

Abstract
Herein, a set of pyridine and pyrimidine derivatives were assessed for their impact on the cell cycle and apoptosis. Human breast cancer (MCF7), hepatocellular carcinoma (HEPG2), larynx cancer (HEP2), lung cancer (H460), colon cancers (HCT116 and Caco2), and hypopharyngeal cancer (FADU), and normal Vero cell lines were used. Compounds 8 and 14 displayed outstanding effects on the investigated cell lines and were further tested for their antioxidant activity in MCF7, H460, FADU, HEP2, HEPG2, HCT116, Caco2, and Vero cells by measuring superoxide dismutase (SOD), malondialdehyde content (MDA), reduced glutathione (GSH), and nitric oxide (NO) content. Besides, Annexin V-FITC apoptosis detection and cell cycle DNA index using the HEPG-2 cell line were established on both compounds as well. Furthermore, compounds 8 and 14 were assessed for their EGFR kinase (Wild and T790M) inhibitory activities, revealing eligible potential. Additionally, molecular docking, ADME, and SAR studies were carried out for the investigated candidates.
Keywords: Pyridine/pyrimidine derivatives, EGFR, in vitro, in silico
Graphical Abstract

Introduction
Being the second leading cause of mortality globally, cancer kills roughly 8 million people each year. Additionally, cancer incidence is expected to elevate regrettably by more than 50% in upcoming years1–3. Besides, different cancer types have developed acquired chemotherapeutic resistance over the last few decades4–6. Furthermore, chemotherapeutics utilised could induce cytotoxicity to other healthy normal cells owing to their poor selectivity. Thus, severe adverse effects may be experienced, such as anaemia, nausea, alopecia, and immunosuppression7,8. As a result, researchers should dedicate their efforts to ice breaking and discovering more appropriate chemotherapeutics, mainly for the most invasive tumours9,10.
Furthermore, cellular functions such as metabolism, survival, apoptosis, and cell proliferation could be regulated by protein kinases (PKs)11,12. Many diseases, including cancer, are caused by disrupting cell signalling cascades via kinase alterations, particularly hyper-activation, or mutations13,14.
Epidermal growth factor receptor (EGFR) is regarded as one of the most outstanding PKs, which play a critical function in cell migration and proliferation15,16. Molecules that may affect the control of cancer cell proliferation are targeted by modern-designed molecular strategies. These strategies are capable of improving cancer therapy efficiency more than conventional chemotherapy. Therefore, EGFRs are regarded as outstanding targets for the design of new anti-tumour agents17,18.
An important factor connecting environmental toxicity to the multistage carcinogenic process is oxidative stress. Responses to both endogenous and external stimuli result in the formation of reactive oxygen species (ROS). An intrinsic antioxidant defence system exists to regulate ROS-mediated harm. However, oxidative stress emerges when oxidation surpasses the regulatory systems. Numerous macromolecular components, including DNA, lipids, and proteins, undergo harmful changes as a result of chronic and cumulative oxidative stress. The increased cellular ROS levels are mediated through an alternative strategy through antioxidant use for the sake of tumour cell depletion from ROS-induced survival signalling pathways. It was revealed that increased intracellular ROS levels may be involved in early events of cancer initiation and progression. These treatments might also have a preventative purpose19.
In comparison to normal healthy cells, cancer cells exhibit a higher rate of ROS generation and a different redox environment. The majority of chemotherapeutic drugs increase intracellular ROS levels and can change cancer cells’ redox balance20.
On the other hand, pyrimidine and pyridine-related compounds are an important class of heterocycles owing to their wide chemical and biological applications. They have been employed widely in the areas of medicine, and material science21–23. They are responsible for various biological significance, such as anti-inflammatory24, antipyretic25, antihypotensive26, anticonvulsant27, antiviral28, antimicrobial29, and antidiabetic activities30. Among this relevance also, a literature survey revealed that a variety of fused pyrimidines have been reported to be extremely potent anticancer activity against various cell lines31–33. Some 2-pyridone derivatives acting as PK inhibitors could exhibit potent anticancer activity34. Besides, cyanopyridines revealed potent PIM‐1 inhibitions and PDE3A inhibition35,36. Hamajima et al. reported pyrazolopyridine as having high PI3Kd inhibitory activity with eligible selectivity and oral availability in mice37. Additionally, the cell lines A431a, HCT116, and SNU638b were employed for assessing the in vitro antiproliferative activity of pyrido[2,3-d]pyrimidine derivatives in addition to its inhibition potential for CDK4/Cyclin D, CDK2-Cyclin A, and EGFR enzyme38. Orlikova et al. reported pyrazolopyridine derivatives to reflect their selective cytotoxic potential against K562 cancer cells upon comparison to normal cells39. Given their significant cytotoxicity against various cell lines, therefore, pyridines, and pyrimidines were incorporated into many FDA-approved anticancer drugs (Figure 1).
Figure 1.
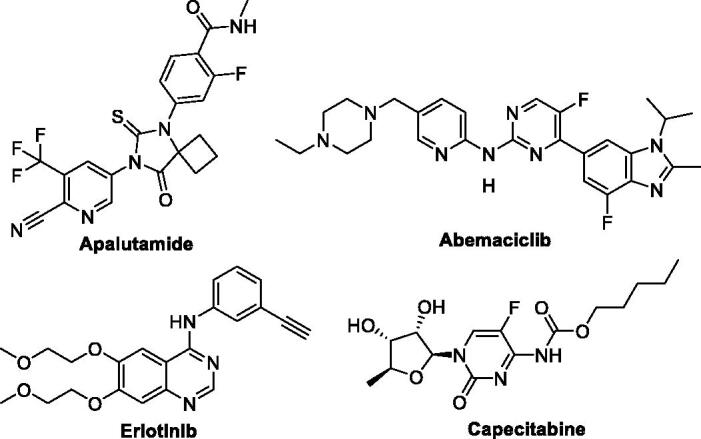
Pyridine and pyrimidines-tethered antitumor drugs.
Because of the previous findings and our ongoing research regarding the synthesis of pharmaceutically important pyridines and pyrimidines40, the impact of the most active compounds was investigated on the cell cycle and apoptosis by the tumour suppressor p53 to put eyes on their effects on cancer biology assuring the proposed mechanism of action. The current work sheds light on the utility of studied compounds as lead compounds for further investigations as anticancer agents.
PKs are one of the most important families contributing to a large number of diseases like inflammation, diabetes, and/or cancer41. PKs constitute one of the apparent and attractive targets for the treatment of many diseases as they regulate a lot of cellular functions, such as apoptosis, proliferation, metabolism, survival, cell cycle, and DNA damage/repair42. The literature revealed some promising pyrazolopyridine derivatives (compounds I–III) reported by Hamajima et al. with inhibitory potential against PI3Kd37. Besides, some promising pyrazolopyridine derivatives (compounds IV–VI) were reported by Orlikova et al. with inhibitory potential against NF-kB39 (Figure 2).
Figure 2.

Some reported pyrazolopyridine derivatives and their IC50 values as anticancer and kinase inhibitors.
Moreover, EGFR is one of the outstanding tyrosine kinase receptors. It regulates several pathways of signal transduction to regulate cell proliferation and apoptosis. Also, it is overexpressed in many cancer types, such as ovarian, colon, and breast by activating the process of angiogenesis43. EGFR inhibitors (EGFRIs), such as erlotinib (Figure 3) were approved by the FDA in 2004 for clinical use as an anticancer drug44.
Figure 3.
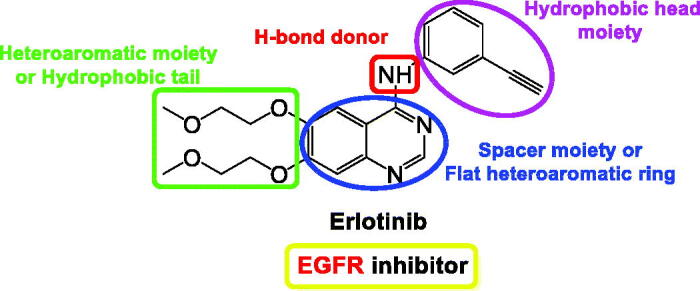
The common pharmacophoric properties of the FDA-approved EGFRI (erlotinib).
The common pharmacophoric properties of EGFRI (erlotinib) are depicted in Figure 3. The first one is the presence of a hydrophobic moiety to act as a head occupying the first hydrophobic region. The second feature is the presence of an H-bond donor in the spacer region occupying the linker region between the adenine binding region and the hydrophobic region I. The third pharmacophoric feature of EGFRIs is required to be a flat heteroaromatic moiety to be able to occupy the binding pocket of adenine (hinge segment). Moreover, a second heteroaromatic or hydrophobic moiety is required to act as a tail, occupying EGFR’s second hydrophobic region45–48.
Rationale-based design
Relying on the basic pharmacophoric properties of EGFRIs represented in Figure 3, we decided to propose the tested pyrimidine and pyridine derivatives as potential EGFRIs.
Guided by the above-discussed pharmacophoric features of EGFRIs which are a hydrophobic moiety to act as a head occupying the first hydrophobic region, an H-bond donor in the spacer region occupying the linker region between the adenine binding region and the hydrophobic region I, a flat heteroaromatic moiety to be able to occupy the binding pocket of adenine (hinge segment), and a second heteroaromatic or hydrophobic moiety to act as a tail and staying at EGFR’s second hydrophobic region45.
Herein, the rationale-based design was based on the presence of a pyrimidine or pyridine ring to be inserted into the binding pocket of adenine and act as a flat heteroaromatic moiety. Also, both the thiophene and furan rings were proposed to act as a head to occupy the first hydrophobic region and a tail to occupy the second hydrophobic region of EGFR, respectively. However, the second pharmacophoric feature, which is an H-bond donor in the spacer region, was observed to be either amino, hydroxy, carboxy, or protonated nitrogen atom (Figure 4).
Figure 4.

Schematic illustration disclosing the pharmacophoric features of the afforded pyrimidine and pyridine derivatives as EGFRIs.
Results and discussion
Chemistry
The pyridine and pyrimidine derivatives (7–21) have been prepared by the general synthetic routes (Schemes 1–3). The chalcone1-(furan-2-yl)-3-(thiophen-2-yl) chalcone 140 was synthesised and utilised as an intermediate to get the desired targets. Initially, compounds (7–17) were prepared through the treatment of 1 with various primary heteroaryl amines attached at the α-site related to the ring nitrogen (1,3-N,N nucleophiles) (2–5), namely: 4-((4-nitrophenyl)diazenyl)-1H-pyrazole-3,5-diamine, 5-amino-1, 2-dihydro-3H-pyrazolo-3-one, 3-amino-1,2,4-triazole, and 5-amino-1,2,3,4-tetrazole monohydride, respectively, in refluxing DMF and in presence of Alc. KOH (Scheme 1). Otherwise, 8 was also prepared in pyridine. Also, compound 1 was cyclised with 2-mercaptobinzemidzole 6 in the same basic conditions. Pyridine derivatives (13 and 14) were synthesised in good yield by further cyclisation of chalcone 4 with 3-cyanoacetyl indole, ethylcyanoacetate, or cyanoacetamide, respectively. Consequently, the nucleophilic substitution of the 2-pyridone derivative 14 with chlorine using POCl3/PCl5 to afford the 2-chloropyridine derivative 15 which by subsequent cyclisation with sodium azide furnished the tetrazolo derivative 16 in a 53% yield. While condensation of 15 with hydrazine hydrate in dioxane yielded the hydrazine derivative 17.
Scheme 1.

The detailed synthesis of the afforded fused pyrimidine derivatives (7–11); Reagents and conditions: i) KOH, DMF, reflux; ii) Pyridine, reflux.
Scheme 2.

The detailed synthesis of the pyridine derivatives (14–17); Reagents and conditions: i) Amm. acetate, Acetic acid, reflux 12 h; ii) Amm. acetate, butanol, reflux 5 h; iii) POCl3, PCl5, heating 10 h; iv) NaN3, DMF, reflux 8 h; ii) NH2NH2, dioxane, reflux 12 h.
Scheme 3.

The detailed synthesis of the afforded pyridine candidates (18–21); Reagents and conditions: i) DMF, K2CO3, reflux 8 h; ii) Ethanol, reflux 6 h; iii) Ethanol, Acetic acid, reflux 2 h (ii) Ethanol, reflux 2 h.
Finally, the alkylation of 14 with ethyl chloroacetate was carried out under alkaline conditions at room temperature, giving the ethyl acetate derivative 18. However, compound 18 was used as an intermediate for the synthesis of Schiff bases 20 and 21 firstly via its condensation with hydrazine hydrate to give the hydrazide derivative 19 and then treatment of 19 with 4-nitrobenzaldehyde and isatin in boiling ethyl alcohol, respectively, as outlined in Scheme 2. Spectral and analytical measurements were used to confirm their structures.
Biological evaluations
Cytotoxicity screening against human cancer cell lines
It was revealed that all investigated cell lines were affected by the afforded pyrimidine and pyridine derivatives at different concentrations (5, 12.5, 25, and 50 μg/mL). Table 1 and Figure 5 show that samples have IC50 on all tested cell lines, which are (MCF7, HEPG2, HEP2, HCT116, Caco2, H460, FaDu, and Vero) after 48 h.
Table 1.
Pyrimidine and pyridine IC50 (µg/mL) on different cell lines.
| Sample | MCF7 |
T47D |
VERO |
H460 |
FADU |
HEP2 |
HEPG2 |
HCT116 |
CaCo2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | IC50 (µg/mL) |
SD | |
| 7 | 13.70 | 1.76 | 44.50 | 0.70 | 12.00 | 1.35 | 3.70 | 0.42 | 8.30 | 0.98 | 35.20 | 0.49 | 12.00 | 1.41 | 15.00 | 1.41 | 21.50 | 0.70 |
| 8 | 4.10 | 0.14 | 7.50 | 0.70 | 3.90 | 0.14 | 11.70 | 1.06 | 5.60 | 0.14 | 7.90 | 0.14 | 3.80 | 0.28 | 7.20 | 0.28 | 12.40 | 0.84 |
| 9 | 17.20 | 0.35 | 11.50 | 0.70 | 32.50 | 0.70 | 43.20 | 1.06 | 19.60 | 0.49 | 14.70 | 0.35 | 18.50 | 2.12 | 24.30 | 0.91 | 23.50 | 0.70 |
| 10 | 18.70 | 1.06 | 18.20 | 0.35 | 28.50 | 2.12 | 2.90 | 0.14 | 4.50 | 0.70 | 22.80 | 0.21 | 7.30 | 0.91 | 33.30 | 0.91 | -- | -- |
| 11 | 15.90 | 0.14 | 19.50 | 0.70 | 24.00 | 1.41 | 4.30 | 0.98 | 18.20 | 1.06 | 19.50 | 0.70 | 18.50 | 2.12 | 42.00 | 2.82 | 24.80 | 0.28 |
| 13 | 7.80 | 0.07 | 21.00 | 1.41 | – | – | – | – | 15.90 | 0.14 | 4.80 | 0.21 | 8.60 | 0.49 | 44.30 | 0.91 | -- | -- |
| 14 | 3.70 | 0.14 | 4.70 | 0.29 | 4.60 | 0.40 | 8.00 | 0.70 | 3.90 | 0.14 | 4.70 | 0.42 | 3.80 | 0.21 | 3.90 | 0.14 | 4.10 | 0.21 |
| 15 | 14.70 | 1.06 | 49.40 | 0.84 | 21.50 | 0.70 | 21.00 | 1.41 | 17.50 | 0.70 | 36.80 | 0.28 | 12.00 | 1.45 | 22.00 | 1.41 | 23.50 | 2.12 |
| 16 | 21.00 | 1.41 | 8.90 | 0.14 | 4.50 | 0.07 | 19.40 | 0.77 | 9.00 | 1.41 | 31.00 | 1.42 | 9.70 | 0.35 | 27.00 | 1.41 | 18.20 | 1.06 |
| 17 | 34.00 | 1.41 | 4.90 | 0.14 | 16.50 | 0.70 | 12.70 | 0.35 | 10.50 | 0.70 | 24.00 | 1.41 | 4.90 | 0.14 | 19.30 | 0.91 | 24.00 | 2.82 |
| 18 | 21.50 | 0.70 | 25.50 | 0.70 | 24.50 | 0.70 | 25.00 | 1.41 | 22.00 | 1.41 | 23.0 | 1.40 | 9.00 | 0.03 | 36.70 | 0.42 | 23.50 | 0.70 |
| 19 | 39.50 | 0.70 | 32.00 | 1.41 | 36.00 | 1.31 | 41.50 | 2.12 | 20.50 | 2.12 | 43.50 | 0.70 | 24.20 | 1.06 | – | – | 10.80 | 0.21 |
| 20 | 12.20 | 0.35 | 49.50 | 0.70 | 10.50 | 0.71 | 11.30 | 0.91 | 8.50 | 2.12 | 31.00 | 1.41 | 11.50 | 0.70 | 24.70 | 0.42 | 41.50 | 2.12 |
| 21 | 9.50 | 0.70 | 17.00 | 1.31 | 38.20 | 0.35 | 23.30 | 0.91 | 10.90 | 1.34 | 19.80 | 0.28 | 5.00 | 1.41 | – | – | 34.00 | 1.41 |
| DOX | 4.50 | 1.50 | 4.00 | 1.10 | 23.00 | 3.00 | 6.50 | 1.40 | 7.00 | 1.60 | 5.60 | 1.20 | 4.58 | 1.20 | 5.50 | 1.60 | 6.00 | 1.80 |
Figure 5.
IC50 (µg/mL) of all molecules on different human cancer cell lines following 48 h.


Hence, the IC50 concentrations of 8 and 14 in all cell lines were employed in all the following mechanistic experiments. Therefore, MCF7 3.80 and 7.00 µg/mL, HEPG2 4.00 and 3.60 µg/mL, HEP2 4.40 and 8.00 µg/mL, HCT116 4.00 and 7.40 µg/mL, Caco2 4.30 and 11.80 µg/mL, H460 8.50 and 12.50 µg/mL, FaDu 3.80 and 5.70 µg/mL, and Vero 4.30 and 4.00 µg/mL of compounds (8 and 14), respectively, were used.
Data are represented as the mean of surviving fraction ± SD of three independent experiments performed in five replicates.
From these findings, both compounds 8 and 14 have been chosen to be tested for their antioxidant activities. On the other hand, the antioxidant activities of 8 and 14 were investigated using the most sensitive cell line.
Data are represented as the mean of surviving fraction ± SD of three independent experiments performed in five replicates.
Antioxidant activity
Consequently, both compounds 8 and 14 were tested for their antioxidant activity in MCF7, H460, FADU, HEP2, HEPG2, HCT 116, Caco2, and Vero cells, and the dose used was the IC50 in all cell lines.
Notably, compound 14 produced a pro-oxidant effect in MCF7, H460, HEP2, HEPG2, HCT, and Vero cells by significantly increasing MDA and NO with an apparent decrease in the levels of SOD and GSH. Moreover, compound 14 achieved an anti-oxidant effect in FADU and Caco2. This was confirmed by the increase in the SOD and GSH levels with the decrease in the levels of MDA and NO. Also, compound 8 showed a pro-oxidant effect in all tested cell lines except Caco2 where it produced an antioxidant effect. It significantly increased the MDA and NO levels with an apparent decrease in the levels of SOD and GSH (pro-oxidant). At the same time, it increased the levels of SOD and GSH and decreased the levels of MDA and NO (antioxidant) in the Caco2 cell line as shown in Figures 6–9.
Figure 6.

Results of compounds 8 and 14 on MDA in cell lyses of all tested cells following 48 h.
Figure 7.

Results of compounds 8 and 14 on NO in culture media of all tested cells following 48 h.
Figure 8.

Results of compounds 8 and 14 on GSH in cell lyses of all tested cells following 48 h.
Figure 9.

Results of compounds 8 and 14 on SOD in cell lyses of all tested cells following 48 h.
Apoptosis activity
Furthermore, both compounds 8 and 14 were evaluated for apoptosis using flow cytometry on the HEPG2 cell line. The % rate of total apoptosis for control, compound 8, and compound 14 were 16.73, 22.44, and 48.36 (%), respectively. Notably, both 8 and 14 derivatives showed an apparent increase in the total, early, and late % rates of apoptosis with respect to the control (p = 0.0001) (Figure 10).
Figure 10.

Results of compounds 8 and 14 on apoptosis in HEPG2 cells following 48 h. (A) Control, (B) Compound 8, and (C) Compound 14.
Cell cycle analysis
Furthermore, both 8 and 14 compounds were investigated for their effects on the cell cycle in HEPG2 cells. There was a significant decrease from control (92.7%) in G0/G1 with derivatives 8 (61.4%) and 14 (69.8%) (p = 0.0001). Briefly, it was clear that an apparent increase from control (5.57%) in the G2/M phase was observed for both 8 (34.4%) and 14 (25.9%) derivatives (p = 0.0001) (Figure 11).
Figure 11.

Results of compounds 8 and 14 on the cell cycle of HEPG2 following 48 h. (A) Control, (B) Compound 8, and (C) Compound 14.
EGFR kinase (wild and T790M) inhibition assay
EGFR plays a pivotal role in tumorigenesis. Hence, cancer treatment targeting the EGFR gene has shown great progress. However, not all cancer patients are sensitive to EGFR-tyrosine kinase inhibitors and that could be attributed to EGFR gene mutation49. So, it is important to reveal the efficacy of our investigated compounds against both non-mutagenic EGFR (EGFR-wild type) and mutagenic EGFR (EGFR-T790M). Consequently, the most active compounds (8 and 14) that displayed outstanding anti-proliferative activities towards the utilised cancer cell lines were employed to assess their EGFRI potential. The reagent, Kinase-Glo MAX, was used50, and luminescence was detected by applying the microplate reader. Erlotinib was used as a reference standard in this experiment as shown in Table 2. Accordingly, considering EGFR kinase wild, it was revealed that the investigated compounds showed less inhibitory potential than erlotinib with IC50 values of 0.131 and 0.203 µM for compounds 8 and 14, respectively, whereas, erlotinib exhibited an IC50 value of 0.042 µM. Hence, it was elicited that compounds 8 and 14 experienced eligible inhibitory potential against EGFR kinase (wild) as depicted in Figure 12(A). However, regarding EGFR kinase T790M, it was disclosed that the assessed compounds displayed less inhibitory potential than erlotinib with IC50 values of 0.027 and 0.156 µM for compounds 8 and 14, respectively, whereas, erlotinib exhibited an IC50 value of 0.009 µM. Hence, we can deduce that compounds 8 and 14 could display feasible inhibitory potential against EGFR kinase (T790M) as depicted in Figure 12(B).
Table 2.
The inhibitory potentials of the investigated 8 and 14 targets towards EGFR (Wild) and EGFR (T790M).
| Compound | EGFR IC50 (µM) |
|
|---|---|---|
| Wild | T790M | |
| 8 | 0.131 ± 0.008 | 0.027 ± 0.002 |
| 14 | 0.203 ± 0.012 | 0.156 ± 0.009 |
| Erlotinib | 0.042 ± 0.003 | 0.009 ± 0.001 |
Figure 12.

The bar chart representation reveals the inhibitory levels of the assessed derivatives against (A): EFGR Wild and (B): EGFR T790.
In silico studies
Docking studies
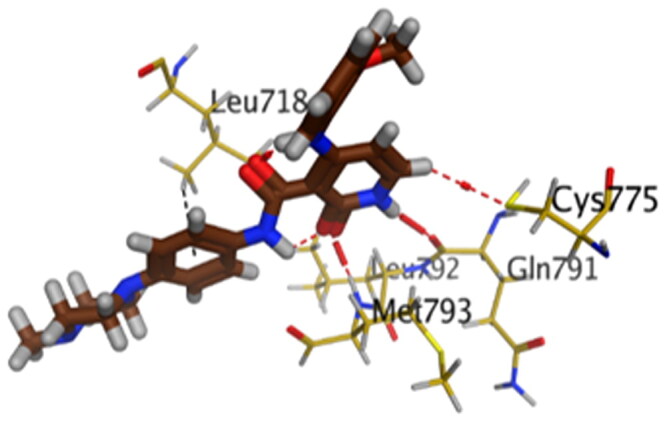
First, the key amino acids required for EGFR-Kinase domain interaction were identified with the aid of co-crystallised downloaded pyridinone ligand (5Q4) interactions. It was revealed that 5Q4 forms four hydrogen bonds with Glu-804, Cys-775, Gln-791, and Met-793, and two ionic bonds with Glu-804 at EGFR-Kinase (Figure 13).
Figure 13.

2D binding interactions of co-crystallised 5Q4 undocked ligand at EGFR-kinase domain.
Accordingly, it was clear that the tested candidates showed diverse binding scores and modes at the EGFR-Kinase domain receptor compared to that of the co-crystallised 5Q4 inhibitor. Hence, concerning its RMSD values and interactions results, synthesised compounds 8 and 14, showed favourable results between tested compounds at the EGFR-Kinase receptor. The synthesised compounds 8 and 14 showed binding interactions nearly similar to that attained by the co-crystallised ligand. The chemically synthesised compound 8 revealed binding interaction to EGFR-Kinase domain through forming two hydrogen bonds with Gln-791 and Met-793, and one pi-H bond with Leu-718 with RMSD = 1.6807, whereas, the chemically synthesised compound 14 interactions revealed its binding with Cys-775 and Met-793 through forming H-bonds, and Gly-796 by a pi-H bond with RMSD = 1.1802. However, the docked 5Q4 was capable of forming three hydrogen bonds with Gln-791, Cys-775, and Met-793, and one pi-H bond with Leu-718 with RMSD = 1.3242 (Table 4).
Table 4.
3D interactions and positioning of the chemically synthesised compounds (8 and 14) and the docked 5Q4 reference at EGFR-Kinase target receptor.
| Comp. | 3D interactions | 3D positioning |
|---|---|---|
| 8 |
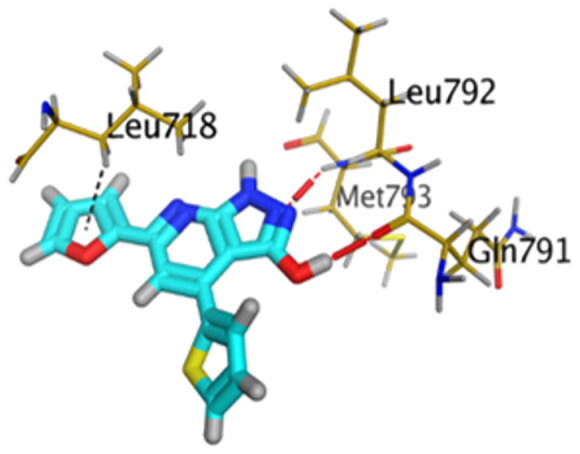
|
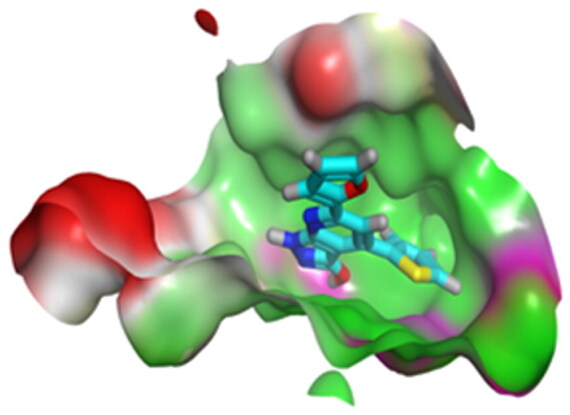
|
| 14 |
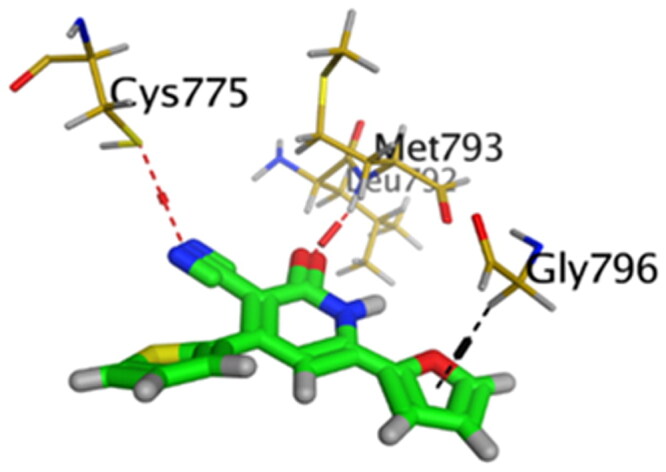
|

|
| 5Q4 |
|
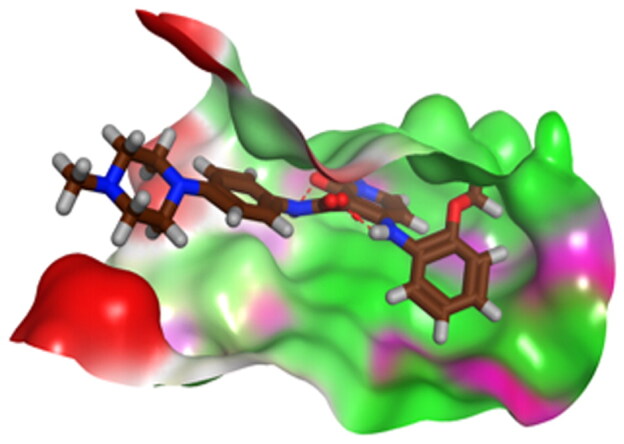
|
Hydrogen bonds are represented in red and H-pi ones are represented in black.
The docking results of compounds 8, 14, and 5Q4 show their interactions and positioning in 3D orientation at the EGFR-kinase domain (Tables 3 and 4). The detailed docking scores, RMSD values, interactions, and visualisation of other candidates are described in the Supplementary data as 2D and 3D interactions, and surface and maps (Tables SI 1–SI 3).
Table 3.
Receptor-binding energies and interactions of compounds 8, 14, and 5Q4 into the 5Q4 pocket of EGFR-kinase.
| Comp. | Sa | Amino acid bond | Distance (Å) |
|---|---|---|---|
| 8 | −5.8756 | Gln-791 (A) H-donor | 2.92 |
| Met-793 (A) H-acceptor | 3.13 | ||
| Leu-718 (A) pi-H | 4.26 | ||
| 14 | −5.5083 | Cys-775 (A) H-donor | 3.52 |
| Met-793 (A) H-acceptor | 3.06 | ||
| Gly-796 (A) pi-H | 4.02 | ||
| 5Q4 | −7.0125 | Gln-791 (A) H-donor | 3.15 |
| Cys-775 (A) H-donor | 4.13 | ||
| Met-793 (A) H-acceptor | 3.04 | ||
| Leu-718 (A) pi-H | 3.95 |
aS: Score of a ligand into the binding pocket of 5Q4 (Kcal/mol).
In silico physicochemical, pharmacokinetic, and ADME studies
The SwissADME website of the Swiss Institute of Bioinformatics (SIB)51 was applied for the estimation and prediction of the physicochemical and pharmacokinetic characteristics of the examined derivatives52–54 (Table 5).
Table 5.
The physicochemical and pharmacokinetic characteristics of the examined derivatives (7–21).
| Comp. | Log Po/w (WLOGP) |
Log S (ESOL) |
Water solubility | GI absorption | BBB permeability | P-gp substrate | Metabolic enzymes inhibition | Drug likeness matching |
|---|---|---|---|---|---|---|---|---|
| 7 | 3.55 | −4.93 | Moderately soluble | Low | No | No | CYP2C19 inhibitor | Lipinski |
| 8 | 3.65 | −3.99 | Soluble | High | No | Yes | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| 9 | 3.11 | −3.52 | Soluble | High | No | No | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| 10 | 1.81 | −3.32 | Soluble | High | No | No | CYP2C19 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| 11 | 5.10 | −5.27 | Moderately soluble | High | No | Yes | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| CYP2C9 inhibitor | ||||||||
| CYP3A4 inhibitor | ||||||||
| 13 | 6.09 | −5.42 | Moderately soluble | Low | No | Yes | CYP1A2 inhibitor | Lipinski, Veber, Muegge |
| CYP2C19 inhibitor | ||||||||
| CYP2C9 inhibitor | ||||||||
| CYP2D6 inhibitor | ||||||||
| CYP3A4 inhibitor | ||||||||
| 14 | 3.24 | −3.10 | Soluble | High | No | No | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| CYP2C9 inhibitor | ||||||||
| CYP2D6 inhibitor | ||||||||
| 15 | 4.60 | −4.40 | Moderately soluble | High | No | Yes | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| CYP2C9 inhibitor | ||||||||
| 16 | 3.90 | −4.42 | Moderately soluble | High | No | No | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| 17 | 3.04 | −3.75 | Soluble | High | No | Yes | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| CYP2C9 inhibitor | ||||||||
| CYP2D6 inhibitor | ||||||||
| CYP3A4 inhibitor | ||||||||
| 18 | 3.88 | −4.11 | Moderately soluble | High | No | No | CYP1A2 inhibitor | Lipinski, Ghose, Veber, Egan, Muegge |
| CYP2C19 inhibitor | ||||||||
| CYP2C9 inhibitor | ||||||||
| CYP3A4 inhibitor | ||||||||
| 19 | 2.31 | −3.04 | Soluble | Low | No | Yes | CYP1A2 inhibitor | Lipinski, Ghose, Muegge |
| CYP2C9 inhibitor | ||||||||
| CYP3A4 inhibitor | ||||||||
| 20 | 4.38 | −5.05 | Moderately soluble | Low | No | No | CYP2C19 inhibitor | Lipinski, Ghose |
| CYP2C9 inhibitor | ||||||||
| 21 | 3.22 | −5.19 | Moderately soluble | Low | No | No | CYP2C19 inhibitor | Lipinski, Ghose |
| CYP2C9 inhibitor |
The aim is to confirm that the most favourable compounds in molecular modelling studies (compounds 8 and 14) are promising candidates regarding their pharmacokinetic properties. Compounds 8 and 14 exhibited predicted wlogP values of 3.65 and 3.24, respectively, with no blood–brain barrier (BBB) permeability and so no CNS side effects are predicted. Both compounds 8 and 14 showed high GIT absorption with reasonable H2O solubility. Moreover, compound 8 is a substrate for P-glycoprotein (PGP+) whereas compound 14 is not a substrate for it (PGP−), so it is not subjected to the efflux mechanism as a drug-resistance mechanism used by many tumour cell lines. Also, compound 8 manifests inhibition for the metabolising enzymes (CYP1A2 and CYP2C19) only. Whereas compound 14 is capable of inhibiting CYP1A2, CYP2C19, CYP2C9, and CYP2D6 metabolising enzymes. Compounds 8 and 14 were in agreement with Lipinski, Veber, Ghose, Muegge, and Egan rules.
Structure–activity relationship (SAR) study
To attain deep insights and get one step closer to understanding the results of chemical modifications on the activity of studied derivatives on their activities towards the EGFR target site. We decided to conclude and analyse a SAR study based on their effects on the different cell lines used as depicted in Figure 14. The following interesting outcomes were unveiled:
Figure 14.

The suggested SAR for the studied pyrimidine and pyridine derivatives.
It was revealed that the best activity towards EGFR was attained by pyridine derivatives. In particular, it was found that the best activity was attained by pyridine derivatives with keto and cyano substitutions at positions 2 and 3, respectively (compound 14), or by fusion with 3-hydroxy pyrazole (compound 8).
Moreover, eligible activities were attained by some pyridine derivatives retaining imidazole and triazole substituents (compounds 7 and 10) and pyrimidine derivatives retaining cyano group at position 3 (compounds 13, 17, and 21) or tetrazole ring (compound 16).
On the other hand, it is worth noting that moderate activities were attained by pyridine derivatives retaining pyrazole (compound 9) and pyridine derivatives retaining fused indole with sulphur atom incorporated in the pyridine ring (compound 11), cyano and chloro groups (compound 15), cyano and oxy ethyl acetate ester (compound 18), cyano and oxy acetohydrazide (compound 19), as well as cyano and bulk group (compound 20).
Conclusion
Briefly, a series of chemically synthesised pyridine and pyrimidine derivatives were screened for their anticancer activities via EGFR inhibition. Among the investigated compounds, both 8 and 14 displayed outstanding effects on the tested cell lines with IC50 concentrations of 3.8 and 7 µg/mL (MCF7), 4 and 3.6 µg/mL (HEPG2), 4.4 and 8 µg/mL (HEP2), 4 and 7.4 µg/mL (HCT116), 4.3 and 11.8 µg/mL (CACO), 8.5 and 12.5 µg/mL (H460), 3.8 and 5.7 µg/mL (FaDu), and 4.3 and 4 µg/mL (VERO), respectively. Besides, compound 14 induced a pro-oxidant state in H460, MCF7, HEP2, HEPG2, HCT, and VERO by significantly increasing the MDA and NO. On the other hand, a significant decrease in SOD and GSH levels was observed. Additionally, compounds 8 and 14 achieved a significant increase in apoptosis percentage (total, early, and late) compared to control. Furthermore, there was a significant decrease in the G0/G1 phase with an apparent increase in the S and G2/M phases with both compounds 8 and 14 (p = 0.0001). Besides, the EGFR kinase (Wild and T790M) inhibitory potential of the most active compounds (8 and 14) assured the rational and the mode of action suggested in this current work. Moreover, the molecular docking study performed ensured the outstanding anticancer activities of the investigated compounds by declaring their binding interactions with the EGFR target receptor. Finally, eligible physicochemical/pharmacokinetics properties, and drug/lead likeness, were obtained.
Materials and methods
Chemistry
The reactions’ progress and the compounds’ purity were checked using thin-layer chromatography (TLC), which were monitored using UV light at 365 and 254 nm. Also, all spectral data were recorded according to our previous study40.
Biological evaluations
Cytotoxicity screening against human cancer cell lines
In this study, a panel of cell lines was examined for their chemosensitivity. Different concentrations of the synthesised pyridines and pyrimidine derivatives were used in this study (5, 12.5, 25, and 50 µg/mL) for all the examined cell lines. In this study, breast tumour cell line (MCF7), hepatocellular carcinoma cell line (HEPG2), larynx cell line (HEP2), lung cancer cell line (H460), colon cancer cell line (HCT116 and Caco2), hypopharyngeal cell line (FADU), and normal Vero cell line were applied. The full methodology is described in the Supplementary data (SI 1).
Oxidative stress assessment
Determination of malondialdehyde content (MDA)
Determination of superoxide dismutase (SOD)
Determination of reduced glutathione (GSH) content
Determination of nitric oxide (NO) content
Cell cycle analysis and apoptosis assay
Liver cancer cells (HEPG2) were seeded in RPMI-1640 media at a density of 250 X 103 cells/mL. Both cell cycle and apoptosis evaluations were performed at 3.8 µg/mL for both compounds 8 and 14. The detailed method is described in the Supplementary data (SI 3).
EGFR kinase (wild and T790M) inhibition assay
The most promising cytotoxic compounds (8 and 14) were finally assessed for their inhibitory potential against both EGFR Wild and EGFR T790M. The assay protocol is fully described in the Supplementary data (SI 4).
In silico studies
Docking studies
The MOE 2019.010255–57 was used to examine the binding affinities of the chemically synthesised derivatives on the EGFR through molecular docking. Accordingly, we could reveal the anticancer inhibitory potential of these compounds as promising EGFRIs. Also, the co-crystallised 5Q4 pyridinone inhibitor was inserted in the docking process as a reference standard.
Examined compounds preparation
The chemical structures of the examined chemically synthesised compounds were drawn by ChemDraw. Using MOE, the previous structures (7–21) were prepared for docking as described earlier58–60. The synthesised compounds under investigation and 5Q4 were saved into the same database for the docking step.
EGFR-Kinase receptor preparation
The Protein Data Bank was searched to give the crystal structure of the EGFR kinase domain (code: 5EM8)61. The preparation process was performed as discussed before62–64. The program default items were followed as before65–67.
Docking of the tested molecules to EGFR-kinase receptor
Docking of the database composed of compounds (7–17) and the reference 5Q4 ligand at the EGFR-Kinase domain was carried out. The applied methodology was performed as discussed in detail68–70. The MOE specifications were modified as previously mentioned71–73. The selected poses were based on their scores and RMSD accordingly74–76.
Furthermore, low RMSD values between the conformations of the redocked and crystal 5Q4 ligand indicated a valid performance in a validation process77–79.
Physicochemical, pharmacokinetic, and ADME studies
The Swiss ADME supplied from the SIB51 was used for the physicochemical, pharmacokinetic, and ADME studies of the target compounds41,80,81.
Statistical analysis
All the previously presented results are the mean ± SD of three separate experiments, which were performed in duplicates. The statistical significance of the results was analysed using one-way ANOVA followed by Tukey’s multiple comparison test. A significantly different from control and p < 0.05.
Supplementary Material
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Small Groups Project under grant number (RGP.1/346/43). The authors extend their appreciation to The Deanship of Scientific Research at Princess Nourah Bint Abdulrahman University Researchers Supporting Project number (PNURSP2022R25), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Taher RF, Al-Karmalawy AA, Abd El Maksoud AI, Khalil H, Hassan A, El-Khrisy E-DA, El-Kashak W.. Two new flavonoids and anticancer activity of Hymenosporum flavum: in vitro and molecular docking studies. J Herbmed Pharmacol. 2021;10(4):443–458. [Google Scholar]
- 2.Ghanem A, Al-Karmalawy AA, Abd El Maksoud AI, Hanafy SM, Emara HA, Saleh RM, Elshal MF.. Rumex Vesicarius L. extract improves the efficacy of doxorubicin in triple-negative breast cancer through inhibiting Bcl2, mTOR, JNK1 and augmenting p21 expression. Inf Med Unlocked. 2022; 29:100869. [Google Scholar]
- 3.Diab RT, Abdel-Sami ZK, Abdel-Aal EH, Al-Karmalawy AA, Abo-Dya NE.. Design and synthesis of a new series of 3,5-disubstituted-1,2,4-oxadiazoles as potential colchicine binding site inhibitors: antiproliferative activity, molecular docking, and SAR studies. New J Chem. 2021;45(46):21657–21669. [Google Scholar]
- 4.Reda R, Al-Karmalawy AA, Alotaibi M, Saleh M.. Quinoxaline derivatives as a promising scaffold for breast cancer treatment. New J Chem. 2022;46:9975–9984. [Google Scholar]
- 5.Khalifa MM, Al-Karmalawy AA, Elkaeed EB, Nafie MS, Tantawy MA, Eissa IH, Mahdy HA.. Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: design, synthesis, anti-proliferative, docking, and in vivo studies. J Enzyme Inhib Med Chem. 2022;37(1):299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Warhi T, Elbadawi MM, Bonardi A, Nocentini A, Al-Karmalawy AA, Aljaeed N, Alotaibi OJ, Abdel-Aziz HA, Supuran CT, Eldehna WM.. Design and synthesis of benzothiazole-based SLC-0111 analogues as new inhibitors for the cancer-associated carbonic anhydrase isoforms IX and XII. J Enzyme Inhib Med Chem. 2022;37(1):2635–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christie E, Bowtell D.. Acquired chemotherapy resistance in ovarian cancer. Ann Oncol. 2017;28(8):viii13–viii15. [DOI] [PubMed] [Google Scholar]
- 8.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7(2):165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munikrishnappa CS, Suresh Kumar GV, Bhandare RR, Konidala SK, Sigalapalli DK, Vaishnav Y, Chinnam S, Yasin H, Al-Karmalawy AA, Shaik AB.. Multistep synthesis and screening of heterocyclic tetrads containing furan, pyrazoline, thiazole and triazole (or oxadiazole) as antimicrobial and anticancer agents. J Saudi Chem Soc. 2022;26(3):101447. [Google Scholar]
- 10.Hammoud MM, Nageeb AS, Morsi MA, Gomaa EA, Abo Elmaaty A, Al-Karmalawy AA.. Design, synthesis, biological evaluation, and SAR studies of novel cyclopentaquinoline derivatives as DNA intercalators, topoisomerase II inhibitors, and apoptotic inducers. New J Chem. 2022;46(23):11422–11436. [Google Scholar]
- 11.El-Helby AGA, Sakr H, Eissa IH, Abulkhair H, Al-Karmalawy AA, El-Adl KJAP.. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors. Arch Pharm Chem Life Sci. 2019;352(10):1900113. [DOI] [PubMed] [Google Scholar]
- 12.El-Helby AGA, Sakr H, Eissa IH, Al-Karmalawy AA, El-Adl K.. Benzoxazole/benzothiazole-derived VEGFR-2 inhibitors: design, synthesis, molecular docking, and anticancer evaluations. Arch Pharm Chem Life Sci. 2019;352(12):1900178. [DOI] [PubMed] [Google Scholar]
- 13.Huang M, Shen A, Ding J, Geng M.. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci. 2014;35(1):41–50. [DOI] [PubMed] [Google Scholar]
- 14.El-Shershaby MH, Ghiaty A, Bayoumi AH, Al-Karmalawy AA, Husseiny EM, El-Zoghbi MS, Abulkhair HS.. From triazolophthalazines to triazoloquinazolines: a bioisosterism-guided approach toward the identification of novel PCAF inhibitors with potential anticancer activity. Bioorg Med Chem. 2021;42:116266. [DOI] [PubMed] [Google Scholar]
- 15.Olayioye MA, Neve RM, Lane HA, Hynes NE.. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19(13):3159–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaki AA, Al-Karmalawy AA, Khodir AE, El-Amier YA, Ashour A.. Isolation of cytotoxic active compounds from Reichardia tingitana with investigation of apoptosis mechanistic induction: in silico, in vitro, and SAR studies. S Afr J Bot. 2022;144:115–123. [Google Scholar]
- 17.Elmaaty AA, Darwish KM, Chrouda A, Boseila AA, Tantawy MA, Elhady SS, Shaik AB, Mustafa M, Al-Karmalawy AA.. In silico and in vitro studies for benzimidazole anthelmintics repurposing as VEGFR-2 antagonists: novel mebendazole-loaded mixed micelles with enhanced dissolution and anticancer activity. ACS Omega. 2022;7(1):875–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Naggar AM, Hassan AMA, Elkaeed EB, Alesawy MSA, Karmalawy AA.. Design, synthesis, and SAR studies of novel 4-methoxyphenyl pyrazole and pyrimidine derivatives as potential dual tyrosine kinase inhibitors targeting both EGFR and VEGFR-2. Bioorg Chem. 2022;123:105770. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs-Tarlovsky V. Role of antioxidants in cancer therapy. Nutrition. 2013;29(1):15–21. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Qi Y, Lai N, Zhang J, Chen Z, Liu M, Zhang W, Luo R, Kang S.. The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res. 2018;37(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohareb R, Fahmy S.. Reaction of malononitrile and ethyl cyanoacetate: a novel synthesis of polyfunctional pyridine derivatives. Zeitschrif Naturforschung B. 1985;40(11):1537–1540. [Google Scholar]
- 22.Khalaf H, Tolan H, El-Bayaa M, Radwan M, El-Manawaty M, El-Sayed W.. Synthesis and anticancer activity of new pyridine-thiophene and pyridine-furan hybrid compounds, their sugar hydrazone, and glycosyl derivatives. Russ J Gen Chem. 2020;90(9):1706–1715. [Google Scholar]
- 23.Sharma V, Chitranshi N, Agarwal AK.. Significance and biological importance of pyrimidine in the microbial world. Int J Med Chem. 2014;2014:202784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maddila S, Gorle S, Seshadri N, Lavanya P, Jonnalagadda SB.. Synthesis, antibacterial and antifungal activity of novel benzothiazole pyrimidine derivatives. Arabian J Chem . 2016;9(5):681–687. [Google Scholar]
- 25.El-Sharkawy KA, AlBratty MM, Alhazmi HA.. Synthesis of some novel pyrimidine, thiophene, coumarin, pyridine and pyrrole derivatives and their biological evaluation as analgesic, antipyretic and anti-inflammatory agents. Braz J Pharm Sci. 2018;54(4). [Google Scholar]
- 26.De Luca M, Ioele G, Ragno G.. 1, 4-Dihydropyridine antihypertensive drugs: Recent advances in photostabilization strategies. Pharmaceutics. 2019;11(2):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaminski K, Obniska J, Zagorska A, Maciag D.. Synthesis, physicochemical and anticonvulsant properties of new N-(pyridine-2-yl) derivatives of 2-azaspiro [4.4] nonane and [4.5] decane-1, 3-dione. Part II. Arch Pharm (Weinheim). 2006;339(5):255–261. [DOI] [PubMed] [Google Scholar]
- 28.Wang N, Yang Q, Deng Z, Mao X, Peng Y.. Rhodium-catalyzed merging of 2-arylquinazolinone and 2, 2-difluorovinyl tosylate: diverse synthesis of monofluoroolefin quinazolinone derivatives. ACS Omega. 2020;5(24):14635–14644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar S, Narasimhan B.. Therapeutic potential of heterocyclic pyrimidine scaffolds. Chem Cent J. 2018;12(1):1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HW, Kim BY, Ahn JB, Kang SK, Lee JH, Shin JS, Ahn SK, Lee SJ, Yoon SS.. Molecular design, synthesis, and hypoglycemic and hypolipidemic activities of novel pyrimidine derivatives having thiazolidinedione. Eur J Med Chem. 2005;40(9):862–874. [DOI] [PubMed] [Google Scholar]
- 31.Davari AS, Abnous K, Mehri S, Ghandadi M, Hadizadeh F.. Synthesis and biological evaluation of novel pyridine derivatives as potential anticancer agents and phosphodiesterase-3 inhibitors. Bioorg Chem. 2014;57:83–89. [DOI] [PubMed] [Google Scholar]
- 32.Prachayasittikul S, Pingaew R, Worachartcheewan A, Sinthupoom N, Prachayasittikul V, Ruchirawat S, Prachayasittikul V.. Roles of pyridine and pyrimidine derivatives as privileged scaffolds in anticancer agents. Mini Rev Med Chem. 2017;17(10):869–901. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Wu H, Song G, Yang D, Huang J, Yao X, Qin H, Chen Z, Xu Z, Xu C.. A novel imidazopyridine derivative exerts anticancer activity by inducing mitochondrial pathway-mediated apoptosis. Biomed Res Int. 2020;2020:4929053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdelaziz ME, El-Miligy MM, Fahmy SM, Mahran MA, Hazzaa AA.. Design, synthesis and docking study of pyridine and thieno [2, 3-b] pyridine derivatives as anticancer PIM-1 kinase inhibitors. Bioorg Chem. 2018;80:674–692. [DOI] [PubMed] [Google Scholar]
- 35.Abouzid KA, Al-Ansary GH, El-Naggar AM.. Eco-friendly synthesis of novel cyanopyridine derivatives and their anticancer and PIM-1 kinase inhibitory activities. Eur J Med Chem. 2017;134:357–365. [DOI] [PubMed] [Google Scholar]
- 36.Cheney IW, Yan S, Appleby T, Walker H, Vo T, Yao N, Hamatake R, Hong Z, Wu JZ.. Identification and structure–activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg Med Chem Lett. 2007;17(6):1679–1683. [DOI] [PubMed] [Google Scholar]
- 37.Hamajima T, Takahashi F, Kato K, Mukoyoshi K, Yoshihara K, Yamaki S, Sugano Y, Moritomo A, Yamagami K, Yokoo K, et al. Discovery and biological evaluation of novel pyrazolopyridine derivatives as potent and orally available PI3Kδ inhibitors. Bioorg Med Chem. 2018;26(9):2410–2419. [DOI] [PubMed] [Google Scholar]
- 38.Ibrahim DA, Ismail NS.. Design, synthesis and biological study of novel pyrido [2, 3-d] pyrimidine as anti-proliferative CDK2 inhibitors. Eur J Med Chem. 2011;46(12):5825–5832. [DOI] [PubMed] [Google Scholar]
- 39.Orlikova B, Chaouni W, Schumacher M, Aadil M, Diederich M, Kirsch G.. Synthesis and bioactivity of novel amino-pyrazolopyridines. Eur J Med Chem. 2014;85:450–457. [DOI] [PubMed] [Google Scholar]
- 40.Radwan MA, Alshubramy MA, Abdel-Motaal M, Hemdan BA, El-Kady DS.. Synthesis, molecular docking and antimicrobial activity of new fused pyrimidine and pyridine derivatives. Bioorg Chem. 2020; 96:103516. [DOI] [PubMed] [Google Scholar]
- 41.Elia SG, Al-Karmalawy AA, Nasr MY, Elshal MF.. Loperamide potentiates doxorubicin sensitivity in triple-negative breast cancer cells by targeting MDR1 and JNK and suppressing mTOR and Bcl-2: In vitro and molecular docking study. J Biochem Mol Tox. 2022;36(1):e22938. [DOI] [PubMed] [Google Scholar]
- 42.El-Azab MF, Al-Karmalawy AA, Antar SA, Hanna PA, Tawfik KM, Hazem RM.. A novel role of Nano selenium and sildenafil on streptozotocin-induced diabetic nephropathy in rats by modulation of inflammatory, oxidative, and apoptotic pathways. Life Sci. 2022;303:120691. [DOI] [PubMed] [Google Scholar]
- 43.Sigismund S, Avanzato D, Lanzetti L.. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12(1):3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R.. FDA drug approval summary: erlotinib (Tarceva®) tablets. Oncologist. 2005;10(7):461–466. [DOI] [PubMed] [Google Scholar]
- 45.Gaber AA, El-Morsy AM, Sherbiny FF, Bayoumi AH, El-Gamal KM, El-Adl K, Al-Karmalawy AA, Ezz Eldin RR, Saleh MA, Abulkhair HS.. Pharmacophore-linked pyrazolo[3,4-d]pyrimidines as EGFR-TK inhibitors: synthesis, anticancer evaluation, pharmacokinetics, and in silico mechanistic studies. Arch Pharm. 2021:e2100258. [DOI] [PubMed] [Google Scholar]
- 46.Gaber AA, Bayoumi AH, El-Morsy AM, Sherbiny FF, Mehany AB, Eissa IH.. Design, synthesis and anticancer evaluation of 1H-pyrazolo [3, 4-d] pyrimidine derivatives as potent EGFRWT and EGFRT790M inhibitors and apoptosis inducers. Bioorg Chem. 2018;80:375–395. [DOI] [PubMed] [Google Scholar]
- 47.Mowafy S, Galanis A, Doctor ZM, Paranal RM, Lasheen DS, Farag NA, Jänne PA, Abouzid KA.. Toward discovery of mutant EGFR inhibitors; design, synthesis and in vitro biological evaluation of potent 4-arylamino-6-ureido and thioureido-quinazoline derivatives. Bioorg Med Chem. 2016;24(16):3501–3512. [DOI] [PubMed] [Google Scholar]
- 48.Othman IM, Alamshany ZM, Tashkandi NY, Gad-Elkareem MA, Anwar MM, Nossier ES.. New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: synthesis, anticancer, antimicrobial evaluation and computational studies. Bioorg Chem. 2021;114:105078. [DOI] [PubMed] [Google Scholar]
- 49.Zheng H, Zhang Y, Zhan Y, Liu S, Lu J, Feng J, Wu X, Wen Q, Fan S.. Prognostic analysis of patients with mutant and wild-type EGFR gene lung adenocarcinoma. CMAR. 2019;11:6139–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura JL. The epidermal growth factor receptor in malignant gliomas: pathogenesis and therapeutic implications. Expert Opin Ther Targets. 2007;11(4):463–472. [DOI] [PubMed] [Google Scholar]
- 51.Daina A, Michielin O, Zoete V.. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elebeedy D, Badawy I, Elmaaty AA, Saleh MM, Kandeil A, Ghanem A, Kutkat O, Alnajjar R, Abd El Maksoud AI, Al-Karmalawy AA.. In vitro and computational insights revealing the potential inhibitory effect of Tanshinone IIA against influenza A virus. Comput Biol Med. 2022;141:105149. [DOI] [PubMed] [Google Scholar]
- 53.El-Demerdash A, Al-Karmalawy AA, Abdel-Aziz TM, Elhady SS, Darwish KM, Hassan AHE.. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021;11(50):31339–31363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El Gizawy HA, Boshra SA, Mostafa A, Mahmoud SH, Ismail MI, Alsfouk AA, Taher AT, Al-Karmalawy AA.. Pimenta dioica (L.) Merr. bioactive constituents exert anti-SARS-CoV-2 and anti-inflammatory activities: molecular docking and dynamics, in vitro, and in vivo studies. Molecules. 2021;26(19):5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salem MA, Aborehab NM, Al-Karmalawy AA, Fernie AR, Alseekh S, Ezzat SM.. Potential valorization of edible nuts by-products: exploring the immune-modulatory and antioxidants effects of selected nut shells extracts in relation to their metabolic profiles. Antioxidants. 2022;11(3):462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hammoud MM, Khattab M, Abdel-Motaal M, Van der Eycken J, Alnajjar R, Abulkhair H, Al-Karmalawy AA.. Synthesis, structural characterization, DFT calculations, molecular docking, and molecular dynamics simulations of a novel ferrocene derivative to unravel its potential antitumor activity. J Biomol Struct Dyn . 2022:1–18. [DOI] [PubMed] [Google Scholar]
- 57.Ezz Eldin RR, Saleh MA, Alotaibi MH, Alsuair RK, Alzahrani YA, Alshehri FA, Mohamed AF, Hafez SM, Althoqapy AA, Khirala SK, et al. Ligand-based design and synthesis of N'-Benzylidene-3,4-dimethoxybenzohydrazide derivatives as potential antimicrobial agents; evaluation by in vitro, in vivo, and in silico approaches with SAR studies. J Enzyme Inhib Med Chem. 2022;37(1):1098–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salem MA, El-Shiekh RA, Aborehab NM, Al-Karmalawy AA, Ezzat SM, Alseekh S, Fernie AR.. Metabolomics driven analysis of Nigella sativa seeds identifies the impact of roasting on the chemical composition and immunomodulatory activity. Food Chem. 2023;398:133906. [DOI] [PubMed] [Google Scholar]
- 59.Elagawany M, Elmaaty AA, Mostafa A, Abo Shama NM, Santali EY, Elgendy B, Al-Karmalawy AA.. Ligand-based design, synthesis, computational insights, and in vitro studies of novel N-(5-Nitrothiazol-2-yl)-carboxamido derivatives as potent inhibitors of SARS-CoV-2 main protease. J Enzyme Inhib Med Chem. 2022;37(1):2112–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kutkat O, Moatasim Y, Karmalawy A, Abulkhair AA, Gomaa HS, El-Taweel MR, Abo Shama AN, GabAllah NM, Mahmoud M, Kayali DB, et al. Robust antiviral activity of commonly prescribed antidepressants against emerging coronaviruses: in vitro and in silico drug repurposing studies. Sci Rep. 2022;12(1):12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bryan MC, Burdick DJ, Chan BK, Chen Y, Clausen S, Dotson J, Eigenbrot C, Elliott R, Hanan EJ, Heald R, et al. Pyridones as highly selective, noncovalent inhibitors of T790M double mutants of EGFR. ACS Med Chem Lett. 2016;7(1):100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaki AA, Ashour A, Elhady SS, Darwish KM, Al-Karmalawy AA.. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: molecular docking, molecular dynamics, and SAR studies. J Tradit Complement Med. 2022;12(1):16–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soltan MA, Eldeen MA, Elbassiouny N, Mohamed I, El-Damasy DA, Fayad E, Abu Ali OA, Raafat N, Eid RA, Al-Karmalawy AA.. Proteome based approach defines candidates for designing a multitope vaccine against the nipah virus. Int J Mol Sci. 2021;22(17):9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soltan MA, Elbassiouny N, Gamal H, Elkaeed EB, Eid RA, Eldeen MA, Al-Karmalawy AA.. In silico prediction of a multitope vaccine against Moraxella catarrhalis: reverse vaccinology and immunoinformatics. Vaccines. 2021;9(6):669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shoala T, Al-Karmalawy AA, Germoush MO, ALshamrani SM, Abdein MA, Awad NS.. Nanobiotechnological approaches to enhance potato resistance against potato leafroll virus (PLRV) using glycyrrhizic acid ammonium salt and salicylic acid nanoparticles. Horticulturae. 2021;7(10):402. [Google Scholar]
- 66.Raslan MA, F. Taher R, Al-Karmalawy AA, El-Ebeedy D, Metwaly AG, Elkateeb NM, Ghanem A, Elghaish RA, Abd El Maksoud AI.. Cordyline fruticosa (L.) A. Chev. leaves: isolation, HPLC/MS profiling and evaluation of nephroprotective and hepatoprotective activities supported by molecular docking. New J Chem. 2021;45(47):22216–22233. [Google Scholar]
- 67.Mahmoud DB, Ismail WM, Moatasim Y, Kutkat O, ElMeshad AN, Ezzat SM, El Deeb KS, El-Fishawy AM, Gomaa MR, Kandeil A, et al. Delineating a potent antiviral activity of Cuphea ignea extract loaded nano-formulation against SARS-CoV-2: In silico and in vitro studies. J Drug Deliv Sci Technol . 2021;66:102845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mahmoud DB, Bakr MM, Al-Karmalawy AA, Moatasim Y, El Taweel A, Mostafa A.. Scrutinizing the feasibility of nonionic surfactants to form isotropic bicelles of curcumin: a potential antiviral candidate against COVID-19. AAPS PharmSciTech. 2021;23(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahmoud A, Mostafa A, Al-Karmalawy AA, Zidan A, Abulkhair HS, Mahmoud SH, Shehata M, Elhefnawi MM, Ali MA.. Telaprevir is a potential drug for repurposing against SARS-CoV-2: computational and in vitro studies. Heliyon. 2021;7(9):e07962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mahmoud A, Kotb E, Alqosaibi AI, Al-Karmalawy AA, Al-Dhuayan IS, Alabkari H.. In vitro and in silico characterization of alkaline serine protease from Bacillus subtilis D9 recovered from Saudi Arabia. Heliyon. 2021;7(10):e08148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma C, Taghour MS, Belal A, Mehany ABM, Mostafa N, Nabeeh A, Eissa IH, Al-Karmalawy AA.. Design and synthesis of new quinoxaline derivatives as potential histone deacetylase inhibitors targeting hepatocellular carcinoma: in silico. Front Chem. 2021;9:725135–725135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khattab M, Al-Karmalawy AA.. Computational repurposing of benzimidazole anthelmintic drugs as potential colchicine binding site inhibitors. Future Med Chem. 2021;13(19):1623–1638. [DOI] [PubMed] [Google Scholar]
- 73.Hamed MI, Darwish A, Soltane KM, Chrouda R, Mostafa A, Abo Shama A, Elhady NM, Abulkhair SS, Khodir HS, Elmaaty AE, et al. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021;11(56):35536–35558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al-Karmalawy AA, Farid MM, Mostafa A, Ragheb AY, Mahmoud SH, Shehata M, Shama NMA, GabAllah M, Mostafa-Hedeab G, Marzouk MM.. Naturally available flavonoid aglycones as potential antiviral drug candidates against SARS-CoV-2. Molecules. 2021;26(21):6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abdallah AE, Alesawy MS, Eissa SI, El-Fakharany EM, Kalaba MH, Sharaf MH, Abo Shama NM, Mahmoud SH, Mostafa A, Al-Karmalawy AA, et al. Design and synthesis of new 4-(2-nitrophenoxy)benzamide derivatives as potential antiviral agents: molecular modeling and in vitro antiviral screening. New J Chem. 2021;45(36):16557–16571. [Google Scholar]
- 76.Abo Elmaaty A, Hamed MIA, Ismail MI, Elkaeed EB, Abulkhair HS, Khattab M, Al-Karmalawy AA.. Computational insights on the potential of some NSAIDs for treating COVID-19: priority set and lead optimization. Molecules. 2021;26(12):3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Belal A, Abdel Gawad NM, Mehany ABM, Abourehab MAS, Elkady H, Al-Karmalawy AA, Ismael AS.. Design, synthesis and molecular docking of new fused 1H-pyrroles, pyrrolo[3,2-d]pyrimidines and pyrrolo[3,2-e][1, 4]diazepine derivatives as potent EGFR/CDK2 inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):1884–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Attia EZ, Khalifa BA, Shaban GM, Amin MN, Akil L, Khadra I, Karmalawy AAA, Alnajjar R, Zaki MYW, Aly OM, et al. Potential topoisomerases inhibitors from Asergillus terreus using virtual screening. S Afr J Bot . 2022;149:632–645. [Google Scholar]
- 79.Hammouda MM, Elmaaty AA, Nafie MS, Abdel-Motaal M, Mohamed NS, Tantawy MA, Belal A, Alnajjar R, Eldehna WM, Al-Karmalawy A, et al. Design and synthesis of novel benzoazoninone derivatives as potential CBSIs and apoptotic inducers: in vitro, in vivo, molecular docking, molecular dynamics, and SAR studies. Bioorg Chem. 2022;127:105995. [DOI] [PubMed] [Google Scholar]
- 80.Khattab M, Al-Karmalawy AA.. Revisiting activity of some nocodazole analogues as a potential anticancer drugs using molecular docking and DFT calculations. Front Chem. 2021;9:628398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Elebeedy D, Elkhatib WF, Kandeil A, Ghanem A, Kutkat O, Alnajjar R, Saleh MA, Abd El Maksoud AI, Badawy I, Al-Karmalawy AA.. Anti-SARS-CoV-2 activities of tanshinone IIA, carnosic acid, rosmarinic acid, salvianolic acid, baicalein, and glycyrrhetinic acid between computational and in vitro insights. RSC Adv. 2021;11(47):29267–29286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.