

ABSTRACT
Cancer immunotherapy approaches target signaling pathways that are highly synonymous between CD4 and CD8 T-cell subsets and, therefore, often stimulate nonspecific lymphocyte activation, resulting in cytotoxicity to otherwise healthy tissue. The goal of our study was to identify intrinsic modulators of basic T lymphocyte activation pathways that could discriminately bolster CD8 anti-tumor effector responses. Using a Tbc1d10c null mouse, we observed marked resistance to a range of tumor types conferred by Tbc1d10c deficiency. Moreover, tumor-bearing Tbc1d10c null mice receiving PD-1 or CTLA-4 monotherapy exhibited a 33% or 90% cure rate, respectively. While Tbc1d10c was not expressed in solid tumor cells, Tbc1d10c disruption selectively augmented CD8 T-cell activation and cytotoxic effector responses and adoptive transfer of CD8 T cells alone was sufficient to recapitulate Tbc1d10c null tumor resistance. Mechanistically, Tbc1d10c suppressed CD8 T-cell activation and anti-tumor function by intersecting canonical NF-κB pathway activation via regulation of Map3k3-mediated IKKβ phosphorylation. Strikingly, none of these cellular or molecular perturbations in the NF-κB pathway were featured in Tbc1d10c null CD4 T cells. Our findings identify a Tbc1d10c-Map3k3-NF-κB signaling axis as a viable therapeutic target to promote CD8 T-cell anti-tumor immunity while circumventing CD4 T cell-associated cytotoxicity and NF-κB activation in tumor cells.
KEYWORDS: Immuno-oncology, squamous cell carcinoma, melanoma, carabin, NF-κB, Map3k3
Introduction
The presence of activated cytotoxic (CD8) T cells within the tumor microenvironment is critical for the eradication of cancer.1 Yet, many tumors circumvent cytotoxic T-cell elimination through mechanisms such as the reduction of tumor antigen expression, the presentation of molecules that engage immune checkpoints (e.g. PD/L-1, CTLA-4), or the secretion of immunoregulatory molecules.2–4 Indeed, some of the most impactful cancer treatments are immunotherapies that counteract these immunosuppressive actions, such as CTLA-4 and PD-1/PDL-1 checkpoint inhibitor antibodies, Chimeric Antigen Receptor (CAR)-T-cell therapy, or adoptive transfer of tumor infiltrating lymphocytes (TILs).5–8 However, these therapies can cause severe toxicity or autoreactivity in patients,9,10 and they are not yet optimal or efficacious for many patients,2,5,6,8,10 necessitating novel therapies to target cancerous malignancies while sparing healthy tissues.
Current FDA-approved immunotherapies designed to increase T-cell effector function target proteins or pathways that are typically present in both CD4 and CD8 T cells. CTLA-4 or PD-1 checkpoint blockade therapies, for instance, stimulate intra-tumoral cytotoxic effector activity but can also stimulate effector autoreactivity in otherwise healthy tissues. As a result, immunotherapy treatments often carry undesirable side effects such as diarrhea, rash, pruritis, colitis, myocarditis, hepatitis, nephritis, encephalitis, thyroiditis, and pneumonitis.10–12 These adverse reactions can occur in up to 60% of patients on CTLA-4 monotherapy treatment, with 10–30% of these cases being potentially fatal.11,13,14 Likewise, PD-1/PDL-1 monotherapy demonstrates a substantial side effect profile that includes fatigue, headache, and nausea as well as similar inflammatory disorders mentioned above,11,15,16 although PDL-1 therapy appears to be less toxic than PD-1 therapy;17 these side effects occur in 5–20% of patients, with serious adverse reactions in up to 10% of patients.11 Combination therapy demonstrates a higher degree of side effects and more commonly leads to treatment discontinuation.11,18 Likewise, CAR-T-cell therapy is associated with neurological toxicity, including headaches, seizures, or loss of consciousness, as well as allergic reactions, increased risk of serious infections, low blood cell counts, and cytokine release syndrome (CRS).10,19,20 A recently developed CAR called Hu19-CD828Z that targets the CD19 receptor shows a dramatic reduction of neurological toxicity and CRS from 50% to 5% of patients.21 In some cases, adverse reactions can be tolerated with the administration of over-the-counter pain medications, lose-dose steroids, or immunosuppressants.11 However, more severe cases require treatment discontinuation or prove fatal to the patient.10,11 As such, the aforementioned clinical phenomena underscore the need for efficacious immunotherapies that target activation pathways selective for CD8 T cells to augment anti-tumor cytotoxic function without impacting CD4 T-cell subsets in healthy tissue, including Th1 or regulatory T cells (Tregs) that inhibit autoreactivity. However, a major impediment to fulfilling this need is that the majority of signaling proteins involved in T-cell activation, such as the canonical T-cell receptor signaling pathway, are largely synonymous between CD4 and CD8 T-cell subsets.22–24
TBC1D10C has been shown to play an immunosuppressive role in human CD4 T-cell activation25 and murine B-cell-mediated autoimmunity.26 These previous findings suggest that TBC1D10C function may play a role in a variety of disease states involving perturbed lymphocyte activation such as cancer. Indeed, previous metadata analyses across multiple cancer types27–31 suggest that TBC1D10C expression may be relevant for tumorigenesis. However, any functional role for TBC1D10C in the immune response to cancer has not been reported. Our work demonstrates that genetic disruption of Tbc1d10c, also referred to as Carabin, selectively confers increased CD8 T-cell-mediated anti-tumor immunity responses with little to no impact on CD4 T cell levels or activation status. Mechanistically, augmented CD8 T-cell activation and cytotoxic function conferred by Tbc1d10c deficiency was due to increased Map3k3 protein stability, which intersects the NF-κB pathway via activation of the IKK complex in untouched and TCR ligated CD8 T cells. However, no perturbations in canonical NF-κB pathway proteins were observed in Tbc1d10c-deficient CD4 T cells. As such, our findings identify a Tbc1d10c-Map3k3 intersection with the canonical NF-κB pathway as a selective therapeutic target to promote anti-tumor immune responses that may also circumvent undesirable cytotoxicity.
Materials and Methods
Animals
The targeting cassettes for Tbc1d10c null (Tbc1d10c−/−) and Tbc1d10c-CreERT2 transgenic mice were generated in our laboratory by bacterial artificial chromosome (BAC) recombineering approaches,32 and chimeric and transgenic founder mice were generated by the CUIMC Transgenic Mouse Core Facility (Supplemental Figures S1 and S2). Experimental Tbc1d10c−/− and Tbc1d10c+/+ (wild-type) mice were generated by backcrossing ten generations onto C57Bl/6 and FVB genetic backgrounds. Tbc1d10c-CreERT2 mice, backcrossed ten generations onto a FVB background, were crossed with FVB-backcrossed Rosa26mT/mG mice33 (Jackson Laboratories) to generate Tbc1d10c-CreERT2;Rosa26mT/mG bigenic mice. Rag2−/−,34 OT-I transgenic,35 and OT-II transgenic36 mice were acquired from Jackson Laboratories. OT-I and OT-II transgenic mice were crossed to Tbc1d10c−/− mice to generate OT-I;Tbc1d10c−/− and OT-II;Tbc1d10c−/− lines. Rag2−/− mice were crossed to Tbc1d10c−/− mice to generate Rag2−/−;Tbc1d10c−/− double null mice. All animal housing and experiments were conducted under an approved Institutional Animal Care and Use Committee protocol (Protocol number AC-AABC3509).
Antibodies and oligonucleotide sequences
Please see Supplemental Table S1 for a complete list of oligonucleotide primers and siRNAs and Supplemental Table S2 for all antibodies used in this study.
Tumor cell lines
Murine squamous cell carcinoma (SCC) cell lines SCC1 and LungMet2 were previously generated in our laboratory from FVB mice.37 Murine B16-F10 melanoma and MC38 colon adenocarcinoma lines were purchased from ATCC. Ovalbumin-expressing B16-F10 melanoma cells (B16-OVA)38 were kindly provided by Dr Charles Drake.
Delayed contact hypersensitivity assay
Contact hypersensitivity assays were performed with 2,4-Dinitro-1-fluorobenzene (DNFB) (Sigma Aldrich) as previously described39 in Tbc1d10c+/+ and Tbc1d10c−/− mice. Mice were sensitized by topical application of 0.5% DNFB as a hapten in a vehicle of acetone/olive oil (4:1 v/v) onto shaved abdominal skin. After 6 days, immune responses were elicited by topical application of 0.2% DNFB onto the ear. Ear thickness was measured on treated and untreated ears prior to and 24 hrs after DNFB with calipers. Epidermal swelling caused by infiltrating lymphocytes was rendered by the change in ear thickness before and after DNFB treatment (normalized to vehicle-treated mice; n = 5 mice per group).
Tamoxifen treatment
Normal or SCC-bearing Tbc1d10c-CreERT2;Rosa26mT/mG bigenic mice were given daily intraperitoneal injections of 1 mg tamoxifen (Sigma Aldrich) in corn oil for four days to induce CreERT2 activity as a means to genetically mark Tbc1d10c-expressing cells with EGFP. 24 hrs after the final tamoxifen treatment, mice were sacrificed and cell suspensions from the spleen, lymph nodes, peritoneal cavity or SCC tumors were isolated and labeled with antibody panels for the specified immune lineages.
Orthotopic tumor assays
Tbc1d10c+/+ and Tbc1d10c−/− mice received sub-cutaneous (s.c.) injections of 4 × 105 SCC1 (FVB background) or 4 × 105 MC38 or 5 × 104 B16-F10 (C57Bl/6 background) cells. Rag2−/−;Tbc1d10c+/+ and Rag2−/−;Tbc1d10c−/− mice on a C57Bl/6 background were administered 4 × 105 MC38 or 5 × 104 B16-OVA cells. All cells were administered in 200 μl of Hanks’s Balanced Salt Solution (Gibco) containing 50% Matrigel (Corning). For immunotherapy SCC studies, Tbc1d10c+/+ and Tbc1d10c−/− mice received intraperitoneal injections of 250 µg PD-1 or IgG2a isotype control or 100 µg CTL-A4 or IgG2b isotype control antibodies (BioXCell) as previously described40 on days 3, 6 and 9 following tumor implantation. In all cases, tumor diameter measurements were recorded every 2 days with calipers. Tumor-bearing mice were designated by the presence of a growth in the flank skin greater than 50 mm in diameter and persistent for 24 hrs. When tumors reached the 2 cm diameter end point, tumors were harvested by surgical excision for cellular and biochemical analyses below. Average tumor size (mm2) and the percentage of tumor-free mice were compared between cohorts.
Tumor proliferation
For proliferation studies, orthotopic SCC tumor-bearing mice received a single intraperitoneal injection of 50 mg/kg 5-ethynyl-2′-deoxyuridine (EdU) (Invitrogen). After 24 hrs, orthotopic tumor single-cell suspensions were generated by enzymatic digestion as previously described.37 EdU incorporation was detected using the Click-iT EdU Alexa Fluor 488 Imaging Kit as per manufacturer’s instruction (Invitrogen) and quantified by flow cytometry using a BD Fortessa scanner (BD Biosciences).
Skin carcinogenesis
Cutaneous SCCs were induced in the skin of Tbc1d10c-CreERT2;Rosa26mT/mG mice as previously described using a 7,12-dimethylbenz[a]anthracene (Acros Organics)/12-O-tetradecanoylphorbo-13-acetate (LC Laboratories) two-step chemical carcinogenesis protocol.37 One week following the emergence of cSCCs, tumor-bearing mice received intraperitoneal injections of 1 mg tamoxifen as described above. 24 hours following tamoxifen administration, cSCCs were harvested for cellular analyses.
Isolation of primary T cells
Mouse splenic and lymph node single-cell suspensions were generated by mechanical dissociation as previously described.41 CD4 or CD8 T cells were purified from single-cell suspensions using EasySep T Cell Negative Isolation Kits as per manufacturer’s instruction (STEMCELL Technologies). We routinely observed 90–95% purity of CD8 T cells by FACS analysis with no differences detected between Tbc1d10c+/+ and Tbc1d10c−/− mice (Supplemental Figure S3). Henceforth, we refer to T cell subsets purified by negative isolation as untouched. For OVA peptide stimulation of OT-I transgenic CD8 T cells, wild-type CD8 T cells were first removed from recovered splenocyte cultures using EasySep T Cell Positive Isolation Kits as per manufacturer’s instruction (STEMCELL Technologies). Primary human CD8 T cells were purified from the peripheral blood mononuclear cell fraction of whole blood (New York Blood Center) using the EasySep Human Naïve CD8 + T Cell Isolation Kit II as per manufacturer’s instruction (STEMCELL Technologies).
Cell Culture
SCC1 and LungMet2 cells were cultured in complete FAD medium.42 MC38, B16, and B16-OVA cells were cultured in DMEM (Gibco) supplemented with 10% defined FBS (HyClone). In some cases, B16-OVA cells were incubated with 10 ng/mL murine recombinant IFN-γ (R&D Systems) for 24 hours to stimulate MHC I expression.43 Murine and human T cells were cultured in RPMI 1640-GlutaMax (Gibco) supplemented with 10% heat-inactivated defined FBS (HyClone) and 55 µM 2-Mercaptoethanol (Gibco).
siRNA Gene Silencing
Primary murine or human CD8 T cells were electroporated with four pooled (SMARTpool) pre-designed siRNAs (Dharmacon) targeting murine Epsti1, Map3k3 or human TBC1D10C or non-targeting siRNA control (Scramble) using Mouse or Human T Cell Nucleofector Kits as per manufacturer’s instruction (Lonza). Electroporated cells were recovered for 24 hrs in complete growth medium prior to downstream experimental use.
T-cell stimulation
For antibody stimulation, CD4 or CD8 T cells were plated in complete RPMI medium at 1.3 × 106 cells/cm2 in wells coated with 1 µg/mL CD3 and soluble (5 µg/mL) CD28 antibodies for either 0, 24, 48 or 72 hrs or 15, 30 or 60 min. Unstimulated control cells were plated without CD3 or CD28 antibodies. For dose response studies, T cells were stimulated on plates coated with a range of 0–10 µg CD3 antibody. For OT-I transgenic CD8 T-cell stimulation, red blood cell-cleared C57Bl/6 splenocytes (2 X 106 cells/cm2) were coated with 10 µg/ml OVA257-264 peptide (SIINFEKL) (GenScript). Co-cultures were incubated in complete RPMI medium for 0, 24, 48 or 72 hours at a cell density of 1 × 106 splenocytes and 5 × 105 OTI CD8 T cells/cm2. OT-II transgenic CD4 T cells were stimulated on plates coated with a range of 0–300 nM OVA323-339 peptide (ISQAVHAAHAEINEAGR) (GenScript). Tbc1d10c+/+ and Tbc1d10c−/− OT-I transgenic CD8 T cells or OT-II transgenic CD4 T cells were loaded with CFSE as per manufacturer’s instruction (BioLegend) and then stimulated with OVA.
T-cell proliferation assay
CD4 or CD8 T cells were pre-loaded with CFSE as per manufacturer’s instruction (BioLegend) and then stimulated with CD3/CD28 antibodies or OVA peptides as described above for up to 72 h. T-cell proliferation as determined by quantification of the percentage of cells exhibiting CFSE dilution was detected by flow cytometry.
Trans-well migration assay
5 × 105 untouched CD8 T cells were loaded in 100 µl of RPMI 1640-GlutaMax media (Gibco) supplemented with 1% heat-inactivated defined FBS (HyClone) into the upper insert of Boyden chambers (Corning). Murine recombinant Cxcl10 (PeproTech) was added to the lower chamber at concentrations of 0, 1, 10, or 100 ng/ml. After 3 h, migratory cells were counted in the media of the lower chamber and the transmigration index was calculated as follows: Index = (experimental cell count) ÷ (0 ng/ml Cxcl10 control cell count).
MC38 tumor killing assay
CFSE-loaded OT-I;Tbc1d10c+/+ and OT-I;Tbc1d10c−/− CD8 T cells were activated for 24 h by OVA peptide stimulation as described above. MC38 cells were pre-incubated with 10 ng/ml murine recombinant Ifn-γ (R&D Systems) and OVA257-264 peptide for 24 hrs and subsequently stained with PKH26 membrane dye as per manufacturer’s protocol (Sigma Aldrich). FACS-sorted CD8 CFSE+ T cells were then plated with OVA-coated MC38 cells for 12 hrs at the following effector-to-target cell ratios: 1:2, 1:1, 2:1, and 4:1. In some cases, siRNA-transduced OT-I CD8 T cells were incubated at a 2:1 effector-to-target ratio. For each ratio, each T cell effector genotype was analyzed in triplicate. Cultures were harvested and stained with Fixable Viability Dye as per manufacturer’s Protocol (Invitrogen), and MC38-specific lysis was quantified by flow cytometry. For control values, MC38 cells were either boiled for 5 minutes (Maximal lysis) or incubated without CD8 T cells (nonspecific lysis). Specific lysis was calculated as follows: Specific Lysis = [(Sample Lysis) – (nonspecific Lysis)] ÷ [(Maximal Lysis) – (nonspecific Lysis)].
Adoptive T-cell transfer
Untouched OT-I;Tbc1d10c+/+ and OT-I;Tbc1d10c−/− CD4 and CD8 T cells were adoptively transferred by tail vein injection into Rag2−/− mice. CD4 and CD8 T cells from the following genotypes were mixed at a 1:1 ratio: OT-I;Tbc1d10c+/+ CD4 and OT-I;Tbc1d10c+/+ CD8; OT-I;Tbc1d10c+/+ CD4 and OT-I;Tbc1d10c−/− CD8; OT-I;Tbc1d10c−/− CD4 and OT-I;Tbc1d10c+/+ CD8; and OT-I;Tbc1d10c−/− CD4 and OT-I;Tbc1d10c−/− CD8. A total of 2 × 106 T cells were injected per mouse. In some cases, OT-I;Tbc1d10c+/+ or OT-I;Tbc1d10c−/− CD8 T cells alone were adoptively transferred into Rag2−/− mice at 1 × 106 per mouse. Control groups included vehicle alone injected mice. Equivalent levels of Tbc1d10c+/+ and Tbc1d10c−/− T-cell uptake were confirmed by FACS analysis of dissociated splenocytes from adoptive transfer mice.
RNA isolation, cDNA synthesis, and Quantitative PCR Analysis
Total RNA extracted from CD8 T cells was subjected to first strand cDNA synthesis as per manufacturer’s instruction using the SuperScript III First-Strand Synthesis SuperMix kit (ThermoFisher). Quantitative PCR (QPCR) analyses for Tbc1d10c, Map3k3, Epsti1 and Gapdh mRNA levels were performed with SYBR® Green Master Mix (Bio-Rad) in a Bio-Rad CFX96 system. Each sample was run in a triplicate. QPCR oligonucleotide primer sequences are listed in Supplemental Table S1.
Immunoblotting
Whole cell protein lysates were generated from CD8 and CD4 T cells in RIPA buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) containing a phosphatase and protease inhibitor cocktail (Sigma-Aldrich). In some cases, nuclear and cytosolic cell fractions were isolated from CD8 T cells using the NE-PER Nuclear and Cytoplasmic Extraction kit as per manufacturer’s protocol (Thermo Scientific). Protein lysates were separated on 10% Tris-Glycine gels (ThermoFisher), transferred to nitrocellulose (Bio-Rad) and probed with primary antibodies followed by chemiluminescence detection. Protein levels were quantified by densitometric analysis using NIH-ImageJ software.
Tbc1d10c expression kinetics in cultured CD8 T cells
Untouched splenic wild-type CD8 T cells were stimulated with CD3/CD28 antibodies as described above for two days followed by expansion in complete RPMI media supplemented with 10 ng/ml recombinant Il-2 protein (R&D Systems) for three days. Cells were then stimulated again with CD3/CD28 antibodies at 2–5 million cells per well over the following time points: 0 min, 30 min, 1 h, 2 h, 4 h, 6 h, 12 h, and 24 h. Cells were harvested at each time point for total RNA or protein extraction (n = 3 replicates for each time point for RNA and protein). Unstimulated control cells were plated in the absence of CD3/CD28 antibodies. In some cases, untouched wild-type CD8 T cells were stimulated as described above, immediately following isolation from the spleen, followed by RNA and protein extraction.
Immunoprecipitation
CD8 T cells for immunoprecipitation were lysed in standard TritonX-100 lysis buffer with phosphatase and protease inhibitor cocktail (Sigma-Aldrich). 1 mM EDTA (Fisher Scientific) and 25 mM N-ethylmaleimide (Sigma-Aldrich) were also added to inhibit deubiquitinating enzymes. Samples were pre-cleaned using agarose-conjugated normal mouse IgG (Santa Cruz Biotechnology), and then protein supernatants were incubated with 5 µg agarose-conjugated Bcl10 primary antibody (Santa Cruz Biotechnology) overnight on a rotator at 4°C. Bead supernatants were boiled in SDS-PAGE protein loading buffer for immunoblot analysis.
Tissue Immunofluorescence
SCC histological tissue sections (7 μm thick) were blocked and probed with primary antibodies followed by detection with species-specific AlexaFluor conjugated secondary antibodies. DAPI counterstain was used to visualize nuclei. Immunofluorescence was captured using a Zeiss LSM 5 Exciter confocal microscope, and reconstructed images were generated using NIH ImageJ software.
Flow cytometry
Single-cell suspensions were generated from mechanically dissociated mouse spleens or enzymatically dissociated xenograft tumors as previously described.30,34 Cell suspensions were labeled with directly conjugated primary antibodies (Supplementary Table S2) and analyzed using a BD LSRFortessa flow cytometer (BD Biosciences). For quantification, all flow cytometry data were analyzed using FlowJo software (BD Biosciences).
Single-Cell RNA sequencing
Live CD8 T cells from Tbc1d10c+/+ and Tbc1d10c−/− spleens were captured on a microfluidic chip at approximately 5000 cells per sample and processed for single-cell RNA sequencing with the 10X Genomics Chromium 3′ Solution platform.44 cDNA synthesized by this method was amplified and sequenced on an Illumina NextSeq. Gene-barcode counts matrices were analyzed with the Seurat R package (version 4.0.3).45 Cells with < 500 or > 42000 genes detected (Doublets) and > 20% mitochondrial gene mapped reads were filtered from downstream analyses. Tbc1d10c+/+ and Tbc1d10c−/− CD8 T-cell cohort samples were merged into one Seurat object. The merged Seurat object was normalized and scaled by regressing out UMI count (104) and percentage of mitochondrial and ribosomal genes. For dimensionality reduction, 2000 highly variable genes were identified based on their average expression and variance and used for clustering analysis. Dimensionality reduction was then performed using PCA, and UMAP plots were generated by the RunUMAP Seurat function with the first 9 PCs as input, determined by visualizing the drop off in PC variance explained using the ElbowPlot function in Seurat. CD8 T Cell Subtypes were classified and annotated using the ProjecTILs R package (version 2.0.0).46 Briefly, the normalized expression matrix of the single-cell scRNA-seq data was provided as input, and non-T cells were filtered and batch corrected. The make.projection function was used to project the query data over the mouse lymphocytes reference data in UMAP space. Cell states were predicted for each cell using a nearest-neighbor algorithm with the cellstate.predict function, and predicted cell states were saved as functional cluster data slot in the Seurat object and plotted on UMAP. Differentially expressed genes within each cluster were filtered by cross-referencing with differentially expressed proteins identified by global proteomic analysis.
LC-MS/MS global proteomic analysis
Tbc1d10c+/+ and Tbc1d10c−/− splenic CD8 T cells (n = 3 replicates per group), were subjected to Data Independent Acquisition (DIA)-based proteomic analysis47 by the Proteomics and Macromolecular Crystallography Shared Resource Core at Columbia University. Peptides were prepared as previously described48 and injected on Orbitrap Fusion™ Tribrid™ mass spectrometer MS/MS analysis. DIA data were analyzed with directDIA 2.0 (Deep learning augmented spectrum-centric DIA analysis) in Spectronaut Pulsar X software (Biognosys). The default settings were used for targeted analysis of DIA data in Spectronaut, and DIA files were searched against the mouse UniProt fasta database. Results obtained from Spectronaut were further analyzed using the Spectronaut statistical package. Significantly changed protein abundance was determined by an unpaired t-test with a threshold for significance of p < .05 (permutation-based FDR correction) and 0.58 log2FC. Data sets can be accessed via the MassIVE repository (MSV000090556).
Experimental replicates and statistical analyses
All orthotopic tumor experiments in Tbc1d10c+/+ and Tbc1d10c−/− mice were repeated at least twice. Adoptive cell transfer experiments were repeated once, and the ICI experiment was conducted once. All T cell activation, siRNA knockdown, tumor cell killing and immunoblotting experiments were repeated at least once. Results are expressed as mean ± SD unless explicitly stated otherwise. Comparisons of two groups were analyzed by unpaired Student’s t-test, and comparisons of more than two groups by one-way ANOVA with Bonferroni post hoc correction (P < .05). Finally, all mandatory laboratory health and safety procedures have been complied within the course of conducting the experimental work outlined in this study.
Results
T-cell mediated tumor resistance conferred by Tbc1d10c deficiency
To investigate the role of Tbc1d10c in tumorigenesis, we generated a Tbc1d10c germline null (Tbc1d10c−/−) mouse model (Supplemental Figure S1A-E). Tbc1d10c−/− mice appear normal and show no overt phenotypic abnormalities (Supplemental Figure S1F-G) or evidence of autoimmunity in animals aged up to one year as determined by differences in skin or colon infiltrate or splenic weight (Supplemental Figure S1H-P). Given the reported roles for TBC1D10C in human and murine lymphocytes,25,26 we examined the steady-state levels of lymphocyte and myeloid lineages in Tbc1d10c−/− mice. No significant changes were observed in lymphocyte composition or maturation or myeloid cell composition between Tbc1d10c+/+ and Tbc1d10c−/− mice (Supplemental Figure S4A-I). In addition, no differences in skin contact hypersensitivity, which is commonly used as a read-out of in vivo CD4 T-cell activation,39 were elicited in Tbc1d10c−/− mice in response to contact allergen treatment (Supplemental Figure S4J). Collectively, these observations suggest that Tbc1d10c disruption does not impact immune homeostasis or dramatically perturb CD4 T-cell activation in vivo.
Next, we examined the sensitivity of Tbc1d10c−/− mice to the growth of orthotopic tumors derived from weakly immunogenic B16 melanoma, highly immunogenic MC38 adenocarcinoma or a cutaneous squamous cell carcinoma (SCC) SCC1 line established in our laboratory from DMBA/TPA-treated FVB mice.37 Across all three tumor types, we observed a 50–75% increase in tumor-free survival as well as a two-to-three-fold decrease in average tumor size in Tbc1d10c−/− mice compared to Tbc1d10c+/+ mice (Figure 1A-F). To elucidate the cellular basis for the observed tumor resistance conferred by Tbc1d10c deficiency, we quantified tumor cell proliferation, as measured by EdU incorporation, in SCCs from Tbc1d10c+/+ and Tbc1d10c−/− mice. No difference in the percentages of EdU+ SCC cells was observed in tumors implanted in Tbc1d10c+/+ versus Tbc1d10c−/− mice (Figure 1G) indicating that the rate of tumor cell proliferation did not underlie the diminished growth of tumors in Tbc1d10c−/− mice. Consistent with the concept that Tbc1d10c does not directly influence tumor cell proliferation, we were unable to detect Tbc1d10c mRNA expression in B16 melanoma cells, two SCC cultures SCC1 and LungMet2,37 or MC38 cells (Figure 1H). TBC1D10C protein was also absent in a panel of five independent human cutaneous SCC cultures previously established in our laboratory49 (Figure 1I). These results suggest that any pro-tumorigenic mechanism elicited by Tbc1d10c is likely mediated by host cells comprising the tumor microenvironment. To test this idea, we compared MC38 and B16 tumor growth in Tbc1d10c+/+ and Tbc1d10c−/− mice crossed onto the immunocompromised Rag2−/− background. The tumor resistance observed in immune competent Tbc1d10c−/− mice (Figure 1B-C,E-F) was completely abolished in the absence of lymphocytes (Figure 1J-M). These data strongly suggest that tumor resistance conferred by Tbc1d10c deficiency is lymphocyte mediated. In addition, since Rag2−/− mice maintain normal levels of myeloid lineages, our findings largely exclude a direct role for myeloid cells in the tumor-resistant phenotype observed Tbc1d10c−/− mice.
Figure 1.
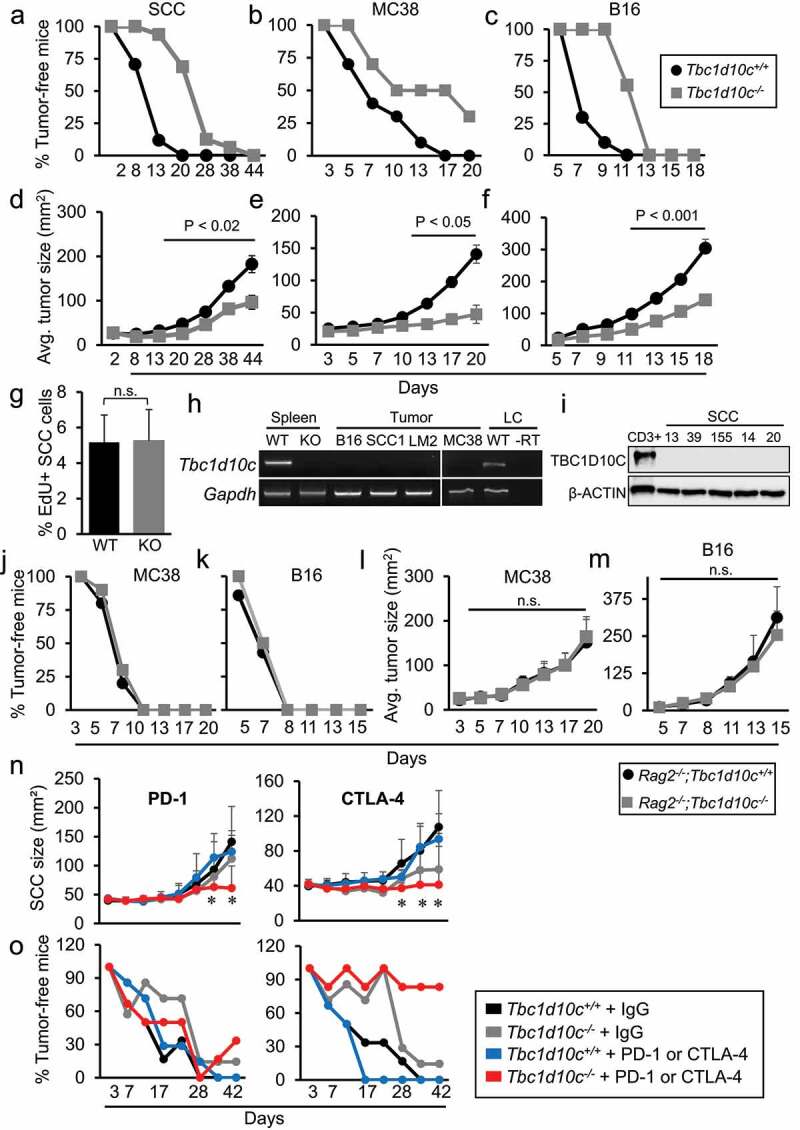
T-cell-mediated tumor resistance conferred by Tbc1d10c deficiency. (A-C) Line graphs showing percentages of tumor-free survival in mice implanted with SCC (A), MC38 (B) and B16 (C) tumors. (D-F) Line graphs showing average tumor size in SCC (D; n = 17 Tbc1d10c+/+ mice and 16 Tbc1d10c−/− mice), MC38 (E; n = 10 mice per group), and B16 (F; n = 10 Tbc1d10c+/+ mice and 9 Tbc1d10c−/− mice) orthotopic mice. (G) Bar graph showing percentages of EdU+ tumor cells in orthotopic SCCs in Tbc1d10c+/+ and Tbc1d10c−/− mice (n = 7 per group). (H) Micrographs showing representative RT-PCR detection of Tbc1d10c mRNA in Tbc1d10c+/+ and Tbc1d10c−/− spleen; SCC1, LungMet2 (LM2), B16 and MC38 tumor cell lines; and splenic lymphocytes (LC). (I) Micrographs showing immunoblot detection of TBC1D10C in human CD3+ PBMCs (positive control) and five independent human SCC cultures SCC13, 39, 155, 14 and 20. µ-ACTIN serves as a loading control. (J-M) Line graphs showing percentage of tumor-free survival (J-K) and average tumor size (L-M) in Rag2−/−;Tbc1d10c+/+ and Rag2−/−;Tbc1d10c−/− mice subjected to MC38 (J,L) (n = 10 per group) or B16 (K,M) (n = 7 per group) xenografts. (N-O) Line graphs showing average SCC size (N) and percentage of tumor-free survival (O) in Tbc1d10c+/+and Tbc1d10c−/− mice receiving injections of PD-1, CTLA-4 or IgG control antibodies at days 3, 6 and 9 following SCC cell implantation (n = 7 mice per group). Statistical differences were determined by one-way ANOVA with Bonferroni post-hoc correction (*, P < .05). All other P values were determined by unpaired Student’s t-test. Abbreviations: WT, Tbc1d10c+/+; KO, Tbc1d10c−/−; RT, reverse transcriptase.
To further explore the therapeutic utility of targeting Tbc1d10c, we performed immunotherapy checkpoint blockade experiments in SCC-bearing Tbc1d10c+/+ and Tbc1d10c−/− mice. Treatment with either PD-1 or CTLA-4 blocking antibodies led to marked reductions in SCC size compared to IgG antibody-treated Tbc1d10c−/− and PD-1- or CTLA-4-treated Tbc1d10c+/+ mice (Figure 1N). In addition, we observed 90% and 30% tumor-free survival in CTLA-4- and PD-1-treated Tbc1d10c−/− mice, respectively (Figure 1O). These results suggest that combinatorial targeting of Tbc1d10c and immune checkpoints, particularly CTLA-4, could dramatically improve tumor outcome.
Tbc1d10c deficiency impacts the levels of tumor-infiltrating CD8 effectors
To identify tumor-infiltrating Tbc1d10c-expressing cell lineages that may be key mediators of Tbc1d10c pro-tumorigenic function, we generated a Tbc1d10c-CreERT2 BAC transgenic mouse that allows for expression the tamoxifen-inducible Cre recombinase (CreERT2)50 under the control of endogenous Tbc1d10c regulatory elements (Supplemental Figure S2A). For validation, Tbc1d10c-CreERT2 mice were crossed with the Cre-inducible EGFP reporter mouse, Rosa26mT/mG to generate Tbc1d10c-CreERT2;Rosa26mT/mG bigenic mice. We observed EGFP induction in splenic, lymph node and peritoneal cavity cells from tamoxifen-treated Tbc1d10c-CreERT2;Rosa26mT/mG mice but not in vehicle-treated mice (Supplemental Figure S2B-D), suggesting that EGFP fluorescence is a reliable read-out for the identification of Tbc1d10c-expressing cells. We observed that virtually all EGFP+ cells expressed the common immune lineage marker CD45 and that EGFP marked most immune lineages, including CD3+, CD19+, CD11b+, CD11c+ and NK1.1+ in the spleen, lymph node and peritoneal cavity (Supplemental Figure S2E-H).
Next, we phenotyped tumor-infiltrating Tbc1d10c-expressing cells (EGFP+) in chemically induced cutaneous SCCs in Tbc1d10c-CreERT2;Rosa26mT/mG mice. SCC-bearing mice were administered tamoxifen to induce EGFP, and tumors were analyzed by tissue immunofluorescence and FACS analyses (Figure 2A; Supplemental Figure S5A-B). In tissue sections, EGFP+ cells were abundantly detected with peri-tumoral and intra-tumoral localization (Figure 2B). However, no EGFP was observed in Krt14+ SCC tumor keratinocytes (Figure 2B), consistent with a lack of Tbc1d10c expression in SCC cultures (Figure 1H-I). Flow cytometric analysis of nine independent SCCs demonstrated that tamoxifen induction of EGFP expression was restricted to CD45+ immune cells and present in a variety of immune lineages (Figure 2C). Interestingly, the vast majority (75%) of the EGFP+ pool in SCCs was allocated to CD11b+ and CD8+ lineages (Figure 2C). Both CD11b+/EGFP+ and CD8+/EGFP+ cells were localized intra-tumoral (Supplemental Figure S5C-D). Next, we assessed whether there may be differences in the proportions of any tumor-infiltrating immune lineages in SCC, B16, and MC38 tumors implanted in Tbc1d10c+/+ and Tbc1d10c−/− mice (Supplemental Figure S5A, E-I). No differences in the levels of CD4 T cells, CD4 Tregs, NK cells, CD11b+, CD11b+Gr-1+ MDSCs, CD11b+F4/80+ macrophages, CD11c+ dendritic cells or CD19 + B cells were observed between tumor cohorts (Supplemental Figure S6A-Q) (n = 10 tumors/group). However, two-to-three-fold increases in total CD8 T cells and two-to-nine-fold increases in Ifn-γ+ CD8 T cells were observed across SCC, B16 and MC38 tumors implanted Tbc1d10c−/− mice (Figure 2D-I; Supplemental Figure S5H-I). Finally, the CD8/CD4 Treg ratio, a critical prognostic factor in human solid tumors, was two-fold and three-fold higher for SCC and B16 tumors, respectively, implanted in Tbc1d10c−/− mice (Figure 2J). The observed increase in cytotoxic effectors in tumors from Tbc1d10c−/− mice provides a cellular basis for the tumor-resistant phenotype conferred by Tbc1d10c deficiency.
Figure 2.
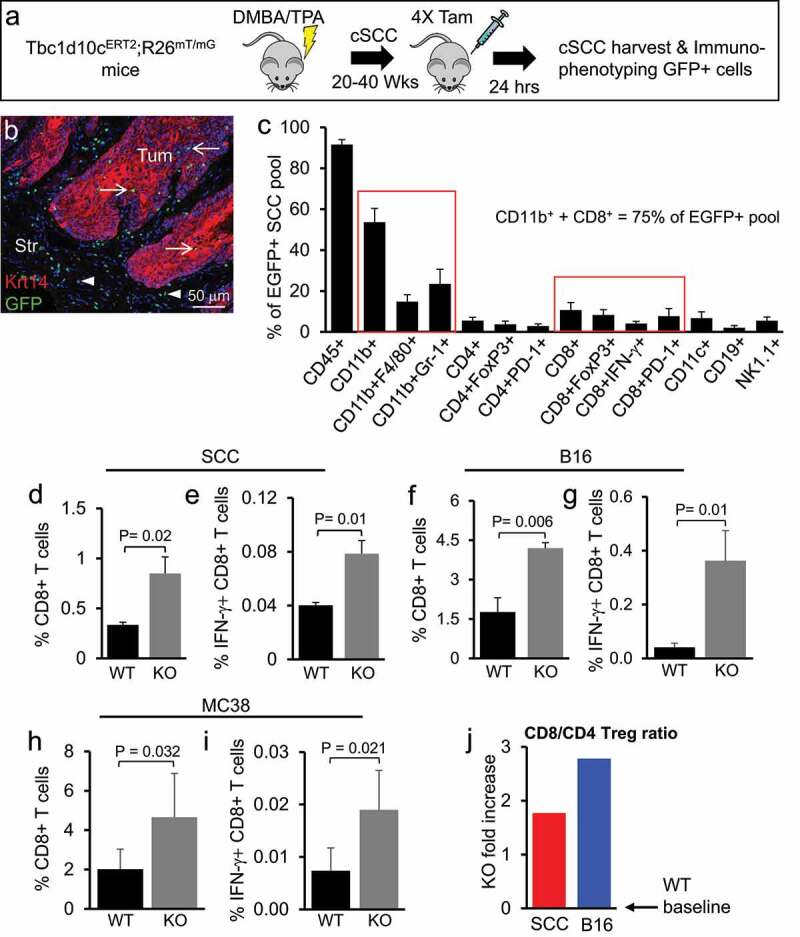
Tbc1d10c deficiency increases tumor-infiltrating CD8 effector cell levels. (A) Schematic of the experimental model for EGFP induction in chemically induced cutaneous SCCs in Tbc1d10c-CreERT2;R26mT/mG bigenic mice. (B) Representative micrograph showing EGFP labeling and immunofluorescence detection of Krt14+ SCC tumor cells in sections of chemically induced cutaneous SCCs from tamoxifen-treated bigenic mice. (C) Bar graph showing average flow cytometric detection of Tbc1d10c-expressing (EGFP+) immune lineages from nine bigenic cutaneous SCCs. Red boxes group the total pool of CD11b+ or CD8+ tumor-infiltrating cells. (D-I) Bar graphs showing the average percentages of total (D,F,H) and activated (E,G,I) CD8 + T cells from SCC (D-E; n = 4 tumors), B16 (F-G; n = 4 tumors) and MC38 (H-I; n = 6 tumors) xenografts. P values were determined by unpaired Student’s t-test. (J) Bar graph showing the fold-change differences in the CD8/CD4 Treg ratio for SCC and B16 tumors in Tbc1d10c−/− mice compared to wild-type (WT ratios set at graph baseline). Abbreviations: WT, Tbc1d10c+/+; KO, Tbc1d10c−/.
Tbc1d10c suppresses CD8 T-cell activation and tumor cell killing
To determine whether the observed increases in the levels of tumor-infiltrating Ifn-γ+ CD8 T cells in Tbc1d10c−/− mice are due to an intrinsic role for Tbc1d10c in the regulation of CD8 T-cell anti-tumor function, we assessed CD8 T-cell activation and proliferation following TCR stimulation and migration in response to chemotactic cues. The Tbc1d10c null allele was crossed onto OT-II TCR transgenic (CD4 T cell analysis) or OT-I TCR transgenic (CD8 T cell analysis) backgrounds maintained in homogeneous C57Bl/6 strains. OT-I;Tbc1d10c+/+ and OT-I;Tbc1d10c−/− CD8 T cells were stimulated on OVA257-264 peptide-coated splenocytes followed by FACS quantification of activation markers and CFSE proliferation (Supplemental Figure S7A-B). We observed two-fold increases in the levels of CD69+, CD25+, Ifn-γ+ CD8 T cells in OVA-stimulated OT-I;Tbc1d10c−/− versus OT-I;Tbc1d10c+/+ cohorts (Figure 3A-C). To establish human relevance for these observations in murine CD8 T cells, we silenced TBC1D10C expression in human CD8 T cells using siRNA pools and assessed its impact on T-cell activation. Human CD8 T cells transfected with TBC1D10C-targeting siRNAs exhibited an approximate 40% decrease in TBC1D10C protein (Supplemental Figure S8A-B) and a 1.3-fold increase in the percentage of IFN-γ + T cells following CD3/CD28 stimulation compared to CD8 T cells transfected with non-targeting control siRNA (Supplemental Figure S8C; Figure 3D). We also observed increases of 30% at 48 h and 6% at 72 h in proliferation of OVA-stimulated OT-I;Tbc1d10c−/− versus OT-I;Tbc1d10c+/+ CD8 T cells (Figure 3E; Supplemental Figure S7). However, the physiological relevance of these small, albeit significant, differences in T-cell proliferation for the tumor-resistant Tbc1d10c−/− phenotype are likely overshadowed by the observed differences in CD8 T-cell activation. Similar levels of CD69+ or Ifn-γ+ CD4 T cells or CFSE proliferation were observed between OVA-stimulated OT-II;Tbc1d10c+/+ and OT-II;Tbc1d10c−/− cohorts (Supplemental Figure S9A-D). We did observe a two-fold increase in Il-2+ CD4 T cells in the OT-II;Tbc1d10c−/− cohort at 72 h of OVA stimulation; however, this difference occurred during the decline of Il-2+ cell levels compared to peak activation at the 24-h time point (Supplemental Figure S9B). For CD3/CD28 stimulation, we observed similar percentages of Il-2+ CD4 T cells and CFSE proliferation between Tbc1d10c+/+ and Tbc1d10c−/− cohorts (Supplemental Figure S9E-F). The similar levels of CD4 T-cell activation between OVA- and CD3/CD28-stimulated Tbc1d10c+/+ and Tbc1d10c−/− CD4 T-cell cohorts are consistent with the similar responses between Tbc1d10c+/+ and Tbc1d10c−/− mice to contact allergens in vivo (Supplemental Figure S4J). Finally, we employed a well-established trans-well migration assay to measure CD8 T-cell migration in response to increased concentration gradients of the chemokine Cxcl10.51 We observed similar levels (approximately 25%) of untouched CD8 T cells expressing the Cxcl10 receptor, Cxcr3 (Supplemental Figure S9G-H) and a Cxcl10 dose-dependent increase in the numbers of migrating Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells (Figure 3F). However, no increased migration was observed in Tbc1d10c−/− CD8 T cells (Figure 3F). Collectively, our findings indicate that Tbc1d10c deficiency primarily impacts CD8 T-cell activation, and not proliferation or migration, following TCR ligation.
Figure 3.
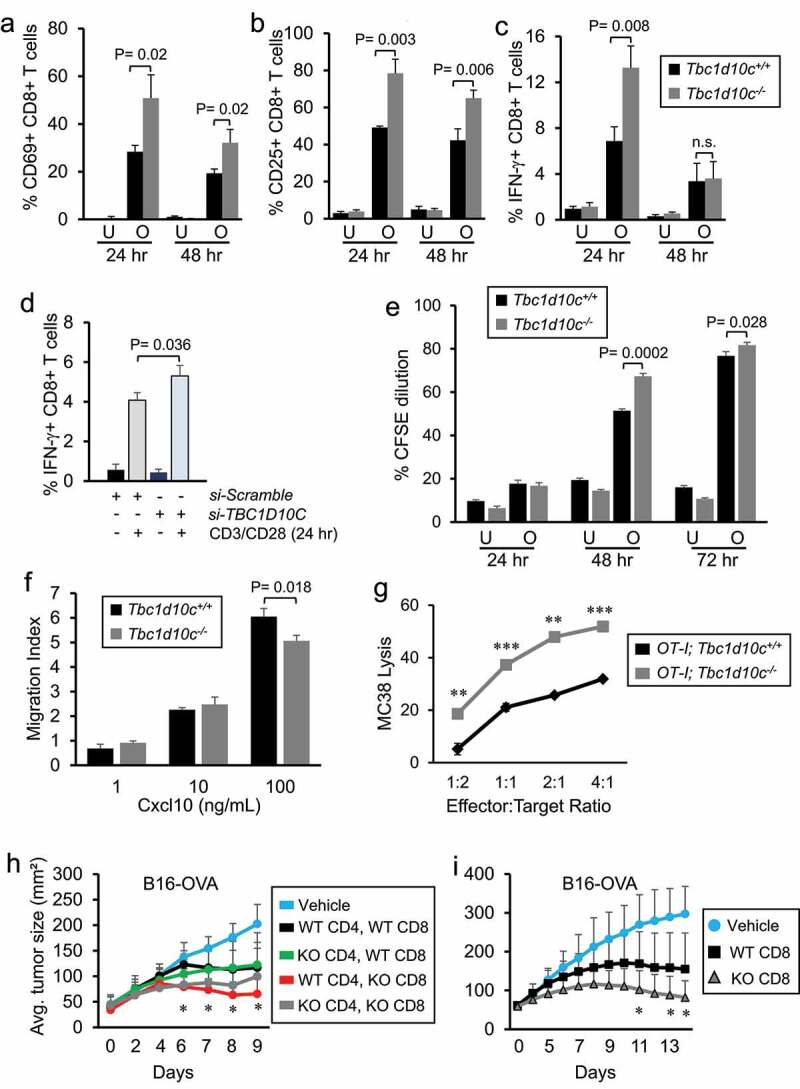
Tbc1d10c is an intrinsic suppressor of CD8 T-cell activation and cytotoxic function. (A-C) Bar graphs showing the average percentages of CD69+ (A), CD25+ (B) and Ifn-γ+ (C) CD8 T cells from OT-I;Tbc1d10c+/+ and OT-I;Tbc1d10c−/− mice either unstimulated (U) or stimulated (O) with OVA peptide for the indicated times (n = 3 replicates per group). (D) Bar graphs showing flow cytometric detection of IFN-γ+ primary human CD8 T cells transfected with non-targeting (Scramble) or TBC1D10C-targeting siRNA pools (n = 3 replicates per group). (E) Bar graphs showing proliferation as measured by CFSE dilution in CD8 T cells from OT-I;Tbc1d10c+/+ and OT-I;Tbc1d10c−/− mice either unstimulated (U) or stimulated (O) with OVA peptide for the indicated times (n = 3 replicates per group). (F) Bar graphs showing the average migration index of CD8 T cells from Tbc1d10c+/+ and Tbc1d10c−/− mice in the presence of 1, 10, and 100 ng/ml Cxcl10 (n = 3 replicates per group). (G) Line graphs showing average MC38 cell lysis by OT-I;Tbc1d10c+/+and OT-I;Tbc1d10c−/− CD8 T cells (n = 3 replicates per ratio group). (H-I) Line graph showing average B16-OVA tumor size in Rag2−/− mice adoptively transferred with OT-I CD4 and CD8 T cells (H; n = 5 mice per group) or OT-I CD8 T cells alone (I; n = 9 mice per group) carrying the indicated (WT or KO) Tbc1d10c genotype. P values were determined by unpaired Student’s t-test. Abbreviation: n.s., not significant.
To assess the relevance of enhanced CD8 T-cell activation conferred by Tbc1d10c deficiency for cytotoxic effector function, we assessed the tumor cell killing capacity of OT-I;Tbc1d10c+/+ and OT-1;Tbc1d10c−/− CD8 T cells against OVA peptide-bearing MC38 tumor cells. We observed an effector cell dose response increase in tumor cell lysis for both OT-1;Tbc1d10c−/− and OT-I;Tbc1d10c+/+ CD8 T cell cohorts (Supplemental Figure S10; Figure 3G). MC38 cell lysis was increased two- to three-fold across all effector-to-target ratios in OT-1;Tbc1d10c−/− compared to OT-I;Tbc1d10c+/+ CD8 T cells (Figure 3G). These results suggest that the augmented CD8 T-cell activation and effector function in Tbc1d10c−/− mice are sufficient to confer the tumor-resistant phenotype. To test this idea, we performed adoptive cellular transfer of CD4 and CD8 T cells into B16-OVA-bearing Rag2−/−;Tbc1d10c+/+ mice. Mice received adoptive transfer combinations of CD4 and CD8 T cells from OT-1;Tbc1d10c+/+ or OT-1;Tbc1d10c−/− mice. In some cases, OT-1 CD4 and CD8 T-cell combinations were mixed from Tbc1d10c+/+ and Tbc1d10c−/− genotypes. B16-OVA-bearing Rag2−/−;Tbc1d10c+/+ mice adoptively transferred with Tbc1d10c−/− CD8 T cells, regardless of the CD4 T-cell Tbc1d10c genotype, exhibited the most resistance to tumor growth (Figure 3H). In a separate set of experiments, B16-OVA-bearing Rag2−/−;Tbc1d10c+/+ mice were adoptively transferred with OT-I;Tbc1d10c+/+ or OT-I;Tbc1d10c−/− CD8 T cells alone. A two-fold reduction in melanoma size was observed in mice transferred with OT-I;Tbc1d10c−/− compared with OT-I;Tbc1d10c+/+ CD8 T cells (Figure 3I), which fully recapitulates the tumor-resistant phenotype observed in immune competent Tbc1d10c−/− mice (Figure 1). Collectively, these observations confirm that tumor resistance conferred by Tbc1d10c deficiency is mediated by CD8 T cells.
RNA and proteomic profiling of Tbc1d10c−/− CD8 T cells
To identify signaling pathways that may be impacted by Tbc1d10c disruption that are also relevant for CD8 T-cell-mediated anti-tumor immunity, we conducted single-cell RNA sequencing (scRNA-Seq) of untouched Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells. Unsupervised clustering of Tbc1d10c+/+ and Tbc1d10c−/− cohorts identified similar distributions of CD8 T-cell clusters and that Tbc1d10c was ubiquitously expressed across all CD8 T-cell subsets (Supplemental Figure S11A-B) suggesting that Tbc1d10c loss does not lead to a spontaneous CD8 T-cell phenotype. To investigate the impact of Tbc1d10c deficiency on functionally relevant CD8 T cell subsets, we utilized ProjecTILs and its mouse lymphocyte reference atlas to interpret CD8 T-cell states46 between Tbc1d10c+/+ and Tbc1d10c−/− cohorts. ProjecTILs classified most CD8 T cells as Naïve cells in both genotypes, and no significant changes in memory and early active subsets between Tbc1d10c+/+ and Tbc1d10c−/− cohorts were observed (Figure 4A-B). scRNA-Seq also demonstrated that Tbc1d10c was ubiquitously expressed across all CD8 T-cell subsets in both genotypes (Figure 4C).
Figure 4.
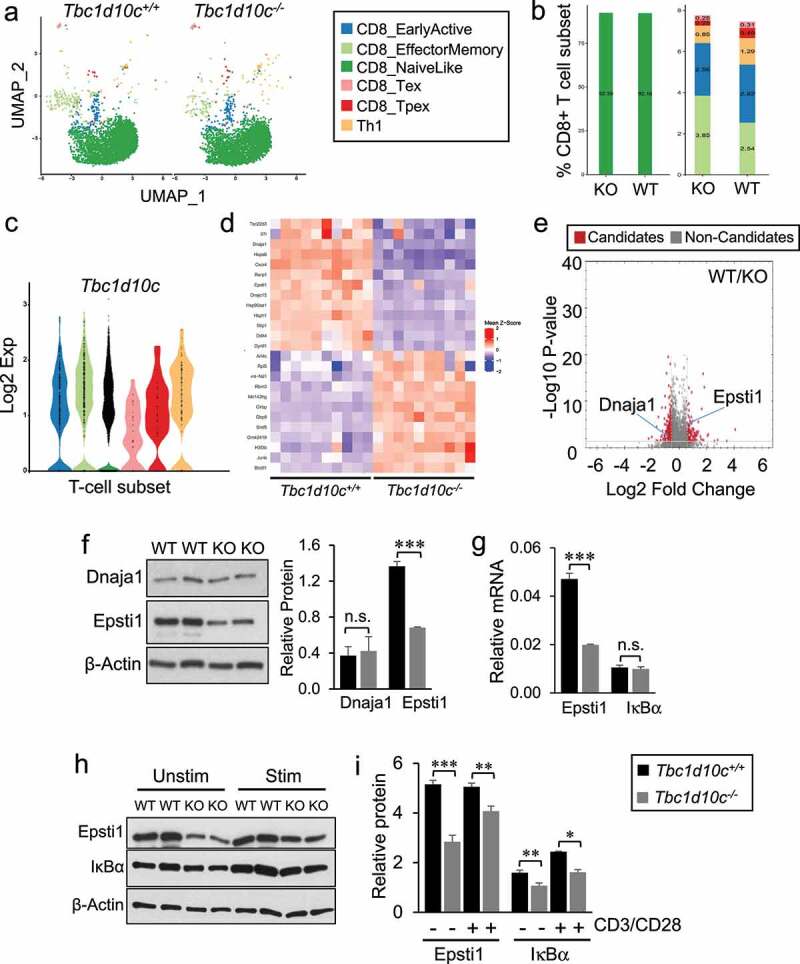
Single-cell RNA sequencing and dual LC-MS/MS proteomic profiling in Tbc1d10c−/− CD8 T cells. (A) Two-dimensional uniform manifold approximation and projection (UMAP) visualization of CD8 T-cell subsets generated by ProjecTILs analysis (cells were projected to the mouse reference TIL atlas) of scRNA-seq Tbc1d10c+/+ and Tbc1d10c−/− data sets. Each point represents a single cell and T-cell subsets are color-coded (middle box). (B) Bar plots comparing the quantified percentages of naïve-like (left) and memory, early active and Th1 (right) CD8 T-cell subsets, according to the ProjecTILs analysis, between Tbc1d10c+/+ and Tbc1d10c−/− cohorts. (C) Violin plot showing mRNA expression of Tbc1d10c across CD8 T-cell subsets. Each black dot represents a cell, and T-cell subtypes are color-coded. (D) Mean-z score heatmap showing the arithmetic mean of the z scores for the top 25 DEGs CD8+ T cells in Tbc1d10c+/+and Tbc1d10c−/− scRNA-Seq integrated data sets (PAdj < 0.05). (E) Volcano plot from LC-MS/MS global proteomic analysis of Tbc1d10c+/+and Tbc1d10c−/− CD8 T cells (n = 3 replicates per group, P values determined by unpaired Student’s t-test with permutation-based FDR correction). (F) Gel images and bar graph (densitometric quantification) showing immunoblot detection of Dnaja1 and Epsti1 in Tbc1d10c+/+and Tbc1d10c−/− CD8 T-cell lysates (n = 2 replicates per group). (G) Bar graphs showing relative mRNA expression of Epsti1 and IκBα as determined by QPCR (n = 3 per replicates per group). (H-I) Gel images (H) and densitometric quantification (I) of relative Epsti1 and IκBα protein levels in unstimulated and CD3/CD28-stimulated cells (n = 2 replicates per group). P values were determined by unpaired Student’s t-test (*, < 0.05; **, < 0.04; ***, < 0.02).
Since no perturbations in specific CD8 T-cell subsets were observed in unsupervised clustering (Supplemental Figure S11A) and Tbc1d10c was broadly expressed in all CD8 T cells (Figure 4C); Supplemental Figure S11B), we opted for a computational strategy that would integrate Tbc1d10c+/+ and Tbc1d10c−/− CD8 T-cell data sets to identify differentially expressed genes (DEGs) that were common across all or most CD8 T-cell clusters. Integrated analysis identified 3023 DEGs across 15 clusters (PAdj < 0.05; Top 25 DEGs shown in Figure 4D). To help prioritize the list of 3023 scRNA-Seq DEGs, we performed global proteomics profiling of Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells, which identified a total of 331 differentially expressed candidates in Tbc1d10c−/− CD8 T cells (PAdj < 0.05; Supplemental Table S3). Next, we employed immunoblot analysis of Tbc1d10c+/+ and Tbc1d10c−/− CD8 T-cell lysates and observed that the following categories of DEGs failed validation by immunoblot analysis: DEGs identified only by scRNA-Seq, such as Hspa8, Syk and Cxcr4, DEGs identified only by global proteomics, such as Stat3, or DEGs identified by both profiling approaches but were unique to an individual cluster by scRNA-Seq analysis, such as Runx3 (Supplemental Figure S11C-D). Since Stat3 was identified by proteomics to be enriched approximately two-fold in Tbc1d10c−/− CD8 T cells (Supplemental Table S3), we analyzed Tbc1d10c−/− CD8 T-cell lysates for Stat1, Stat3 and Stat5 expression levels given the well-established role for the JAK-STAT pathway in T-cell activation. However, highly analogous protein levels for all Stat proteins were observed between CD8 T-cell cohorts (Supplemental Figure S11E).
Given the broad expression of Tbc1d10c across CD8 T cells (Figure 4C; Supplemental Figure S11B), we posited that DEGs specific to an individual CD8 T-cell cluster may not be validated at the protein expression level, thereby diminishing the relevance as a key mediator of Tbc1d10c signaling. As such, to facilitate the identification of critical DEGs in Tbc1d10c−/− CD8 T cells, we cross-referenced DEG lists from the integrated scRNA-Seq and global proteomics analyses and identified nine candidates that were common to both profiling assays. Of these nine candidates, only two, Dnaja1 and Epsti1, were significantly different across all scRNA-Seq CD8 T-cell clusters (Figure 4D-E). While Epsti1 was consistently downregulated at mRNA and protein levels in Tbc1d10c−/− CD8 T cells, Dnaja1 protein and mRNA differences were inconsistent (Figure 4D-E). Immunoblot analysis confirmed a 50% decrease in Epsti1 protein levels in Tbc1d10c−/− compared to Tbc1d10c+/+ CD8 T cells, but no difference in DnaJa1 protein levels (Figure 4F). As such, we focused on a potential role for Epsti1 as an effector of Tbc1d10c signaling in CD8 T cells.
QPCR analysis confirmed a 50% reduction in Epsti1 mRNA expression in Tbc1d10c−/− CD8 T cells (Figure 4G). Epsti1 protein downregulation was maintained following CD3/CD28 TCR stimulation for 24 h (Figure 4H-I), suggesting that the perturbed expression of Epsti1 may also be relevant for the observed augmented activation of Tbc1d10c−/−CD8 T cells. Epsti1 regulates the 26S proteasomal degradation of IκBα,52 which is a key suppressor of p65 transactivation in the NF-κB pathway.53 Therefore, we also assessed IκBα mRNA and protein levels in Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells. While no difference in IκBα mRNA expression was observed in Tbc1d10c−/− CD8 T cells (Figure 4G), we identified a 40% decrease in total IκBα protein in unstimulated and TCR-stimulated Tbc1d10c−/− CD8 T cells (Figure 4H-I). The observed downregulation of IκBα protein in Tbc1d10c−/− CD8 T cells is inconsistent with the established role for Epsti1 in promoting IκBα protein degradation in B cells.52 This observation suggests that either Epsti1 does not regulate IκBα protein levels in CD8 T cells or that additional mechanisms are involved in IκBα protein homeostasis in the context of Tbc1d10c disruption. To address these issues and whether Epsti1 downregulation is responsible for the CD8 T-cell activation phenotype due to Tbc1d10c deficiency, we assessed the impact of silencing Epsti1 mRNA in Tbc1d10c+/+ CD8 T cells on IκBα protein levels and T-cell activation. Epsti1 siRNA-transfected cells exhibited a 50% reduction in protein expression compared to si-Scramble-transfected cells (Figure 5A-B), which closely resembles Epsti1 downregulation in Tbc1d10c−/− CD8 T cells (Figure 4). As reported in B cells,52 Epsti1 knockdown in wild-type CD8 T cells led to a two-fold increase in IκBα protein levels in CD3/CD28 TCR-stimulated cells (Figure 5A-B). However, Epsti1-knockdown cells actually exhibited reduced levels of activation markers, including CD69, Ifn-γ and Tnf-α, following TCR stimulation (Supplemental Figure S12A; Figure 5C). These observations indicate that IκBα protein downregulation and augmented activation levels observed in Tbc1d10c−/− CD8 T cells are likely not due to Epsti1 downregulation. As such, Epsti1 is not the primary effector responsible for the Tbc1d10c null phenotype. However, IκBα is a known inhibitor of NF-κB pathway activity,53 which is a crucial component of TCR activation.54 Our data suggest that Tbc1d10c signaling mediates post-translational regulation of IκBα and thus may suppress downstream NF-κB activity.
Figure 5.
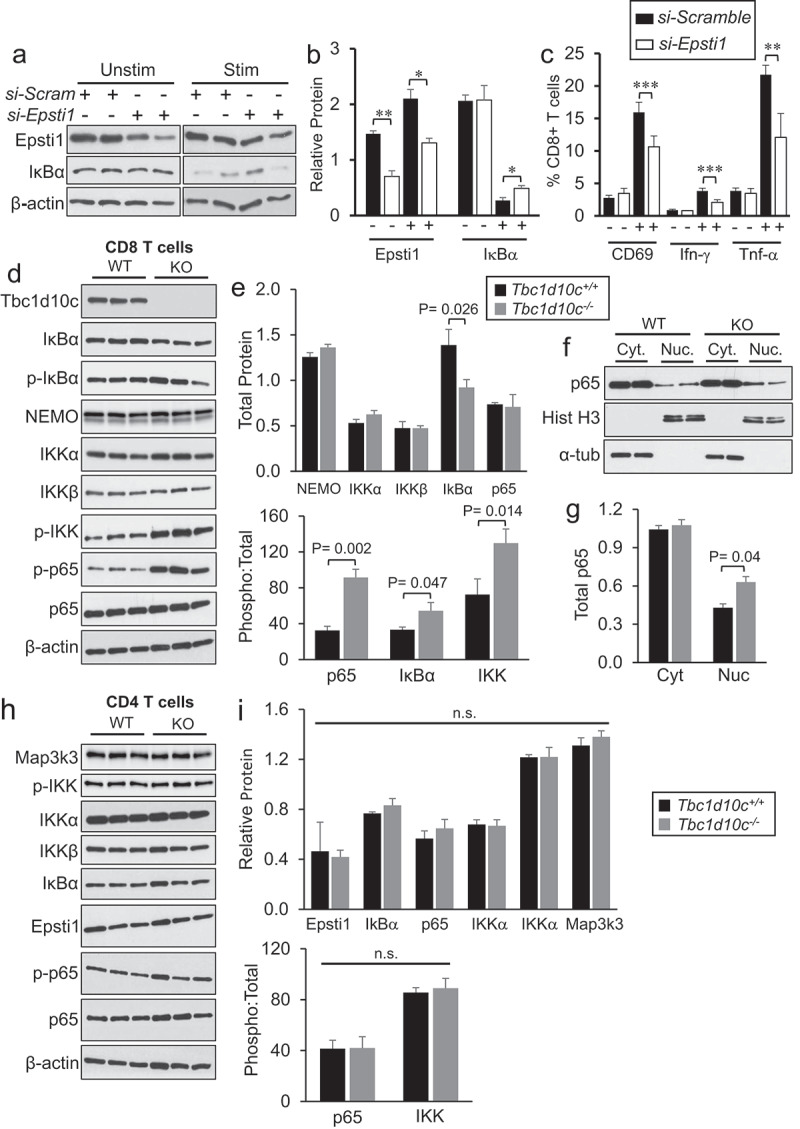
NF-κB pathway activation in CD8 T cells. (A-B) Gel images (A) and densitometric quantification bar graphs (B) showing relative protein levels for Epsti1 and IκBα in wild-type CD8 T cells after siRNA knockdown of Epsti1 (n = 2 replicates per group). (C) Bar graphs showing the average percentages of si-Scramble-or si-Epsti1-transduced CD8 T cells positive for CD69 (A), Ifn-γ (B) or Tnf-α (C) following 24 hr co-stimulation with CD3/CD38 antibodies. (D-I) Gel images and densitometric quantification (bar graphs) showing relative protein levels for CD8 T-cell lysates (D-G) or CD4 T-cell lysates (H-I) of total and phosphorylated protein levels of canonical NF-κB pathway members (n = 3 per replicates per group). Abbreviations: Cyt, cytoplasmic fraction; Nuc, nuclear fraction. P values were determined by unpaired Student’s t-test.
To test this assertion, we quantified the total and activated (phosphorylated) levels of members of the canonical NF-κB pathway downstream of IκBα in Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells by immunoblot analysis. As expected, we observed a 50% decrease in total IκBα protein in Tbc1d10c−/− CD8 T cells; however, no differences in the total levels of IKK complex members, IKKα, IKKβ or Nemo, or p65 were detected (Figure 5D-E). Strikingly, the phosphorylated levels of IκBα, IKK and p65 were dramatically higher in Tbc1d10c−/− CD8 T cells (Figure 5D-E) culminating in two-to-three-fold increases in the ratio of phosphorylated-to-total protein for these downstream NF-κB pathway members (Figure 5E). Next, we analyzed the nuclear and cytosolic fractions of Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells to determine whether the observed increase in p65 phosphorylation (Figure 5D-E) correlated with increased nuclear localization. Nuclear p65 levels were approximately two-fold higher in untouched Tbc1d10c−/− CD8 T cells without a discernible change in cytosolic p65 levels (Figure 5F-G). The canonical NF-κB pathway downstream of TCR activation is generally thought to be analogous between CD4 and CD8 T cells; however, our Tbc1d10c−/− T-cell activation phenotype is highly skewed toward CD8 T cells. Therefore, we also analyzed the total and phosphorylated protein levels of NF-κB pathway members in CD4 T cells. Interestingly, we observed no quantifiable differences in total or phosphorylated forms of Epsti1 or any NF-κB pathway proteins in Tbc1d10c−/− CD4 T cells (Figure 5H-I). Finally, a significant, but modest increase (25%) in Tbc1d10c protein levels was observed in CD4 compared to CD8 T cells (Supplemental Figure S12B). These observations further strengthen the mechanistic relevance for Tbc1d10c suppression of NF-κB pathway activity specifically in CD8 T-cell activation.
Our findings above correlate downregulation of the NF-κB pathway inhibitor IκBα with increased activation of canonical NF-κB pathway proteins including IKKα/β and p65. To determine whether these observations in untouched CD8 T cells hold functional relevance for the increased activation levels in Tbc1d10c−/− CD8 T cells following TCR stimulation, we assessed total and phosphorylated IKK and p65 levels during a time course of CD3/CD28 TCR stimulation. While no differences in total protein levels were observed, the average percentages of phosphorylated p65 and IKKβ were significantly higher in Tbc1d10c−/− compared to Tbc1d10c+/+ CD8 T cells at 15, 30 and 60 min after CD3/CD28 TCR stimulation (Figure 6A-B). Importantly, these observations provide a mechanistic link between perturbed NF-κB pathway activation and the augmented response of Tbc1d10c−/− CD8 T cells to TCR stimulation. To bridge our observations of upregulated NF-κB pathway activation and CD8 T-cell activation following TCR stimulation to the increased levels of activated tumor-infiltrating CD8 T cells and tumor resistance phenotype in Tbc1d10c−/− mice, we quantified the activated (phosphorylated) levels of IKK proteins in tumor-infiltrating CD8 T cells from SCCs implanted in Tbc1d10c+/+ and Tbc1d10c−/− mice by Phosflow analysis. We observed a 2.5-fold increase in p-IKK+ CD8 T cells in SCCs from Tbc1d10c−/− mice (Figure 6C-D), indicating that augmented NF-κB signaling observed in Tbc1d10c−/− CD8 T cells from healthy spleen is mechanistically relevant for the augmented anti-tumor response in tumor-infiltrating Tbc1d10c−/− CD8 T cells. Importantly, p-IKK levels were also significantly increased in primary human CD8 T cells transfected with si-TBC1D10C compared to non-targeting transfected cells in both unstimulated and CD3/CD28 TCR-stimulated cohorts (Figure 6E-F).
Figure 6.
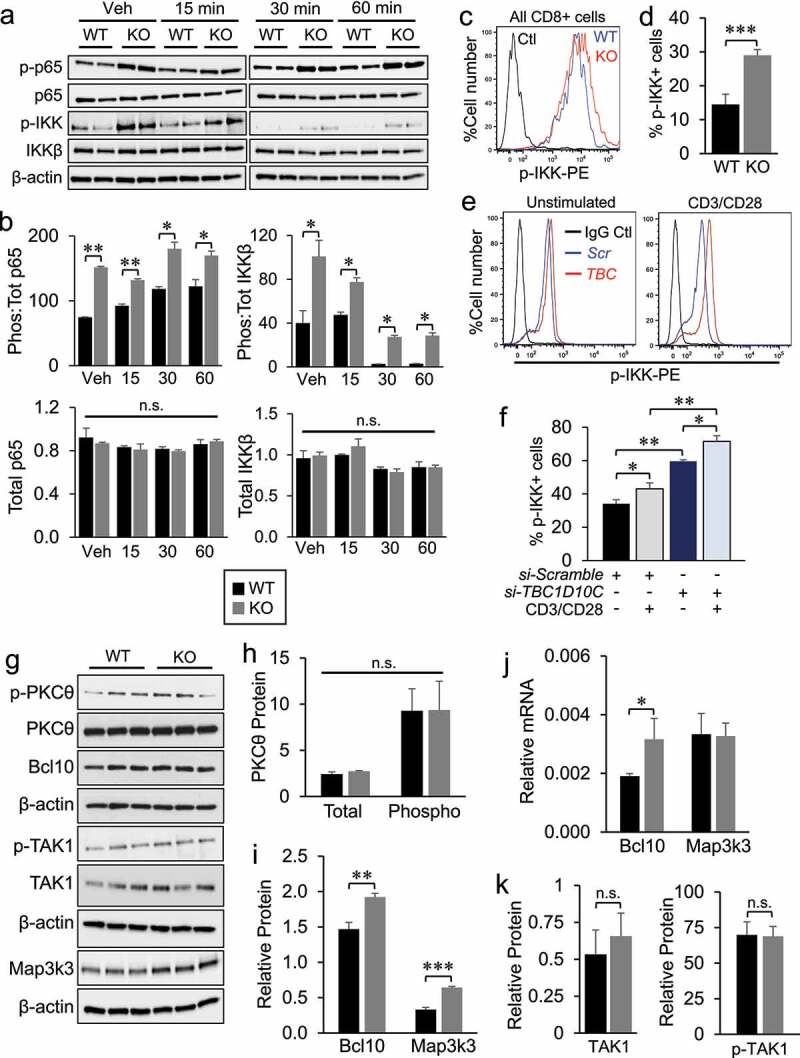
Assessment of IκBα and Map3k3 expression in Tbc1d10c−/− CD8 T cells. (A-B) Gel images (A) and densitometric quantification (B) of total and phosphorylated p65 and IKKβ protein levels in Tbc1d10c+/+and Tbc1d10c−/− CD8 T cells following CD3/CD28 activation at the indicated time points (n = 2 replicates per group). (C-D) Representative dot plots of flow cytometric detection of p-IKK levels of SCC-infiltrating CD3+ CD8 + T cells and bar graph (D) showing average percentage of p-IKK+ CD8 T cells (n = 4 tumors per group). Histogram shows an overlay of representative p-IKK+ cell numbers in Tbc1d10c+/+and Tbc1d10c−/− cohorts. (E) Representative dot plots of flow cytometric detection of p-IKK+ human CD8 T cells subjected to TBC1D10C siRNA or control (si-Scramble) silencing and vehicle (unstimulated) or CD3/CD28 stimulation. (F) Bar graph showing the average percentage of p-IKK+ human CD8 T cells subjected to TBC1D10C siRNA or control (si-Scramble) silencing and CD3/CD28 stimulation (n = 5 replicates per group). (G-I) Gel images and densitometric quantification (bar graphs) showing relative total and phosphorylated protein levels of indicated proteins in Tbc1d10c+/+and Tbc1d10c−/− CD8 T cells (n = 3 replicates per group). (J) Bar graphs showing relative mRNA expression of Bcl10 and Map3k3 as determined by QPCR (n = 3 per replicates per group). (K) Bar graphs showing relative total and phosphorylated TAK1 protein levels in Tbc1d10c+/+and Tbc1d10c−/− CD8 T cells from panel G (n = 3 replicates per group). P values were determined by unpaired Student’s t-test.
In both murine and human CD8 T cells, Tbc1d10c deficiency leads to increased activation levels of canonical NF-κB pathway members even in untouched murine and unstimulated human cells. These observations primarily implicate Tbc1d10c as a constitutive suppressor of the NF-κB pathway in CD8 T cells. Tbc1d10c was previously classified as a feedback inhibitor of TCR activation in human CD4 T cells based on its low expression in unstimulated and rapid induction in TCR-stimulated CD4 T cells and its ability to bind and inhibit Calcineurin and Ras signaling.25 In both murine and human CD8 T cells, we observed robust Tbc1d10c mRNA and protein expression in untouched or unstimulated cells and up to eight-fold downregulation of Tbc1d10c mRNA and three-fold downregulation of protein following TCR stimulation (Supplemental Figure S13). We also observed similar levels of nuclear NFAT1, as a read-out of Calcineurin signaling activity, and phosphorylated ERK1/2 following TCR ligation in Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells (Supplemental Figure S14).
Tbc1d10c deficiency perturbs NF-κB signaling via Map3k3 stability
Our findings above illustrate a mechanistic role for Tbc1d10c in suppressing NF-κB pathway activation following TCR ligation in CD8 T cells. To further explore the mechanism by which Tbc1d10c signaling suppresses NF-κB pathway activation, we analyzed the status of TCR signaling proteins known to be upstream of IKB phosphorylation. First, we performed CD3/CD28 TCR stimulation experiments with CD3 alone, CD28 alone, or CD3/CD28 co-stimulation in order to determine whether Tbc1d10c may intersect one of these pathways individually upstream of the merge with PKC-θ.55 CD3/CD28 co-stimulation led to the expected increases in Tbc1d10c−/− CD8 T-cell activation markers CD69, Ifn-γ, and Tnf-α (Supplemental Figure S15A-D), corroborating our findings with OVA peptide stimulation of OT-I CD8 T cells (Figure 3). However, there were no differences in the residual levels of these activation markers in CD8 T cells in the presence of CD3 or CD28 alone (Supplemental Figure S15A-D) indicating that Tbc1d10c does not directly impact either of these pathways upstream of PKC-θ. No difference in total or phosphorylated levels of PKC-θ was observed between Tbc1d10c+/+ and Tbc1d10c−/− CD8 T cells (Figure 6G-H). The CARMA1-Bcl10-MALT1 (CBM) signalosome complex is directly downstream of PKC-θ and serves to recruit and activate the IKK complex upon TCR ligation.56 Within the CBM, Bcl10 upregulation is sufficient for downstream NF-κB transactivation.57 We observed a modest (20%) increase in total Bcl10 protein levels (Figure 6G,I), which was likely due to a 40% upregulation of Bcl10 mRNA expression (Figure 6J) in Tbc1d10c−/− CD8 T cells. No perturbations were observed in Bcl10 protein ubiquitination (Supplemental Figure S15E-F), suggesting that the increased Bcl10 mRNA and protein expression likely resulted from a p65-mediated positive feedback loop on Bcl10 regulation58,59 rather than changes in protein stabilization. No differences in total TAK1 or phosphorylated TAK1 protein levels were observed in Tbc1d10c−/− CD8 T cells (Figure 6G, K), which largely excludes a direct role for TAK1 in the observed two-to-three-fold increases in p-IKK levels (Figure 5). Collectively, these findings indicate that increased activation of the IKK complex is the furthest upstream target in the canonical NF-κB pathway impacted by Tbc1d10c deficiency. As such, our results suggest that Tbc1d10c likely intersects canonical NF-κB signaling via a mediator of IKK complex phosphorylation from a heterologous pathway.
The serine/threonine kinase Map3k3 is known to directly phosphorylate IKK complex members and mediate downstream NF-κB activity.60,61 In untouched Tbc1d10c−/− CD8 T cells, we observed a two-fold increase in Map3k3 protein (Figure 6G, I) that was not due to a change in Map3k3 mRNA expression (Figure 6J). To investigate the relevance of this observation for CD8 T cell activation, we quantified Map3k3 protein levels following TCR ligation. Similar to untouched CD8 T cells, Map3k3 protein was increased two- to three-fold in Tbc1d10c−/− CD8 T cells at 15, 30 and 60 min following TCR ligation (Figure 7A-B). These data suggest that i) Tbc1d10c activity negatively impacts Map3k3 protein stability and ii) Tbc1d10c may suppress canonical NF-κB signaling by regulating Map3k3 protein levels. To address this idea, we performed siRNA knockdown of Map3k3 and assessed its impact on activation levels of canonical NF-κB pathway members known to be perturbed in Tbc1d10c−/− CD8 T cells (Figure 5). Map3k3 siRNA knockdown led to a two-fold decrease in the phosphorylation levels of IKKβ protein with no impact on total protein levels of IKKα or IKKβ in Tbc1d10c−/− CD8 T cells (Figure 7C-E), indicating that the increased activation levels of canonical NF-κB pathway members in Tbc1d10c−/− CD8 T cells are Map3k3-dependent. Importantly, Map3k3 siRNA knockdown rescued the activation and cytotoxic effector phenotype in Tbc1d10c−/− CD8 T cells but had no effect on the activation or effector function of Tbc1d10c+/+ CD8 T cells (Figure 7F-I; Supplemental Figure S16A). Collectively, these observations support the conclusion that Tbc1d10c intersects the canonical NF-κB pathway by negatively regulating Map3k3 protein levels and that Map3k3 is the key mediator of the Tbc1d10c null phenotype in CD8 T cells (Supplemental Figure S16B).
Figure 7.
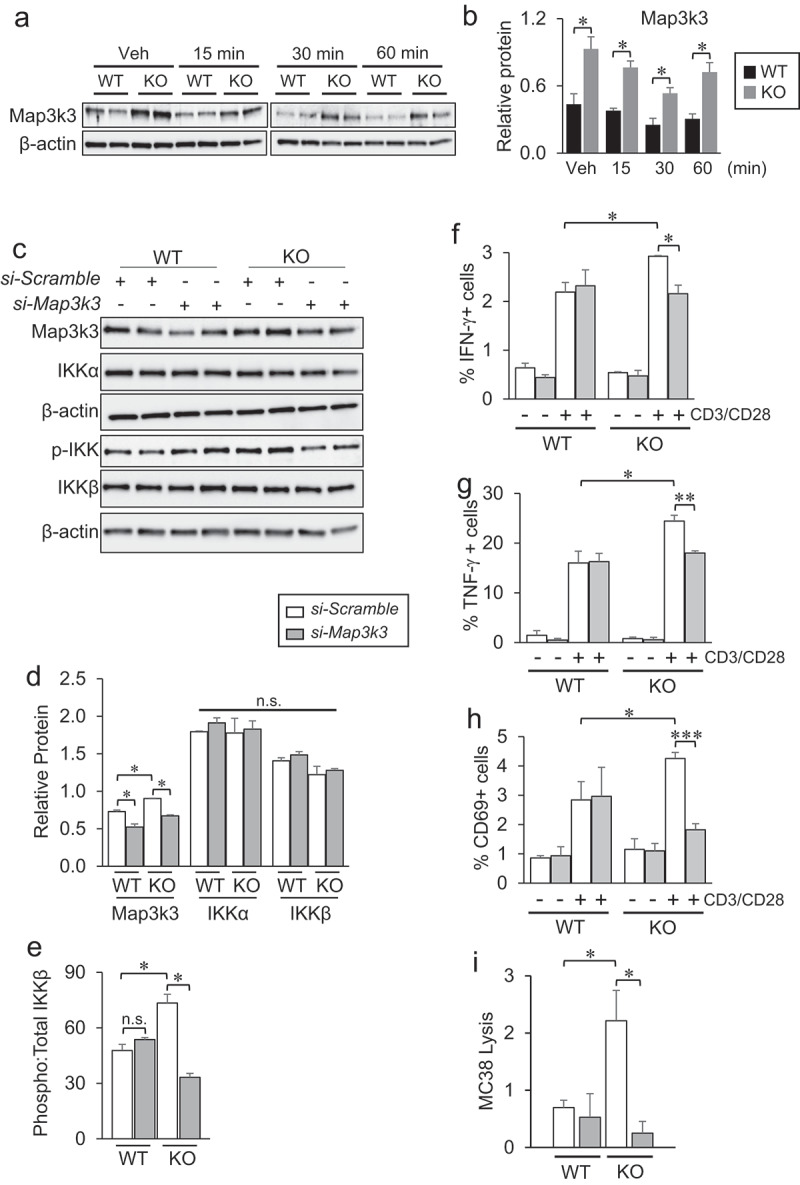
Map3k3 knockdown rescues the Tbc1d10c null phenotype in CD8 T cells. (A-B) Gel images (A) and densitometric quantification (B) of total Map3k3 protein levels in Tbc1d10c+/+and Tbc1d10c−/− CD8 T cells following CD3/CD28 activation at the indicated time points (n = 2 replicates per group). (C-E) Gel images and densitometric quantification (bar graphs) showing relative total and phosphorylated protein levels of indicated proteins in Map3k3 siRNA knockdown Tbc1d10c−/− CD8 T cells (n = 2 per replicates per group). (F-H) Percent of Ifn-γ+ (F), Tnf-α+ (G) and CD69+ (H) CD8 T cells following siRNA silencing of Map3k3 and CD3/CD28 stimulation (n = 3 replicates per group). (I) Bar graph showing MC38 lysis levels by OT-I;Tbc1d10c+/+and OT-I;Tbc1d10c−/− CD8 T cells following Map3k3 siRNA silencing at a 2:1 effector-to-target cell ratio (n = 3 per replicates per group). P values were determined by unpaired Student’s t-test (*, P < .05; **, P < .005; ***, P < .0005).
Discussion
We identified Tbc1d10c to be a selective and constitutive suppressor of CD8 T-cell activation and cytotoxic effector function. Mechanistically, our findings indicate that Tbc1d10c promotes tumor growth and incidence by tempering phosphorylation-dependent activation of IKKβ via regulation of Map3k3 steady-state protein levels. While a tumor cell-specific role for Tbc1d10c is reported in large B cell lymphoma,62 our findings are the first to elaborate a cellular and molecular basis for Tbc1d10c signaling in solid malignancies. As such, our findings highlight the Tbc1d10c-Map3k3-NF-κB signaling axis as a viable therapeutic target to promote anti-tumor immunity.
The NF-κB pathway is a pleiotropic regulator of tumorigenesis.63 In a variety of tumor cell types, the pro-tumorigenic effects of NF-κB signaling are primarily levied during tumor promotion through enhancement of cancer cell proliferation and survival.64 In addition to tumor cell-intrinsic mechanisms, NF-κB signaling modulates the function of a variety of cellular constituents of the tumor microenvironment including macrophages, MDSCs, NK cells, lymphocytes, fibroblasts and endothelial cells.65,66 NF-κB signaling also plays a role in T cell-mediated anti-tumor immunity. Canonical activation of NF-κB in T cells increases the number of tumor-specific IFN-γ producing CD8 T cells and is required for tumor elimination.67–69 Collectively, this large body of published work underscores the importance and broad reach of the NF-κB pathway in inflammation and cancer, while, in doing so, also implies the complexity associated with preventive or therapeutic approaches designed to block or activate NF-κB signaling. In doing so, it stresses the importance of identifying NF-κB pathway effectors or modifiers that discretely function in select immune lineages in their response to cancer.70 Our findings show that Tbc1d10c blocks canonical NF-κB (p65) transactivation downstream of TCR ligation in CD8 T cells but not in CD4 T cells. To our knowledge, there are no other published findings demonstrating discriminative mechanistic regulation of TCR-mediated canonical NF-κB activation between CD4 and CD8 T-cell subsets as the downstream constituents of the TCR pathways in each subset are largely thought to be analogous. A previous study in regulatory CD4 Tregs showed that selective inhibition of c-Rel, but not p65, blocked Treg immunosuppressive function and augmented the efficacy of PD-1 immune checkpoint inhibitor therapy while marginalizing cytotoxic side effects to normal tissue.71 However, conditional ablation of the non-canonical NF-κB pathway component p100, a suppressor of RelB, in CD4 Tregs, manifests in RelB-mediated autoimmunity.72 While not involving Tbc1d10c, this work illustrated the impact of identifying and targeting T-cell subset-specific mechanisms that regulate NF-κB activation on tumor outcome following immunotherapy.
Map3k3 regulates canonical NF-κB pathway activation in T cells downstream of TCR ligation via direct phosphorylation of IKKβ.60,61 Map3k3 expression knockdown completely rescued the Tbc1d10c null phenotype with respect to i) CD8 T cell activation and effector function and ii) NF-κB pathway activation, indicating that Tbc1d10c intersects the NF-κB pathway via Map3k3. As there is no precedence for interactions between these three proteins, our future work will focus on a potential role for Tbc1d10c GTPase function in the regulation of Map3k3 protein stability and the mechanistic basis for the discriminative interplay of these proteins in CD8 versus CD4 T cells.
Supplementary Material
Acknowledgments
We thank Charles Drake for the generous gift of B16-OVA cells and Wendy Mao for technical assistance with in vitro tumor cell killing assays. Pierre Chambon provided the pCreERT2 expression vector. Chyuan-Sheng Lin provided technical assistance with generation of Tbc1d10c null and Tbc1d10c-CreERT2 mice. We thank Ching-Yuan Chen, Caisheng Lu and Wei Wang for assistance with FACS sorting. We also thank Annemieke de Jong and Ioanna Karantza for assistance with isolating human CD8 T cells from whole blood. We are grateful to Donna Farber, Pawel Muranski and Arnold Han for their helpful advice on our study. The Genetically Modified Mouse Models, Genomics and High Throughput Screening, and Flow Cytometry Shared Resources within the HICCC are supported by NCI 5P30CA013696-44. Some Flow cytometry experiments were also performed in the CCTI Flow Cytometry Core, supported by NIH awards S10RR027050 and S10OD020056. A.O.C. was supported by NYDOH DOH01-C32582GG. S.H.W. was supported by NIH R01CA114014. J.C. and J.G. were supported by a Cancer Research Institute CLIP Award. R.D. and O.Y. were supported by NIH P30AR069632. M.A.B. was supported by NIH F32AR055007. D.M.O. was supported by a Cancer Research Institute CLIP Award, NIH P30AR069632 and NYDOH DOH01-C32582GG.
Funding Statement
The author(s) reported that there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
D.M.O. contributed to conceptualization; D.M.O., A.O.C., M.S.H., S.H.W., and M.A.B. contributed to methodology; A.O.C., J.Z., M.O.F., and R.K.S. contributed to formal analysis; A.O.C., S.H.W., J.Z., J.C., M.E.R., J.G., R.D., R.K.S., O.Y., D.Z.J., and M.A.B. contributed to investigation; A.O.C. and D.M.O. contributed to writing—original draft; A.O.C., J.Z. and D.M.O. contributed to writing—reviewing and editing; A.O.C., R.K.S. and D.M.O. contributed to visualization; D.M.O. contributed to supervision; D.M.O. contributed to funding acquisition.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2141011
References
- 1.Waldman AD, Fritz JM, Lenardo MJ.. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–19. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muenst S, Laubli H, Soysal SD, Zippelius A, Tzankov A, Hoeller S. The immune system and cancer evasion strategies: therapeutic concepts. J Intern Med. 2016;279(6):541–562. doi: 10.1111/joim.12470. [DOI] [PubMed] [Google Scholar]
- 3.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its component phases-elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:S185–198. doi: 10.1016/j.coi.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giroux Leprieur E, Dumenil C, Julie C, Giraud V, Dumoulin J, Labrune S, Chinet T. Immunotherapy revolutionizes non-small-cell lung cancer therapy: results, perspectives and new challenges. Eur J Cancer. 2017;78:16–23. doi: 10.1016/j.ejca.2016.12.041. [DOI] [PubMed] [Google Scholar]
- 7.Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68(1):139–152. doi: 10.1186/s13287-020-02128-1. [DOI] [PubMed] [Google Scholar]
- 8.Hopewell EL, Cox C, Pilon-Thomas S, Kelley LL. Tumor-infiltrating lymphocytes: streamlining a complex manufacturing process. Cytotherapy. 2019;21(3):307–314. doi: 10.1016/j.jcyt.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teply BA, Lipson EJ. Identification and management of toxicities from immune checkpoint-blocking drugs. Oncology. 2014;28(S3):30–38. PMID:25384885 [PubMed] [Google Scholar]
- 10.Kalaitsidou M, Kueberuwa G, Schutt A, Gilham DE. CAR T-cell therapy: toxicity and the relevance to preclinical models. Immunotherapy. 2015;7(5):487–497. doi: 10.2217/imt.14.123. [DOI] [PubMed] [Google Scholar]
- 11.Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 12.Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, Zhao S, Das S, Beckermann KE, Ha L, et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 2018;4(12):1721–1728. doi: 10.1001/jamaoncol.2018.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eggermont AMM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845–1855. doi: 10.1056/NEJMoa1611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–2697. doi: 10.1200/JCO.2012.41.6750. [DOI] [PubMed] [Google Scholar]
- 15.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 16.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, Hermes B, Cay Senler F, Csoszi T, Fulop A, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379(21):2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 17.Khunger M, Rakshit S, Pasupuleti V, Hernandez AV, Mazzone P, Stevenson J, Pennell NA, Velcheti V. Incidence of pneumonitis with use of programmed death 1 and programmed death-ligand 1 inhibitors in non-small cell lung cancer: a systematic review and meta-analysis of trials. Chest. 2017;152(2):271–281. doi: 10.1016/j.chest.2017.04.177. [DOI] [PubMed] [Google Scholar]
- 18.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, Xue A, Goff SL, Yang JC, Sherry RM, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. 2017;35(16):1803–1813. doi: 10.1200/JCO.2016.71.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, Xue A, Bot A, Scholler N, Mikkilineni L, et al. Safety and feasibility of anti-CD19 CAR T cells with fully huma binding domains in patients with B-cell lymphoma. Nat Med. 2020;26(2):270–280. doi: 10.1038/s41591-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27(1):591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia E, Ismail S. Spatiotemporal regulation of signaling: focus on T cell activation and the immunological synapse. Int J Mol Sci. 2020;21(9):3283. doi: 10.3390/ijms21093283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang JR, Byeon Y, Kim D, Park SG. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. 2020;52(5):750–761. doi: 10.1038/s12276-020-0435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan F, Sun L, Kardian DB, Whartenby KA, Pardoll DM, Liu JO. Feedback inhibition of Calcineurin and Ras by a dual inhibitory protein Carabin. Nature. 2007;445(7126):433–436. doi: 10.1038/nature05476. [DOI] [PubMed] [Google Scholar]
- 26.Schickel JN, Pasquali JL, Soley A, Knapp AM, Decossas M, Kern A, Fauny JD, Marcellin L, Korganow AS, Martin T, et al. Carabin deficiency in B cells increases BCR-TLR9 costimulation-induced autoimmunity. EMBO Mol Med. 2012;4(12):1261–1275. doi: 10.1002/emmm.201201595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Y, Pan Y, Liu J. The relevance between the immune response-related gene module and clinical traits in head and neck squamous cell carcinoma. Cancer Manag Res. 2019;11(21):7455–7472. doi: 10.2147/CMAR.S201177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng H, Song X, Ji J, Chen L, Liao Q, Ma X. HPV infection related immune infiltration gene associated therapeutic strategy and clinical outcome in HNSCC. BMC Cancer. 2020;20(1):796. doi: 10.1186/s12885-020-07298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang L, Peng C, Zhu SS, Zhou Z, Liu H, Cheng Q, Chen X, Chen X-P. Tre2-Bub2-Cdc16 family proteins based nomogram serve as a promising prognosis predicting model for melanoma. Front Oncol. 2020;10:579625. doi: 10.3389/fonc.2020.579625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai F, Jin Y, Zhang P, Chen H, Fu Y, Zhang M, Weng Z, Wu K. Bioinformatic profiling of prognosis-related genes in the breast cancer immune microenvironment. Aging (Albany NY). 2019;11:9328–9347. doi: 10.18632/aging.102373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choy CT, Wong CH, Chan SL. Embedding of genes using cancer gene expression data: biological relevance and potential application on biomarker discovery. Front Genet. 2019;9:682. doi: 10.3389/fgene.2018.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutation. Genome Res. 2003;13(3):476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 34.Hao H, Rajewsky K. Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J Exp Med. 2001;194(8):1151–1164. doi: 10.1084/jem.194.8.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 36.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta- chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 37.Stumpfova M, Ratner D, Desciak EB, Eliezri YD, Owens DM. The immunosuppressive surface ligand CD200 augments the metastatic capacity of squamous cell carcinoma. Cancer Res. 2010;70(7):2962–2972. doi: 10.1158/0008-5472.CAN-09-4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bellone M, Cantarella D, Castiglioni P, Crosti MC, Ronchetti A, Moro M, Garancini MP, Casorati G, Dellabona P. Relevance of the tumor antigen in the validation of three vaccination strategies for melanoma. J Immunol. 2000;165(5):2651–2656. doi: 10.4049/jimmunol.165.5.2651. [DOI] [PubMed] [Google Scholar]
- 39.Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980;124(1):445–453. PMID:6153101 [PubMed] [Google Scholar]
- 40.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107(9):4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan IZ, Del Guzzo CA, Shao A, Cho J, Du R, Cohen AO, Owens DM. The CD200-CD200R axis promotes squamous cell carcinoma metastasis via regulation of cathepsin K. Cancer Res. 2021;81(19):5021–5032. doi: 10.1158/0008-5472.CAN-20-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6(3):331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Xiang Z, Ertl HC, Wilson JM. Upregulation of class I major histocompatibility complex antigens by interferon gamma is necessary for T-cell-mediated elimination of recombinant adenovirus-infected hepatocytes in vivo. Proc Natl Acad Sci USA. 1995;92(16):7257–7261. doi: 10.1073/pnas.92.16.7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(1):14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andreatta M, Corria-Osorio J, Müller S, Cubas R, Coukos G, Carmona SJ. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat Commun. 2021;12(1):2965. doi: 10.1038/s41467-021-23324-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruderer R, Bernhard OM, Gandhi T, Miladinovic SM, Cheng LY, Messner S, Ehrenberger T, Zanotelli V, Butscheid Y, Escher C, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to Acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteomics. 2015;14(5):1400–1410. doi: 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11(3):319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 49.Bachelor MA, Lu Y, Owens DM. L-3-Phosphoserine phosphatase (PSPH) regulates cutaneous squamous cell carcinoma proliferation independent of L-serine biosynthesis. J Dermatol Sci. 2011;63:164–1672. doi: 10.1016/j.jdermsci.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Comm. 1997;237(3):752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 51.Campanella GSV, Luster AD. Chapter 18. A chemokine-mediated in vivo T-cell recruitment assay. Methods Enzymol. 2009;461:397–412. doi: 10.1016/S0076-6879(09)05418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li T, Lu H, Shen C, Lahiri SK, Wason MS, Mukherjee D, Yu L, Zhao J. Identification of epithelial stromal interaction 1 as a novel effector downstream of Krüppel-like factor 8 in breast cancer invasion and metastasis. Oncogene. 2013;33(39):4746–4755. doi: 10.1038/onc.2013.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosh S, Baltimore D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature. 1990;344(6267):678–682. doi: 10.1038/344678a0. [DOI] [PubMed] [Google Scholar]
- 54.Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor κB. Immunity. 2006;25(5):701–715. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 55.Isakov N, Altman A. PKC-theta mediated signal delivery from the TCR/CD28 surface receptors. Front Immunol. 2012;3:273. doi: 10.3389/fimmu.2012.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gehring T, Seeholzer T, Krappmann D. Bcl10 – bridging CARDs to immune activation. Front Immunol. 2018;9:1539. doi: 10.3389/fimmu.2018.01539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou H, Wertz I, O’Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature. 2004;427(6970):167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 58.Stelzer G, Rosen R, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et al. The GeneCards Suite: from Gene Data Mining to Disease Genome Sequence Analysis. Curr Protoc Bioinformatics. 2016;54(1):1.30.1–1.30.33. doi: 10.1002/cpbi.5. [DOI] [PubMed] [Google Scholar]
- 59.Becnel LB, Ochsner SA, Darlington YF, McOwiti A, Kankanamge WH, Dehart M, Naumov A, McKenna NJ. Discovering relationships between nuclear receptor signaling pathways, genes and tissues in Transcriptomine. Sci Signal. 2017;10(476):eaah6275. doi: 10.1126/scisignal.aah6275. [DOI] [PubMed] [Google Scholar]
- 60.Di Y, Li S, Wang L, Zhang Y, Dorf ME. Homeostatic interactions between MEKK3 and TAK1 involved in NF-κB signaling. Cell Signal. 2008;20(4):705–713. doi: 10.1016/j.cellsig.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samanta AK, Huang HJ, Bast RC, Liao WSL. Overexpression of MEKK3 confers resistance to apoptosis through activation of NFkappaB. J Biol Chem. 2004;279(9):7576–7583. doi: 10.1074/jbc.M311659200. [DOI] [PubMed] [Google Scholar]
- 62.Jiang W, Zhou X, Li Z, Liu K, Wang W, Tan R, Cong X, Shan J, Zhan Y, Cui Z, et al. Prolyl 4-hydroxylase 2 promotes B-cell lymphoma progression via hydroxylation of Carabin. Blood. 2018;131(12):1325–1336. doi: 10.1182/blood-2017-07-794875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taniguchi K, Karin M. NF-□B, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18(5):309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 64.Karin M, Greten FR. NF-□B: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 65.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerondakis S, Siebenlist U. Roles of NF-□B pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol. 2010;2(5):a000182. doi: 10.1101/cshperspect.a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gerondakis S, Fulford TS, Messina NL, Grumont RJ. NF-□B control of T cell development. Nat Immunol. 2014;15(1):15–25. doi: 10.1038/ni.2785. [DOI] [PubMed] [Google Scholar]
- 69.Evaristo C, Spranger S, Barnes SE, Miller ML, Molinero LL, Locke FL, Gajewski TF, Alegre ML. Cutting edge: engineering active IKK□ in T cells drives tumor rejection. J Immunol. 2016;196(7):2933–2938. doi: 10.4049/jimmunol.1501144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mortezaee K, Najafi M, Farhood B, Ahmadi A, Shabeeb D, Musa AE. NF-□B targeting for overcoming tumor resistance and normal tissues toxicity. J Cell Physiol. 2019;234(10):17187–17204. doi: 10.1002/jcp.28504. [DOI] [PubMed] [Google Scholar]
- 71.Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, Schmid RM, Klein U, Hayden MS, Ghosh S. NF-κB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell. 2017;170(6):1096–1108. doi: 10.1016/j.cell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grinberg-Bleyer Y, Caron R, Seeley JJ, De Silva NS, Schindler CW, Hayden MS, Klein U, Ghosh S. The alternative NF-κB pathway in regulatory T cell homeostasis and suppressive function. J Immunol. 2018;200(7):2362–2371. doi: 10.4049/jimmunol.1800042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.