 87369) (Blum et al., 2015). Where indicated, cells were transfected for 72 h with Venus BiFC vectors that encoded Tau, in which all 14 S/P or T/P amino acid residues were mutated to alanine (Tau-VN-AP and Tau-VC-AP). The dimerization of Tau protein resulted in the reconstitution of functional fluorescent Venus protein, whose fluorescence was analyzed using a fluorescence microplate reader (Ex/Em 488/528) (Synergy H1 Hybrid Multi-Mode Microplate Reader; BioTek) and a Zeiss LSM700 confocal microscope. Fluorescence was normalized to transfection efficiency that was verified by immunoblotting.
BiFC plasmid construction using the SLiCE method
Venus BiFC vectors that encoded Tau (Tau-AP), in which all 14 S/P or T/P amino acid residues (T111, T153, T175, T181, S199, S202, T205, T212, T217, T231, S235, S396, S404, and S422; numbering based on the longest 441-amino-acid brain isoform of human Tau [hTau]) were mutated to alanine (Tau-VN-AP, Tau-VC-AP), were constructed on the basis of the following plasmids: VN-Tau (wt) plasmid (Addgene; catalogue no. 87368), Tau (wt)-VC plasmid (Addgene; catalogue no. 87369) (Blum et al., 2015), and pRK5-EGFP-Tau AP plasmid (Addgene; catalogue no. 46905) (Hoover et al., 2010). To perform Tau-AP subcloning from the pRK5-EGFP-Tau AP plasmid to BiFC vectors, the seamless ligation cloning extract (SLiCE) method was used (Zhang et al., 2012, 2014). The SLiCE method used bacterial cell extracts for DNA assembly into recombinant DNA molecules in a single in vitro recombination reaction. To amplify the Tau-AP insert, complementary primers were designed that contained additional homology arms for the following BiFC vectors: Tau_VN (forward, GCTCTGGAGGTGGTGGGTCCCTTAAGATGGCTGAGCCCCGCCA; reverse, TTTAAACGGGCCCTCTAGACTCGAGTCACAAACCCTGCTTGGCCAG), Tau_VC (forward, AAGCTGGCTAGCGTTTAAACTTAAGATGGCTGAGCCCCGCCA; reverse, TGGCCTTGATGCCGTTCTTCTCGAGCAAACCCTGCTTGGCCAGGG). pRK5-EGFP-Tau AP plasmid was used as a template, and the insert (Tau-AP) was amplified by PCR using DNA polymerase with proofreading activity (PfuPlus DNA Polymerase; EURx; catalogue no. E1118). The samples were then treated with DpnI to remove residual plasmid template DNA. The insert was then cleaned using the GeneMATRIX PCR/DNA Clean-Up Purification Kit (EURx; catalogue no. E3520). Venus Tau BiFC plasmids (VN-Tau [wt], Tau [wt]-VC) were digested with restriction enzymes (AflII and XhoI) to cut off wild-type Tau inserts and linearize the vectors. Venus BiFC plasmids were then purified by extraction from agarose gel using the GeneJET Gel Extraction Kit (Thermo Scientific; catalogue no. K0692). The SLiCE reaction was then performed at 37°C for 30 min. The mixture contained the following ingredients: 50 ng of linearized plasmid, 200 ng insert, 1 μl of SLiCE extract, and 1 μl of SLiCE buffer (500 mM Tris/Cl [pH 7.4], 100 mM MgCl2, 10 mM adenosine triphosphate, and 10 mM dithiothreitol [DTT]). Double-distilled H2O was added to reach a total volume of 10 μl. Afterward, the samples were cooled on ice and used for bacterial transformation. The transformed cells were plated onto agar plates that contained appropriate antibiotics.
Immunofluorescence
HEK293T wild-type and HEK293T NDUFA11 knockout cells were transfected with BiFC plasmids that encoded Tau for 72 h, washed twice with PBS, and fixed with 3.7% formaldehyde for 10 min at 4°C. The cells were then washed twice with PBS and permeabilized for 5 min by treatment with 0.1% Triton X-100 in PBS. They were then rinsed once with PBS and once with water, and the samples were mounted in ProLong Diamond Antifade Mountant with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific; catalogue no. P36962). The analysis was performed using a Zeiss LSM700 confocal microscope.
ROS measurements
To analyze levels of ROS production, HEK293T wild-type and HEK293T NDUFA11 knockout cells were cultured for 96 h in DMEM that contained 4.5 g/l glucose supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 50 μg/ml uridine. HEK293T cells were treated for 68 h with 100 nM rotenone and for the last 24 h before the experiment with NAC or MitoQ as indicated. HEK293T NDUFA11 knockout cells were treated with NAC or MitoQ for 24 h before the ROS measurements where indicated. On the day of the experiment, the cells were incubated on 6 cm dishes for 20 min at 37°C in a CO2 incubator in a culture medium that contained 5 μM red mitochondrial superoxide indicator MitoSOX (Thermo Fisher Scientific; catalogue no. M36008) or 10 μM general oxidative stress indicator CM-H2DCFDA dye (Thermo Fisher Scientific; catalogue no. C6827). Cells were then collected, washed twice, and resuspended in cold PBS at 4°C. Fluorescent signals were corrected for autofluorescence. Fluorescence was measured at an excitation wavelength of 510 nm and emission wavelength of 580 nm for MitoSOX or an excitation wavelength of 495 nm and emission wavelength of 527 nm for CM-H2DCFDA using a fluorescence microplate reader (Synergy H1 Hybrid Multi-Mode Microplate Reader; BioTek).
Protein glutathionylation analysis
HEK293T cells treated with rotenone for 68 h and HEK293T NDUFA11-deficient cells were treated for the last 24 h with NAC, and then protein glutathionylation was analyzed in these cells and in untreated HEK293T cells. On the day of the experiment, cells were collected, washed twice with PBS, and fixed with 3.7% formaldehyde for 10 min at 4°C. Next, cells were washed twice with PBS, permeabilized for 5 min in 0.1% Triton X-100 solution in PBS, washed again with PBS, and incubated for 30 min in 10% FBS in PBS. After that, cells were incubated for 1 h at room temperature with an anti-glutathione antibody (1:250; Abcam; catalogue no. ab19534). After two washes with PBS, the cells were incubated for 1 h with Alexa Fluor 568 goat anti-mouse antibody (1:250; Invitrogen; catalogue no. A-11031) and then washed twice, resuspended in ice-cold PBS, and analyzed using a flow cytometer (BD LSRFortessa).
Quantitative real-time PCR
Total RNA from cells was purified using the RNeasy Plus Mini Kit (Qiagen; catalogue no. 74134). cDNA was synthesized from 500 ng of total RNA using the Cloned AMV First-Strand cDNA Synthesis Kit (Invitrogen; catalogue no. 12328-032) according to the manufacturer’s instructions. Endogenous mRNA was measured by quantitative real-time PCR (RT-PCR) using the LightCycler 480 Instrument II (Roche) and SensiFAST SYBR Hi-ROX Kit (Bioline; catalogue no. BIO-92005). The following primer sequences were used for quantitative RT-PCR: (GADD34; PPP1R15A; forward, 5′-AGGAAGAGGAAGCTGCTGAG-3′; reverse, 5′-AATGGACAGTGACCTTCTCG-3′), actin cytoplasmic 1 (ACTB; forward, 5′-GCCGGGACCTGACTGACTAC-3′; reverse, 5′-TTCTCCTTAATGTCACGCACGAT-3′). ACTB was used as the internal control.
Determination of PP2A activity by malachite green phosphatase assay
To determine PP2A phosphatase activity in HEK293T wild-type and HEK293T NDUFA11 knockout cells, we used the Ser/Thr Phosphatase Assay Kit 1 (K-R-pT-I-R-R) (Merck; catalogue no. 17-127). The activity was based on dephosphorylation of the phosphopeptide (K-R-pT-I-R-R). Total cellular protein extracts were prepared in a buffer that contained 20 mM imidazole-HCl, 2 mM EDTA, pH 7.0, with 10 μg/ml each of aprotinin, leupeptin, bestatin, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were homogenized on ice using a syringe and needle (10 strokes) and centrifuged at 2000 × g for 5 min at 4°C. After the protein concentration measurements, the supernatants were used to assay phosphatase activity. For the enzymatic reaction, 50 μg of protein was used, and the volume of the protein extract was brought to 16.7 μl with lysis buffer. Afterward, 6.3 μl of 1 mM phosphopeptide was added to obtain a final concentration of 250 μM. After the addition of 2 μl of Ser/Thr assay buffer (50 mM Tris-HCl [pH 7.0] and 100 mM CaCl2), the final volume of the enzyme reaction was 25 μl. The sample was then incubated for 10 min at 30°C in a shaking incubator, briefly centrifuged, and transferred to a 96-well plate. Malachite green phosphate detection solution (100 μl) was added, and then the solution was left for 15 min to allow color development. The plate was read at 650 nm with a microplate reader. The amount of phosphate released was calculated from a standard curve (0–2000 pmol). Sample absorbance values were compared with those of negative controls that contained no enzyme.
Miscellaneous
Total cellular protein extracts were prepared in RIPA buffer (65 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% IGEPAL CA-630 [NP-40], 0.25% sodium deoxycholate, and 1 mM EDTA) that contained 2 mM PMSF (Sigma; catalogue no. P7626) and phosphatase inhibitor cocktail (PhosSTOP; Roche; catalogue no. 04 906 837 001). Laemmli sample buffer that contained 50 mM DTT was added, and proteins were denatured at 65°C for 15 min. Total protein extracts were separated by SDS–PAGE on 12% gels. The following commercially available antibodies against mammalian proteins were used: Tau (TAU-5) (Merck; catalogue no. 577801), phospho-Tau (Ser-202, Thr-205) (AT8) (Thermo Fisher Scientific; catalogue no. MN1020), eIF2α (Cell Signaling Technology; catalogue no. 9722), phospho-eIF2α (Ser-51) (Cell Signaling Technology; catalogue no. 9721), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology; catalogue no. sc-47724), ATF4 (Cell Signaling Technology; catalogue no. 11815), and glutathione (GSH) (D8) (Abcam; catalogue no. ab19534). Protein bands were visualized using secondary anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase and chemiluminescence. Chemiluminescence signals were detected using Amersham Imager 600 RGB or x-ray films. The images were digitally processed using Adobe Photoshop CS4 software. ImageJ software was used to quantify the Western blots and confocal microscopy images. The represented fold changes are means of fold changes that were obtained from independent biological replicates ± SEM. VN-Tau (wt) was a gift from Tiago Outeiro, University Medical Center Goettingen, Goettingen, Germany (Addgene plasmid no. 87368; http://n2t.net/addgene:87368; RRID:Addgene_87368; Blum et al., 2015). Tau (wt)-VC was a gift from Tiago Outeiro; Addgene plasmid no. 87369; http://n2t.net/addgene:87369; RRID:Addgene_87369; Blum et al., 2015). pRK5-EGFP-Tau AP was a gift from Karen Ashe (University of Minnesota, MI; Addgene plasmid no. 46905; http://n2t.net/addgene:46905; RRID:Addgene_46905; Hoover et al., 2010). The experiments were replicated at least three times.
Statistical analysis
Statistical significance was tested using Student’s t test or one-way analysis of variance (ANOVA) as indicated. The statistical tests were performed using GraphPad Prism. Values of p < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank David Stroud and Michael Ryan for providing the HEK293T NDUFA11 knockout cell line and Minji Kim and Agnieszka Chacin´ska for valuable comments and discussions. This research was funded by the National Science Centre, Poland (NCN; Grant 2019/33/B/NZ3/00533). Figure 8E (the mitochondrion image) was created using a template (Mitochondria) from Servier Medical Art, which is licensed under a Creative Commons Attribution 3.0 Unported License (http://smart.servier.com/).
Abbreviations used:
- ACTB
actin cytoplasmic 1
- AD
Alzheimer’s disease
- AFG3L2
AFG3-like protein 2
- ATF4
activating transcription factor 4
- BiFC
bimolecular fluorescence complementation
- CReP
constitutive repressor of eIF2α phosphorylation
- DTT
dithiothreitol
- eIF2α
eukaryotic translation initiation factor 2α
- ER
endoplasmic reticulum
- GADD34
growth arrest and DNA damage–inducible protein 34
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- hTau
human Tau
- ISR
integrated stress response
- ISRIB
ISR inhibitor
- MIA40
mitochondrial intermembrane space import and assembly protein 40
- mitoCPR
mitochondrial compromised protein import response
- mitoRQC
mitochondria-associated ribosome quality control
- mitoTAD
mitochondrial protein translocation–associated degradation
- mROS
mitochondrial reactive oxygen species
- mtDNA
mitochondrial DNA
- NAC
N-acetyl- l-cysteine
- NDUFA11
NADH:ubiquinone oxidoreductase subunit A11
- NFT
neurofibrillary tangle
- PBS
phosphate-buffered saline
- PERK
PKR-like ER kinase
- PHB2
prohibitin-2
- PKR
protein kinase R
- PMSF
phenylmethylsulfonyl fluoride
- PP1
protein phosphatase 1
- ROS
reactive oxygen species
- RT-PCR
real-time PCR
- SLiCE
seamless ligation cloning extract
- YFP
yellow fluorescent protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-11-0553) on April 21, 2022.
REFERENCES
- Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K (1994). Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA 91, 5562–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM (2012). Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet 8, e1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger I, Hershkovitz E, Shaag A, Edvardson S, Saada A, Elpeleg O (2008). Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol 63, 405–408. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Blum D, Herrera F, Francelle L, Mendes T, Basquin M, Obriot H, Demeyer D, Sergeant N, Gerhardt E, Brouillet E, et al. (2015). Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum Mol Genet 24, 76–85. [DOI] [PubMed] [Google Scholar]
- Boos F, Labbadia J, Herrmann JM (2020). How the mitoprotein-induced stress response safeguards the cytosol: a unified view. Trends Cell Biol 30, 241–254. [DOI] [PubMed] [Google Scholar]
- Bruderek M, Jaworek W, Wilkening A, Rub C, Cenini G, Fortsch A, Sylvester M, Voos W (2018). IMiQ: a novel protein quality control compartment protecting mitochondrial functional integrity. Mol Biol Cell 29, 256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral-Costa JV, Kowaltowski AJ (2020). Neurological disorders and mitochondria. Mol Aspects Med 71, 100826. [DOI] [PubMed] [Google Scholar]
- Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze-Specking A, Truscott KN, Guiard B, Meisinger C, Pfanner N (2004). Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J 23, 3735–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS (2014). Mitochondria as signaling organelles. BMC Biol 12, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe YJ, Park SH, Hassemer T, Korner R, Vincenz-Donnelly L, Hayer-Hartl M, Hartl FU (2016). Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature 531, 191–195. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Walter P (2020). The integrated stress response: from mechanism to disease. Science 368, eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, Sigurdardottir A, Bertolotti A (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, Tsvetkov AS, Moore AN (2015). Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma 32, 1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres C, Byrne C, Qi H, Cantrelle FX, Huvent I, Chambraud B, Baulieu EE, Jacquot Y, Landrieu I, Lippens G, et al. (2017). Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci USA 114, 9080–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfawy HA, Das B (2019). Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: etiologies and therapeutic strategies. Life Sci 218, 165–184. [DOI] [PubMed] [Google Scholar]
- Guha P, Kaptan E, Gade P, Kalvakolanu DV, Ahmed H (2017). Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mTORC1. Oncotarget 8, 68191–68207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasi M, Wang M, Chavan TS, Gaponenko V, Hay N, Gartel AL (2013). ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem J 454, 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Khersonsky S, Marciniak S, Scheuner D, Kaufman RJ, Javitt N, Chang YT, Ron D (2005). Bioactive small molecules reveal antagonism between the integrated stress response and sterol-regulated gene expression. Cell Metab 2, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Hefti MM, Kim S, Bell AJ, Betters RK, Fiock KL, Iida MA, Smalley ME, Farrell K, Fowkes ME, Crary JF (2019). Tau phosphorylation and aggregation in the developing human brain. J Neuropathol Exp Neurol 78, 930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innos J, Hickey MA (2021). Using rotenone to model Parkinson’s disease in mice: a review of the role of pharmacokinetics. Chem Res Toxicol 34, 1223–1239. [DOI] [PubMed] [Google Scholar]
- Izawa T, Park SH, Zhao L, Hartl FU, Neupert W (2017). Cytosolic protein Vms1 links ribosome quality control to mitochondrial and cellular homeostasis. Cell 171, 890–903.e818. [DOI] [PubMed] [Google Scholar]
- Jiang HQ, Ren M, Jiang HZ, Wang J, Zhang J, Yin X, Wang SY, Qi Y, Wang XD, Feng HL (2014). Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 277, 132–138. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Bobrovskaya L (2015). An update on the rotenone models of Parkinson’s disease: their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 46, 101–116. [DOI] [PubMed] [Google Scholar]
- Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivela R, Pessia A, Velagapudi V, Suomalainen A (2017). mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab 26, 419–428.e415. [DOI] [PubMed] [Google Scholar]
- Kondadi AK, Wang S, Montagner S, Kladt N, Korwitz A, Martinelli P, Herholz D, Baker MJ, Schauss AC, Langer T, et al. (2014). Loss of the m-AAA protease subunit AFG(3)L(2) causes mitochondrial transport defects and tau hyperphosphorylation. EMBO J 33, 1011–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A, Larsson NG (2017). Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 6, e30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Park YH, Min H, Kim YR, Seok YJ (2019). Determination of protein phosphorylation by polyacrylamide gel electrophoresis. J Microbiol 57, 93–100. [DOI] [PubMed] [Google Scholar]
- Lee DY, Lee KS, Lee HJ, Kim DH, Noh YH, Yu K, Jung HY, Lee SH, Lee JY, Youn YC, et al. (2010). Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS One 5, e10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Haque MM, Kim D, Kim DJ, Kim YK (2014). Cell-based models to investigate Tau aggregation. Comput Struct Biotechnol J 12, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B, et al. (2019). Mitochondrial protein translocation-associated degradation. Nature 569, 679–683. [DOI] [PubMed] [Google Scholar]
- Meraz-Rios MA, Lira-De Leon KI, Campos-Pena V, De Anda-Hernandez MA, Mena-Lopez R (2010). Tau oligomers and aggregation in Alzheimer’s disease. J Neurochem 112, 1353–1367. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, Rugarli EI, Langer T (2012). Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet 8, e1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina F, Cecconi F, Rodolfo C (2020). Do you remember mitochondria? Front Physiol 11, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraj K, Nowicka U, Chacinska A (2020). Mitochondrial control of cellular protein homeostasis. Biochem J 477, 3033–3054. [DOI] [PubMed] [Google Scholar]
- Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C, Loeffler T, Niederkofler V, Daum G, Attems J, Hutter-Paier B (2018). Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka U, Chroscicki P, Stroobants K, Sladowska M, Turek M, Uszczynska-Ratajczak B, Kundra R, Goral T, Perni M, Dobson CM, et al. (2021a). Cytosolic aggregation of mitochondrial proteins disrupts cellular homeostasis by stimulating the aggregation of other proteins. eLife 10, e65484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka U, Kim MJ, Chacinska A (2021b). Suppressing toxic aggregates: let MIA do it! EMBO J 40, e109001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM (2016). The integrated stress response. EMBO Rep 17, 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta S, Pinto M, Arguello T, Garcia S, Diaz F, Moraes CT (2020). Metformin delays neurological symptom onset in a mouse model of neuronal complex I deficiency. JCI Insight 5, e141183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, Auwerx J (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek CR, Chandel NS (2015). ROS-dependent signal transduction. Curr Opin Cell Biol 33, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijonen S, Putkonen N, Norremolle A, Lindholm D, Korhonen L (2008). Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp Cell Res 314, 950–960. [DOI] [PubMed] [Google Scholar]
- Rodenburg RJ (2016). Mitochondrial complex I-linked disease. Biochim Biophys Acta 1857, 938–945. [DOI] [PubMed] [Google Scholar]
- Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z, Florens L, Li R (2017). Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samluk L, Chroscicki P, Chacinska A (2018). Mitochondrial protein import stress and signaling. Curr Opin Physiol 3, 41–48. [Google Scholar]
- Samluk L, Urbanska M, Kisielewska K, Mohanraj K, Kim MJ, Machnicka K, Liszewska E, Jaworski J, Chacinska A (2019). Cytosolic translational responses differ under conditions of severe short-term and long-term mitochondrial stress. Mol Biol Cell 30, 1864–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagowski AM, Knoringer K, Morlot S, Sanchez Vicente A, Flohr T, Kramer L, Boos F, Khalid N, Ahmed S, Schramm J, et al. (2021). Increased levels of mitochondrial import factor Mia40 prevent the aggregation of polyQ proteins in the cytosol. EMBO J 40, e107913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT (2003). Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci 23, 10756–10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Kukreti R, Saso L, Kukreti S (2019). Oxidative stress: a key modulator in neurodegenerative diseases. Molecules 24, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag JM, Sontag E (2014). Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M (2013). Tau pathology and neurodegeneration. Lancet Neurol 12, 609–622. [DOI] [PubMed] [Google Scholar]
- Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, et al. (2016). Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126. [DOI] [PubMed] [Google Scholar]
- Tak H, Haque MM, Kim MJ, Lee JH, Baik JH, Kim Y, Kim DJ, Grailhe R, Kim YK (2013). Bimolecular fluorescence complementation; lighting-up tau-tau interaction in living cells. PLoS One 8, e81682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, et al. (2011). Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topf U, Suppanz I, Samluk L, Wrobel L, Boser A, Sakowska P, Knapp B, Pietrzyk MK, Chacinska A, Warscheid B (2018). Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat Commun 9, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyama T, Tsubouchi A, Usui T, Imamura H, Uemura T (2017). Mitochondrial dysfunction induces dendritic loss via eIF2alpha phosphorylation. J Cell Biol 216, 815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842, 1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zuo X, Kucejova B, Chen XJ (2008). Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat Cell Biol 10, 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, Robinson AP, Brugarolas P, Miller RH, Miller SD, et al. (2015). Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun 6, 6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Amon A (2018). MitoCPR—a surveillance pathway that protects mitochondria in response to protein import stress. Science 360, eaan4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidling IW, Swerdlow RH (2020). Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp Neurol 330, 113321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Takano Y, Satrialdi, Abe J, Hibino M, Harashima H (2020). Therapeutic strategies for regulating mitochondrial oxidative stress. Biomolecules 10, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Werling U, Edelmann W (2012). SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res 40, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Werling U, Edelmann W (2014). Seamless Ligation Cloning Extract (SLiCE) cloning method. Methods Mol Biol 1116, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.