

SUMMARY
Noradrenergic afferents to hypothalamic corticotropin releasing hormone (CRH) neurons provide a major excitatory drive to the hypothalamic-pituitary-adrenal (HPA) axis via α1 adrenoreceptor activation. Noradrenergic afferents are recruited preferentially by somatic, rather than psychological, stress stimuli. Stress-induced glucocorticoids feed back onto the hypothalamus to negatively regulate the HPA axis, providing a critical autoregulatory constraint that prevents glucocorticoid overexposure and neuropathology. Whether negative feedback mechanisms target stress modality-specific HPA activation is not known. Here, we describe a desensitization of the α1 adrenoreceptor activation of the HPA axis following acute stress in male mice that is mediated by rapid glucocorticoid regulation of adrenoreceptor trafficking in CRH neurons. Glucocorticoid-induced α1 receptor trafficking desensitizes the HPA axis to a somatic but not a psychological stressor. Our findings demonstrate a rapid glucocorticoid suppression of adrenergic signaling in CRH neurons that is specific to somatic stress activation, and they reveal a rapid, stress modality-selective glucocorticoid negative feedback mechanism.
In brief
Physical and psychological stressors activate neuroendocrine secretion of corticosteroid. Noradrenaline circuits are critical to the neuroendocrine response to physical but not psychological stress. Jiang et al. show that stress-induced corticosteroids decrease noradrenaline sensitivity in the brain, which suppresses the response to subsequent physical stressors but leaves the psychological stress response intact.
Graphical Abstract
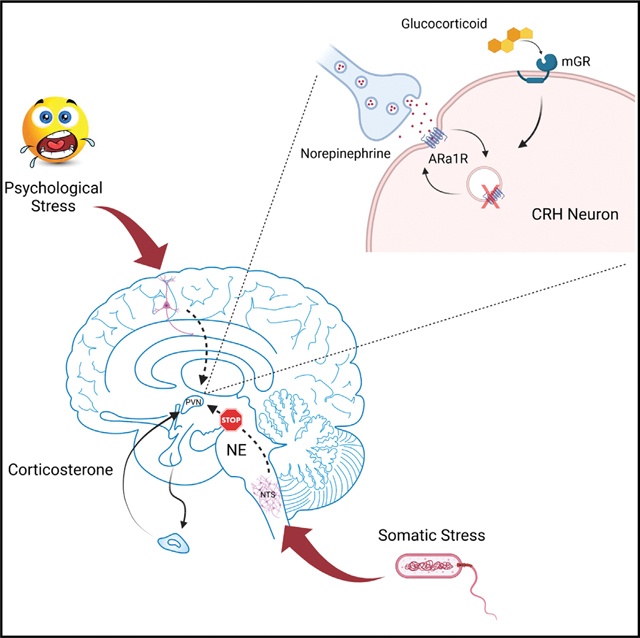
INTRODUCTION
Activation of the hypothalamic-pituitary-adrenal (HPA) axis is part of Selye’s generalized stress response described over 70 years ago (Selye, 1950). How this response that stimulates adrenal secretion of glucocorticoids discriminates between different stress modalities is not known. Here, we targeted the noradrenergic excitatory drive to the HPA axis, which is activated preferentially by somatic stress stimulation (Banihashemi and Rinaman, 2006; Bienkowski and Rinaman, 2008; Sawchenko and Swanson, 1982). The corticotropin releasing hormone (CRH) neurons of the hypothalamic paraventricular nucleus (PVN), the primary effector cells of the HPA axis, are strongly innervated by noradrenergic afferents from the nucleus of the solitary tract (NTS) (Cunningham and Sawchenko, 1988; Füzesi et al., 2007; Wittmann, 2008). Multiple studies have suggested that this afferent pathway provides the main excitatory drive to the HPA axis during somatic stress such as glycemic and immune challenges (Banihashemi and Rinaman, 2006; Bienkowski and Rinaman, 2008; Cole and Sawchenko, 2002; Plotsky, 1987). We recently characterized the α1 adrenoreceptor-mediated stimulation of PVN CRH neurons in male mice via activation of local glutamate circuits following postsynaptic α1 receptor activation, dendritic peptide release, and retrograde glial-neuronal signaling. We also found evidence for the monosynaptic co-release of glutamate from the noradrenergic afferents (Chen et al., 2019).
Glucocorticoid negative feedback inhibits the activation of the HPA axis (de Kloet et al., 2008), which protects the organism from the damaging effects of sustained corticosteroid exposure. Previous studies showed that elevated plasma corticosterone decreases the PVN neuronal response to norepinephrine (NE) and HPA axis activation. Systemic pretreatment with the synthetic glucocorticoid dexamethasone inhibited the adrenocorticotropic hormone (ACTH) and corticosterone responses to noradrenergic afferent stimulation in a dose-dependent manner (Weidenfeld et al., 1997). Manipulating corticosterone levels by adrenalectomy, corticosterone pellet supplementation, and acute stress demonstrated that plasma corticosterone levels are inversely correlated with PVN α1b adrenoreceptor mRNA expression (Day et al., 1999). The norepinephrine excitation of a subset of PVN parvocellular neurons was blocked by cortisol pretreatment (Kasai and Yamashita, 1988). In addition, adrenalectomy increased the α1 adrenoreceptor-mediated excitation of PVN parvocellular neurons, which was reversed by corticosterone replacement (Yang et al., 2007). These findings suggest an inhibitory effect of glucocorticoids on the NE-induced excitation of hypothalamic PVN neurons and that the NE-induced excitation of the HPA axis is highly sensitive to corticosteroid levels, suggesting it is a target of glucocorticoid negative feedback.
The mechanisms of glucocorticoid suppression of NE signaling to the HPA axis represent a potential pharmacotherapy target due to the association of HPA dysfunction with stress disorders, but these mechanisms are unknown. Here, we investigated these mechanisms using ex vivo patch-clamp recordings, live-cell imaging, and in vivo viral gene transfer and physiological analyses. We found that stress desensitizes PVN CRH neurons to NE via glucocorticoid regulation of α1-adrenoceptor trafficking. The stress/glucocorticoid-induced desensitization of CRH neurons to NE causes a suppression of HPA activation by somatic but not psychological stress, revealing a stress modality-selective glucocorticoid negative feedback regulation of the HPA axis.
RESULTS
Stress-state dependence of the NE α1 adrenoreceptor response in CRH neurons
Norepinephrine elicits a concentration-dependent excitation of PVN CRH neurons from male mice by stimulating local glutamate circuits via activation of postsynaptic α1 adrenoreceptors in the CRH neurons, dendritic peptide release, and retrograde activation of a local glial-neuronal circuit (Chen et al., 2019) (Figure S1A). Here, we tested for the stress-state dependence of the NE response in male mouse PVN CRH neurons by subjecting the mice to an acute stress before performing whole-cell recordings in acute slices. In PVN CRH neurons from control (unstressed) mice, bath application of NE (100 μM) caused a robust increase in the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) (335.2% ± 61.5% of baseline, p < 0.001, n = 17 cells from 10 mice, Wilcoxon signed-rank test; in this and subsequent electrophysiological experiments, the baseline was calculated from the last 3 min of recorded sEPSCs before drug application and was used for a within-cell comparison) (Figures 1A–1C); NE had no effect on the sEPSC amplitude or decay time (Chen et al., 2019). In CRH neurons from mice subjected to a prior 30-min restraint stress, the NE response was suppressed, resulting in a non-significant increase in sEPSC frequency (145.9% ± 39.4% of baseline, p = 0.29, n = 7 cells from five mice, paired t test, t(6) = −1.164) (Figures 1A–1C). No difference was detected between CRH neurons from unstressed mice and stressed mice in the basal sEPSC frequency (unstressed: 1.56 ± 0.10 Hz, n = 47 cells; stressed: 1.36 ± 0.16 Hz, n = 12, p = 0.46, ANOVA on ranks), amplitude (unstressed: 19.12 ± 0.50 pA, n = 47 cells; stressed: 17.64 ± 1.03 pA, n = 12, p = 0.13, ANOVA on ranks), or decay time (unstressed: 2.18 ± 0.06 ms, n = 47 cells; stressed: 2.05 ± 0.12 ms, n = 12, p = 0.38, ANOVA on ranks).
Figure 1. The NE-induced facilitation of excitatory synaptic input to CRH neurons is sensitive to stress.
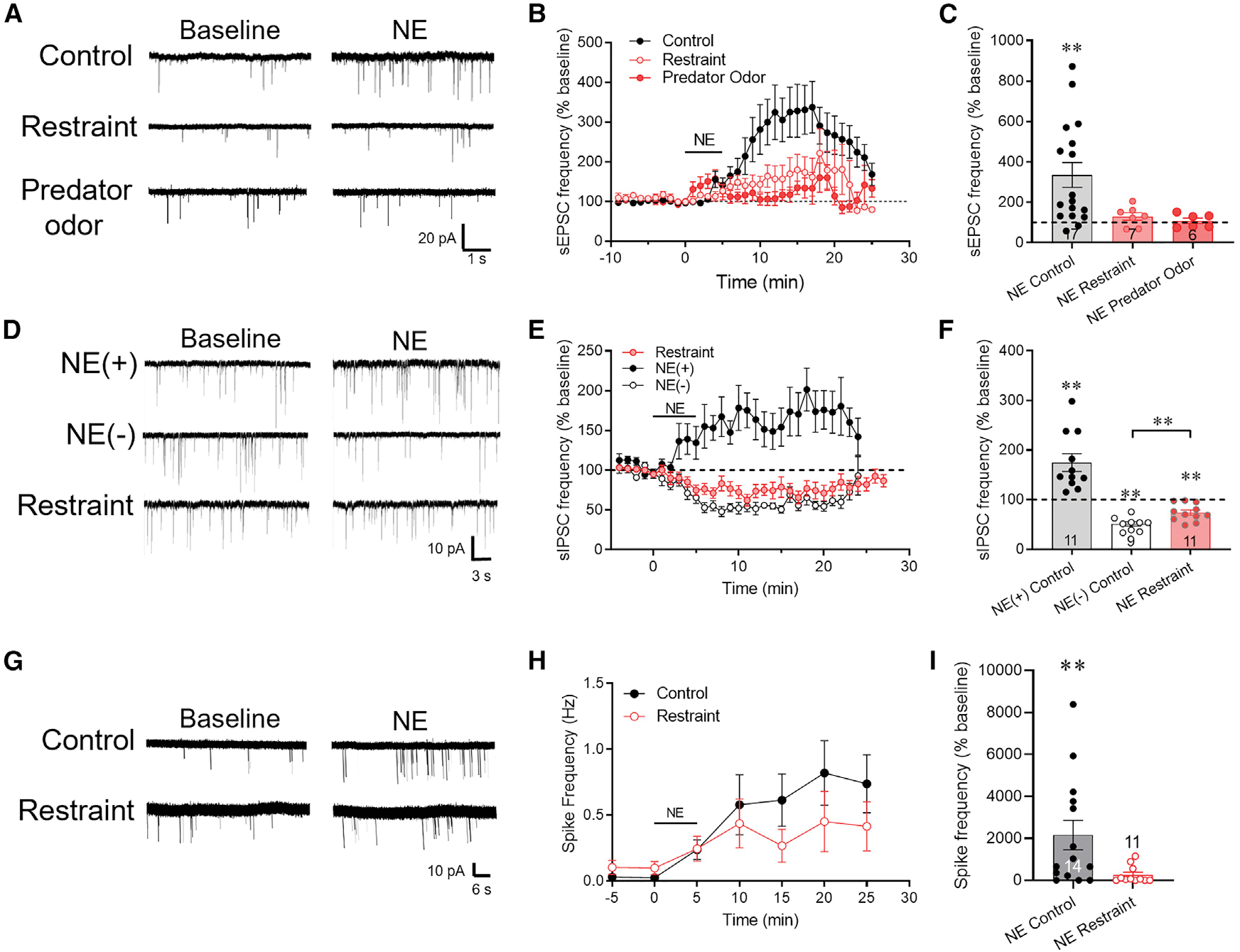
(A) Representative traces of the effect of NE on sEPSCs recorded in CRH neurons from control mice and mice exposed to 30-min restraint or 30-min predator odor.
(B) Time course of mean normalized sEPSC frequency response (percent of baseline) to NE (100 μM) in CRH neurons from control, restraint-exposed, and predator odor-exposed mice. Baseline frequency = mean sEPSC frequency during 3 min prior to NE application.
(C) Summary of normalized mean sEPSC frequency response to NE in CRH neurons from control, restraint-exposed, and predator odor-exposed mice.
(D) Representative traces of the dual effect of activation and suppression of sIPSCs by NE in control PVN CRH neurons (NE(+) and NE(−), respectively), and of the NE response in a CRH neuron from a restraint-stressed mouse (Restraint). NE increased sIPSC frequency (NE(+)) in ~50% and decreased sIPSC frequency (NE(−)) in ~50% of control CRH neurons (Chen et al., 2019), but it only decreased sIPSC frequency in CRH neurons following restraint.
(E) Time course of the mean normalized sIPSC frequency responses (percent of baseline) to NE in CRH neurons from control mice (NE(+) and NE(−)) and mice subjected to a 30-min restraint. Restraint stress abolished the NE facilitation of sIPSCs (NE(+)) but not the NE suppression of sIPSCs (NE(−)).
(F) Summary of the normalized mean sIPSC frequency responses to NE showing the dual response in CRH neurons from control mice (NE(+) and NE(−)) and the block of the NE facilitation of sIPSCs by restraint (NE Restraint).
(G) Representative loose-seal patch-clamp recordings of the NE effects on CRH neuron spiking in slices from control and restraint-stressed mice.
(H) Time course of the NE-induced increase in normalized mean spiking frequency in CRH neurons from control and restraint-stressed mice.
(I) Summary of the normalized mean spiking frequency response to NE (percent of baseline). Data are represented as mean ± SEM; numerals in bar graphs represent the numbers of recorded neurons. **p < 0.01.
See also Figure S1.
To determine whether the acute stress-induced desensitization of the CRH neurons to α1 adrenoreceptor activation extends to other types of acute stressors, we also tested a 30-min exposure to a predator odor (Kondoh et al., 2016). Male mice were exposed to bobcat urine for 30 min prior to sacrifice. The predator odor suppressed completely the NE-induced facilitation of sEPSC frequency in CRH neurons (108.3% ± 13.82% of baseline, p = 0.57, n = 6 cells from two mice, paired Student’s t test, t(5) = 0.36) (Figures 1A–1C).
Norepinephrine has a dual effect on inhibitory synaptic inputs to PVN CRH neurons in male mice: (1) a postsynaptic α1 receptor-mediated retrograde activation of local GABA circuits that facilitates inhibitory inputs, and (2) a presynaptic α2 receptor-mediated suppression of GABA release that suppresses inhibitory inputs (Figure S1B); approximately half of the CRH neurons respond with an α1 receptor-mediated retrograde activation of GABAergic synaptic inputs, while all the CRH neurons respond with a presynaptic α2 receptor-dependent suppression of GABAergic synaptic inputs, which is masked in the neurons showing the α1 receptor-mediated activation of GABAergic synaptic inputs (Chen et al., 2019). The NE activation, but not suppression, of local inhibitory synaptic inputs to CRH neurons is also mediated by retrograde neuronal-glial signaling (Figure S1B) (Chen et al., 2019), so we also tested for stress desensitization of the α1 receptor-mediated facilitation of spontaneous inhibitory postsynaptic currents (sIPSCs). Following restraint stress, none of the CRH neurons responded to NE (100 μM) with an increase in sIPSC frequency, and all 11 cells responded with a decrease in sIPSC frequency (74.23% ± 5.24% of baseline, n = 11 cells from six mice, p < 0.01, paired Student’s t test, t(10) = 3.15) (Figures 1D–1F). Therefore, prior stress exposure suppressed the CRH neuron response to postsynaptic α1 adrenoreceptor activation.
Norepinephrine stimulates an increase in spiking activity in CRH neurons that is driven by the NE-induced increase in excitatory synaptic inputs (Chen et al., 2019). We tested whether the stress-induced suppression of excitatory synaptic inputs reduces the NE activation of the CRH neurons with loose-seal patch-clamp recordings. Slices were incubated in high-[K+] solution (10 mM) to elicit spontaneous spiking; the mean baseline spiking frequency in CRH neurons from stressed mice (0.10 ± 0.05 Hz, n = 11 cells from five mice) was ~4-fold higher than in CRH neurons from control mice (0.03 ± 0.02 Hz, n = 14 cells from five mice), but this difference did not reach statistical significance (p = 0.15, Mann-Whitney rank-sum test). Bath application of NE (100 μM, 5 min) caused a 25-fold, significant increase in the spiking frequency in CRH neurons from control mice (from 0.03 ± 0.02 Hz to 0.74 ± 0.22 Hz, n = 14 cells from five mice, p < 0.01, RM ANOVA) (Figures 1G–1I), whereas it caused a 4-fold increase in the spiking frequency in CRH neurons from restraint-stressed mice that did not reach statistical significance (from 0.10 ± 0.05 Hz to 0.42 ± 0.19 Hz, n = 11 cells from five mice, p = 0.067) (Figures 1G–1I).
Glucocorticoid suppression of NE excitation of CRH neurons
Several studies have shown that acute glucocorticoid pretreatment suppresses stress-induced HPA activation in male rats (Ginsberg et al., 2003; Osterlund and Spencer, 2011; Weiser et al., 2011). Here, we tested whether the acute stress-induced suppression of the NE activation of excitatory synaptic inputs to PVN CRH neurons in male mice is due to increased plasma glucocorticoids.
The 11-β-hydroxylase inhibitor metyrapone suppresses stress-induced corticosterone synthesis (Figure S2). As before, the NE-induced increase in sEPSC frequency was abolished in CRH neurons in slices from intraperitoneally (i.p.) vehicle-injected, restraint-stressed mice (128.5% ± 21.0%, n = 8 cells from four mice, p = 0.22, paired t test, t(7) = −1.36). The NE response was partially restored in CRH neurons from mice treated 30 min prior to restraint stress with metyrapone (100 mg/kg i.p.) (204.2% ± 32.0% of baseline, p = 0.012, n = 9 cells from six mice, paired t test, t(8) = −3.252) (Figures 2A and 2B), suggesting that the stress-induced suppression of NE activation of excitatory synaptic inputs to the CRH neurons is dependent on circulating corticosterone levels.
Figure 2. Acute stress and glucocorticoid inhibit the NE-induced increase in excitatory synaptic inputs to CRH neurons.
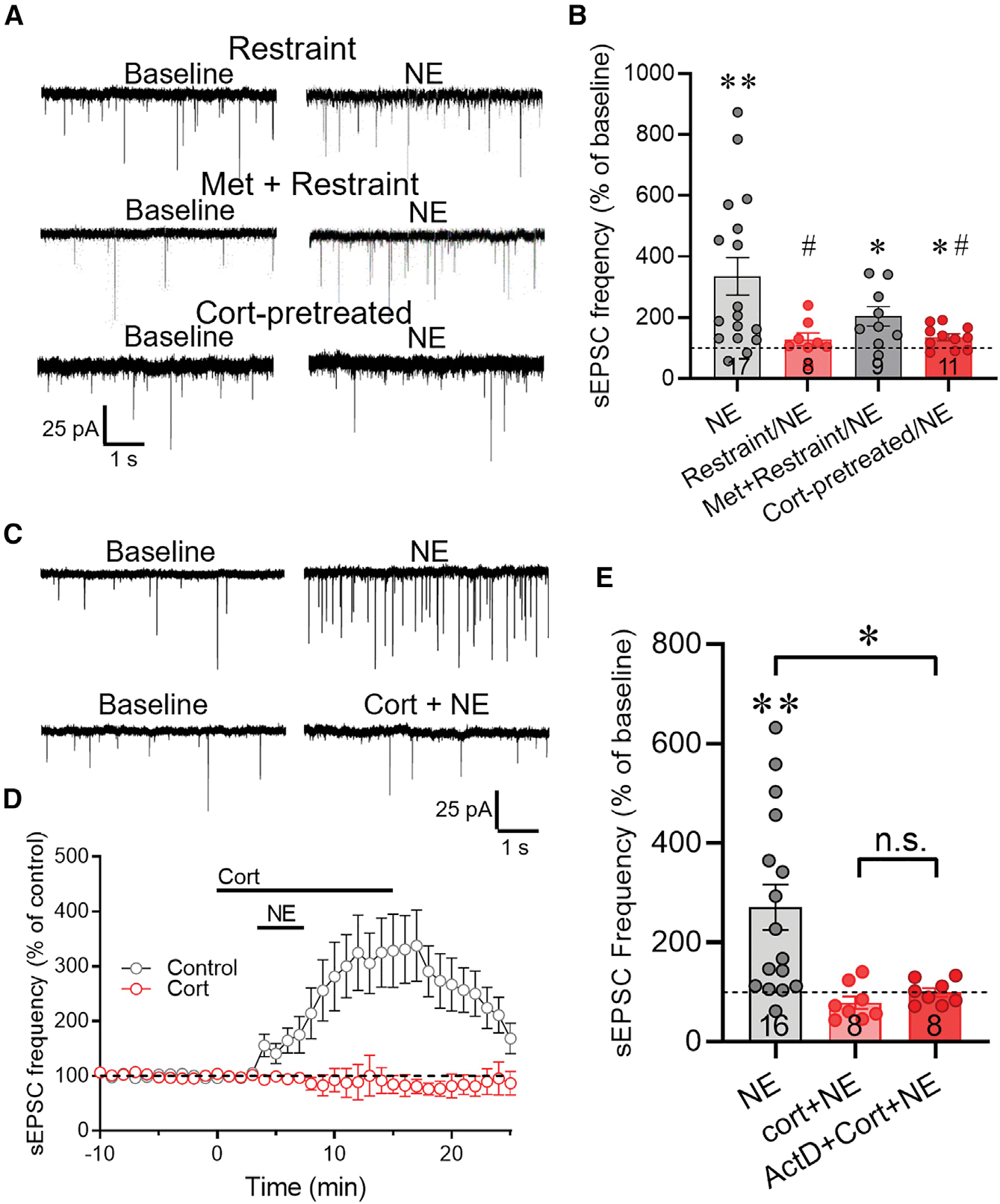
(A) Representative recordings of sEPSCs at baseline and following NE application in CRH neurons in slices from restraint-stressed mice after i.p. saline injection (Restraint) or metyrapone injection (Met + Restraint), and in slices from control mice that were preincubated for 5 min in corticosterone 2 h prior to NE application (Cort-pretreated).
(B) Summary of the NE-induced change in normalized mean sEPSC frequency (percent of baseline) in CRH neurons from unstressed control mice (NE), mice subjected to restraint stress following i.p. saline injection (Restraint/NE), and mice subjected to restraint stress following i.p. metyrapone injection (Met + Restraint/NE), and from slices from control mice preincubated for 30 min, R2 h earlier in corticosterone (Cort-pretreated/NE). The NE-induced increase in sEPSC frequency was blocked in CRH neurons following restraint in vivo and ex vivo preincubation in corticosterone, and it was partially rescued with metyrapone treatment prior to in vivo restraint.
(C) Representative recordings of the sEPSC response to NE in CRH neurons in the absence (NE) and presence of corticosterone (Cort + NE).
(D) Time course of the change in the normalized mean sEPSC frequency (% of baseline) in CRH neurons in response to NE (Control) and to NE and corticosterone co-application (Cort). The NE-induced increase in sEPSC frequency was abolished in corticosterone.
(E) Summary of the changes in normalized mean sEPSC frequency elicited by NE in the absence of corticosterone (NE), in the presence of corticosterone (Cort + NE), and in corticosterone following preincubation in the transcription inhibitor actinomycin D (ActD + Cort + NE). Data are represented as mean ± SEM; numerals in bar graphs represent numbers of recorded neurons. *p < 0.05, **p < 0.01 compared to baseline sEPSC frequency; #p < 0.05 compared to NE effect in CRH neurons from unstressed mice. n.s., not significant.
See also Figure S2.
To test for direct glucocorticoid suppression of NE signaling, we simulated the in vivo stress-induced corticosterone exposure in vitro by incubating slices from untreated mice in corticosterone (2 μM) for 30 min prior to returning them to regular aCSF for 2–6 h. We selected this concentration of corticosterone to correspond to the stress levels we measured in the blood using radioimmunoassay, which reached up to ~560 ng/mL, or ~1.6 μM, in our experiments (Figure S2). Basal sEPSC frequency (aCSF control: 1.56 ± 0.10 Hz vs. corticosterone: 1.88 ± 0.32 Hz, n = 47 cells from 22 mice and 16 cells from 11 mice, respectively, p = 0.70, ANOVA on ranks) and amplitude (aCSF control: 19.13 ± 0.50 pA vs. corticosterone: 19.11 ± 1.32 pA, p = 0.49, ANOVA on ranks) were unaffected by preexposure of slices to corticosterone 2–6 h prior to recordings. However, like acute stress, in vitro corticosterone exposure suppressed the NE-induced increase in sEPSC frequency 2–6 h later (136.1% ± 11.2% of baseline, n = 11 cells from eight mice, p < 0.05 compared with vehicle-treated slices, rank-sum test) (Figures 2A and 2B). These findings together suggest that the acute restraint stress-induced desensitization of CRH neurons to NE is due to stress-induced circulating corticosterone.
Corticosterone activates both nuclear glucocorticoid receptors to exert genomic actions and membrane-associated receptors to exert both genomic (Rainville et al., 2019) and nongenomic (Di et al., 2003, 2005; Osterlund and Spencer, 2011; Sapolsky et al., 2000) actions. Increasing evidence suggests that neural plasticity can be rapidly induced non-genomically by membrane glucocorticoid receptors (Di et al., 2003, 2005; Groeneweg et al., 2011; Harrison and Tasker, 2022). To determine whether the corticosterone suppression of NE-induced excitation of CRH neurons is mediated by a rapid glucocorticoid action, we preincubated hypothalamic slices in corticosterone (2 μM) for 5 min prior to co-application of corticosterone and NE (100 μM) for 5 min. Corticosterone alone caused a significant decrease in sEPSC frequency (to 88.3% ± 3.2% of baseline, t(6) = −3.62, p < 0.05, n = 7 cells from six mice, paired Student’s t test), which we showed previously to be due to retrograde endocannabinoid suppression of glutamate release (Di et al., 2003; Malcher-Lopes et al., 2006; Nahar et al., 2015). Corticosterone preincubation for 5 min completely blocked the NE-induced increase in sEPSC frequency and resulted in a significant decrease in sEPSC frequency (81.9% ± 16.8% of baseline; p < 0.05, n = 8 cells from five mice, signed-rank test) (Figures 2C–2E). Next, to determine whether the rapid glucocorticoid desensitization of the NE effect is dependent on a transcription-dependent mechanism, we preincubated the slices in the transcription inhibitor actinomycin D (25 μM) for 30 min prior to corticosterone (2 μM) and NE (100 μM) application. Blocking transcription failed to reverse the glucocorticoid suppression of the NE facilitation of sEPSC frequency (99.37% ± 8.53% of baseline; p = 0.28 compared with NE effect in corticosterone, n = 8 cells from four mice, rank-sum test) (Figure 2E). However, inhibiting translation with cycloheximide (100 μM) for 30 min blocked the corticosterone suppression and restored the NE facilitation of sEPSCs (223.10% ± 60.36% of baseline; p = 0.02 compared with NE effect in corticosterone, n = 8 cells from five mice, rank-sum test) (Figure S3). The sensitivity of the corticosterone suppression of the NE effect to blocking protein synthesis but not to blocking gene transcription suggested a dependence of the NE desensitization on constitutive protein synthesis. The stress desensitization of CRH neurons to NE excitation, therefore, is mediated by a rapid non-genomic corticosteroid signaling mechanism.
To test whether the nuclear glucocorticoid receptor participates in the corticosteroid-induced desensitization of NE excitation of CRH neurons, we used a transgenic mouse model in which the glucocorticoid receptor (GR) is conditionally knocked out in PVN neurons, the CRH-eGFP; sim1GR−/− (GRKOPVN) mouse (Laryea et al., 2013). Previous studies showed that these mice display increased ACTH and corticosterone secretion in response to acute stress due to an impaired glucocorticoid negative feedback (Solomon et al., 2015), and that PVN CRH neurons in these mice do not respond to rapid glucocorticoid suppression of excitation (Nahar et al., 2015). In CRH neurons from GRKOPVN mice, preincubation of slices for 10 min in corticosterone (1 μM) failed to abolish the NE-induced increase in sEPSC frequency (303.11% ± 20.22% of baseline, p < 0.01, n = 5 cells from five mice, paired Student’s t test, t(4) = 3.09) (Figure S2C). Similarly, prior restraint stress (30 min) in vivo did not block the NE-induced increase in sEPSC frequency recorded subsequently in CRH neurons in slices from GRKOPVN mice (208.97% ± 33.88% of baseline, p < 0.01, n = 5 cells from three mice, paired Student’s t test, t(4) = 4.55) (Figure S2C). These data indicate that depletion of GR prevents the corticosteroid desensitization of CRH neurons to excitation by NE, suggesting that the rapid glucocorticoid desensitization of α1 adrenoreceptors is dependent on intact nuclear GR signaling.
Glucocorticoid desensitization of CRH neurons to NE excitation is mediated by ligand-dependent endocytosis
Corticosterone has been shown to regulate glutamate receptor trafficking in pyramidal neurons (Conboy and Sandi, 2010; Groc et al., 2008; Yuen et al., 2011). Here, we tested whether the rapid glucocorticoid desensitization of CRH neurons to NE is caused by dynamin-dependent α1 receptor endocytosis. Blocking endocytosis with a non-competitive dynamin GTPase inhibitor, dynasore (80 μM) (Macia et al., 2006; Newton at al., 2006) applied in the bath for 10 min had no effect on baseline sEPSC frequency (t(9) = 0.88, p = 0.875, n = 10 cells from four mice, paired Student’s t test), but it prevented the corticosterone (2 μM) suppression of the NE-induced increase in sEPSC frequency (259.4% ± 61.3% of baseline, p < 0.05, n = 10 cells from four mice, signed-rank test) (Figures 3A–3C).This suggested that the rapid glucocorticoid desensitization of the CRH neurons to NE is mediated by dynamin-dependent internalization of α1 adrenoceptors.
Figure 3. The corticosterone-induced desensitization of CRH neurons to NE excitation is dependent on ligand-mediated endocytosis.
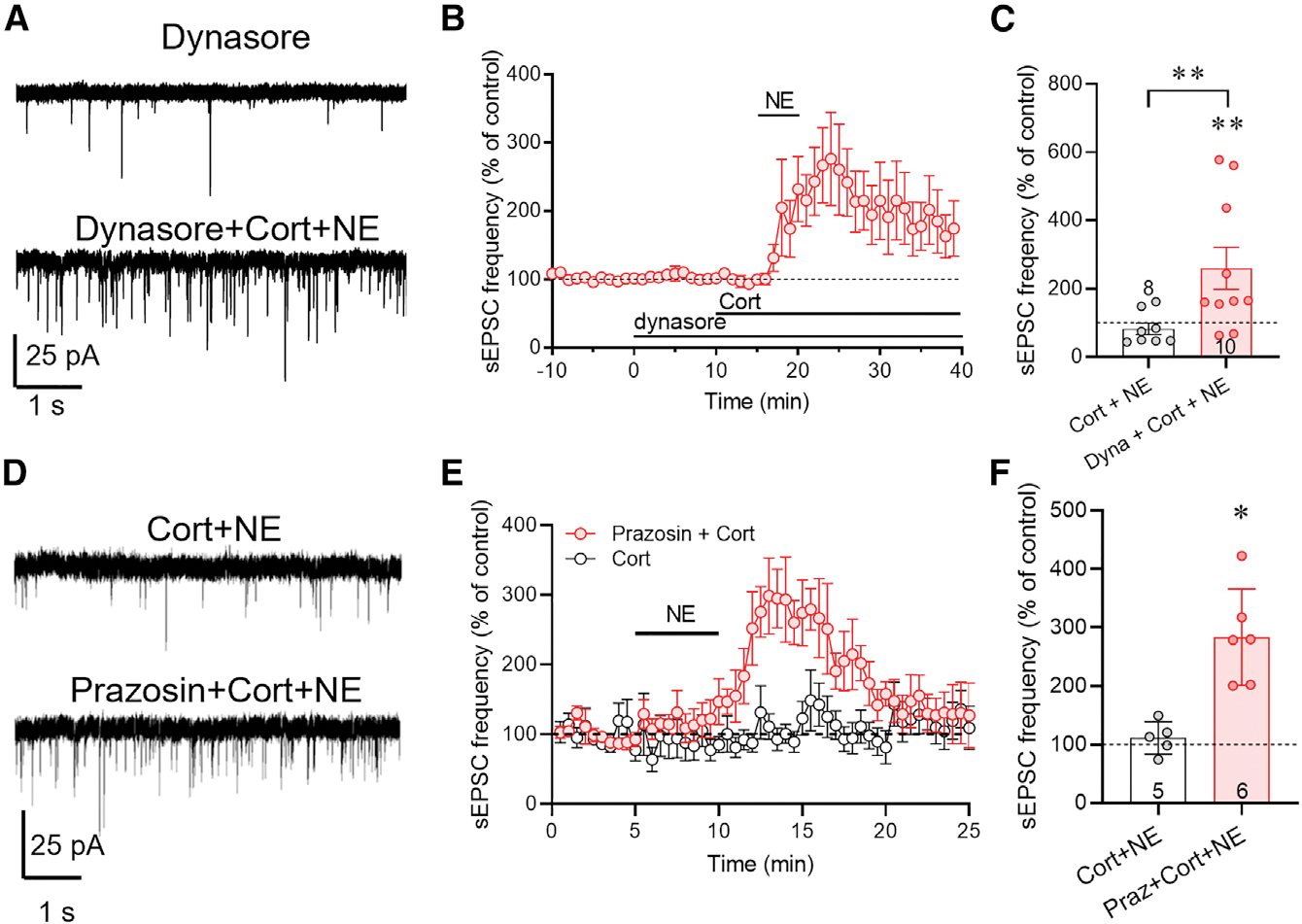
(A) Representative recordings of sEPSCs recorded in CRH neurons at baseline in the dynamin inhibitor dynasore and after NE application following dynasore and corticosterone co-application (dynasore + Cort + NE).
(B) Time course of the change in normalized mean sEPSC frequency showing restoration of the NE-induced increase in sEPSC frequency by dynasore preapplication prior to co-application of Cort and NE.
(C) Summary of NE effect on normalized mean sEPSC frequency in corticosterone alone (Cort + NE) and in dynasore and corticosterone (Dyna + Cort + NE). Inhibiting endocytosis reversed the corticosterone blockade of the NE-induced increase in sEPSC frequency.
(D) Representative recordings of the sEPSC response to NE 3 h following preincubation of slices in corticosterone (Cort) and in the α1 adrenoreceptor antagonist prazosin and corticosterone (Prazosin + Cort).
(E) Time course of the normalized mean sEPSC frequency response to NE following corticosterone preapplication alone (Cort) and prazosin and corticosterone (Prazosin + Cort).
(F) Summary of the sEPSC frequency response to NE following pretreatment with corticosterone (Cort + NE) or with prazosin and corticosterone (Praz + Cort + NE). The corticosterone desensitization of the NE response was reversed by blocking α1 adrenoreceptors during corticosterone exposure. Data are represented as mean ± SEM; numerals in bar graphs represent numbers of recorded neurons. *p < 0.05, **p < 0.01.
See also Figure S2D.
G protein-coupled receptors undergo ligand-dependent internalization via β-arrestin-triggered endocytosis. α1 adrenoreceptors in PVN CRH neurons are tonically activated by ambient extracellular NE levels in our slices (Figure S2D). To determine whether glucocorticoid desensitization of NE signaling in CRH neurons depends on basal ligand-mediated α1 receptor internalization by ambient NE, we tested for the dependence of the corticosterone suppression of the NE response on tonic adrenoreceptor activation by pre-incubating slices in the α1 adrenoreceptor antagonist prazosin (10 μM) for 10 min prior to a 5-min co-incubation in prazosin and corticosterone (1 μM) for 5 min and 30 min of washout of both. We reasoned that a 30-min washout is sufficient to reverse the prazosin blockade of α1 receptors but not to reverse the glucocorticoid desensitization of the α1 receptors, since the corticosterone desensitization of CRH neurons to NE lasts for over 2 h (e.g., see Figures 2A and 2B). Norepinephrine application (100 μM) 30 min after corticosterone (1 μM) and prazosin (10 μM) co-application caused a robust increase in sEPSC frequency (283% ± 33% of baseline, p < 0.05, n = 6 cells from four mice, Student’s t test, t(4) = 4.43) (Figures 3D–3F). This indicated that blocking α1 receptor binding during corticosterone exposure prevented the glucocorticoid-induced desensitization of the NE response and suggests that corticosterone facilitates endogenous NE-dependent internalization and/or trafficking of the α1 adrenoreceptors.
Corticosterone facilitates NE-induced ARα1b internalization
The dominant form of α1 adrenoreceptor in PVN CRH neurons is the α1b receptor (Day et al., 1999). To test directly whether the glucocorticoid-induced desensitization to NE is due to α1 adrenoreceptor internalization, we performed an imaging experiment using the overexpression of a GFP-tagged α1b adrenoreceptor (ARα1b) in an embryonic hypothalamic cell line, MHYPOE N42 (N42) cells, which express both CRH and membrane-associated GRs (Belsham et al., 2004; Rainville et al., 2019; Weiss et al., 2019). A 20-min treatment of the cells with corticosterone (2 μM) had no effect on the membrane localization of ARα1b receptors in the N42 cells (Figures 4A and 4B), which suggests that corticosterone alone does not induce α1 adrenoreceptor internalization. We next tested whether corticosterone facilitates NE-dependent ARα1b internalization. Norepinephrine alone (1 μM) caused a decrease in the membrane localization of the ARα1b-GFP, indicating internalization of membrane α1b receptors (Figures 4A and 4B). Co-application of NE and corticosterone (2 μM) caused a ~2-fold decrease (ANOVA, F = 59.66, p < 0.0001, Tukey’s post-hoc, NE vs. NE + Cort, p < 0.001) in the ARα1b membrane localization compared with NE alone (Figures 4A and 4B). These data indicate that corticosterone does not directly induce α1 adrenoreceptor internalization but facilitates the internalization of the receptors induced by NE binding.
Figure 4. Corticosterone facilitates ligand-mediated α1b adrenoreceptor internalization.
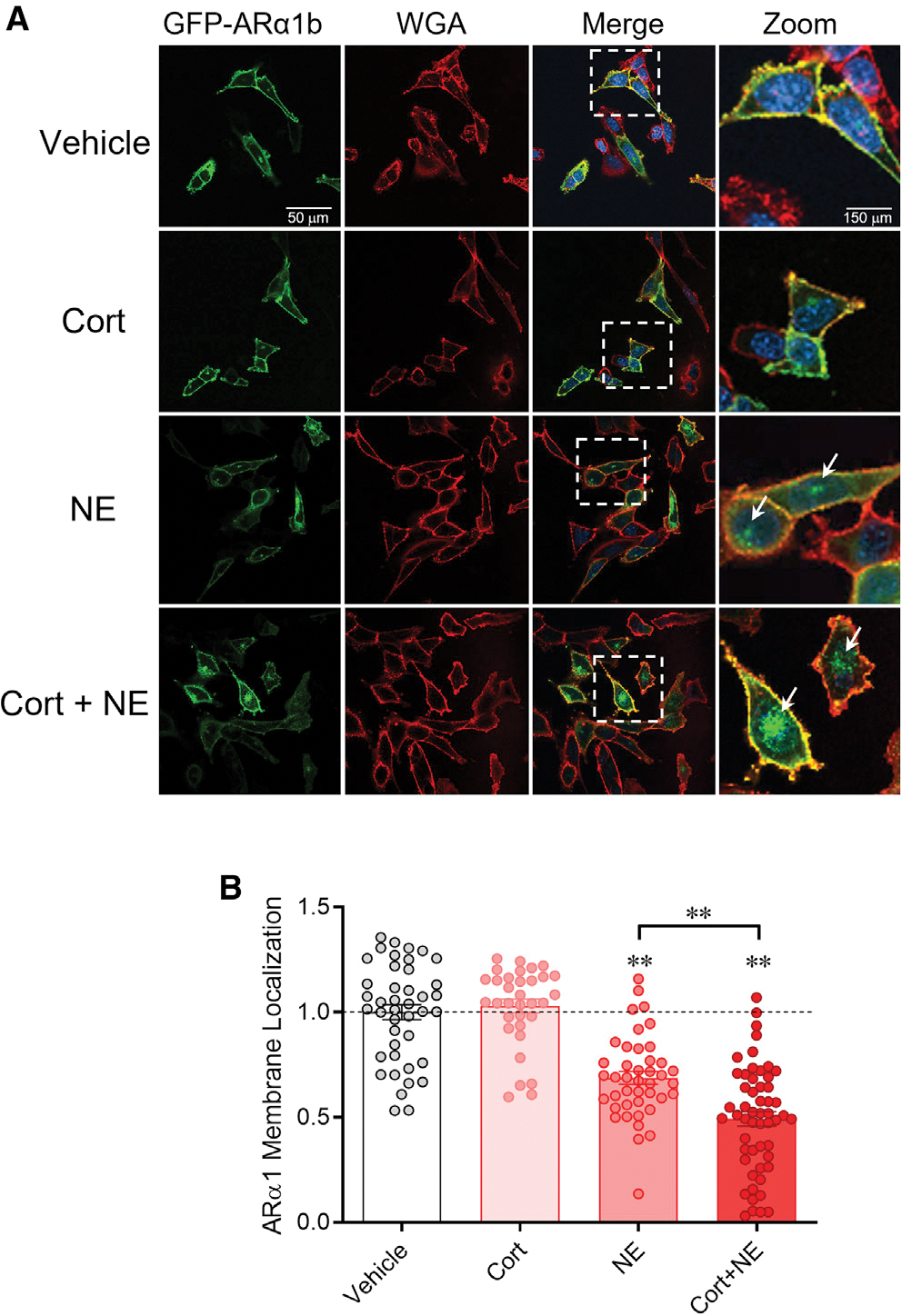
(A) Serum-stripped N42 hypothalamic cells were transiently transfected with ARα1b-eGFP (green) and co-stained with the membrane marker wheatgerm agglutinin (WGA-594, red) and the chromatin marker DAPI (blue). Vehicle: Expression of ARα1b-eGFP is localized largely in the plasma membrane under baseline conditions. Cort: ARα1b-eGFP localization is maintained in the membrane following corticosterone treatment. NE: NE treatment increased ARα1b-eGFP localization in the cells, seen as eGFP staining in intracellular “hotspots” (white arrows) and a decrease in membrane co-localization with WGA-594. Cort + NE: Co-application of corticosterone with NE increased the ARα1b-eGFP concentrated in intracellular hotspots (white arrows) and decreased ARα1b-eGFP and WGA-594 membrane co-localization.
(B) Summary of the relative ARα1b membrane localization in N42 cells in vehicle and after incubation in corticosterone alone (Cort), NE alone (NE), and corticosterone and NE (Cort + NE), normalized to vehicle. Co-localization of ARα1b-eGFP with WGA was calculated as a Pearson’s coefficient comparing pixel intensity normalized to vehicle. Data are represented as mean ± SEM. **p < 0.01.
Glucocorticoid suppression of the NE excitatory drive is specific to somatic stress activation
Previous studies have reported that noradrenergic afferents to the PVN mediate the somatic stress-induced excitation, but not psychological stress-induced excitation, of the PVN CRH neurons in male rats (Schiltz and Sawchenko, 2007). Since immune activation is considered a stressor that is mainly systemic (i.e., somatic), without a strong psychological component (Reyes et al., 2003), we used an endotoxin immune challenge as a somatic stressor and compared it to a psychological stressor, predator odor, to test whether a previous “priming” stress exposure (restraint) would have a differential effect on HPA activation by a subsequent “test” somatic versus psychological stressor. The same timeline was used for all subjects in which the priming stressor, 20-min restraint, was administered between 9:00 a.m. and 10:00 a.m. and the subsequent test stressor was applied between 12:00 p.m. and 1:00 p.m. (Figure 5A). Mice that received no prior priming restraint stress and only the test somatic immune stress (lipopolysaccharide [LPS] injection [0100 μg/kg i.p.]) or psychological stress (predator odor [30 min]) showed an increase in corticosterone in blood collected by submandibular vein puncture compared with non-stressed controls (LPS: 189% ± 22.7% compared with i.p. saline injection (n = 10); psychological stress: 123% ± 22.5% compared with no odor (n = 11), ANOVA, F = 20.31, Tukey’s post-hoc: p < 0.0001) (Figure 5B). Prior exposure to the priming restraint stress caused a significant reduction in the LPS-induced increase in corticosterone (−40.7% ± 9.4%, p < 0.001, n = 11) but had no effect on the corticosterone response to predator odor stress (n = 11) (Figure 5B). Non-stressed, handled mice (n = 11) were used to show baseline corticosterone levels (Figure 5B).
Figure 5. Prior stress exposure differentially suppresses the HPA response in a stress modality-specific manner via desensitization to noradrenergic activation.
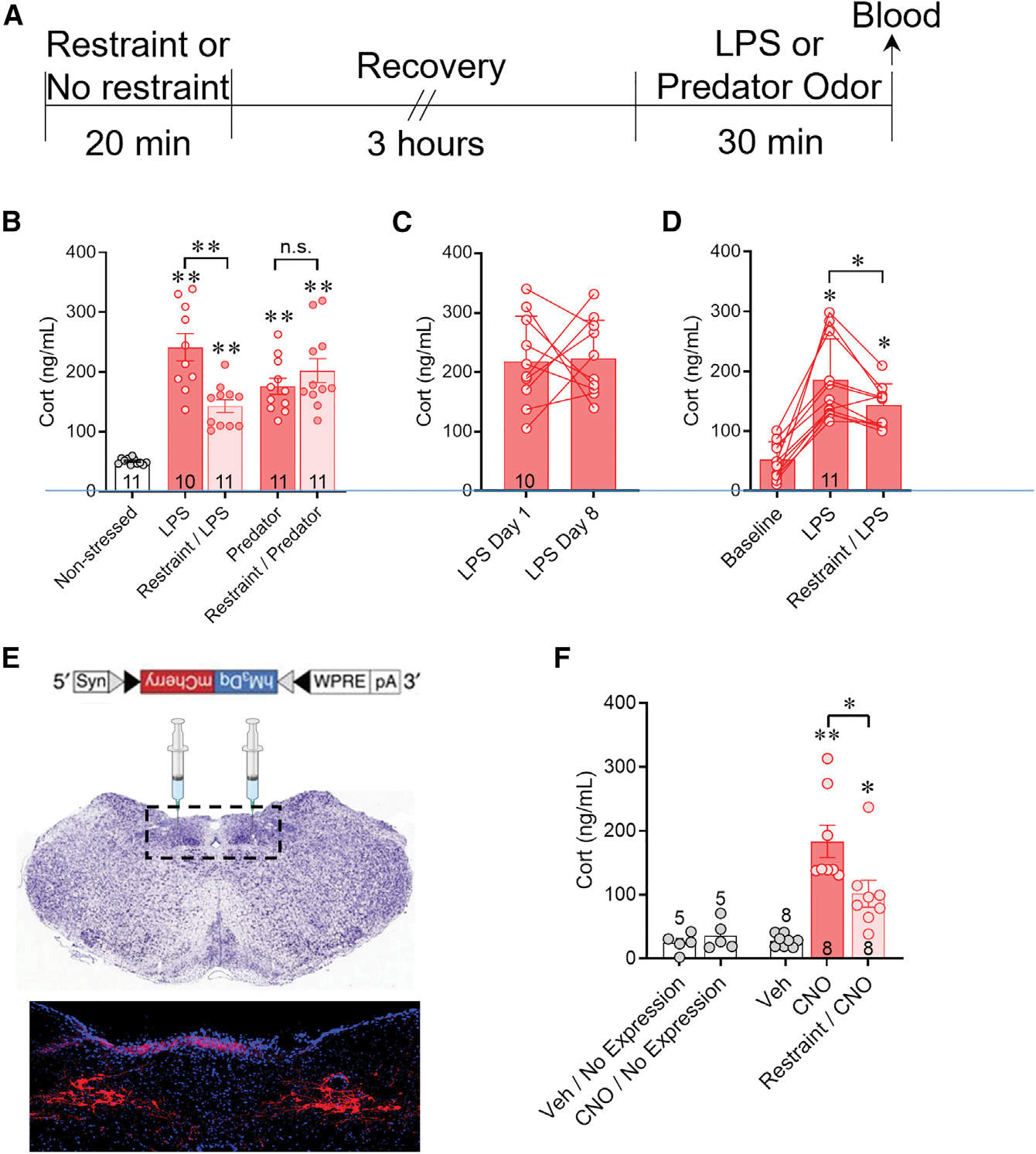
(A) Experimental protocol used for sequential stress exposures shown in (B). An initial restraint stress (Restraint) or home cage control (No restraint) was followed 3 h later by i.p. LPS injection or predator odor exposure for 30 min and plasma corticosterone was measured by ELISA.
(B) The LPS-induced increase in corticosterone (LPS) was suppressed when preceded by a restraint stress (Restraint/LPS). The predator odorinduced increase in corticosterone (Predator) was unaffected by prior restraint stress (Restraint/Predator). Handled mice were used to measure baseline corticosterone (Non-stressed).
(C) The mean level of plasma corticosterone in LPS-treated mice (LPS Day 8) was unchanged in a within-subject comparison with the mean corticosterone level evoked by a previous LPS treatment in the same mice 7 days earlier (LPS Day 1).
(D) Plasma corticosterone levels measured using a within-subject analysis after i.p. saline injection (Baseline), i.p. LPS injection (LPS) alone, and LPS injection 3 h after a 30-min restraint (restraint/LPS). The corticosterone response to LPS injection following restraint was significantly smaller than that following LPS injection without prior restraint.
(E) Cre-dependent Gq-coupled DREADD ((hM3Dq)/mCherry)-expressing AAV8 injected bilaterally into the NTS of a TH-Cre mouse expressed mCherry in neurons of the A2 noradrenergic region of the solitary tract nucleus after 3 weeks.
(F) In mice in which Gq-DREADD expression was detected in the NTS in post hoc histological controls, CNO injection stimulated an HPA corticosterone response (CNO) compared to vehicle injection (Veh), and this was suppressed by prior restraint stress (Restraint/CNO). Vehicle injections and CNO injections in mice in which no virus was detected in post hoc brain sections (Veh/No Expression and CNO/No Expression) failed to elicit an increase in corticosterone. Data are represented as mean ± SEM; numerals in bar graphs represent the numbers of animals tested. **p < 0.01, *p < 0.05, n.s., not significant, Tukey’s multiple comparisons following one-way ANOVA.
See also Figure S3.
We also tested for an acute stress-induced desensitization of the HPA axis response to LPS injection using a within-subject, repeated-measures design to eliminate individual, litter, or cohort variability. First, to test for an order effect of LPS exposure, mice were subjected to LPS injections (100 μg/kg i.p.) twice in succession with a 7-day interval between injections. There was considerable variability in the relative corticosterone levels collected by submandibular vein puncture from the first to the second LPS injections, but no significant difference in the mean serum corticosterone levels between the two responses (paired t test, t(9) = 0.1379, p = 0.89, n = 10) (Figure 5C). Because of the high variability in the serial LPS-induced corticosterone responses, we reran the same experiment in a second cohort of mice and found similar equal numbers of increased and decreased corticosterone responses within subjects between the first and second LPS injections, which also resulted in no difference in the population means (n = 9) (Figure S3). While this experiment did not rule out the possibility of an interaction between the two LPS injections in individual subjects, it indicated that there was no overall order effect on the population response and that there was equal variability of the response in both directions, which justified the within-subject design of the following experiment.
To test the effect of acute stress on subsequent LPS-induced corticosterone release within subjects, mice were restrained for 20 min (priming stress), allowed to recover for 3 h, then administered a test injection of LPS (100 μg/kg, i.p.), and blood was collected by submandibular vein puncture 30 min later for corticosterone assay. Seven days later, the same mice were reinjected with LPS (100 μg/kg, i.p.) without prior priming restraint stress and submandibular blood was collected 30 min later. Finally, after another 7 days the mice were i.p. injected with saline without the priming restraint stress, and submandibular blood was again collected 30 min later. While both LPS injections elicited a corticosterone response compared to saline injection, significantly lower corticosterone levels were seen in response to LPS injection following a priming restraint stress than in the same mice injected with LPS without prior restraint (−42.39% ± 13.23% repeated-measures ANOVA, F = 34.54, Tukey’s post-hoc: p < 0.05, n = 11) (Figure 5D).
To test for the effect of prior stress exposure on immune activation, we also assayed circulating cytokines in groups of mice injected with LPS (100 μg/kg, i.p.) with and without priming restraint stress exposure 3 h prior to LPS injection. Using an ELISA immunoassay, we measured levels of TNFα, IL-1β, IFNγ, IL-6, IGF-1, MCP-1, and TGFβ in the blood collected by submandibular vein puncture 45 min after LPS or saline injection. Four groups were compared using a one-way ANOVA: i.p. saline-injected without prior stress (no-restraint/saline), LPS-injected without prior stress (no-restraint/LPS), saline-injected with prior stress (restraint/saline), and LPS-injected with prior stress (restraint/LPS). Of the seven cytokines analyzed, prior restraint stress blunted the LPS-induced increases in circulating IL-6 and TGFβ (Figure S4).
The LPS-induced stress response activates noradrenergic inputs from the NTS (Schiltz and Sawchenko, 2007). We next used a chemogenetic strategy to test whether the acute stress-induced blunting of the HPA axis response to LPS is due to desensitization to NTS noradrenergic input. We expressed a Gq-coupled, excitatory designer receptor exclusively activated by designer drug (Gq-DREADD) in A2 noradrenergic neurons by injecting Cre-dependent AAV8-hSyn-DIO-hM3Dq-mCherry into the NTS of male tyrosine hydroxylase (TH)-Cre mice (Figure 5E). Following a 2- to 3-week recovery, the mice underwent 30-min restraint, followed 3 h later by systemic clozapine N-oxide (CNO) injection (5 mg/kg, i.p.) to activate the Gq-DREADD, and then trunk blood was collected 30 min later at sacrifice. The mice subjected to a prior priming restraint stress responded to CNO with significantly lower levels of corticosterone (−44.5% ± 12.9%, repeated measures ANOVA, F = 17.10, Tukey’s post-hoc: p < 0.05, n = 8) compared with the stress-naive mice (n = 8) (Figure 5F). The priming stress-induced suppression of the HPA response to NTS noradrenergic neuron activation was comparable to the stress-induced suppression of the HPA response to systemic LPS injection. These data suggest that acute stress causes specific desensitization of the NE-induced HPA activation by somatic stress but not by psychological stress.
DISCUSSION
Norepinephrine causes a robust activation of glutamatergic synaptic inputs and a less robust activation of GABAergic synaptic inputs to PVN CRH neurons via a postsynaptic α1 adrenoreceptor-dependent retrograde neuronal-glial signaling mechanism, resulting in a net excitation of the CRH neurons (Chen et al., 2019). Here, we show that acute stress-induced glucocorticoid feedback rapidly desensitizes CRH neurons in male mice to NE activation of presynaptic glutamate and GABA circuits by regulating ligand-dependent α1 adrenoreceptor trafficking, and that this specifically suppresses the HPA response to somatic but not psychological stress exposure. Our model of rapid glucocorticoid-induced desensitization of α1 adrenoreceptors via receptor internalization is supported by the following findings: (1) the NE response in CRH neurons is restored by inhibiting glucocorticoid synthesis during acute stress in vivo; (2) the NE response is preserved by inhibiting endocytosis during corticosterone exposure in vitro; and (3) corticosterone facilitates the ligand-induced internalization of the α1b receptor. That corticosterone by itself did not induce changes in membrane receptor trafficking in cultured cells and that the NE desensitization by corticosterone was blocked by blocking α1 adrenoreceptors during corticosterone exposure in brain slices suggest that corticosterone per se does not induce α1 receptor internalization, but rather that it facilitates ligand-induced α1 receptor internalization or regulates α1 receptor trafficking following internalization. The CRH neurons were almost entirely desensitized to bath-applied NE within 5 min of corticosterone preexposure, which suggests that the membrane α1 adrenoreceptor undergoes nearly complete internalization and recycling within 5 min, and that corticosterone acts rapidly to prevent α1 receptor trafficking back to the membrane once it has been internalized.
The rapid regulation of α1 adrenoreceptor trafficking by corticosterone suggests a non-genomic mechanism of action of the steroid, which was supported by the ineffectiveness of the transcription blocker actinomycin D to reverse the corticosterone effect. Nevertheless, the effect of the steroid was lost in CRH neurons from GR knockout mice, which suggests that it is dependent on the nuclear GR. We have reported a rapid glucocorticoid-induced endocannabinoid suppression of excitatory synapses in PVN CRH neurons and magnocellular neurons that is also dependent on the GR, but does not require gene transcription and is mediated by an intracellular G protein-dependent signaling cascade (Di et al., 2003; Harris et al., 2019; Malcher-Lopes et al., 2006; Nahar et al., 2015). The rapid glucocorticoid suppression of excitatory synaptic inputs was also observed here and was also reversed in CRH neurons from GR knockout mice. It is reasonable to envision the membrane transduction mechanism responsible for the rapid glucocorticoid regulation of α1 receptor trafficking in CRH neurons as the same as or as a branch of the membrane GR signaling mechanism that leads to endocannabinoid synthesis at glutamate synapses (Harris et al., 2019), nitric oxide synthesis at GABA synapses (Di et al., 2009), and ligand-independent nuclear translocation of the GR (Rainville et al., 2019), although further experiments are required to test this (Harrison and Tasker, 2022). An unexpected finding here was that the corticosterone suppression of the NE excitatory effect was reversed by inhibiting translation with cycloheximide (Figure S2), since this suggests that a component of the glucocorticoid regulation of α1 receptor trafficking may depend on constitutive protein synthesis.
Corticosterone has been shown to facilitate glutamate neurotransmission by regulating alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking in pyramidal neurons of the hippocampus and prefrontal cortex (Conboy and Sandi, 2010; Groc et al., 2008; Yuen et al., 2011). Serum- and glucocorticoid-inducible kinase (SGK), guanosine nucleotide dissociation inhibitor (GSI), and Rab family small-molecule G proteins were reported to mediate the corticosterone regulation of AMPA receptor trafficking in the prefrontal cortex (Liu et al., 2010; Yuen et al., 2011). In hippocampal cell cultures, corticosterone stimulated the tyrosine kinases Pyk2 and Src to cause a rapid increase in PSD95 clustering and N-methyl-D-aspartate receptor surface expression by activating a membrane receptor and downstream signaling (Yang et al., 2013). It is reasonable, therefore, to envision that corticosterone may also regulate α1-adrenoceptor trafficking by triggering intracellular kinase cascades and/or destabilizing scaffolding proteins, although this remains to be determined.
Several lines of evidence support different stress modalities signaling through distinct neural circuits to the PVN CRH neurons. Psychosocial stressors and immune stressors, for example, have additive effects on plasma corticosterone levels (Gibb et al., 2008). The innate response to predator odor activates CRH neurons in the PVN through the amygdalo-piriform transition area and cortical amygdala, which does not require activation of brainstem noradrenergic neurons (Kondoh et al., 2016; Masini et al., 2009; Root et al., 2014). The innate response to a predator usually happens within seconds (Daviu et al., 2020; De Franceschi et al., 2016; Yilmaz and Meister, 2013), presumably because instinctive and immediate reaction to danger is critical to survival. How systemic LPS activates CRH neurons is less well characterized, although it has been shown to be dependent on p44/42 mitogen-activated protein kinase activation (Singru et al., 2008). LPS increases norepinephrine release in the PVN dose dependently both in vivo and ex vivo, suggesting that LPS can act directly on the hypothalamus to increase NE release, which subsequently activates CRH neurons (Francis et al., 2000, 2001). Therefore, previous stress exposure that desensitizes the α1 adrenoceptors would be expected to make mice less responsive to LPS but have no impact on the innate response to predator odor, which is supported by our findings.
To our knowledge, this is the first direct demonstration of a stress modality-specific feedback inhibition of the HPA axis, which takes somatic stress signaling to the HPA axis largely offline and leaves psychological stress activation of the HPA axis intact. The somatic stress activation of the HPA axis is not entirely abolished, with about 50% of the response remaining following stress inactivation. We reported previously the possible co-release of glutamate with norepinephrine from NTS-derived noradrenergic afferents to the PVN CRH neurons (Chen et al., 2019). The residual, stress-insensitive component of the response is likely to be mediated by these ascending monosynaptic glutamatergic inputs.
The question remains as to the functional significance of the selective suppression of the somatic stress response and sparing of the psychological stress response by prior stress exposure. Ascending noradrenergic afferents carry signals generated by somatic stress stimuli, such as immune system, glycemic, and blood pressure signals, whereas descending limbic afferents to the HPA axis carry emotional and cognitive signals largely from the amygdala, hippocampus, and prefrontal cortex. The ascending somatic afferents, therefore, signal long-lasting physiological/internal states of the organism. It is likely that desensitization of the noradrenergic afferents prevents the tonic activation of the HPA axis by protracted somatic stress signals, protecting the organism from the damaging effects of sustained glucocorticoid exposure, while maintaining the phasic activation of the HPA axis by descending limbic signals. The organism is thus able to continue to mount normal transient HPA responses to anticipated or programmed threats to its survival while limiting its exposure to sustained elevated glucocorticoids induced by somatic homeostatic perturbations. Note that systemic corticosterone injection failed to suppress the HPA response to the somatic stress induced by a salt load (Thrivikraman et al., 2000). Further study is required, therefore, to determine the physiological significance of the stress modality-specific regulation of the HPA axis in the context of susceptibility/resilience to somatic stressors. This is especially critical in light of the fact that prior stress exposure can sensitize, rather than desensitize, the HPA axis to a heterotypic stressor (Schmidt et al., 1995). It will also be important to ascertain whether the dependence of the sensitivity of the HPA axis on the prior stress experience of the organism generalizes to other somatic stressors, such as glycemic, hydromineral, and pain stimuli.
Limitations of the study
Our study was limited to CRH neurons from male mice. PVN CRH neurons recorded in slices from female mice did not respond to NE (100 μM) with an increase in excitatory synaptic inputs (unpublished observation). Therefore, future studies need to be performed to characterize the noradrenergic modulation of PVN CRH neurons and to determine whether glucocorticoid feedback alters noradrenergic regulation of the CRH neurons in females. Additionally, some experiments could have been powered more by increasing the cell and/or animal sample size, although the effects were robust enough that we do not think this affected the interpretation of the results.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey Tasker (tasker@tulane.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The published article includes all data generated or analyzed during this study.
No code was used or generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
We used 6–10-week-old male mice for these experiments; the mice were genotyped at 2–3 weeks using the primer sets provided in Table S1. All animals and procedures were approved by the Tulane Institutional Animal Care and Use Committee (IACUC) according to National Institutes of Health (NIH) guidelines. Mice were housed in an AALAAC-accredited animal facility on a 12:12 light/dark cycle (lights on at 7:00 AM) under controlled temperature and humidity and received food and water ad libitum.
Three mouse models were used in this study, a CRH-eGFP mouse, a triple transgenic sim1-cre::loxP-GR::CRH-eGFP mouse, and a tryrosine hydroxylase (TH)-cre mouse that expresses cre recombinase in catecholaminergic neurons. CRH-eGFP mice were obtained from the Mutant Mouse Resource and Research Center (MMRRC) (stock: Tg(Crh-eGFP)HS57Gsat/Mm, RRID:MMRRC_017058-UCD) at the University of California at Davis; they were used to visualize individual CRH neurons as targets for patch-clamp recordings. CRH-eGFP mice were bred on a C57BL/6 background from BAC transgenic mice expressing enhanced green fluorescent protein (eGFP) controlled by the CRH promoter (CRH-eGFP mice). A previous study reported that a different strain of the CRH-eGFP BAC transgenic mouse from the MMRRC expresses eGFP ectopically (Chen et al., 2015). We validated the eGFP expression in PVN CRH neurons in our mouse line by immunofluorescence in a previous study using rabbit anti-CRH (1:2000, T4037, Peninsula Laboratories (Chen et al., 2019) and reconfirmed the eGFP expression in CRH neurons in the current study using a different primary antibody to CRH, the X antibody (Figure S4). The immunofluorescence procedure performed for this purpose is described below.
Mice with a conditional knockout of GR in the PVN were generated by crossing a sim1-cre BAC transgenic mouse on a C57BL/6J background (Sim1-Cre, kindly provided by Dr Bradford Lowell, Beth Israel Deaconess Medical Center, Boston, MA) (Balthasar et al., 2005) with a loxP-GR transgenic mouse (kindly provided by Dr. Louis Muglia, University of Cincinnati Children’s Hospital, Cincinnati, OH) (Laryea et al., 2013; Nahar et al., 2015; Solomon et al., 2015). Sim1-cre:loxP-GR (GRKO) mice show a deletion of GR in the PVN (Nahar et al., 2015; Solomon et al., 2015). We crossed the sim1-cre/loxP-GR mouse with the CRH-eGFP mouse to produce a sim1-cre::loxP-GR::CRH-eGFP triple transgenic mouse in order to visualize CRH neurons in our brain slices.
Tyrosine hydroxylase (TH)-Cre 1 mice (B6.Cg-7630403G23RikTg(Th-cre)1Tmd/J) were purchased from Jackson Laboratory (Strain # 008601) and bred in-house on a C57BL/6J background. They were genotyped at 2–3 weeks of age using the supplier’s suggested primer set (see Table S1).
METHOD DETAILS
Acute stress paradigms
Restraint stress
Mice were immobilized for 20–30 min between 9 AM and 10 AM by placing them in a transparent, pliable tapered plastic cone open at the tip to allow for breathing (DecapiCones, Braintree Scientific). Immediately after the 30 min restraint stress, mice were decapitated in the plastic cone without anesthetic and brain slices were prepared. For experiments to block corticosteroids synthesis, metyrapone was administered (100 mg/kg, IP) 30 min prior to the restraint stress.
Predator odor stress
Mice were placed in a clean cage between 9 AM and 10 AM and a sponge soaked with bobcat (Lynx rufus) urine (Maine Outdoor Solutions) was placed in a dish in the cage’s water bottle holder. Mice had no direct access to or contact with the sponge. Following a 30-min exposure to the bobcat urine, they were then placed immediately into a decapicone and decapitated without anesthetic for brain slice preparation.
Immune stress/LPS
All animals receiving LPS injections were habituated to injection or handling for 7–10 days before beginning the experiments. A single lot of lipopolysaccharides (LPS) from Escherichia coli O26:B6 (≥10,000 EU/mg, Sigma-Aldrich) was used for all immune stress experiments. LPS powder was dissolved in sterile H2O at 5 mg/mL and frozen until use. On the day of injection, the stock solution was diluted in sterile saline to yield the appropriate working concentration. LPS was administered via I.P. injection at a dose of 100 μg/kg in an injection volume of 10 mL/kg.
Serial stressors
Mice were first immobilized for 30 min between 9 AM and 10 AM and then returned to their home cage for 3 hours of recovery. They were then exposed to either predator odor for 30 min (see above) or they were injected I.P. with 100 μg/kg LPS 45 min prior to blood collection for corticosterone measurement. Mice not undergoing restraint stress were exposed to predator odor or injected with LPS between 12:00 PM and 1:00 PM to align with the animals that experienced restraint stress. Baseline corticosterone levels were taken from uninjected, naïve mice between 12:00 PM and 1:00 PM for the between-subjects experiments. In the repeated-measures experiments, the same general timeline was used. A control group received only LPS injection between 12:00 PM and 1:00 PM on two separate days, one week apart. The experimental group underwent restraint stress the first week in the morning between 9 AM and 10 AM, followed by LPS injection the same day between 12 and 1 PM, then received an LPS injection I.P. between 12 PM and 1 PM seven days later, and finally received a saline injection I.P. between 12 PM and 1 PM seven days after that.
Brain slice preparation and electrophysiology
Acute brain slice preparation
Electrophysiological experiments were conducted in acutely prepared hypothalamic slices from 6–9-week-old male CRH-eGFP or Sim1-cre::loxP-GR::CRH-eGFP mice. Mice were gently removed from their home cage to a transfer cage and transported to an adjacent room, where they were immobilized in a DecapiCone (Braintree Scientific) and decapitated without anesthesia. They were decapitated rapidly, within less than 2 min of removal from their home cage, to avoid stress-elevated circulating corticosterone levels reaching the brain. Mice were killed without anesthesia because anesthetics activate the HPA axis and increase circulating ACTH and corticosterone (Vahl et al., 2005). Blood corticosterone levels remain low in our hands for >3 min from the start of handling.
Following decapitation, the brain was quickly removed from the skull and cooled in oxygenated, ice-cold artificial cerebrospinal fluid (aCSF) containing (in mM): 140 NaCl, 3 KCl, 1.3 MgSO4, 11 Glucose, 5 HEPES, 1.4 NaH2PO4, 3.25 NaOH, and 2.4 CaCl2, with a pH of 7.2–7.4 and an osmolarity of 290–300 mOsm. The base of the brain was then blocked and the caudal face of the block was glued to the chuck of a vibratome (Leica or Vibratome). Two or three 300-mm coronal slices containing the PVN were sectioned in cooled aCSF and the slices were bisected down the midline and transferred to an incubation chamber. They were maintained in oxygenated aCSF at room temperature for at least 1 h prior to beginning recording sessions to allow for recovery.
Whole-cell and loose-seal patch clamp recordings
Hemi-slices were transferred one-at-a-time from the incubation chamber to the recording chamber, where they were submerged and perfused with aCSF at a rate of ~2 mL/min. eGFP-expressing CRH neurons in the PVN were located under fluorescence illumination and subsequently targeted for whole-cell patch clamp recordings using infrared light and differential interference contrast optics at room temperature on a fixed-stage, upright microscope (Olympus BXW51) equipped with a long working distance, water-immersion 40x objective. Patch pipettes with a resistance of 3–6 MΩ were fabricated from borosilicate glass (ID 1.2 mm, OD 1.65 mm; Garner Glass) on a horizontal puller (P-97, Sutter Instr.) and filled with an internal patch solution containing (in mM): 120 potassium gluconate, 10 KCl, 1 NaCl, 1 MgCl2, 0.1 CaCl2, 5.5 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP; the pH was adjusted to 7.3 with KOH and the osmolarity was adjusted to 300 mOsm with D-sorbitol. The GABAA receptor antagonist picrotoxin (PTX, 50 μM) was applied via bath perfusion to isolate sEPSCs; glutamate receptor antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX, 15 μM) and DL-2-amino-5-phosphonopentanoic acid (AP5, 50 μM) were applied in the bath perfusion to isolate sIPSCs.
For loose-seal, cell-attached patch clamp recordings, glass pipettes with a resistance of 1–2 MU filled with aCSF were used. Cells were recorded at resting potential with minimal current transfer and no exchange of intracellular ions (Haam et al., 2012, 2014). An extracellular aCSF with high [K+] (10 mM) was used to stimulate spontaneous spiking activity (in mM): 133 NaCl, 10 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 2.4 CaCl2, 11 glucose, and 5 HEPES; pH was adjusted to 7.2–7.4 with NaOH.
Only one cell was recorded from in each slice. Multiple cells from the same mouse were sometimes recorded, but they were each recorded in different slices. A minimum of three mice were used for all experiments. Cell numbers are designated as ‘n’ and mouse numbers as ‘N’.
Cell imaging
Immortalized embryonic mouse hypothalamic cells that express CRH and glucocorticoid receptors (mHypoE-N42, Cedar Lane, Cellutions Biosystems) (Dalvi et al., 2011; Rainville et al., 2019) were transiently transfected with pEGFP-ARα1b (generous gift from Dr. Chris Hague, University of Washington, Seattle, WA) using the Neon® Transfection System (Life Technologies) with 2 pulses, 10 ms, 1600 mV. Cells were placed on coverslips and the media was replaced with media containing charcoal-stripped BSA to eliminate basal steroid content. The cells were treated with 1 μM NE, 2 μM corticosterone:HBC, or both (Sigma Aldrich) for 20 min. Cells were then fixed immediately with 4% paraformaldehyde before staining for 30 s with WGA Alexa Fluor 594 (ThermoFisher) to produce a plasma membrane-specific label. Image analysis was performed on a Nikon A1+ laser confocal microscope (Nikon Instruments, Inc) with a 28 μm pinhole to capture an intracellular focal plane. The average Pearson’s coefficient was calculated in each transfected cell using Cell Profiler (Broad Institute, Cambridge, MA).
Stereotaxic injection
TH-cre mice (~ 6 wks old) were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (8 mg/kg) and placed in a stereotaxic frame. Each mouse received bilateral stereotaxic injections of excitatory DREADD AAVs (AAV8-hSyn-DIO-hM3D(Gq)-mCherry, Addgene, 4 × 1012 vg/mL) in the NTS (400 nL at 100 nL/min) (coordinates: AP: 7.6 mm caudal to bregma, ML: ± 0.5 mm from the midline, DV: −4.6 mm from the brain surface) using a Hamilton syringe with 10 μL in the beveled tip connected to a microsyringe pump (UMP2; WPI) and controller (Micro4; WPI). The injection needle was maintained in place for 10 min following injections to minimize virus spread up the needle track. Mice were allowed to recover for 2–3 weeks prior to assays.
Plasma corticosterone assay
To measure serum corticosterone concentration, blood (~200 μL) was collected by decapitation (for terminal corticosterone measurement) or by submandibular vein puncture (for serial corticosterone measurement). The blood was allowed to coagulate at room temperature for 90 min and was centrifuged at 2000 × g for 15 min. Serum was collected and samples were stored at −20°C until they were shipped to the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core (terminal experiments), where corticosterone levels were measured by 125I corticosterone radioimmunoassay, or until they were assayed in-house using a corticosterone ELISA (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer’s instructions (for serial measurement experiments).
Immunohistochemistry
To confirm that eGFP was expressed in CRH neurons in the CRH-eGFP transgenic mice, immunohistochemistry was performed on brains of four 5-week-old CRH-eGFP mice. Each mouse received an intracerebroventricular stereotaxic injection of colchicine (20 μg in 2 μL saline, Cayman Chemical Company) into the lateral ventricle under isoflurane anesthesia 24 h before sacrifice. Mice were then anesthetized with ketamine/xylazine and transcardially perfused with ice-cold phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Brains were dissected from the cranium and post-fixed with 4% PFA at 4°C overnight and then submerged in 30% sucrose/4% PFA/PBS. Forty μm-thick coronal sections of the hypothalamus containing the PVN were cut on a freezing microtome. The sections were pre-incubated in a blocking solution containing 1.5% BSA and 0.4% Triton X-100 in PBS for 1 h and then incubated with rabbit anti-human/rat CRF primary antibody (1:20,000, #PBL rC68; RRID:AB_2650435, Salk Institute for Biological Studies) at 4°C overnight followed by incubation in a secondary antibody Alexa Fluor 647 donkey anti-rabbit (1:500, Abcam) at room temperature for 1 h. Sections were mounted on glass slides, coverslipped, and imaged on a laser scanning confocal microscope (Nikon A1).
Serum cytokine assay
Using a mouse ER stress colorimetric ELISA assay (Signosis, Cat # EA-1131), we conducted quantitative profiling and measures of the following cytokines: TNFα, IL-1β, IFNγ, IL-6, IGF-1, MCP-1, and TGFβ. Forty-five min following injections of LPS or saline, blood was collected from the submandibular vein. After clotting of blood samples for 90 min at room temperature, serum was isolated by centrifugation at 4000 rpm for 20 min followed by removal of supernatant. The ELISA was run in duplicate on samples diluted 1:12. Absorbance was read at 450 nm on a SpectraMax iD3 microplate reader (Molecular Devices) and background values were subtracted from sample values.
Drugs used
The following drugs were kept at −20°C as stock solutions and dissolved in aCSF to their final concentrations on the day of experiments: L-(−)-norepinephrine (+)-bitartrate salt monohydrate (NE, 1 μM, 100 μM, Sigma-Aldrich), picrotoxin (PTX, 50 μM, Tocris), corticosterone (2 μM, Tocris), 3-hydroxynaphthalene-2-carboxylic acid (3,4-dihydroxybenzylidene) hydrazide (Dynasore, 80 μM, Tocris). 2-Methyl-1,2-di-3-pyridinyl-1-propa-none (metyrapone, Tocris) was dissolved in sterile saline, and injected intraperitoneally at a dose of 100 mg/kg body weight. Clozapine N-oxide (CNO, Sigma-Aldrich) was made fresh daily by dissolving in DMSO (100 mg/50 μL) and then diluting in saline to a working concentration of 5 mg/mL.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the mean ± standard error of the mean. Postsynaptic currents were selected and analyzed for changes in frequency, amplitude, and decay time with Minianalysis 6.0 (Synaptosoft Inc.). Baselines were calculated from the 3 min of recording prior to drug application. Statistical significance was determined with the two-tailed, paired Student’s t-test for within-cell drug effects and the two-tailed, unpaired Student’s t-test for between-group drug effects (Prism 7 and Prism 8, Graphpad, and SigmaPlot 11.0, Systat Software, Inc.). A one-way analysis of variance (ANOVA) with post-hoc Tukey’s tests were used for comparing corticosterone levels in vivo with multiple group comparisons. Animal numbers are designated as the “N” and cell numbers as the “n”. A minimum of 3 mice were used in all experiments.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry(AAV8) | Addgene | RRID: Addgene_44361 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| L-(−)-norepinephrine (+)-bitartrate salt monohydrate (NE) | Sigma-Aldrich | A9512, CAS 108341-18-0 |
| Picrotoxin | Tocris | Cat# 1128, CAS 124-87-8 |
| Corticosterone | Tocris | Cat# 3685, CAS 50-22-6 |
| Bobcat urine | Maine Outdoor Solutions | 91412-1 |
| Lipopolysaccharides from Escherichia coli O26:B6 | Sigma-Aldrich | Cat# L2654 |
| 2-Methyl-1,2-di-3-pyridinyl-1-propa-none(metyrapone) | Tocris | Cat# 3292, CAS 54-36-4 |
| 3-hydroxynaphthalene-2-carboxylic acid (3,4-dihydroxybenzylidene) hydrazide (Dynasore) | Tocris | Cat# 2897, CAS 304448-55-3 |
| corticosterone:HBC | Sigma-Aldrich | C174, MDL MFCD00163376 |
| WGA Alexa Fluor 594 | ThermoFisher | W11262 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Corticosterone RIA | University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core | RRID:SCR_004318 |
| corticosterone ELISA | Enzo Life Sciences | ADI-900-097 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Hypothalamic cell line mHypoE-N42 | Cellutions Biosystems, Inc. | RRID:CVCL_D443 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Tg(CRH-EGFP)HS57Gsat/Mm | MMRRC | RRID:MMRRC_017058-UCD |
| Mouse: Sim1-cre | Jackson Laboratory | RRID:IMSR_JAX:006451 |
| Mouse: Loxp-GR | (Laryea et al., 2013) | N/A |
| Mouse: B6.Cg-7630403G23RikTg(Th-cre) 1Tmd/J | Jackson Lab | RRID:IMSR_JAX:008601 |
|
| ||
| Oligonucleotides | ||
|
| ||
| For genotyping primer sequences, please see Table S1 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Mini Analysis | Synaptosoft, Inc. | http://www.synaptosoft.com/MiniAnalysis/ |
| Graphpad Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| SigmaPlot 11.0 | Systat Software | https://systatsoftware.com/products/sigmaplot/ |
Highlights.
Stress-induced glucocorticoid suppresses norepinephrine activation of CRH neurons
The rapid glucocorticoid effect is mediated by α1 adrenoreceptor desensitization
The glucocorticoid effect is specific to somatic, not psychological, stress
This reveals a stress modality-specific rapid glucocorticoid feedback mechanism
ACKNOWLEDGMENTS
This study was supported by NIH grants 2R01 MH066958 and R01 MH119283, Merit Review Award 1 I01 BX005118 from the U.S. Department of Veterans Affairs Biomedical Laboratory Research, the Catherine and Hunter Pierson Chair in Neuroscience, and the Tulane Undergraduate Research in Neuroscience Program.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111509.
REFERENCES
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, et al. (2005). Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123, 493–505. 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Banihashemi L, and Rinaman L (2006). Noradrenergic inputs to the bed nucleus of the stria terminalis and paraventricular nucleus of the hypothalamus underlie hypothalamic-pituitary-adrenal axis but not hypophagic or conditioned avoidance responses to systemic yohimbine. J. Neurosci. 26, 11442–11453. 10.1523/JNEUROSCI.3561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham DD, Cai F, Cui H, Smukler SR, Salapatek AMF, and Shkreta L (2004). Generation of a phenotypic array of hypothalamic neuronal cell models to study complex neuroendocrine disorders. Endocrinology 145, 393–400. 10.1210/en.2003-0946. [DOI] [PubMed] [Google Scholar]
- Bienkowski MS, and Rinaman L (2008). Noradrenergic inputs to the paraventricular hypothalamus contribute to hypothalamic-pituitary-adrenal axis and central Fos activation in rats after acute systemic endotoxin exposure. Neuroscience 156, 1093–1102. 10.1016/j.neuroscience.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jiang Z, Fu X, Yu D, Huang H, and Tasker JG (2019). Astrocytes amplify neuronal dendritic volume transmission stimulated by norepinephrine. Cell Rep. 29, 4349–4361.e4. 10.1016/j.celrep.2019.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Molet J, Gunn BG, Ressler K, and Baram TZ (2015). Diversity of reporter expression patterns in transgenic mouse lines targeting corticotropin-releasing hormone-expressing neurons. Endocrinology 156, 4769–4780. 10.1210/en.2015-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RL, and Sawchenko PE (2002). Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. J. Neurosci. 22, 959–969. https://www.ncbi.nlm.nih.gov/pubmed/11826124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy L, and Sandi C (2010). Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology 35, 674–685. 10.1038/npp.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham ET, and Sawchenko PE (1988). Anatomical specificity of noradrenergic inputs to the paraventricular and supraoptic nuclei of the rat hypothalamus. J. Comp. Neurol. 274, 60–76. 10.1002/cne.902740107. [DOI] [PubMed] [Google Scholar]
- Dalvi PS, Nazarians-Armavil A, Tung S, and Belsham DD (2011). Immortalized neurons for the study of hypothalamic function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R1030–R1052. 10.1152/ajpregu.00649.2010. [DOI] [PubMed] [Google Scholar]
- Daviu N, Füzesi T, Rosenegger DG, Rasiah NP, Sterley TL, Peringod G, and Bains JS (2020). Paraventricular nucleus CRH neurons encode stress controllability and regulate defensive behavior selection. Nat. Neurosci. 23, 398–410. 10.1038/s41593-020-0591-0. [DOI] [PubMed] [Google Scholar]
- Day HE, Campeau S, Watson SJ Jr., and Akil H (1999). Expression of alpha(1b) adrenoceptor mRNA in corticotropin-releasing hormone-containing cells of the rat hypothalamus and its regulation by corticosterone. J. Neurosci. 19, 10098–10106. https://www.ncbi.nlm.nih.gov/pubmed/10559417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franceschi G, Vivattanasarn T, Saleem AB, and Solomon SG (2016). Vision guides selection of freeze or flight defense strategies in mice. Curr. Biol. 26, 2150–2154. 10.1016/j.cub.2016.06.006. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Karst H, and Joëls M (2008). Corticosteroid hormones in the central stress response: quick-and-slow. Front. Neuroendocrinol. 29, 268–272. 10.1016/j.yfrne.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, and Tasker JG (2003). Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J. Neurosci. 23, 4850–4857. https://www.ncbi.nlm.nih.gov/pubmed/12832507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, and Tasker JG (2005). Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology 146, 4292–4301. 10.1210/en.2005-0610. [DOI] [PubMed] [Google Scholar]
- Francis J, MohanKumar PS, and MohanKumar SM (2001). Lipopolysaccharide stimulates norepinephrine efflux from the rat hypothalamus in vitro: blockade by soluble IL-1 receptor. Neurosci. Lett. 308, 71–74. 10.1016/s0304-3940(01)01903-6. [DOI] [PubMed] [Google Scholar]
- Di S, Maxson M, Franco A, and Tasker, Jeffrey G (2009). Glucocorticoids regulate glutamate and GABA synapse-specific retrograde transmission via divergent non-genomic signaling pathways. Journal of Neuroscience 29, 393–401. 10.1523/JNEUROSCI.4546-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis J, MohanKumar SM, and MohanKumar PS (2000). Correlations of norepinephrine release in the paraventricular nucleus with plasma corticosterone and leptin after systemic lipopolysaccharide: blockade by soluble IL-1 receptor. Brain Res. 867, 180–187. 10.1016/s0006-8993(00)02311-8. [DOI] [PubMed] [Google Scholar]
- Füzesi T, Wittmann G, Liposits Z, Lechan RM, and Fekete C (2007). Contribution of noradrenergic and adrenergic cell groups of the brainstem and agouti-related protein-synthesizing neurons of the arcuate nucleus to neuropeptide-y innervation of corticotropin-releasing hormone neurons in hypothalamic paraventricular nucleus of the rat. Endocrinology 148, 5442–5450. 10.1210/en.2007-0732. [DOI] [PubMed] [Google Scholar]
- Gibb J, Hayley S, Gandhi R, Poulter MO, and Anisman H (2008). Synergistic and additive actions of a psychosocial stressor and endotoxin challenge: circulating and brain cytokines, plasma corticosterone and behavioral changes in mice. Brain Behav. Immun. 22, 573–589. 10.1016/j.bbi.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Ginsberg AB, Campeau S, Day HE, and Spencer RL (2003). Acute glucocorticoid pretreatment suppresses stress-induced hypothalamic-pituitary-adrenal axis hormone secretion and expression of corticotropin-releasing hormone hnRNA but does not affect c-fos mRNA or fos protein expression in the paraventricular nucleus of the hypothalamus. J. Neuroendocrinol. 15, 1075–1083. 10.1046/j.1365-2826.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- Groc L, Choquet D, and Chaouloff F (2008). The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat. Neurosci. 11, 868–870. 10.1038/nn.2150. [DOI] [PubMed] [Google Scholar]
- Groeneweg FL, Karst H, de Kloet ER, and Joëls M (2011). Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol. 209, 153–167. 10.1530/JOE-10-0472. [DOI] [PubMed] [Google Scholar]
- Haam J, Halmos KC, Di S, and Tasker JG (2014). Nutritional state-dependent ghrelin activation of vasopressin neurons via retrograde transneuronal-glial stimulation of excitatory GABA circuits. J. Neurosci. 34, 6201–6213. 10.1523/JNEUROSCI.3178-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haam J, Popescu IR, Morton LA, Halmos KC, Teruyama R, Ueta Y, and Tasker JG (2012). GABA is excitatory in adult vasopressinergic neuroendocrine cells. J. Neurosci. 32, 572–582. 10.1523/JNEUROSCI.3826-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C, Weiss GL, Di S, and Tasker JG (2019). Cell signaling dependence of rapid glucocorticoid-induced endocannabinoid synthesis in hypothalamic neuroendocrine cells. Neurobiol. Stress 10, 100158. 10.1016/j.ynstr.2019.100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai M, and Yamashita H (1988). Cortisol suppresses noradrenalineinduced excitatory responses of neurons in the paraventricular nucleus; an in vitro study. Neurosci. Lett. 91, 65–70. 10.1016/0304-3940(88)90250-9. [DOI] [PubMed] [Google Scholar]
- Kondoh K, Lu Z, Ye X, Olson DP, Lowell BB, and Buck LB (2016). A specific area of olfactory cortex involved in stress hormone responses to predator odours. Nature 532, 103–106. 10.1038/nature17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laryea G, Schütz G, and Muglia LJ (2013). Disrupting hypothalamic glucocorticoid receptors causes HPA axis hyperactivity and excess adiposity. Mol. Endocrinol. 27, 1655–1665. 10.1210/me.2013-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison LM, and Tasker JG (2022). Multiplexed membrane signaling by glucocorticoids. Curr. Opin. Endocr. Metab. Res. 10.1016/j.coemr.2022.100390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yuen EY, and Yan Z (2010). The stress hormone corticosterone increases synaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors via serum- and glucocorticoid-inducible kinase (SGK) regulation of the GDI-Rab4 complex. J. Biol. Chem. 285, 6101–6108. 10.1074/jbc.M109.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, and Kirchhausen T (2006). Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850. 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, and Tasker JG (2006). Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J. Neurosci. 26, 6643–6650. 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masini CV, Sasse SK, Garcia RJ, Nyhuis TJ, Day HEW, and Campeau S (2009). Disruption of neuroendocrine stress responses to acute ferret odor by medial, but not central amygdala lesions in rats. Brain Res. 1288, 79–87. 10.1016/j.brainres.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahar J, Haam J, Chen C, Jiang Z, Glatzer NR, Muglia LJ, Dohanich GP, Herman JP, and Tasker JG (2015). Rapid nongenomic glucocorticoid actions in male mouse hypothalamic neuroendocrine cells are dependent on the nuclear glucocorticoid receptor. Endocrinology 156, 2831–2842. 10.1210/en.2015-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AJ, Kirchhausen T, and Murthy VN (2006). Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc. Natl. Acad. Sci. USA 103, 17955–17960. 10.1073/pnas.0606212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund C, and Spencer RL (2011). Corticosterone pretreatment suppresses stress-induced hypothalamic-pituitary-adrenal axis activity via multiple actions that vary with time, site of action, and de novo protein synthesis. J. Endocrinol. 208, 311–322. 10.1530/JOE-10-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotsky PM (1987). Facilitation of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation after activation of catecholaminergic pathways or central norepinephrine injection. Endocrinology 121, 924–930. 10.1210/endo-121-3-924. [DOI] [PubMed] [Google Scholar]
- Rainville JR, Weiss GL, Evanson N, Herman JP, Vasudevan N, and Tasker JG (2019). Membrane-initiated nuclear trafficking of the glucocorticoid receptor in hypothalamic neurons. Steroids 142, 55–64. 10.1016/j.steroids.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes TM, Walker JR, DeCino C, Hogenesch JB, and Sawchenko PE (2003). Categorically distinct acute stressors elicit dissimilar transcriptional profiles in the paraventricular nucleus of the hypothalamus. J. Neurosci. 23, 5607–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root CM, Denny CA, Hen R, and Axel R (2014). The participation of cortical amygdala in innate, odour-driven behaviour. Nature 515, 269–273. 10.1038/nature13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, and Munck AU (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 21, 55–89. 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, and Swanson LW (1982). The organization of noradrenergic pathways from the brainstem to the paraventricular and supraoptic nuclei in the rat. Brain Res. 257, 275–325. 10.1016/0165-0173(82)90010-8. [DOI] [PubMed] [Google Scholar]
- Schiltz JC, and Sawchenko PE (2007). Specificity and generality of the involvement of catecholaminergic afferents in hypothalamic responses to immune insults. J. Comp. Neurol. 502, 455–467. 10.1002/cne.21329. [DOI] [PubMed] [Google Scholar]
- Schmidt ED, Janszen AWJW, Wouterlood FG, and Tilders FJH (1995). Interleukin-1 induced long-lasting changes in hypothalamic corticotropin-releasing hormone (CRH) -neurons and hyperresponsiveness of the hypothalamus–pituitary–adrenal-axis. Journal of Neuroscience 15, 7417–7426. 10.1523/JNEUROSCI.15-11-07417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selye H (1950). Stress and the general adaptation syndrome. Br. Med. J. 1, 1383–1392. 10.1136/bmj.1.4667.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singru Praful S., Sanchez E, Acharya R, Fekete C, and Lechan Ronald M. (2008). Mitogen-activated protein kinase contributes to lipopolysaccharide-induced activation of corticotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus. Endocrinology 149, 2283–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MB, Loftspring M, de Kloet AD, Ghosal S, Jankord R, Flak JN, Wulsin AC, Krause EG, Zhang R, Rice T, et al. (2015). Neuroendocrine function after hypothalamic depletion of glucocorticoid receptors in male and female mice. Endocrinology 156, 2843–2853. 10.1210/en.2015-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrivikraman KV, Nemeroff CB, and Plotsky PM (2000). Sensitivity to glucocorticoid-mediated fast-feedback regulation of the hypothalamic-pituitary-adrenal axis is dependent upon stressor specific neurocircuitry. Brain Res. 870, 87–101. 10.1016/s0006-8993(00)02405-7. [DOI] [PubMed] [Google Scholar]
- Vahl TP, Ulrich-Lai YM, Ostrander MM, Dolgas CM, Elfers EE, Seeley RJ, D’Alessio DA, and Herman JP (2005). Comparative analysis of ACTH and corticosterone sampling methods in rats. Am. J. Physiol. Endocrinol. Metab. 289, E823–E828. 10.1152/ajpendo.00122.2005. [DOI] [PubMed] [Google Scholar]
- Weidenfeld J, Itzik A, and Feldman S (1997). Effect of glucocorticoids on the adrenocortical axis responses to electrical stimulation of the amygdala and the ventral noradrenergic bundle. Brain Res. 754, 187–194. 10.1016/s0006-8993(97)00078-4. [DOI] [PubMed] [Google Scholar]
- Weiser MJ, Osterlund C, and Spencer RL (2011). Inhibitory effects of corticosterone in the hypothalamic paraventricular nucleus (PVN) on stress-induced adrenocorticotrophic hormone secretion and gene expression in the PVN and anterior pituitary. J. Neuroendocrinol. 23, 1231–1240. 10.1111/j.1365-2826.2011.02217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss GL, Rainville JR, Zhao Q, and Tasker JG (2019). Purity and stability of the membrane-limited glucocorticoid receptor agonist dexamethasone-BSA. Steroids 142, 2–5. 10.1016/j.steroids.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann G (2008). Regulation of hypophysiotrophic corticotrophinreleasing hormone- and thyrotrophin-releasing hormone-synthesising neurones by brainstem catecholaminergic neurones. J. Neuroendocrinol. 20, 952–960. 10.1111/j.1365-2826.2008.01748.x. [DOI] [PubMed] [Google Scholar]
- Yang JH, Li LH, Lee S, Jo IH, Lee SY, and Ryu PD (2007). Effects of adrenalectomy on the excitability of neurosecretory parvocellular neurones in the hypothalamic paraventricular nucleus. J. Neuroendocrinol. 19, 293–301. 10.1111/j.1365-2826.2007.01531.x. [DOI] [PubMed] [Google Scholar]
- Yang S, Roselli F, Patchev AV, Yu S, and Almeida OFX (2013). Non-receptor-tyrosine kinases integrate fast glucocorticoid signaling in hippocampal neurons. J. Biol. Chem. 288, 23725–23739. 10.1074/jbc.M113.470146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz M, and Meister M (2013). Rapid innate defensive responses of mice to looming visual stimuli. Curr. Biol. 23, 2011–2015. 10.1016/j.cub.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, and Yan Z (2011). Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol. Psychiatry 16, 156–170. 10.1038/mp.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during this study.
No code was used or generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.