

Abstract
Bacteriophages, infecting bacterial hosts in every environment on our planet, are a driver of adaptive evolution in bacterial communities. At the same time, the host range of many bacteriophages—and thus one of the selective pressures acting on complex microbial systems in nature—remains poorly characterized. Here, we computationally inferred the putative host ranges of 40 cluster P mycobacteriophages, including members from 6 subclusters (P1–P6). A series of comparative genomic analyses revealed that mycobacteriophages of subcluster P1 are restricted to the Mycobacterium genus, whereas mycobacteriophages of subclusters P2–P6 are likely also able to infect other genera, several of which are commonly associated with human disease. Further genomic analysis highlighted that the majority of cluster P mycobacteriophages harbor a conserved integration-dependent immunity system, hypothesized to be the ancestral state of a genetic switch that controls the shift between lytic and lysogenic life cycles—a temperate characteristic that impedes their usage in antibacterial applications.
Keywords: mycobacteriophages, cluster P, phylogenomics, comparative genomics, host range
Introduction
Less than 1% of the virosphere on our planet has been characterized to date (Geoghegan and Holmes 2017). An important part of this virosphere is bacteriophages (i.e. bacteria-infecting viruses), which are impacting bacterial genome evolution and community dynamics in every environment (Howard-Varona et al. 2017).
Bacteriophages can establish lytic or lysogenic infections—the former leading to cell destruction while the latter being “dormant,” with bacteriophages replicating as prophages within the host without the production of virions (Howard-Varona et al. 2017). Temperate bacteriophages can switch between lytic and lysogenic life cycles, for example through the usage of integration-dependent immunity systems that establish lysogeny by suppressing lytic growth through an interplay between 3 proteins: integrase (Int), repressor (Rep), and Cro [for an in-depth discussion on these and other genetic switches, see the commentary by Broussard and Hatfull (2013)]. In integration-dependent immunity systems, the decision on whether lytic or lysogenic growth will take place depends by and large on the activity of Int as modulated by targeted proteolysis (Broussard et al. 2013). Under conditions where integrases are broken down (i.e. in the presence of a C-terminal ssrA-like protease degradation tag in Int), integration fails to occur. Instead, the viral form of Rep is generated and subsequently degraded due to the presence of its own C-terminal ssrA-like tag. The lytic protein Cro is freely expressed and stops repressor function (Hochschild et al. 1986). Conversely, when integrases escape proteolysis due to either decreased levels of proteases (such as ClpXP) or high multiplicity of infection (i.e. a high ratio of bacteriophages to infection targets), integration of bacteriophage genetic material will occur. This leads to the expression of an active (truncated) form of Rep that lacks the ssrA-like tag, causing a downregulation of Cro expression, which ultimately leads to lysogenic establishment and prophage induction. Thereby, the integration into the host genome is mediated by recombination between the bacteriophage attachment site (attP) and the bacterial attachment site (attB) in the host genome. Attachment sites are recognized by Int—an integral part of the attP–Int cassette required for integrase-mediated site-specific recombination (Singh et al. 2013). Thereby, Int is either a tyrosine recombinase (which requires additional host cofactors such as the one present in Mycobacterium smegmatis; Pedulla et al. 1996; Peña et al. 1999; Lewis and Hatfull 2003; Chen et al. 2019) or a serine recombinase (which functions without any cofactors but recognizes shorter attP sequences than the tyrosine recombinase; Groth and Calos 2004).
Mycobacteriophages are a group of both lytic and temperate bacteriophages that infect mycobacterial hosts—including the causative agents for several human diseases such as tuberculosis (M. tuberculosis) or leprosy (M. leprae), separated into 31 clusters (A–Z and AA–AE) based on their nucleotide similarity and genomic architecture (Pope et al. 2011). Out of these, temperate cluster P bacteriophages are of particular interest to the scientific community to, for example study the evolution of genetic switches as several members of this cluster have been shown to harbor an unusual switch in which the bacteriophage attachment site is located within the repressor gene (e.g. Broussard et al. 2013; Doyle et al. 2017).
Interestingly, many mycobacteriophages have the ability to broaden their host range to infect either different strains or completely new mycobacterial species (Jacobs-Sera et al. 2012). In contrast to lytic bacteriophages, which are frequently exploited as antimicrobial agents (Sharma et al. 2017), the life cycle of temperate bacteriophages often impedes their usage, particularly with regard to bacteriophage therapy, due to the risk of transferring virulence factors through genomic pathogenicity islands (Malachowa and Deleo 2010; Xia and Wolz 2014). Thus, host ranges of many temperate bacteriophages remain poorly characterized, despite their important impact on bacterial evolution. To advance our knowledge on the topic, and as part of a course-based undergraduate research experience at Arizona State University, we analyzed the genomes and computationally inferred the host ranges of 40 cluster P mycobacteriophages.
Materials and methods
Comparative genomic analyses
A multiple sequence alignment of 40 cluster P mycobacteriophages previously isolated in M. smegmatis mc2155 (Supplementary Table 1) was generated via MAFFT v.7.407 (Katoh and Standley 2013) and subsequently used to construct a neighbor-joining tree in MEGA X (Kumar et al. 2018) using a bootstrap test of phylogeny with 10,000 replicates. Additional whole-genome and gene-specific trees were generated, including 16 bacteriophages from clusters G1, I1, and N for which integration-dependent immunity systems had previously been identified (either experimentally or through the computational identification of an attP site within the repressor gene; Supplementary Table 2). Trees were visualized using FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/; last accessed 2022 April 24) and the Interactive Tree Of Life (Letunic and Bork 2019). Sequence relatedness was determined using pairwise average nucleotide identity scores calculated using the DNA Master “Genome Comparison” tool v.5.23.6 and plotted using the ggplot2 function (Wickham 2016) in R v.4.0.2. All software were executed using default settings.
Identification of attP and attB sites
Following Pham et al. (2007), NCBI BLASTn (Altschul et al. 1990) was used to compare the 300-bp region surrounding the 5′-end of the immunity repressor gene in each cluster P mycobacteriophage (Supplementary Table 1) against the genomes of 14 putative mycobacterial host species (Supplementary Table 3) to determine the plausibility of attP/attB sites. In addition, Tandem Repeats Finder v.4.09 (Benson 1999) was used to search for integrase binding sites near the attP common core.
Host prediction
Following the best practices suggested by Versoza and Pfeifer (2022), both exploratory and confirmatory methods were used to computationally predict host ranges for 40 closely related cluster P mycobacteriophages (Supplementary Table 1). First, the exploratory tool PHERI v.0.2 (Baláž et al. 2020) was used to predict bacterial host genera. Among the currently available exploratory host range prediction tools, PHERI was the most user-friendly and well-documented, making it ideally suited for course-based undergraduate research experiences. Next, WIsH v.1.1 (Galiez et al. 2017)—a bacterial host range predictor that compares virus and host sequence composition—was used to estimate the likelihood of these 40 cluster P bacteriophages to infect 14 putative mycobacterial host species with particular relevance to human health and disease (Supplementary Table 3). WIsH was selected as the representative for confirmatory host range prediction tools as it was an easily applicable alternative to alignment-based tools which frequently underpredict phage–host interactions (Zielezinski et al. 2021). Lastly, following Crane et al. (2021), PHASTER (Arndt et al. 2016) was used to search the genome of these putative host species for prophages to determine whether cluster P mycobacteriophages might be able to integrate into the host.
Results and discussion
Comparative genomic analyses between 40 cluster P mycobacteriophages (32 subcluster P1, 1 subcluster P2, 1 subcluster P3, 2 subcluster P4, 2 subcluster P5, and 1 subcluster P6; Supplementary Table 1) demonstrated a close relatedness at the sequence level (Fig. 1a), with cluster assignments supported by pairwise average nucleotide identities between the bacteriophages (Supplementary Fig. 1). With the exception of Tortellini (P2), Xavia (P3), and ThulaThula (P5), cluster P bacteriophage genomes harbor a conserved integration-dependent immunity system, comprised of an immunity repressor flanked by a tyrosine integrase, an excise gene, and an antirepressor (Supplementary Fig. 2) that governs the transition from the lytic to lysogenic state by binding and inactivating the lysogenic repressor (Lemire et al. 2011; Kim and Ryu 2013). It has previously been hypothesized that conserved integration-dependent immunity systems form the ancestral state of more complex genetic switches (Broussard and Hatfull 2013), such as those present in λ bacteriophages (Oppenheim et al. 2005). Interestingly, a neighbor-joining tree generated from whole-genome sequences of 16 cluster G1, I1, and N bacteriophages containing an integration-dependent immunity system (Supplementary Table 2) places cluster P4–P6 bacteriophages as sister taxa to the G1, I1, and N subclusters (Fig. 1b)—a tree topology supported by the gene-specific tree based on the immunity repressor sequences (Fig. 1c).
Fig. 1.
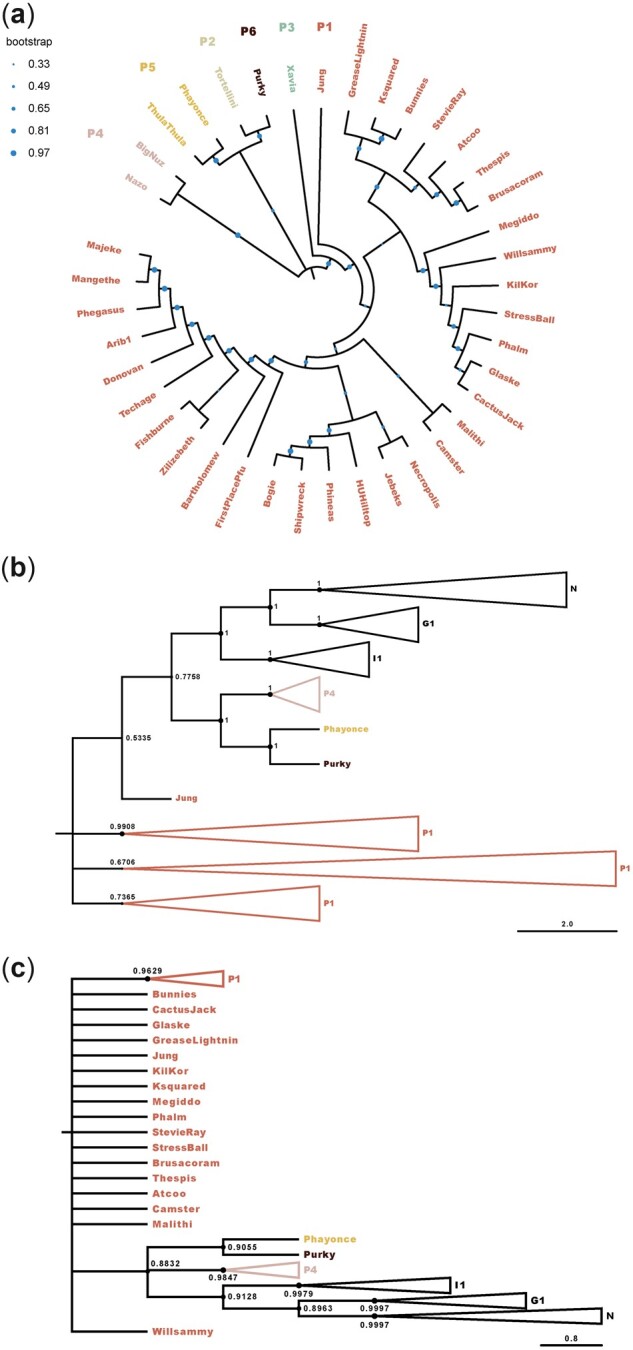
Neighbor-joining trees. Neighbor-joining trees generated in MAFFT (Katoh and Standley 2013) using the multiple-sequence alignment of (a) 40 cluster P mycobacteriophages (Supplementary Table 1) and (b) 16 cluster G1, I1, and N bacteriophages with a previously identified integration-dependent immunity system (Supplementary Table 2), with 10,000 bootstrap replicates. c) Gene-specific tree based on the immunity repressor sequences of the bacteriophages included in (b). Colors highlight membership in subclusters P1–P6.
To explore the impact of cluster P mycobacteriophages on bacterial communities, their host ranges were computationally predicted using a combination of exploratory and confirmatory tools, together with 14 putative mycobacterial host species relevant to human health and disease. Using the exploratory method, all but 1 P1 bacteriophages (Donovan) appear restricted to the Mycobacterium genus (Table 1). In contrast, bacteriophages of subclusters P2–P6 are likely also able to infect the nonpathogenic microbes Gordonia and Rhizobium as well as hosts of the genera Clostridiodes, Clostridium, and Corynebacterium, frequently associated with human disease, including diphtheria (Corynebacterium diphtheriae) as well as several hospital-acquired infections (see reviews by Bernard 2012 and Mangutov et al. 2021). As the ability to bind to new receptors is a key step in host-range evolution (Meyer et al. 2012), mutations within tail protein genes might explain the predicted expanded host range of subclusters P2–P6. At the species level, confirmatory results (Fig. 2) suggest that, in addition to M. smegmatis mc2155 used to isolate the bacteriophages, subcluster P1 mycobacteriophages are likely able to infect Mycobacterium fortuitum—which can cause infections in the skin, lymph nodes, and joints of immunocompromised individuals (Sethi et al. 2014), as well as Mycobacterium gilvum, and Mycobacterium intracellulare—which can cause pulmonary infections and lymphadenitis in immunocompromised individuals (Han et al. 2005). In contrast, bacteriophages of subclusters P2–P6 displayed low likelihoods of infection for all tested hosts.
Table 1.
Exploratory host range prediction.
| Phage | Subcluster | Mycobacterium | Gordonia | Clostridioides | Corynebacterium | Rhizobium | Clostridium |
|---|---|---|---|---|---|---|---|
| Arib1 | P1 | ✓ | |||||
| Atcoo | P1 | ✓ | |||||
| Bartholomew | P1 | ✓ | |||||
| Bogie | P1 | ✓ | |||||
| Brusacoram | P1 | ✓ | |||||
| Bunnies | P1 | ✓ | |||||
| CactusJack | P1 | ✓ | |||||
| Camster | P1 | ✓ | |||||
| Donovan | P1 | ✓ | ✓ | ||||
| FirstPlacePfu | P1 | ✓ | |||||
| Fishburne | P1 | ✓ | |||||
| Glaske | P1 | ✓ | |||||
| GreaseLightnin | P1 | ✓ | |||||
| HUHilltop | P1 | ✓ | |||||
| Jebeks | P1 | ✓ | |||||
| Jung | P1 | ✓ | |||||
| KilKor | P1 | ✓ | |||||
| Ksquared | P1 | ✓ | |||||
| Majeke | P1 | ✓ | |||||
| Malithi | P1 | ✓ | ✓ | ||||
| Mangethe | P1 | ✓ | |||||
| Megiddo | P1 | ✓ | |||||
| Necropolis | P1 | ✓ | |||||
| Phalm | P1 | ✓ | |||||
| Phegasus | P1 | ✓ | |||||
| Phineas | P1 | ✓ | |||||
| Shipwreck | P1 | ✓ | |||||
| StevieRay | P1 | ✓ | |||||
| StressBall | P1 | ✓ | |||||
| Techage | P1 | ✓ | |||||
| Thespis | P1 | ✓ | |||||
| Willsammy | P1 | ✓ | |||||
| Zilizebeth | P1 | ✓ | |||||
| Tortellini | P2 | ✓ | ✓ | ✓ | ✓ | ||
| Xavia | P3 | ✓ | ✓ | ✓ | ✓ | ||
| BigNuz | P4 | ✓ | ✓ | ||||
| Nazo | P4 | ✓ | ✓ | ||||
| Phayonce | P5 | ✓ | ✓ | ||||
| ThulaThula | P5 | ✓ | ✓ | ✓ | |||
| Purky | P6 | ✓ | ✓ |
Putative host genera of the 40 cluster P bacteriophages included in this study (Supplementary Table 1) as predicted by PHERI (Baláž et al. 2020).
Fig. 2.
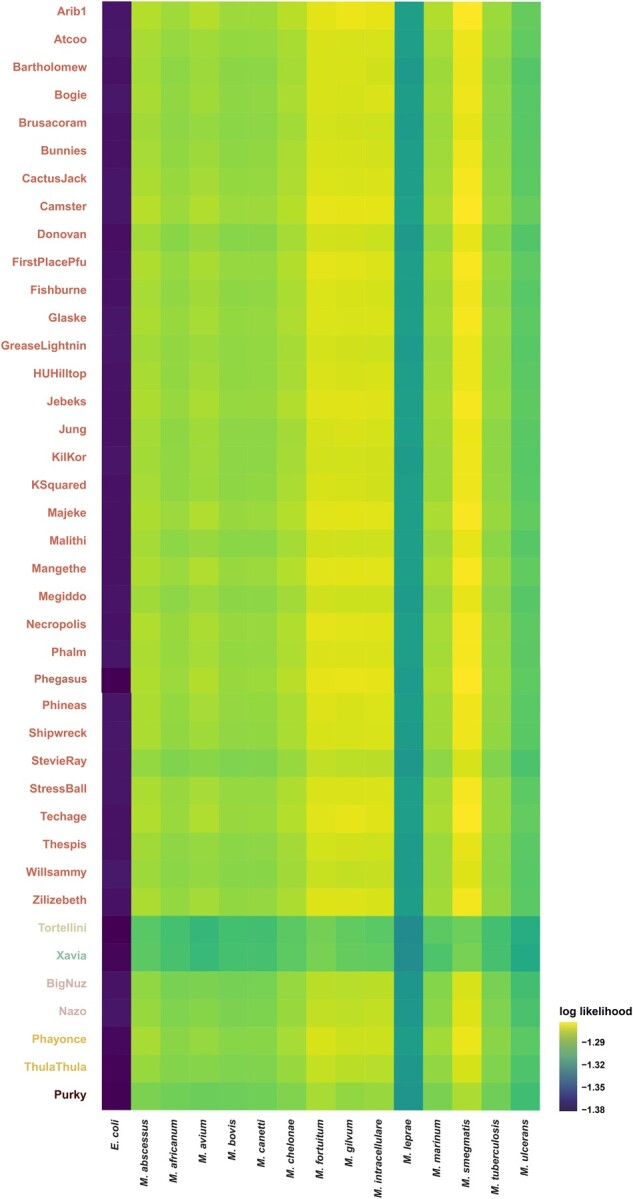
Confirmatory host range prediction. Putative bacteriophage–host interactions as predicted by WIsH (Galiez et al. 2017), using 40 cluster P mycobacteriophages (Supplementary Table 1), together with 14 potential bacterial hosts and Escherichia coli as a negative control (Supplementary Table 2). The higher the reported value, the more likely a bacteriophage is able to infect a putative host.
To investigate the temperate nature of cluster P mycobacteriophages, prophage sequences were computationally predicted within the putative host genomes. Three putative hosts (Mycobacterium abscessus, Mycobacterium marinum, and M. smegmatis) contain intact prophages—however, none of them correspond to prophages that stem from the integration of cluster P mycobacteriophages. In addition, incomplete prophages from the integration of cluster P mycobacteriophages were detected in both M. abscessus and M. marinum (Fig. 3)—2 opportunistic pathogens known to inflict pulmonary (Winthrop and Roy 2020) and cutaneous (Aubry et al. 2000) infections in humans—indicating that these hosts are at risk of incorporating virulence factors from these bacteriophages. Interestingly, the 2 partial prophages within M. abscessus and M. marinum were predicted to stem from the integration of 2 (out of only 3) cluster P bacteriophages that lack an integration-dependent immunity system (ThulaThula and Xavia, respectively).
Fig. 3.

Prophage prediction. Complete (green) and incomplete (red) prophages from the integration of bacteriophages were detected in both M. abscessus (left) and M. marinum (right). Incomplete prophages from the integration of cluster P mycobacteriophages are displayed at the bottom (region 2 in M. abscessus and region 1 in M. marinum), together with the protein-coding genes contained in these regions. Phage-like proteins on forward and reverse strands (indicated by orange arrows) are displayed above and below the ruler for each region, respectively.
For temperate bacteriophages, the risk of transfer of virulence factors depends (at least in part) on the presence of an attP region in the bacteriophage as well as a corresponding attB attachment site in the host genome (Pham et al. 2007). Putative attP sites in cluster P bacteriophages are similar in length to those previously reported in other mycobacteriophages (Pham et al. 2007; Morris et al. 2008) and the lack of arm-type integrase binding sites flanking the attP common core—known to be present in nonintegration-dependent immunity system bacteriophages such as λ (Landy 1989) and L5 (Peña et al. 1997) but notably absent in integration-dependent immunity system bacteriophages (Broussard et al. 2013)—is further evidence of a functional integration-dependent immunity system in these bacteriophages. To identify putative attachment sites, attP sites were compared against the genomes of 14 mycobacteria. Out of the 14 mycobacterium species tested, only 3 (M. smegmatis, Mycobacterium chelonae, and Mycobacterium leprae) contained a homologous attB bacterial attachment site, overlapping with the 3′-end of a tRNAThr gene (Supplementary Table 4), indicating that these hosts are at risk of incorporating virulence factors from bacteriophages that utilize tyrosine integrases in their integration-dependent immunity systems. Yet, despite the presence of an attB attachment site, 2 out of these 3 species (M. chelonae and M. leprae) were not predicted as potential hosts for any cluster P bacteriophage. However, it is important to note that WIsH evaluates host likelihood on the basis of oligonucleotide frequency similarity between the virus and host genomes. Consequently, more sophisticated approaches that rely on several distinct genomic features to predict the success of phage infection (such as advanced machine learning-based methods) may be able to provide a more complete picture of the putative host ranges.
Taken together, our computational predictions indicate that cluster P bacteriophages harboring a conserved integration-dependent immunity system likely exhibit similar host ranges. An important future endeavor will be the experimental validation of the presented computational results by phenotypic studies in order to lend further credence to the hypothesis that the type of genetic switch used to induce lysogeny plays an important role in host range evolution.
Supplementary Material
Contributor Information
Abigail A Howell, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA; Biodesign Institute, Arizona State University, Tempe, AZ 85281, USA.
Cyril J Versoza, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA; Center for Evolution and Medicine, Arizona State University, Tempe, AZ 85281, USA.
Gabriella Cerna, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA; Biodesign Institute, Arizona State University, Tempe, AZ 85281, USA; School of Molecular Sciences, Arizona State University, Tempe, AZ 85281, USA.
Tyler Johnston, School of Molecular Sciences, Arizona State University, Tempe, AZ 85281, USA.
Shriya Kakde, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Keith Karuku, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Maria Kowal, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Jasmine Monahan, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Jillian Murray, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA; School of Molecular Sciences, Arizona State University, Tempe, AZ 85281, USA.
Teresa Nguyen, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Aurely Sanchez Carreon, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA; Biodesign Institute, Arizona State University, Tempe, AZ 85281, USA; School of Molecular Sciences, Arizona State University, Tempe, AZ 85281, USA.
Abigail Streiff, School of Molecular Sciences, Arizona State University, Tempe, AZ 85281, USA.
Blake Su, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA; School of Politics and Global Studies, Arizona State University, Tempe, AZ 85281, USA.
Faith Youkhana, School of Molecular Sciences, Arizona State University, Tempe, AZ 85281, USA.
Saige Munig, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Zeel Patel, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Minerva So, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Makena Sy, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Sarah Weiss, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA.
Susanne P Pfeifer, School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA; Center for Evolution and Medicine, Arizona State University, Tempe, AZ 85281, USA.
Data Availability
Genomic data for all 40 cluster P mycobacteriophages, 16 cluster G1, I1, and N bacteriophages with a previously identified integration-dependent immunity system, and 14 putative bacterial host species can be downloaded from the NCBI Sequence Read Archive using the accession numbers provided in Supplementary Tables 1–3, respectively. Supplementary Table 4 lists the mycobacteriophage integration systems and putative integration sites of cluster P mycobacteriophages in M. chelonae, M. leprae, and M. smegmatis.Supplementary Fig. 1 displays the pairwise average nucleotide identities of the 40 cluster P bacteriophages. Supplementary Fig. 2 displays the Phamerator map of the regions encoding the tyrosine integrase, immunity repressor, and excise genes in cluster P mycobacteriophages.
Supplemental material is available at G3 online.
Funding
This work was supported by a National Science Foundation CAREER grant to SPP (DEB-2045343).
Conflicts of interest
None declared.
Literature cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS.. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry A, Jarlier V, Escolano S, Truffot-Pernot C, Cambau E.. Antibiotic susceptibility pattern of Mycobacterium marinum. Antimicrob Agents Chemother. 2000;44(11):3133–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baláž A, Kajsík M, Budiš J, Szemeš T, Turňa J.. 2020. PHERI—phage host exploration pipeline. In pre-print. BioRxiv. doi: 10.1101/2020.05.13.093773. [DOI] [PMC free article] [PubMed]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard K. The genus Corynebacterium and other medically relevant coryneform-like bacteria. J Clin Microbiol. 2012;50(10):3152–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard GW, Hatfull GF.. Evolution of genetic switch complexity. Bacteriophage. 2013;3(1):e24186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF.. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell. 2013;49(2):237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhan Z, Zhang H, Bi L, Zhang X-E, Fu YV.. Kinetic analysis of DNA compaction by mycobacterial integration host factor at the single-molecule level. Tuberculosis (Edinb). 2019;119:101862. [DOI] [PubMed] [Google Scholar]
- Crane A, Versoza CJ, Hua T, Kapoor R, Llyod L, Mehta R, Menolascino J, Morais A, Munig S, Patel Z, et al. Phylogenetic relationships and codon usage bias amongst cluster K mycobacteriophages. G3 (Bethesda). 2021;11(11):jkab291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle EL, Fillman CL, Reyna NS, Tobiason DM, Westholm DE, Askins JL, Backus BP, Baker AC, Ballard HS, Bisesi PJ, et al. Genome sequences of four cluster P mycobacteriophages. Genome Announc. 2017;6:e01101-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiez C, Siebert M, Enault F, Vincent J, Söding J.. WIsH: who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics. 2017;33(19):3113–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan JL, Holmes EC.. Predicting virus emergence amid evolutionary noise. Open Biol. 2017;7(10):170189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth AC, Calos MP.. Phage integrases: biology and applications. J Mol Biol. 2004;335(3):667–678. [DOI] [PubMed] [Google Scholar]
- Han XY, Tarrand JJ, Infante R, Jacobson KL, Truong M.. Clinical significance and epidemiologic analyses of Mycobacterium avium and Mycobacterium intracellulare among patients without AIDS. J Clin Microbiol. 2005;43(9):4407–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochschild A, Douhan J 3rd, Ptashne M.. How lambda repressor and lambda Cro distinguish between OR1 and OR3. Cell. 1986;47(5):807–816. [DOI] [PubMed] [Google Scholar]
- Howard-Varona C, Hargreaves KR, Abedon ST, Sullivan MB.. Lysogeny in nature: mechanisms, impact, and ecology of temperate phages. ISME J. 2017;11(7):1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Modlin RL, et al. ; Science Education Alliance Phage Hunters Advancing Genomics And Evolutionary Science Sea-Phages Program. On the nature of mycobacteriophage diversity and host preference. Virology. 2012;434(2):187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Ryu S.. Antirepression system associated with the life cycle switch in the temperate Podoviridae phage SPC32H. J Virol. 2013;87(21):11775–11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K.. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landy A. Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu Rev Biochem. 1989;58:913–949. [DOI] [PubMed] [Google Scholar]
- Lemire S, Figueroa-Bossi N, Bossi L.. Bacteriophage crosstalk: coordination of prophage induction by trans-acting antirepressors. PLoS Genet. 2011;7(6):e1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P.. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Hatfull GF.. Control of directionality in L5 integrase-mediated site-specific recombination. J Mol Biol. 2003;326(3):805–821. [DOI] [PubMed] [Google Scholar]
- Malachowa N, Deleo FR.. Mobile genetic elements of Staphylococcus aureus. Cell Mol Life Sci. 2010;67(18):3057–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangutov EO, Georgievna Kharseeva G, Alutina EL.. Coryneebacterium spp.—problematic pathogens of the human respiratory tract. Klin Lab Diagn. 2021;66(8):502–508. [DOI] [PubMed] [Google Scholar]
- Meyer JR, Dobias DT, Weitz JS, Barrick JE, Quick RT, Lenski RE.. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science. 2012;335(6067):428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris P, Marinelli LJ, Jacobs-Sera D, Hendrix RW, Hatfull GF.. Genomic characterization of mycobacteriophage Giles: evidence for phage acquisition of host DNA by Illegitimate recombination. J Bacteriol. 2008;190(6):2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim AB, Kobiler O, Stavans J, Court DL, Adhya S.. Switches in bacteriophage lambda development. Annu Rev Genet. 2005;39:409–429. [DOI] [PubMed] [Google Scholar]
- Pedulla ML, Lee MH, Lever DC, Hatfull GF.. A novel host factor for integration of mycobacteriophage L5. Proc Natl Acad Sci U S A. 1996;93(26):15411–15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña CEA, Kahlenberg JM, Hatfull GF.. Protein-DNA complexes in mycobacteriophage L5 integrative recombination. J Bacteriol. 1999;181(2):454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña CEA, Lee MH, Pedulla ML, Hatfull GF.. Characterization of the mycobacteriophage L5 attachment site. J Mol Biol. 1997;266(1):76–92. [DOI] [PubMed] [Google Scholar]
- Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF.. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology (Reading). 2007;153(Pt 8):2711–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope WH, Jacobs-Sera D, Russell DA, Peebles CL, Al-Atrache Z, Alcoser TA, Alexander LM, Alfano MB, Alford ST, Amy NE, et al. Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution. PLoS One. 2011;6(1):e16329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Arora S, Gupta V, Kumar S.. Cutaneous Mycobacterium fortuitum infection: successfully treated with Amikacin and Ofloxacin combination. Indian J Dermatol. 2014;59(4):383–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Chatterjee S, Datta S, Prasad R, Dubey D, Prasad RK, Vairale MG.. Bacteriophages and its applications: an overview. Folia Microbiol. (Praha). 2017;62(1):17–55. [DOI] [PubMed] [Google Scholar]
- Singh S, Ghosh P, Hatfull GF.. Attachment site selection and identity in Bxb1 serine integrase-mediated site-specific recombination. PLoS Genet. 2013;9(5):e1003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versoza CJ, Pfeifer SP.. Computational prediction of bacteriophage host ranges. Microorganisms. 2022;10(1):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. ggplot2: elegant graphics for data analysis. New York (NY: ): Springer-Verlag; 2016. ISBN 978-3-319–24277-4. [Google Scholar]
- Winthrop KL, Roy EE.. Mycobacteria and immunosuppression. In: Atzeni F, Galloway JB, Gomez-Reino JJ, Galli M, editors. Handbook of Systemic Autoimmune Diseases, Vol. 16. Elsevier Ltd; 2020. p. 83–107. [Google Scholar]
- Xia G, Wolz C.. Phages of Staphylococcus aureus and their impact on host evolution. Infect Genet Evol. 2014;21:593–601. [DOI] [PubMed] [Google Scholar]
- Zielezinski A, Barylski J, Karlowski WM.. Taxonomy-aware, sequencing similarity ranking reliably predicts phage-host relationships. BMC Biol. 2021;19(1):223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic data for all 40 cluster P mycobacteriophages, 16 cluster G1, I1, and N bacteriophages with a previously identified integration-dependent immunity system, and 14 putative bacterial host species can be downloaded from the NCBI Sequence Read Archive using the accession numbers provided in Supplementary Tables 1–3, respectively. Supplementary Table 4 lists the mycobacteriophage integration systems and putative integration sites of cluster P mycobacteriophages in M. chelonae, M. leprae, and M. smegmatis.Supplementary Fig. 1 displays the pairwise average nucleotide identities of the 40 cluster P bacteriophages. Supplementary Fig. 2 displays the Phamerator map of the regions encoding the tyrosine integrase, immunity repressor, and excise genes in cluster P mycobacteriophages.
Supplemental material is available at G3 online.