

Abstract
While studying indolylthio glycosides, previously we determined their activation profile that required large excess of activators. This drawback was partially addressed in the present study of N-alkylated SInR derivatives. The activation process was studied by NMR and the increased understanding of the mechanism led to a discovery of different activation pathways taking place with SIn versus SInR derivatives. Also investigated was orthogonality of the SInR leaving groups versus thioglycosides and selective activation of thioimidates over SInR glycosides.
Keywords: Activation, Carbohydrates, Glycosylation, Mechanism, Oligosaccharides
Introduction
Chemical synthesis of glycans and glycoconjugates remains challenging to chemists, and even advanced strategies and technologies can address this challenge only in part.[1] While most syntheses of glycans are accomplished using thioglycosides or O-imidates as glycosyl donors, the development of new efficient methods for chemical glycosylation remains a vibrant area of scientific quest. Following early work by Zinner, Mukaiyama, and Hanessian, our lab became interested in glycosyl thioimidates, glycosyl donors equipped with the SCR1=NR2 leaving group.[2] Among these, S-benzoxazolyl (SBox),[3] S-thiazolinyl (STaz),[4] and S-benzimidazolyl (SBiz)[5] imidates showed great levels of versatility and efficacy, both for single step glycosylations and expeditious strategies for glycan assembly.[6]
Our recent endeavor was dedicated to studying (1H-indol-2-yl)thio (S-indolyl, SIn) glycosides.[7] These compounds are structurally similar both to thioglycosides and to thioimidates, but in terms of reactivity act as neither due to their unusual activation profile. In fact, SIn glycosides were found to be fully orthogonal in respect to ethylthio glycosides.[7] This is because of very different reaction conditions required for their glycosidation and/or activation pathways taking place with the SIn glycosides in respect to other structurally similar compounds. The activation of the SIn leaving group for glycosylation persistently demanded large amounts of reagents, regardless of the activation method employed. For example, the activation required 5.0 equiv. of AgBF4 versus typical 2—3 equiv. used for the activation of thioimidates.[8] Also N-iodosuccinimide (NIS, 4.5 equiv) and TMSOTf (1.0 equiv) or TfOH were needed for the SIn leaving group activation versus a typical NIS (2.0 equiv) and TMSOTf or TfOH (0.2 equiv) needed for the activation of alkyl/aryl thioglycosides.[9] Cooperative silver catalysis required Ag2CO3 (1.0 equiv) and TMSOTf (3.0—4.0 equiv), whereas only catalytic TMSOTf was needed in all of our previous applications of this reaction.[10]
These results were indicative of side processes that were taking place leading to the unusually high consumption of reagents. Reaction monitoring showed that in the presence of NIS, SIn glycosides only start to react upon consumption of 3.0 equiv. of NIS, and the formation of intermediates A and B shown in Scheme 1A has been proven by NMR. A similar study performed in the presence of the silver salt and TMSOTf showed that the SIn leaving group should first tautomerize into a thioimidate C, and then be activated via intermediate D (Scheme 1B). This stepwise pathway was used to explain the demand for multiple equivalents of TMSOTf. With acquiring preliminary understanding of the mechanistic pathways, we wondered whether blocking the nitrogen atom of the SIn leaving group would help to decrease the amounts of reagents required for the activation and hence allow for a more benign glycosylation reaction. Reported herein is our first attempt to modify the SIn leaving group by introducing a stable N-alkyl substituent.
Scheme 1.

A summary of previous mechanistic studies with SIn glycosyl donors.
Results and Discussion
We have selected atom economical methyl and allyl protecting groups for this study. As shown in Scheme 2, N-substituted thioindoles were prepared from the respective N-substituted oxindoles[11] via thionation with P2S5 in tetrahydrofuran (THF).[12] N-Methyl thioindole and N-allyl thioindole were obtained in 88% and 65% yield, respectively. The leaving groups were then introduced by reactions of the respective N-alkylated thioindoles with glycosyl bromides in the presence of potassium carbonate and 18-crown-6 (18-C-6) in dry acetone. Thus, per-O-benzoylated glucosyl bromide 1[13] was converted into SinMe glucosyl donor 2 in 88% yield. Similarly, per-O-acetylated SinMe derivative 4 was prepared from acetobromoglucose 3 in 60% yield. Per-O-benzylated SinMe glucosyl donor 5 was prepared from acetylated derivative 4 using conventional deacetylation — benzylation sequence. This two-step reprotection accomplished in 86% yield served as an indication that the new N-alkylated leaving groups are sufficiently stable to withstand strongly basic reaction conditions associated with these transformations. SinAll glucosyl donors 6 and 8 were prepared in 78% and 61% (α/β=1.6:1) from glucosyl bromides 1 and 7, respectively. SinAll galactosyl donor 10 and mannosyl donor 12 were synthesized from the corresponding per-O-benzoylated glycosyl bromides 9 and 11, in 88% (separable α,β-mixture) and 67% (α-anomer), respectively (Scheme 2).[13] We do not know what triggered the formation of compound 10 as a mixture of anomers. Such factors as relatively higher reactivity of compounds of the D-galacto series and/or post S-glycosylation anomerization could have been driving forces of this process.
Scheme 2.

Synthesis of protected SInMe and SInAll glycosides 2, 4–6, 8, 10, and 12.
With the novel SinR derivatives in hand, we probed their potential as glycosyl donors with primary glycosyl acceptor 13.[14] The key results of this preliminary study are summarized in Table 1. We chose to explore the same activators as those used for regular (NH) thioindole glycosyl donors, i.e. NIS/TfOH, NIS/TMSOTf and Ag2CO3/TMSOTf.[7] Glycosidation of donor 2 with acceptor 13 in the presence of 3.0 equiv. NIS and 0.8 equiv. TfOH was somewhat sluggish (19 h) and disaccharide 14[15] was obtained in a modest yield of 60% (entry 1). Reaction performed in the presence of 4.0 equiv. of NIS and 1.0 equiv. of TfOH rapidly (15 min) afforded disaccharide 14 in 75% yield (entry 2). Reaction in the presence of 4.5 equiv. of NIS and 1.0 equiv. of TMSOTf was even faster (10 min), and disaccharide 14 was obtained in 83% yield (entry 3). Glycosidation of donor 2 in the presence of 0.8 equiv. of Ag2CO3 and 4.0 equiv. of TMSOTf led to the formation of disaccharide 14 in 70% yield in 3.5 h (entry 4).
Table 1.
Optimization of glycosidations of SInMe donor 2 and SInAll donor 6 with 6-OH acceptor 13.

| ||
|---|---|---|
| Entry | Conditions [equiv.] | Yield of 14 |
| 1 | 2, NIS (3.0), TfOH (0.8), rt, 19 h | 60% |
| 2 | 2 , NIS (4.0), TfOH (1.0), rt, 15 min | 75% |
| 3 | 2, NIS (4.5), TMSOTf (1.0), rt, 10 min | 83% |
| 4 | 2, Ag2CO3 (0.8), TMSOTf (4.0), rt, 3.5 h | 70% |
| 5 | 6, NIS (2.0), TMSOTf (0.2), rt,16 h | 29% |
| 6 | 6, NIS (3.0), TMSOTf (0.8), rt, 2 h | 68% |
| 7 | 6, NIS (3.0), TMSOTf (1.0), rt, 1 h | 89% |
| 8 | 6, NIS (3.0), TfOH (0.4), rt, 24 h | 52% |
| 9 | 6, NIS (3.0), TfOH (0.8), rt, 24 h | 69% |
| 10 | 6, NIS (3.0), TfOH (1.0), rt, 24 h | 73% |
| 11 | 6 , NIS (3.2), TfOH (1.0), rt, 1 h | 94% |
| 12 | 6, Ag2CO3 (1.0), TMSOTf (4.0), rt, 1 h | 79% |
It should be noted that while the results of these reactions were generally good, we saw no significant difference between the new N-methylated leaving group and the previously investigated SIn glycosides. A similar level of reagent consumption was observed, and all our attempts to perform reaction in the presence of lower amounts of activators resulted in sluggish reactions and/or low yields. Therefore, we turned our attention to studying glycosidation of N-allylated donor 6 with glycosyl acceptor 13 (Table 1). Starting with glycosylation in the presence of 2.0 equiv. of NIS and 0.2 equiv. of TMSOTf, typical conditions for the activation of alkyl/aryl thioglycosides, the activation of the SinAll leaving groups was sluggish and inefficient. Disaccharide 14 was obtained in only 29% yield in 16 h (entry 5). A steady increase of the amount of reagents led to improved outcomes of these reactions. Thus, reactions performed with 3.0 equiv. of NIS and 0.8 or 1.0 equiv. of TMSOTf rapidly (1—2 h) afforded disaccharide 14 in 68% or 89% yield (entries 6 and 7). We then attempted glycosylation in the presence of 3.0 equiv. of NIS and 0.4 equiv. of TfOH. In this case, disaccharide 14 was obtained in 52% yield in 24 h (entry 8). Keeping the amount of NIS constant (3.0) and increasing the amount of TfOH to 0.8 and 1.0 equiv. allowed us to improve yields of disaccharide 14 to 69% and 73%, respectively (entries 9 and 10). These somewhat slow reactions were stopped after 24 h. The optimal conditions were found to be 3.2 equiv. of NIS and 1.0 equiv. of TfOH which afforded disaccharide 14 in 94% in 1 h (entry 11). Glycosidation of donor 6 in the presence of 0.8 equiv. of Ag2CO3 and 4.0 equiv. of TMSOTf led to the formation of disaccharide 14 in 79% yield in 1 h (entry 12).
Based on the preliminary experimentation, we chose NIS/TfOH activation conditions for both donors 2 and 6 in the ratios depicted in entries 2 and 11 (Table 1), respectively, to perform further studies. This selection was made solely based on the amount of NIS needed, which was lower in case of TfOH versus TMSOTf (compare entries 2 and 3). For expanding the scope of this reaction, we chose glycosyl donors 2, 5, 6, 8, 10, and 12 (Scheme 2) and standard primary acceptor 13 as well as secondary glycosyl acceptors 15—17[14] depicted in Figure 1. The results of this study are summarized in Table 2.
Figure 1.

Standard glycosyl acceptors used in this study.
Table 2.
Expanding the scope of glycosylation with SInR donors: synthesis of disaccharides 18–32.Yields and product ratios are invisible in the PDF file for all entries with the figure

| ||
|---|---|---|
| Entry | Donor+Acceptor, amount of NIS, time | Product (Yield, α/β ratio) |
| 1 | 2 (SInMe) + 15, NIS (4.0 equiv), 45 min |

|
| 2 | 2 + 16, NIS (4.0 equiv), 45 min |
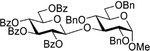
|
| 3 | 2 + 17, NIS (4.0 equiv), 15 min |

|
| 4 | 5 (SInMe) +13, NIS (4.0 equiv), 10 min |

|
| 5 | 5 + 13, NIS (3.0 equiv), 20 min | 21 (83%, 1.1/1) |
| 6 | 5 +15, NIS (4.0 equiv), 15 min |

|
| 7 | 5 + 16, NIS (4.0 equiv), 15 min |

|
| 8 | 5 + 17, NIS (4.0 equiv), 15 min |
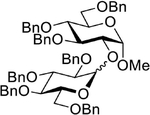
|
| 9 | 6 (SInAll) +15, NIS (3.5 equiv), 3 h | 18 (68%, β only) |
| 10 | 6 + 16, NIS (3.5 equiv), 1 h | 19 (75%, β only) |
| 11 | 6 + 17, NIS (3.5 equiv), 45 min | 20 (87%, β only) |
| 12 | 8 (SInMe) +13, NIS (3.2 equiv), 10 min | 21 (84%, 1.3/1) |
| 13 | 8 + 15, NIS (3.2 equiv), 20 min | 22 (75%, 1/1.3) |
| 14 | 8 + 16, NIS (3.2 equiv), 10 min | 23 (76%, 1.1/1) |
| 15 | 8 + 17, NIS (3.2 equiv), 10 min | 24 (83%, 1.5/1) |
| 16 | 10 (SInAll) + 13, NIS (3.2 equiv), 1h |

|
| 17 | 10 + 15, NIS (3.5 equiv), 1 h |

|
| 18 | 10 + 16, NIS (3.5 equiv), 1 h |

|
| 19 | 10 + 17, NIS (3.5 equiv), 1 h |
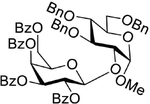
|
| 20 | 12 (SInAll) +13, NIS (3.2 equiv), 1.5 h |

|
| 21 | 12 + 15, NIS (3.5 equiv), 2.5 h |

|
| 22 | 12 + 16, NIS (4.0 equiv), 2.5 h |

|
| 23 | 12 + 17, NIS (3.5 equiv), 2.5 h |

|
Glycosidation of per-O-benzoylated SinMe glucosyl donor 2 with glycosyl acceptors 15—17 gave the respective β-linked disaccharides 18—20[14–16] in 79—85% yield (entries 1—3). Glycosylation of acceptor 13 with per-O-benzylated SinMe donor 5 in the presence of NIS (4.0 equiv) and TfOH (1.0) was swift (10 min) and efficient. As a result, disaccharide 21 was obtained in 92% yield in 10 min (entry 4). The high reactivity of per-O-benzylated (armed) donor 5 allowed us to decrease the amount of NIS to 3.0 equiv. In this case, a slower reaction (20 min) and a reduced yield (83%) for the formation of disaccharide 21 were recorded (entry 5). Glycosidation of SinMe donor 5 with secondary glycosyl acceptors 15—17 gave the respective disaccharides 22—24 in 61—69% yields and poor stereoselectivity (entries 6—8). The low yields were attributed to the formation of unidentified side products. The reactions with donor 5 lack stereoselectivity due to a non-participating benzyl substituent at C-2.
Glycosidation of per-O-benzoylated SinAll glucosyl donor 6 with acceptors 15—17 was conducted in the presence of NIS (3.5 equiv) and TfOH (1.0 equiv). These reactions required 45 min—3 h to produce the respective β-linked disaccharides 18—20 in 68—87% yields (entries 9—11). Glycosidations of per-O-benzylated SinAll glucosyl donor 8 with acceptors 13 and 15—17 were conducted in the presence of a reduced amount of NIS (3.2 equiv) and TfOH (1.0 equiv). There reactions required only 10—20 min to afford the respective β-linked disaccharides 21—24 in 75—84% yields (entries 12—15). The lack of stereoselectivity in these reactions was also due to a non-participating benzyl ether substituent at C-2 of the donor.
We then turned to investigating glycosylations with benzoylated N-allyl galactosyl and mannosyl donors 10 and 12, respectively. Glycosidation of galactosyl donor 10 with acceptor 13 in the presence of 3.2 equiv. of NIS and 1.0 equiv. of TfOH afforded β-linked disaccharide 25[17] in 85% yield in 1 h (entry 16). Moving towards glycosylations with less reactive, secondary acceptors 15—17 we decided to increase the amount of NIS to 3.5 equiv. keeping the amount of TfOH constant. This slight modification paid off, and the corresponding β-linked disaccharides 26—28[18] were swiftly (1 h) obtained in excellent yields of 92—96% (entries 17—19). Glycosidation of mannosyl donor 12 with acceptor 13 in the presence of 3.2 equiv. NIS and 1.0 equiv. TfOH afforded α-linked disaccharide 29[17] in 88% yield in 1.5 h (entry 20). Like in case of the galactosyl donor, we decided to increase the amount of NIS to 3.5 equiv. keeping the amount of TfOH constant for glycosylations of less reactive secondary glycosyl acceptors 14—16. As a result, the corresponding α-linked disaccharides 30—32[17,19] were obtained in 89—93% yields in 2.5 h (entries 21—23).
Next, we advanced our studies toward testing possible orthogonality of SinR donors with glycosyl donors of other series. For this experimentation, we chose ethylthio glycoside 33[20] and Sbox imidate 34.[21] In such comparative study, 1.0 equiv. of a thioindolyl glycoside (SinMe 5 or SinAll 8) were set to complete with 1.0 equiv. of other donors (33 or 34) for 2.0 equiv. of the primary glycosyl acceptor 13. First competitive reaction was set between donor 5 and ethylthio glycoside 33 in the presence of NIS (1.2 equiv) and TfOH (0.2 equiv), conditions that are suitable for the activation of thioglycosides. Indeed, SEt donor 33 was consumed completely in 5 h while SinMe donor 5 was recovered as its SEt adduct 35 in 86% yield (Scheme 3). The formation of this adduct is a result of a capturing the departed leaving group. Disaccharide 21 was obtained in 95% yield that was calculated based on the acceptor recovery. A similar reaction of SinAll donor 8 and SEt donor 33 with glycosyl acceptor 13 led to a complete activation of SEt donor 33 while donor 8 was recovered as its SEt adduct 36 in 91% yield (Scheme 3). Disaccharide 21 was obtained in 98% yield. It should be noted that excellent yields obtained for the formation of disaccharide 21 is attributed, in part, to excess of donors used in these reactions.
Scheme 3.

Competition experiments between SInR and SEt that led to preferential activation of thioglycoside 33.
Another competitive set of reactions was performed in the presence of Ag2CO3 and TMSOTf promoter system. Thioglycoside donor 33 was expected to remain unreactive under these reaction conditions. Indeed, the competitive reaction between donors 5 and 33 led to complete consumption of donor 5 in 15 min. SEt donor 33 was recovered in 92% yield, and disaccharide 21 was isolated in 98% yield (Scheme 4). A similar competition between donors 8 and 33 led to complete consumption of donor 8 in 15 min. As a result, donor 33 was recovered in 94% yield and disaccharide 21 was isolated in 99% yield (Scheme 4). These experiments signified that the thioindole analogs show entire orthogonality in respect to common ethylthio glycosides.
Scheme 4.

Competition experiments between SInR and SEt that led to the preferential activation of SInMe 5 or SInAll 8.
We also conducted competition experiments between thioindolyl glycosides and Sbox imidate 34 in the presence of 2.0 equiv. of AgOTf as promoter. These reaction conditions are standard for the activation of Sbox glycosides,[22] but unmodified SIn glycosides reacted very slowly, even in the presence of a large access of AgOTf.[7] Thus, a competition reaction between SinMe donor 5 and Sbox donor 34 resulted in a complete consumption of donor 34 in 5 min. As a result, donor 5 was recovered in 91% yield, and disaccharide 21 was obtained in 98% yield (Scheme 5). A similar competition reaction between SinAll donor 8 and Sbox donor 34 led to complete consumption of donor 34 in 5 min. Unreacted donor 8 was recovered in 95% yield, and disaccharide 21 was isolated in 99% yield (Scheme 5).
Scheme 5.

Competition experiments between SInR and SBox building blocks that led to the preferential activation of SBox donor 34.
To gain understanding of the glycosylation reaction mechanism we performed a set of experiments monitored by NMR. First, we investigated the activation in the presence of NIS. For this purpose, SinAll donor 6 was dissolved in CDCl3, the solution was transferred into a standard 5 mm NMR tube, and 1H NMR spectrum was recorded (Scheme 6). NIS (1.0 equiv) was added and 1H NMR spectrum was recorded in 25 min showing the formation of iodine adduct 37 as the only product. A similar experiment performed with SinMe donor 2 cleanly produced analogous adduct 38 (structure shown in Scheme 7, see the SI for the experiment details). Intermediates 37 and 38 are stable compounds, can be purified by column chromatography, were fully characterized by NMR, and their identity was confirmed by mass spectrometry.
Scheme 6.

NMR experiment with SInAll donor 6 in the presence of NIS (1.0 equiv) to observe the formation of 37.
Scheme 7.

Plausible mechanistic pathways for the activation of SInR glycosyl donors.
Corrected Scheme 7 has been provided as a chemdraw file. The changes in part A, wherein all Bx have been replaced with Bz
To uncover the mechanism of glycosylation reaction proceeding under the cooperative silver salt-Lewis acid catalysis we also set up a series of NMR experiments. For this purpose, SinAll donor 6 was dissolved in CDCl3, the solution was transferred into a standard 5 mm NMR tube, and 1H NMR spectrum was recorded. TMSOTf (1.0 equiv) was added and 1H NMR spectrum was recorded in 10 min showing the formation of TMS-adduct 39 (structure shown in Scheme 7, see the SI for the experiment details). A similar experiment performed with SinMe donor 2 produced analogous adduct 40 (structure shown in Scheme 7). Intermediates 39 and 40 are relatively unstable compounds that produced very elusive NMR spectra. Upon attempt to purify them by column chromatography, they reverted to the respective starting materials 6 and 2.
Based on this NMR monitoring our current understanding of the NIS-promoted reaction mechanism for the activation of SinR derivatives is as follows. Upon addition of NIS and TfOH to the reaction mixture containing SinAll donor 6 or SinMe donor 2 in 1,2-DCE, the first equivalent of NIS gets consumed by the electrophilic addition of the iodonium ion at the C-3 position of the SinR aglycone to afford stable intermediates 37 or 38, respectively (Scheme 7A). The presence of TfOH facilitates the formation of the iodonium ion from NIS. The additional NIS is needed to convert 37/38 into the activated species B’ that results in the leaving group departure followed by glycosylation.
Note the key difference between intermediate B’ and intermediate B that was observed with unprotected SIn derivatives in our previous work (Scheme 1). This difference explains the reduced amount of NIS that was typically required for the activation of SinR donors (3.2 equiv) versus their unprotected SIn counterparts (4.5 equiv). While glycosidation of benzoylated SinR donors proceeds via the intermediacy of the acyloxonium intermediate leading to complete stereoselectivity, glycosidations of benzylated SinR donors proceed via oxacarbenium ion intermediate. The formation of the latter intermediate explains poor stereoselectivity of many uncontrolled glycosylations taking place without the assistance of the neighboring group at C-2.
Based on NMR monitoring our current understanding of the reaction mechanism of glycosidation of SinR donors 6 or 2 in the presence of TMSOTf and Ag2CO3 is as follows. Upon addition of the promoters to the reaction mixture containing SinAll donor 6 or SinMe donor 2 in 1,2-DCE, the first equivalent of TMSOTf gets consumed by the electrophilic addition of TMS at the C-3 position of the SinR aglycone to afford elusive intermediates 39 or 40, respectively (Scheme 7B). This step represents the key difference from the activation of unprotected SIn donors that were first tautomerized into its thioimidate counterpart (refer to Scheme 1). Silver carbonate then complexes with adducts 39/40 to produce respective silver complexes, which become strongly ionized in the presence of access TMSOTf. In should be noted that the resulting intermediate D’ signifies a drastic activation difference from the analogous intermediate D observed with SIn derivatives (see Scheme 1). Whereas SinR donors are activated via the anomeric sulfur, following the direct activation pathway, like in thioglycosides, SIn donors are activated via the endocyclic nitrogen atom of the leaving group, which represents the remote activation pathway, like in thioimidates.
Conclusions
We presented an extended study of a new class of glycosyl donors, indolylthio glycosides. Previously we observed that while the activation profile of the Sin leaving group is interesting, most reactions required large excess of reagents. This drawback was partially addressed in the present study of N-alkylated SInR derivatives. The activation process was studied by NMR, and the increased understanding of the mechanism led to a discovery of different activation pathways taking place with SIn versus SInR derivatives. We also investigated orthogonality of the SInR leaving groups versus thioglycosides. Further investigation of the leaving groups of this class in application to the synthesis of glycans is currently underway in our laboratory.
Experimental Section
General.
The reactions were performed using commercial reagents and the ACS-grade solvents used for reactions were purified and dried in accordance with standard procedures. Column chromatography was performed on silica gel 60 (70–230 mesh), reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 5% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40°C. CH2Cl2 and ClCH2CH2Cl (1,2-DCE) were distilled from CaH2 directly prior to application. Molecular sieves (3 Å), used for reactions, were crushed and activated in vacuo at 390°C for 8 h in the first instance and then for 2–3 h at 390°C directly prior to application. Optical rotations were measured at ‘Jasco P-2000’ polarimeter. 1H NMR spectra were recorded in CDCl3 at 300 MHz, 13C NMR spectra were recorded at 75 MHz. The 1H NMR chemical shifts were referenced to tetramethyl silane (TMS, δ H=0 ppm) or CDCl3 (δ H=7.26 ppm) for 1H NMR spectra for solutions in CDCl3. The 13C NMR chemical shifts were referenced to the central signal of CDCl3 (δ C=77.00 ppm) for solutions in CDCl3. HR FAB-MS determinations were made with the use of JEOL MStation (JMS-700) Mass Spectrometer.
Synthesis of HSInR aglycones
1-Methylindoline-2-one (N-methyl oxindole).
The title compound was prepared from Isatin as previously described.[11] Analytical data for N-methyl oxindole were in accordance with that previously reported.[11]
1-Methylindoline-2-thione (N-methyl thioindole, HSInMe).
The title compound was obtained following the previously described procedure,[12] which was modified as follows. N-Methyl oxindole (4.0 g, 27.2 mmol) was dissolved in dry tetrahydrofuran (100 mL) under argon. P2S5 (7.37 g, 33.16 mmol) was added followed by a portionwise addition of sodium bicarbonate (4.57 g, 54.6 mmol), and the resulting mixture was stirred under argon for 16 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in ethyl acetate (400 mL) and washed with water (3×50 mL). The organic phase was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane 10% gradient elution) to afford the title compound in 88% yield. Analytical data for HSInMe: Rf=0.45 (ethyl acetate/hexane, 2/3, v/v); 1H NMR (300 MHz): δ 3.57 (s, 3H, NCH3), 4.02 (s, 2H, CH2), 6.93–7.34 (m, 4H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 30.9, 48.7, 109.3, 123.6, 124.0, 127.7, 128.7, 146.2, 200.6 ppm; ESI-TOF [M+H]+: calcd for [C9H10NS]+ 164.0528; found: 164.0532.
1-Allylindolin-2-one (N-allyl oxindole).
The title compound was prepared from Isatin using previously described procedure.[11] Analytical data for N-allyl oxindole were in accordance with that previously reported.[11]
1-Allylindolin-2-thione (N-allyl thioindole, HSInAll).
N-allyl oxindole (2.0 g, 11.54 mmol) was dissolved in dry tetrahydrofuran (40 mL) under argon. P2S5 (3.12 g, 14.08 mmol) was added followed by a portionwise addition of sodium bicarbonate (1.93 g, 23.08 mmol), and the resulting mixture was stirred under argon for 16 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in ethyl acetate (300 mL) and washed with water (3×40 mL). The organic phase was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane 10% gradient elution) to afford the title compound in 65% yield. Analytical data for HSInAll: Rf=0.50 (ethyl acetate-hexane, 1/4, v/v); 1H NMR (300 MHz): δ 4.06 (s, 2H, CH2C=S), 4.81 (d, 2H, NCH2), 5.19–5.25 (m, 2H, =CH2), 5.79–5.91 (m, 1H, CH=), 6.92–7.30 (m, 4H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 46.2, 48.6, 109.8, 118.0, 123.5, 123.8, 127.4, 128.5, 129.2, 145.2, 200.5 ppm. ESI-TOF [M+H]+: calcd for [C11H12NS]+ 190.0685; found: 190.0690.
Synthesis of SInR Glycosides
1-Methylindolyl 2,3,4,6-tetra-O-benzoyl-1-thio-β-D-glucopyranoside (2).
HSInMe (0.33 g, 2.02 mmol), anhydrous K2CO3 (0.28 g, 2.02 mmol), and 18-crown-6 (0.10 g, 0.37 mmol) were added to a solution of 2,3,4,6-tetra-O-benzoyl-α-D-glucopyranosyl bromide[23] (1, 1.21 g, 1.83 mmol) in dry acetone (32 mL), and the resulting mixture was stirred under argon at 50°C for 3 h. After that, the solids were filtered off through a pad of Celite, rinsed successively with CH2Cl2, and the combined filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (~150 mL), washed with 10% aq. NaHCO3 (25 mL), and water (2×25 mL). The organic phase was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (toluene - ethyl acetate 1% gradient elution) to afford the title compound as a white amorphous solid in 88% yield (1.17 g, 1.615 mmol). Analytical data for 2: Rf=0.3 (ethyl acetate/diethyl ether, 4/6, v/v); [α]D22 −20.0 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 3.73 (s, 3H, NCH3), 4.06–4.12 (m, 1H, J5,6a=5.3, J5,6b=2.4 Hz, H-5), 4.43 (dd, 1H, J6a,6b=12.2 Hz, H-6a), 4.59 (dd, 1H, H-6b), 4.89 (d, 1H, J1,2=9.9 Hz, H-1), 5.51–5.59 (m, 2H, J2,3=9.5 Hz, H-2, 4), 5.91 (dd, 1H, J3,4=9.5 Hz, H-3), 6.89 (s, 1H, =CH), 7.11–7.56 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 30.1, 62.8, 68.9, 70.4, 74.0, 76.3, 86.7, 109.9, 112.3, 112.4, 119.7, 120.8, 122.9, 125.1 (x2), 126.9 (x2), 128.2, 128.3, 128.4 (x3), 128.5 (x2), 128.6 (x2), 129.0, 129.3, 129.5, 129.6 (x2), 129.7 (x2), 129.8 (x2), 133.1, 133.2, 133.4, 138.6, 164.9 (x2), 165.7, 166.0 ppm; ESI-TOF [M+Na]+: calcd for [C43H35NO9SNa]+ 764.1925; found: 764.1916.
1-Methylindolyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (4).
HSInMe (1.3 g, 8.02 mmol), anhydrous K2CO3 (1.11 g, 8.02 mmol), and 18-crown-6 (0.38 g, 1.46 mmol) were added to a solution of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide[23] (3, 3.00 g, 1.83 mmol) in dry acetone (50 mL), and the resulting mixture was stirred under argon at rt for 4 h. After that, the solids were filtered off through a pad of Celite, rinsed successively with CH2Cl2 and the combined filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (300 mL), washed with 10% aq. NaHCO3 (30 mL) and water (2×30 mL). The organic phase was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (hexane - ethyl acetate 10% gradient elution) to afford the title compound as a white amorphous solid in 60% yield (2.15 g, 4.36 mmol). Analytical data for 4: Rf=0.45 (ethyl acetate/hexane, 3/7, v/v); [α]D22 −36.2 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 1.92–2.15 (m, 12 H, 4×CH3CO), 3.57–3.62 (m, 1H, H-5), 3.78 (s, 3H, NCH3) 4.10–4.11 (m, 2H, H-6a, 6b), 4.51 (d, 1H, J1,2=9.9 Hz, H-1), 4.90–5.05 (m, 2H, H-2, 4), 5.18 (dd, 1H, J3,4=9.3 Hz, H-3), 6.86 (s, 1H, =CH, indole), 7.09–7.62 (m, 4H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 20.4 (x2), 20.5, 20.7, 30.1, 61.6, 67.7, 69.7, 73.7, 75.6, 85.9, 109.8, 112.2 (x2), 119.7, 120.8, 122.9, 124.8, 126.8, 138.5, 169.2 (x2), 170.1, 170.3 ppm; ESI-TOF [M+Na]+: calcd for [C23H27NO9SNa]+ 516.1299; found: 516.1308.
1-Methylindolyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside (5).
Compound 4 (2.0 g, 4.05 mmol) was dissolved in minimum amount of methanol (~25 mL), a 1 M solution of NaOMe in MeOH was added until pH >9, and the resulting mixture was stirred for 1 h at rt. The reaction mixture was neutralized with Dowex (H+), the resin was filtered off, and rinsed successively with MeOH. The combined filtrate (~75 mL) was concentrated under reduced pressure, and the residue was dried in vacuo for 2 h. The crude residue was dissolved in dry DMF (24 mL), NaH (1.94 g, 48.66 mmol) was added portionwise, followed by the dropwise addition of benzyl bromide (3.61 mL, 30.41 mmol), and the resulting mixture was stirred for 16 h at rt. The reaction mixture was poured into ice-water (50 mL) and stirred for 30 min. The mixture was extracted with ethyl acetate/diethyl ether (1:1, v/v, 3×100 mL), and the combined organic extract was washed with water (2×50 mL). The organic phase was separated, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane, 5% gradient elution) to afford the title compound as a white amorphous solid in 86% yield (2.4 g, 3.49 mmol). Analytical data for 5: Rf =0.55 (ethyl acetate/hexane, 3/7, v/v); [α]D22 −10.9 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 3.33 (dd, 1H, J6a,6b =10.1 Hz, H-6a), 3.46–3.74 (m, 5H, H-2, 3, 4, 5, 6b), 3.80 (s, 3H, NCH3), 4.41–4.53 (m, 4H, H-1, 3×CHPh), 4.43 (d, 1H, 2J=10.8 Hz, CHPh), 4.81–4.89 (m, 3H, 3×CHPh), 5.02 (d, 2J=10.3 Hz, CHPh), 6.87 (s, 1H, =CH, indole), 7.03–7.31 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 30.3, 68.6, 73.2 (x2), 74.8, 74.4, 75.2, 78.6, 80.4, 86.5, 88.2 (x2), 109.7, 110.9, 119.5, 120.5, 122.5, 126.9 (x2), 127.1 (x2), 127.5 (x3), 127.6 (x2), 127.8 (x2), 128.0 (x2), 128.3 (x3), 137.8 (x4), 137.9 (x2), 138.2 (x2), 138.4 (x2), ppm; ESI-TOF [M+H]+: calcd for [C43H43NO5SNa]+ 708.2754; found: 708.2748.
1-Allyllindolyl 2,3,4,6-tetra-O-benzoyl-1-thio-β-D-glucopyranoside (6).
HSInAll (0.47 g, 2.5 mmol), anhydrous K2CO3 (0.35 g, 2.5 mmol), and 18-crown-6 (0.12 g, 0.46 mmol) were added to a solution of 2,3,4,6-tetra-O-benzoyl-α-D-glucopyranosyl bromide[13] (1, 1.50 g, 2.27 mmol) in dry acetone (40 mL), and the resulting mixture was stirred under argon for 3 h at 50°C. After that, the solids were filtered off through a pad of Celite, rinsed successively with CH2Cl2, and the combined filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (~150 mL), washed with 10% aq. NaHCO3 (25 mL) and water (2×25 mL). The organic phase was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (toluene - ethyl acetate 1% gradient elution) to afford the title compound as a white amorphous solid in 78% yield (1.33 g, 1.71 mmol). Analytical data for 6: Rf =0.6 (ethyl acetate/toluene, 0.5/9.5, v/v); [α]D22 −4.3 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 4.00–4.10 (m, 1H, J5,6a=5.5, J5,6b=2.3 Hz, H-5), 4.45 (dd, 1H, J6a,6b=12.2 Hz, H-6a), 4.61 (dd, 1H, H-6b), 4.66–5.00 (m, 5H, H-1, NCH2, =CH2), 5.56–5.62 (m, 2H, H-2, 4), 5.73–5.83 (m, 1H, CH=), 5.90 (dd, 1H, J3,4=9.5 Hz, H-3), 6.90 (s, 1H, =CH, indole), 7.07–8.01 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 45.7, 62.8, 68.9, 70.4, 73.9, 76.3, 86.8, 110.4, 112.5 (x2), 115.9, 119.9, 120.8, 123.0, 125.1, 127.1, 128.2, 128.3 (x2), 128.4 (x4), 128.5 (x2), 128.9, 129.3, 129.5 (x2), 129.6 (x2), 129.7 (x2), 129.8 (x2), 133.1, 133.2, 133.4 (x2), 133.5, 137.8, 164.9 (x2), 165.6, 165.9 ppm; ESI-TOF [M+Na]+: calcd for [C45H37NO9SNa]+ 790.2081; found: 790.2074.
1-Allylindolyl 2,3,4,6-tetra-O-benzyl-1-thio-α-D-glucopyranoside (8).
Ethyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside[20] (33, 1.10 g, 1.88 mmol) and freshly activated molecular sieves (3 Å, 0.75 g) in CH2Cl2 (50 mL) were stirred under argon for 1 h at rt. Bromine (0.12 mL, 2.26 mmol) was added, and the resulting mixture was stirred for 20 min at rt. After that, the volatiles were removed under reduced pressure, the residue was co-evaporated with dry toluene (2×20 mL), and dried in vacuo for 2 h. The residue containing crude 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl bromide 7 was dissolved in CH2Cl2 (10 mL), HSInAll (0.39 g, 2.07 mmol), anhydrous K2CO3 (0.29 g, 2.068 mmol), and 18-crown-6 (0.1 g, 0.37 mmol) were added, and the resulting mixture was stirred under argon for 8 h at rt. The solids were filtered off through a pad of Celite and rinsed successively with CH2Cl2. The combined filtrate (~150 mL) as washed with water (30 mL), 10% aq. NaHCO3 (25 mL), and water (2×30 mL). The organic phase was separated, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene 2% gradient elution) to afford the title compound as a white amorphous solid in 61% yield (0.82 g, 1.15 mmol, α/β=1.6:1). A small sample of α-8 was separated for characterization purposes. Analytical data for α-8: Rf=0.50 (ethyl acetate/hexane, 3/7, v/v); [α]D22 68.8 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 3.59–3.71 (m, 3H, H-3, 5, 6b), 3.84–3.97 (m, 2H, H-2, 4), 4.38–4.42 (m, 1H, H-6a), 4.46–5.07 (m, 12H, =CH2, NCH2, 8×CHPh), 5.49 (d, 1H, J1,2=4.4 Hz, H-1), 5.82–5.91(m, 1H, CH=), 6.79 (s, 1H, =CH indole), 7.07–7.51 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 45.6, 45.8, 67.9, 68.2, 68.6, 71.7, 72.4, 73.3 (x2),74.8, 75.0. 75.3, 75.6, 75.7, 76.0, 77.1, 78.8, 79.4, 80.7, 82.2, 86.6, 88.0, 88.5, 110.0, 110.1, 110.4, 110.5, 111.5, 115.8, 116.1, 119.7, 120.2, 120.3, 120.5, 120.6, 122.2, 122.5, 127.2, 127.6 (x4), 127.8 (x7), 128.2 (x4), 128.3 (x7), 133.4, 133.5, 133.7, 133.8, 137.4, 137.5, 137.7, 137.8, 137.9, 138.0, 138.2, 138.4 ppm; ESI-TOF [M+H]+: calcd for [C45H45NO5SNa]+ 734.2916; found: 734.2927. Selected 1H NMR (300 MHz) data for β-8: 3.33 (dd, H-5), 3.48–3.58 (m, H-2, 6a, 6b), 3.37–3.71 (m, H-3, 4), 4.39 (d, H-1), 5.82–5.91 (m, CH=), 6.89 (s, =CH indole), 7.07–7.51 (m, aromatic) ppm.
1-Allyllindolyl 2,3,4,6-tetra-O-benzoyl-1-thio-α- and β-D-galactopyranoside (α-10and β-10).
HSInAll (0.79 g, 4.20 mmol), anhydrous K2CO3 (0.58 g, 4.20 mmol) and 18-crown-6 (0.20 g, 0.76 mmol) were added to a solution of 2,3,4,6-tetra-O-benzoyl-α-D-galactopyranosyl bromide[13] (9, 2.50 g, 3.80 mmol) in dry acetone (66 mL), and the resulting mixture was stirred under argon for 3 h at 50°C. After that, the solids were filtered off through a pad of Celite, rinsed successively with CH2Cl2, and the combined filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (~250 mL), washed with 10% aq. NaHCO3 (30 mL) and water (2×30 mL). The organic phase was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (toluene - ethyl acetate 1% gradient elution) to afford separate anomers of the title compound as white amorphous solid. Eluted first was α-10 (18% yield, 0.53 g, 0.685 mmol) followed by elution of β-10 (70% yield, 2.04 g, 2.66 mmol). Analytical data for α-10: Rf=0.55 (ethyl acetate/toluene, 0.5/9.5, v/v); [α]D22 −4.20 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 4.48 (dd, 1H, J6a,6b=11.6 Hz, H-6a), 4.61–4.68 (m, 3H, H-6b, NCH2), 4.77–5.02 (m, 2H, =CH2), 5.27 (dd, 1H, J5,6a =5.2 Hz, H-5), 5.65–5.77 (m, 1H, =CH), 5.88–6.02 (m, 3H, CH=, H-1, 2, 3), 6.13 (m, 1H, =CH, H-4), 6.78 (s, 1H, =CH indole), 7.00–8.07 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 45.5, 62.6, 68.3, 68.5, 68.8, 38.9, 86.8, 109.9, 111.8, 116.3, 119.8, 120.5, 122.7, 125.7, 127.3, 128.2, 128.4 (x2), 128.5 (x2), 128.6, 128.8 (x4), 128.9, 129.3, 129.7, 129.8 (x2), 129.9 (x4), 132.9, 133.0, 133.2 (x2), 133.6 (x2), 137.6, 165.3, 165.4, 165.5, 166.1 ppm; ESI-TOF [M+Na]+: calcd for [C45H37NO9SNa]+ 790.2081; found: 790.2033. Analytical data for β-10: Rf=0.50 (ethyl acetate/toluene, 0.5/9.5, v/v); [α]D22 −6.3 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 4.30–4.36 (m, 2H„ J5,6b=8.5, J6a,6b=13.3 Hz, H-5, 6a), 4.57 (m, 1H, H-6b), 4.73–4.94 (m, 3H, H-1, NCH2), 5.00–5.07 (m, 2H, =CH2), 5.59 (dd, 1H, H-3), 5.77–5.97 (m, 3H, J2,3=6.9 Hz, H-2, 4, CH=), 7.05–8.03 (m, 25H, =CH indole, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 45.8, 62.3, 67.7, 67.9, 72.9, 75.1, 85.8, 110.6, 113.3, 115.9, 119.9, 121.2, 123.0, 124.3, 127.2, 128.2, 128.4 (x5), 128.5 (x2), 128.6 (x2), 129.1, 129.2, 129.6 (x5), 129.7 (x4), 133.2, 133.3 (x2), 133.6, 138.1, 165.0, 165.1, 165.4, 165.9 ppm; ESI-TOF [M+Na]+: calcd for [C45H37NO9SNa]+ 790.2081; found: 790.2076.
1-Allyllindolyl 2,3,4,6-tetra-O-benzoyl-1-thio-α-D-mannopyranoside (12).
HSInAll (0.79 g, 4.20 mmol), anhydrous K2CO3 (0.58 g, 4.20 mmol) and 18-crown-6 (0.20 g, 0.76 mmol) were added to a solution of 2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl bromide[13] (11, 2.50 g, 3.80 mmol) in dry acetone (66 mL), and the resulting mixture was stirred under argon for 3 h at 50°C. After that, the solids were filtered off through a pad of Celite, rinsed successively with CH2Cl2, and the combined filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (~250 mL), washed with 10% aq. NaHCO3 (30 mL) and water (2×30 mL). The organic phase was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (toluene - ethyl acetate 1% gradient elution) to afford the title compound as a white amorphous solid in 67% yield (1.91 g, 2.54 mmol). Analytical data for 12: Rf=0.55 (ethyl acetate/toluene, 0.5/9.5, v/v); [α]D22 −49.0 (c=1.0, CHCl3); 1H NMR (300 MHz): 4.00–4.03 (m, 1H, J5,6a=5.1 Hz, H-5), 4.49 (dd, 1H, J6a,6b=12.2 Hz, H-6a), 4.71–4.90 (m, 3H, H-6b, NCH2), 5.01–5.09 (m, 3H, H-1, =CH2), 5.56 (dd, 1H, J3,4=3.0 Hz, H-3), 5.88–6.04 (m, 3H, H-4, =CH2), 6.19 (dd, 1H, J2,3=2.7 Hz, H-2), 6.92 (s, 1H, =CH indole), 7.08–8.07 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 45.7, 62.9, 66.2, 71.1 72.6, 86.7, 110.3, 111.5 (x2), 115.9, 119.9, 120.8, 123.0, 126.9, 127.2, 127.7, 128.2, 128.4 (x2), 128.5 (x3), 128.6 (x3), 128.8, 129.6 (x3), 129.7 (x4), 129.9 (x2), 133.1, 133.2, 133.4 (x2), 133.7, 133.8, 137.8, 165.2, 165.3, 165.5, 166.0 ppm; ESI-TOF [M+Na]+: calcd for [C45H37NO9SNa]+ 790.2081; found: 790.2073.
Synthesis of Disaccharides
General procedure for glycosylation in the presence of NIS and TfOH or TMSOTf.
A mixture of a glycosyl donor (0.040–0.044 mmol), a glycosyl acceptor (0.036–0.039 mmol), and molecular sieves (3 Å, 80 mg) in 1,2-DCE or DCM (1.0 mL) was stirred under argon for 1 h at rt. After that, NIS (0.122–0.182 mmol) and TfOH (0.040 mmol) or TMSOTf (0.040 mmol) were added, and the resulting mixture was stirred for the time indicated in Tables at rt. The solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~30 mL) was washed with 10% aq. Na2S2O3 (2×10 mL) and H2O (2×10 mL). The organic phase was separated, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution). If necessary, further purification was accomplished by size-exclusion column chromatography on Sephadex LH-20.
General procedure for glycosylation in the presence of Ag2CO3 and TMSOTf.
A mixture of a glycosyl donor (0.040–0.044 mmol), a glycosyl acceptor (0.036–0.039 mmol), and molecular sieves (3 Å, 80 mg) in 1,2-DCE or DCM (1.0 mL) was stirred under argon for 1 h at rt. Ag2CO3 (0.035–0.042 mmol) and TMSOTf (0.168–0.175 mmol) were added, and the resulting mixture was for the time indicated in Tables at rt. The solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~30 mL) was washed with H2O (2×10 mL). The organic phase was separated, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford the corresponding disaccharide. If necessary, further purification was accomplished by size-exclusion column chromatography on Sephadex LH-20.
Methyl 6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (17).
The title compound was obtained from donor 2 and acceptor 13[14] by general glycosylation procedure in 75% yield as a white amorphous solid. Analytical data for 17 was in accordance with that previously reported.[15]
Methyl 4-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (18).
The title compound was obtained from donor 2 and acceptor 15[14] by the general glycosylation procedure in 79% yield as a clear syrup. Analytical data for 18 was in accordance with that previously reported.[15]
Methyl 3-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (19).
The title compound was obtained from donor 2 and acceptor 16[14] by the general glycosylation procedure in 80% yield as a clear syrup. Analytical data for 19 was in accordance with that previously reported.[14]
Methyl 2-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (20).
The title compound was obtained from donor 2 and acceptor 17[14] by the general glycosylation procedure in 85% yield as a clear syrup. Analytical data for 20 was in accordance with that previously reported.[16]
Methyl 6-O-(2,3,4,6-tetra-O-benzyl-D-glucopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (21).
The title compound was obtained from donor 5 and acceptor 13 by the general glycosylation procedures in 92% yield (α/β=1.2/1) as a colorless foam. Analytical data for 21 was in accordance with that previously reported.[24]
Methyl 4-O-(2,3,4,6-tetra-O-benzyl-D-glucopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (22).
The title compound was obtained from donor 5 and acceptor 15 by the general glycosylation procedure in 61% yield (α/β=1/1.2) as a clear syrup. Analytical data for 22 was in accordance with that previously reported.[25]
Methyl 3-O-(2,3,4,6-tetra-O-benzyl-D-glucopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (23).
The title compound was obtained from donor 5 and acceptor 16 by the general glycosylation procedure in 69% yield (α/β=1.2/1) as a clear syrup. Analytical data for 23 was in accordance with that previously reported.[26]
Methyl 2-O-(2,3,4,6-tetra-O-benzyl-D-glucopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (24).
The title compound was obtained from donor 5 and acceptor 17 by the general glycosylation procedure in 67% yield (α/β=1.2/1) as a colorless foam. Analytical data for 24 was in accordance with that previously reported.[27]
Methyl 6-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (25).
The title compound was obtained from donor 10 and acceptor 13 by general glycosylation procedure in 85% yield as a white amorphous solid. Analytical data for 25 was in accordance with that previously reported.[17]
Methyl 4-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (26).
The title compound was obtained from donor 10 and acceptor 15 by the general glycosylation procedure in 92% yield as a colorless amorphous solid. Analytical data for 26 was in accordance with that previously reported.[18]
Methyl 3-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (27).
The title compound was obtained from donor 10 and acceptor 16 by the general glycosylation procedure in 96% yield as a colorless amorphous solid. Analytical data for 27: Rf=0.5 (ethyl acetate/toluene, 0.5/9.5, v/v); [α]D22 5.1 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 3.25 (s, 3H, CH3), 3.36 (dd, 1H, J2,3=3.5 Hz, H-2), 3.56–3.70 (m, 4H, H-4, 5, 6a, 6b), 4.16 (d, 1H, 2J=12.1 Hz, CHPh), 4.29–4.46 (m, 9H, H-1, 3, 5’, 6a’, 6b’, 4×CHPh), 5.28 (d, 1H, 2J=10.4 Hz, CHPh), 5.50 (d, 1H, J1’,2’=7.8 Hz, H-1’), 5.67 (dd, 1H, J3’,4’=7.5 Hz, H-3’), 5.87 (dd, 1H, J2’,3’=9.1 Hz, H-2’), 5.99 (dd, 1H, H-4’), 7.06–8.00 (m, 35H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 55.0, 61.4, 68.0, 68.3, 69.5, 70.5, 70.8, 71.6, 73.5, 73.7, 74.8, 75.1, 78.5, 80.6, 97.6, 97.7, 100.9, 127.5, 127.7 (x4), 128.0 (x4), 128.3 (x3), 128.4 (x5), 128.5 (x2), 128.7, 128.9, 129.3, 129.4, 128.8, 133.2, 137.8, 137.9, 138.5, 165.2, 165.4, 165.5, 165.6 ppm; ESI-TOF [M+Na]+: calcd for [C62H58O15Na]+ 1065.3668; found: 1065.3629.
Methyl 2-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (28).
The title compound was obtained from donor 10 and acceptor 17 by general glycosylation procedure in 96% yield as a colorless amorphous solid. Analytical data for 28: Rf=0.50 (ethyl acetate/toluene, 0.5/9.5, v/v); [α]D22 53.3 (c=1.0, CHCl3); 1H NMR (300 MHz): δ 3.42 (s, 3H, CH3), 3.60–3.74 (m, 4H, H-4, 5, 6a, 6b), 3.97 (dd, 1H, J3,4=9.2 Hz, H-3), 4.33–4.67 (m, 9H, H-5’, 6a’, 6b’, 6×CHPh), 5.09 (d, 1H, J1,2=2.3 Hz, H-1), 5.15 (d, 1H, J1’,2’=8.1 Hz, H-1’), 5.58 (dd, 1H, J3’,4’=2.8 Hz, H-3’), 5.95–5.99 (m, 2H, H-2’, 4’), 6.97–8.08 (m, 35H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 55.3, 55.4, 62.2, 68.1, 68.4, 69.6, 69.9, 71.4, 72.1, 73.4, 74.9, 75.2, 80.8, 82.2, 99.4, 102.7, 127.0 (x3), 127.5, 127.6, 127.7 (x3), 127.8, 128.0 (x4), 128.1 (x9), 128.5 (x6), 128.8, 128.9, 129.1, 129.7 (x5), 130.0 (x2), 133.2, 137.9, 138.0, 138.4, 165.0, 165.5, 165.6, 166.0 ppm; ESI-TOF [M+Na]+: calcd for [C62H58O15Na]+ 1065.3668; found: 1065.3627.
Methyl 6-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (29).
The title compound was obtained from donor 12 and acceptor 13 by general glycosylation procedure in 70% yield as a white amorphous solid. Analytical data for 29 was in accordance with that previously reported.[17]
Methyl 4-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (30).
The title compound was obtained from donor 12 and acceptor 15 by the general glycosylation procedure in 80% yield as a clear syrup. Analytical data for 30 was in accordance with that previously reported.[17]
Methyl 3-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-2,4,6-tri-O-benzyl-α-D-glucopyranoside (31).
The title compound was obtained from donor 12 and acceptor 16 by the general glycosylation procedure in 93% yield as a clear syrup. Analytical data for 31 was in accordance with that previously reported.[19a]
Methyl 2-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-3,4,6-tri-O-benzyl-α-D-glucopyranoside (32).
The title compound was obtained from donor 12 and acceptor 17 by the general glycosylation procedure in 92% yield as a clear syrup. Analytical data for 32 was in accordance with that previously reported.[19b]
Competition experiments
Experiment A.
A mixture of ethylthio glucoside 33 (26.0 mg, 0.044 mmol), methyl thioindolyl glucoside 5 (30.0 mg, 0.044 mmol), acceptor 13 (40.6 mg, 0.087 mmol), and 3 Å molecular sieves (80 mg) in dichloroethane (1.0 mL) was stirred under argon for 1 h at rt. After that, triflic acid (0.78 μL, 0.0087 mmol) was added followed by the addition of NIS (11.8 mg, 0.0524 mmol), and the resulting mixture was stirred for 5 h at rt. The solid was filtered off through a pad of Celite, rinsed successively with dichloromethane, and the combined filtrate (~30 mL) was washed with 10% aq. Na2S2O3 (2×10 mL). The organic phase was separated, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane 5% gradient elution). The unreacted donor 5 was isolated as SEt adduct 35 in 86% yield. Analytical data for 35: Rf=0.55 (ethyl acetate/hexane, 3/7, v/v); 1H NMR (300 MHz): δ 1.14 (t, 3H, CH2CH3), 2.71–2.80 (m, 2H, CH2CH3), 3.26 (m, 1H, H-5), 3.54–3.66 (m, 5H, H-2, 3, 4, 6a, 6b), 3.86 (s, 3H, NCH3), 4.39–4.57 (m, 4H, H-1, 3×CHPh,), 4.79 (d, 1H, 2J=11.5 Hz, CHPh), 4.86–4.93 (m, 3H, 3×CHPh), 5.17 (d, 1H, 2J=10.4 Hz, CHPh), 7.14–7.81 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 15.1, 30.8, 68.8, 73.9, 75.0, 75.6, 75.8, 78.7, 81.7, 86.6, 89.9, 110.1, 113.7, 114.1, 120.1, 120.2, 120.3, 120.4, 123.5, 127.6 (x4), 127.7 (x4), 127.9 (x4), 128.3 (x4), 128.4 (x6), 133.8, 137.9, 138.0, 138.4 ppm; ESI-TOF [M+H]+: calcd for [C45H47NO5S2Na]+ 768.2788; found: 768.2743.
A competition experiment with allyl thioindolyl glucoside 8 (30 mg, 0.042 mmol) instead of donor 5 was performed in a similar manner. The unreacted donor 8 was obtained as SEt adduct 36 in 91% yield. Analytical data for 36: Rf=0.50 (ethyl acetate/hexane, 3/7, v/v); 1H NMR (300 MHz): δ 3.54–3.70 (m, 2H, H-3, 5), 3.83–3.98 (m, 3H, H-2, 4, 6a), 4.31–4.38 (m, 4H, H-6b, 3×CHPh), 4.69–4.86 (m, 5H, NCH2, 3×CHPh), 5.00–5.10 (m, 4H, =CH2, 2×CHPh), 5.82–5.91 (m, 2H, J1,2=4.7 Hz, H-1, CH=), 7.15–7.76 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 15.1, 29.7, 30.7, 46.7, 68.3, 68.8, 72.5, 72.6, 73.5, 75.3, 75.8, 79.6, 82.3, 89.2, 110.6, 113.4, 116.5, 119.8, 120.5, 123.2, 127.6, 127.9, 128.0, 128.1, 128.3, 128.4, 130.2, 133.3, 133.8, 137.5, 137.8, 137.9, 138.1, 138.7 ppm; ESI-TOF [M+H]+: calcd for [C47H49NO5S2Na]+ 794.2944; found: 794.2945.
Experiment B.
A mixture of ethylthio glucoside 33 (26.0 mg, 0.044 mmol), methyl thioindolyl glucoside 5 (30 mg, 0.044 mmol), glucosyl acceptor 13 (40.6 mg, 0.087 mmol), and 3 Å molecular sieves (80 mg) in dichloroethane (1.0 mL) was stirred under argon for 15 min at rt. After that, Ag2CO3 (9.6 mg, 0.035 mmol) and TMSOTf (31.6 μL, 0.175 mmol) were added, and the resulting mixture was stirred under argon for 15 min at rt. The reaction was quenched with triethyl amine (~0.2 mL), the solid was filtered off through a pad of Celite, rinsed successively with dichloromethane, and the combined filtrate (~30 mL) was washed with 10% aq. Na2S2O3 (2×10 mL). The organic phase was separated, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane 5% gradient elution). The unreacted donor 33 was recovered in 92% yield and its identity was confirmed by 1H NMR and mass spectroscopy. A competition experiment with allyl thioindolyl glucoside 8 (30 mg, 0.042 mmol) instead of donor 5 was performed in a similar manner. The unreacted donor 33 was recovered in 94% yield and its identity was confirmed by 1H NMR and mass spectroscopy.
Experiment C.
A mixture of S-benzoxazolyl glucoside 34 (29.5 mg, 0.044 mmol), methyl thioindolyl glucoside 5 (30.0 mg, 0.044 mmol), glucosyl acceptor 11 (40.6 mg, 0.087 mmol), and 3 Å molecular sieves (80 mg) in dichloroethane (1.0 mL) was stirred under argon for 1 h at rt. After that, freshly activated AgOTf (22.5 mg, 0.087 mmol) was added, and the resulting mixture was stirred for 5 min at rt. The solid was filtered off through a pad of Celite, rinsed successively with dichloromethane, and the combined filtrate (~ 30 mL) was washed with H2O (2×10 mL). The organic phase was separated, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane 5% gradient elution). The unreacted donor 5 was recovered in 91% yield and its identity was confirmed by 1H NMR and mass spectroscopy. A competition experiment with allyl thioindolyl glucoside 8 (30 mg, 0.042 mmol) instead of donor 5 was performed in a similar manner. The unreacted donor 8 was recovered in 95% yield and its identity was confirmed by 1H NMR and mass spectroscopy.
1H NMR monitoring experiments
A typical experiment in the presence of NIS.
A solution of SInMe donor 2 (0.0404 mmol) or SInAll donor 6 (0.0391 mmol) and NIS (0.0391–0.1173 mmol) in CDCl3 (1.0 mL) was stirred in a RB flask under argon for 30 min at rt. The resulting solution was transferred into a standard 5 mm NMR tube and 1H NMR spectra recorded at 10 min, 1 h, 16 h timepoints or as needed. The recorded spectra are presented in the SI. Analytical data for intermediate 37: Rf=0.6 (ethyl acetate/toluene, 0.5/9.5, v/v); 1H NMR (300 MHz): δ 3.95–3.98 (m, 1H, J5,6a=5.7 Hz, H-5), 4.42 (dd, 1H, J6a,6b=11.9 Hz, H-6a), 4.56 (dd, 1H, H-6b), 4.69–5.22 (m, 5H, H-1, NCH2, =CH2), 5.62–5.72 (m, 2H, H-2, 4), 5.77–5.86 (m, 1H, CH=), 5.92 (dd, 1H, J3,4=9.4 Hz, H-3), 7.09–7.94 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 47.8, 62.8, 69.1, 70.6, 73.1, 74.5, 76.3, 88.5, 111.3, 116.6, 121.2, 122.5, 124.6, 128.3 (x2), 128.4 (x2), 128.5 (x2), 128.6 (x2), 128.9 (x2), 129.3 (x2), 129.7 (x4), 129.9 (x2), 130.0 (x2), 130.2 (x2), 133.2 (x2), 133.3 (x2), 133.4, 137.9, 165.1, 165.3, 165.7, 165.9 ppm; ESI-TOF [M+Na]+: calcd for [C45H36INO9SNa]+ 916.1048; found: 916.0990. Analytical data for intermediate 38: Rf=0.3 (ethyl acetate/diethyl ether, 2/3, v/v); 1H NMR (300 MHz): δ 3.85 (s, 3H, NCH3), 3.96–4.00 (m, 1H, J5,6a=5.9, H-5), 4.41 (dd, 1H, J6a,6b=12.2 Hz, H-6a), 4.56 (dd, 1H, H-6b), 4.93 (d, 1H, J1,2=10.1 Hz, H-1), 5.62–5.70 (m, 2H, H-2, 4), 5.92 (dd, 1H, J3,4=9.5 Hz, H-3), 7.09–7.95 (m, 24H, aromatic) ppm; 13C{1H} NMR (75 MHz): δ 32.1, 63.0, 69.1, 70.6, 71.9, 73.7, 76.2, 88.9, 110.5, 120.8, 121.9, 124.3, 128.3 (x2), 128.4 (x4), 128.5 (x2), 128.6 (x2), 128.9 (x2), 129.3 (x2), 129.6 (x2), 129.8 (x2), 130.2 (x2), 130.4 (x2), 133.3, 133.5 (x2), 138.5, 165.1, 165.3, 165.7, 165.9 ppm; ESI-TOF [M+Na]+: calcd for [C43H34INO9SNa]+ 890.0896; found: 890.0832.
A typical experiment in the presence of TMSOTf.
A solution of SInMe donor 2 (0.0404 mmol) or SInAll donor 6 (0.0391 mmol) and TMSOTf (0.040–0.1564 mmol) in CDCl3 (1.0 mL) was stirred in a RB flask under argon for 5 min at rt. The resulting solution was transferred into a standard 5 mm NMR tube and 1H NMR spectra recorded at 10 min, 30 min, 1 h, 16 h timepoints or as needed. The recorded spectra are presented in the SI.
Supplementary Material
Acknowledgements
This work was supported by awards from the NIGMS (GM111835) and the NSF (USA, CHE-1800350).
Footnotes
The authors declare no competing financial interests.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is available on the WWW under https://doi.org/10.1002/ejoc.202200300
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Panza M, Pistorio SG, Stine KJ, Demchenko AV, Chem. Rev 2018, 118, 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hasty SJ, Demchenko AV, Chem. Heterocycl. Compd 2012, 48, 220–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Demchenko AV, Malysheva NN, De Meo C, Org. Lett 2003, 5, 455–458. [DOI] [PubMed] [Google Scholar]
- [4].Demchenko AV, Pornsuriyasak P, De Meo C, Malysheva NN, Angew. Chem. Int. Ed 2004, 43, 3069–3072; Angew. Chem. 2004, 116, 3131–3134. [DOI] [PubMed] [Google Scholar]
- [5].Hasty SJ, Kleine MA, Demchenko AV, Angew. Chem. Int. Ed 2011, 50, 4197–4201; Angew. Chem. 2011, 123, 4283–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yasomanee JP, Demchenko AV, Trends Glycosci. Glycotechnol 2013, 25, 13–42. [Google Scholar]
- [7].Shrestha G, Panza M, Singh Y, Rath NP, Demchenko AV, J. Org. Chem 2020, 85, 15885–15894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaeothip S, Pornsuriyasak P, Demchenko AV, Tetrahedron Lett. 2008, 49, 1542–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhong W and Boons G-J in Glycoside synthesis from 1-sulfur/selenium-substituted derivatives: thioglycosides in oligosaccharide synthesis, (Ed. Demchenko AV), Wiley-VCH, Weinheim, Germany, 2008, pp. 261–303. [Google Scholar]
- [10].Singh Y, Demchenko AV, Chem. Eur. J 2020, 26, 1042–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang QB, Jia WL, Ban YL, Zheng Y, Liu Q, Wu LZ, Chem. Eur. J 2016, 22, 2595–2598. [DOI] [PubMed] [Google Scholar]
- [12].Pedras MSC, Jha M, J. Org. Chem 2005, 70, 1828–1834. [DOI] [PubMed] [Google Scholar]
- [13].Lemieux RU in Acylglycosyl halides. Tetra-O-benzoyl-α-D-glucopyranosyl bromide, Vol. 2 Eds.: Whistler RL and Wolform ML), Academic Press Inc., New York and London, 1963, pp. 226–228. [Google Scholar]
- [14].Ranade SC, Kaeothip S, Demchenko AV, Org. Lett 2010, 12, 5628–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Garcia BA, Gin DY, J. Am. Chem. Soc 2000, 122, 4269–4279. [Google Scholar]
- [16].Pornsuriyasak P, Demchenko AV, Chem. Eur. J 2006, 12, 6630–6646. [DOI] [PubMed] [Google Scholar]
- [17].Nigudkar SS, Parameswar AR, Pornsuriyasak P, Stine KJ, Demchenko AV, Org. Biomol. Chem 2013, 11, 4068–4076. [DOI] [PubMed] [Google Scholar]
- [18].Brennan S, Finan PA, J. Chem. Soc C 1970, 1742–1744. [Google Scholar]
- [19].a) Baek JY, Lee B-Y, Pal R, Lee W-Y, Kim KS, Tetrahedron Lett. 2010, 51, 6250–6254; [Google Scholar]; b) Singh Y, Wang T, Geringer SA, Stine KJ, Demchenko AV, J. Org. Chem 2018, 83, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Andersson F, Fugedi P, Garegg PJ, Nashed M, Tetrahedron Lett. 1986, 27, 3919–3922. [Google Scholar]
- [21].Kamat MN, Rath NP, Demchenko AV, J. Org. Chem 2007, 72, 6938–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Demchenko AV, Kamat MN, De Meo C, Synlett 2003, 1287–1290. [Google Scholar]
- [23].Lemieux RU in Acylglycosyl halides. Tetra-O-acetyl-α-D-glucopyranosyl bromide, Vol. 2 Eds.: Whistler RL and Wolform ML), Academic Press Inc., New York and London, 1963, pp. 221–222. [Google Scholar]
- [24].Eby R, Schuerch C, Carbohydr. Res 1975, 39, 33–38. [DOI] [PubMed] [Google Scholar]
- [25].Pougny JR, Nassr MAM, Naulet N, Sinay P, Nouveau J. Chem 1978, 2, 389–395. [Google Scholar]
- [26].Chiba H, Funasaka S, Mukaiyama T, Bull. Chem. Soc. Jpn 2003, 76, 1629–1644. [Google Scholar]
- [27].Ito Y, Ogawa T, Numata M, Sugimoto M, Carbohydr. Res 1990, 202, 165–175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.