

Abstract
Transglutaminase 2 (TG2) is a highly expressed mammalian enzyme whose biological function is unclear, although its catalytic activity in the small intestine appears necessary for celiac disease (CeD) pathogenesis. While TG2 activity is reversibly regulated by multiple allosteric mechanisms, their roles under fluctuating physiological conditions are not well understood. Here we demonstrate that extracellular TG2 activity is competitively controlled by the mutually exclusive binding of a high-affinity Ca2+ ion or the formation of a strained disulfide bond. Binding of Ca2+ at the high-affinity site does not activate TG2 per se, but it protects against oxidative enzyme deactivation while preserving the ability of Ca2+ ions to occupy weaker binding sites capable of allosteric TG2 activation. In contrast, disulfide bond formation competitively occludes the high-affinity Ca2+ site while resulting in complete TG2 inactivation. Because both outcomes are comparably favorable under typical extracellular conditions, subtle changes in the availability of redox catalysts or promoters in the extracellular matrix can dramatically alter steady-state TG2 activity. Thus, TG2 harbors a molecular “OR” gate that determines its catalytic fate upon export from cells.
Graphical Abstract
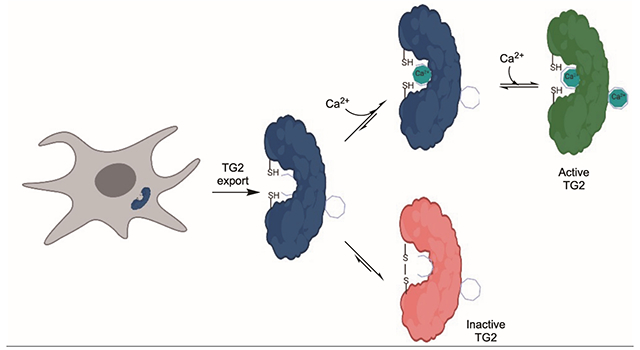
Transglutaminase 2 (TG2) is an enigmatic protein that is abundant in both the intracellular and extracellular environments of mammalian organ systems. In its namesake reaction, this multifunctional protein catalyzes Ca2+-dependent post-translational modification of selected glutamine residues in its peptide and protein substrates. In the context of celiac disease (CeD), an autoimmune disorder triggered by the ingestion of gluten proteins from wheat, barley or rye, TG2 is responsible for regio-specifically deamidating gluten peptides in the extracellular environment of the small intestine, which renders them highly immunogenic and leads to a T cell-driven inflammatory response.1
Mammalian TG2 activity is controlled by multiple allosteric regulatory mechanisms2 including Ca2+ ion binding3 and the formation of a disulfide bond.4 Given the abundance of free Ca2+ in the extracellular matrices of most organs5, one would expect TG2 to be active in this environment. However, extracellular TG2 is predominantly inactive due to the presence of a relatively strained disulfide bond between C370 and C3716 whose formation is preceded by a transient disulfide bond between C230 and C370.7 Formation of this disulfide bond is promoted by the protein cofactor ERp57 via thiol oxidation8, and its cleavage is promoted by the protein cofactor thioredoxin-1 (TRX) via disulfide reduction.9,10 However, until now, the logic of how a balance is maintained between calcium-dependent activity and oxidative inactivation of extracellular TG2 has remained a mystery. Here we show that this balance is competitively achieved through mutually exclusive structural changes at a single allosteric regulatory site in the protein.
While human TG2 has six Ca2+ binding sites, their relative affinity varies by orders of magnitude (< 1 μM to > 1 mM).3 Of particular interest in this study is the tightest binding site, whose affinity for Ca2+ is comparable to that of EDTA (KD~0.1 μM). Based on our observation that C230 is nestled within this Ca2+ binding site (Fig. 1A,1B)3, mutants of both human TG2 (hTG2) and murine TG2 (mTG2) were cloned, expressed and purified. C230S (human TG2 numbering) was predicted to be resistant to oxidation7, whereas N231S was anticipated to attenuate Ca2+ affinity to this site by decreasing Ca2+ complexation.3 TG2-catalyzed deamidation was monitored using a synthetic dipeptide substrate CBz-Gln-Gly (ZQG)12,13, whereas TG2-catalyzed transamidation was measured by the incorporation of 5-biotinamido pentylamine (5BP) into N,N-dimethylcasein.14 As expected, the activities of wild-type and C230S TG2 showed similar Ca2+ concentration dependence (Fig. 1C, 1D). On the other hand, the N231S mutant showed reduced sensitivity to Ca2+, consistent with an earlier report of some cooperativity between the high-affinity Ca2+ binding site and the other Ca2+ binding sites.3 Similar results were obtained with mTG2 mutants (data not shown; see Fig. S3 for relevance).
Figure 1.
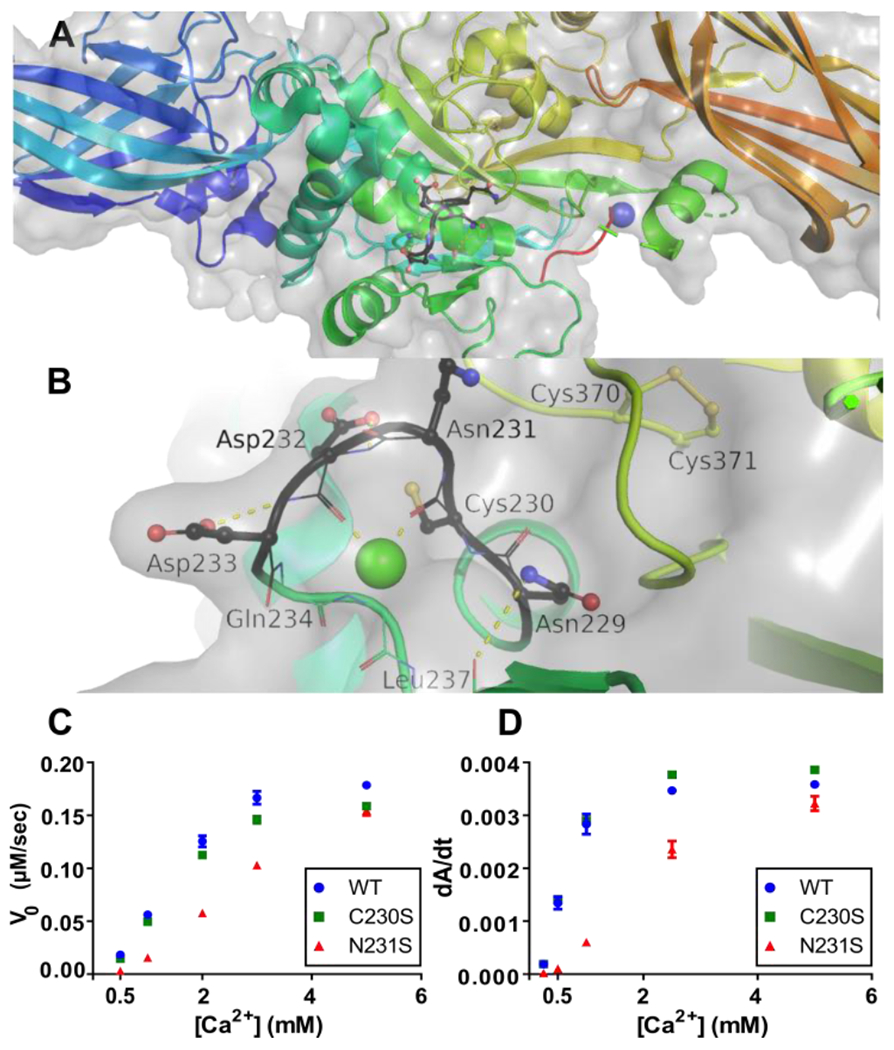
Dependence of TG2 structure and activity on its high-affinity Ca2+ binding site. (A) Zoomed out view of human TG2 (PDB 2Q3Z). The high-affinity Ca2+ ion at the S1 binding site has been modeled based on the known structure of human transglutaminase 3 (TG3)11, as described elsewhere.3 (See also Fig. S1.) (B) Zoomed in view with C230 and the allosteric disulfide bond between C370 and C371, which are visible in 2Q3Z. Activity of wild-type (WT) human TG2 and its C230S, and N231S mutants in a (C) deamidation assay (1 μM TG2) involving a soluble peptide substrate, and (D) transamidation assay (25 nM TG2) in which transwell-coated casein is the substrate. All data are the means of 3 replicates with errors bars depicting standard deviation.
As an initial test of whether the high-affinity Ca2+ site is protective against TG2 oxidation, the rate of oxidation of the N231S mutant was compared against wild-type TG2 by incubation of each recombinant protein with 100 μM cystamine at 22 °C for varying durations, followed by subjecting the protein to a TG2-catalyzed deamidation assay (Fig. 2A).4 Alternatively, each protein was incubated with 1 μM ERp57 at 37 °C for varying durations, followed by evaluation in the transamidation assay (Fig. 2B).4 Both experiments, which were performed in the absence of exogenously added Ca2+ during the oxidative phases of the assays, revealed a markedly increased rate of oxidative inactivation of the N231S mutant compared to wild-type TG2 as evidenced by a sharp decrease in enzyme activity.
Figure 2.
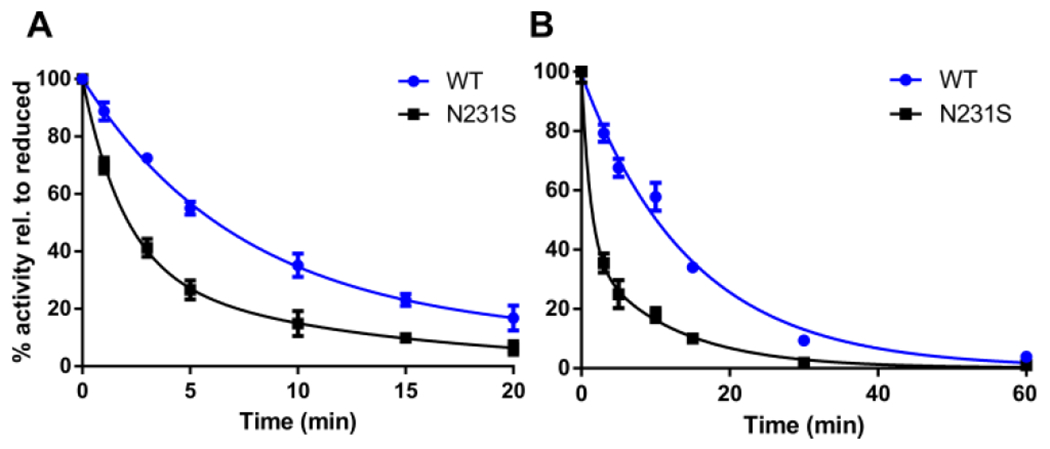
Kinetics of oxidative inactivation of wild-type (WT) human TG2 and its N231S mutant via activity assays. (A) Oxidation of 10 μM TG2 by 100 μM cystamine. Residual activity at individual time-points was measured by a kinetic assay for TG2-catalyzed deamidation of the dipeptide ZQG.4 (B) Oxidation of 25 nM TG2 by 1 μM ERp57. Residual activity at individual time-points was measured by a kinetic assay for TG2-catalyzed transamidation of N,N-dimethylcasein with 5BP.8,14 Data shown are the means of 3 replicates with errors bars depicting standard deviation. Data were normalized to wells without oxidant and fit to two-phase exponentials.
All deactivation curves were better fit to a double-exponential decay equation than a single-exponential decay equation (Table 1). Because Ca2+ persists at sub-stoichiometric levels in recombinant TG2 preparations notwithstanding extensive purification3, the higher decay rate constant is presumably a measure of the oxidation rate of the calcium-free protein fractions, whereas the lower rate constant reflects the off-rate of the high-affinity Ca2+ binding site. Similar trends were observed with mTG2, though mTG2 required more cystamine concentrations and longer incubation times to be fully inactivated (data not shown; see Fig. S3 for relevance).
Table 1.
Oxidation rate constants of wild-type (WT) TG2 or its N231S mutant
| Oxidant | k’fast, k’slow (min−1) | |
|---|---|---|
| WT | N231S | |
| cystamine | 0.14 ± 0.07 0.01 ± NDa |
0.51 ± 0.08 0.08 ± 0.04 |
| ERp57 | 0.07 ± NDa 0.07 ± NDa |
0.83 ± 0.27 0.09 ± 0.02 |
Not determined
To definitively establish that Ca2+ protects TG2 against inactivation by disulfide bond formation, wild-type TG2 and its C230S and N231S mutants were subjected to cystamine-promoted inactivation, as above, at varying [Ca2+]. To protect against inactivation via self-crosslinking in the presence of Ca2+, a saturating concentration (30 mM) of the ZQG substrate was added to assay mixtures and allowed to equilibrate for 10 min before Ca2+ addition. Increasing Ca2+ concentration enhances the resistance of both wild-type TG2 and the N231S mutant to cystamine-mediated oxidation. While the N231S mutant is much more susceptible to oxidation at lower Ca2+ concentrations, it approaches the behavior of the wild-type enzyme as Ca2+ concentration is increased (Fig. 3A, 3B). In contrast to both wild-type and N231S TG2, the C230S mutant is resistant to oxidation across all Ca2+ concentrations (Fig. 3C).
Figure 3.
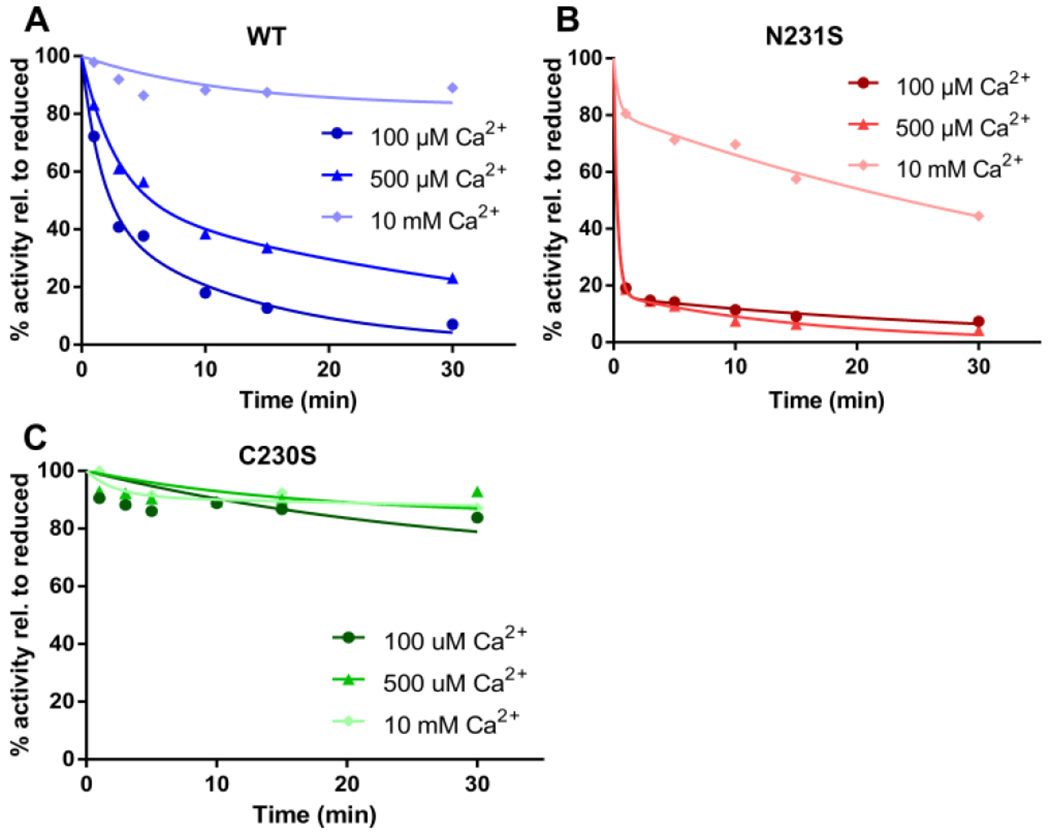
Ca2+ dependence of oxidative inactivation of TG2 by cystamine. (A) Wild-type TG2; (B) N231S; (C) C230S. Proteins were pre-incubated with 30 mM ZQG before Ca2+ addition to prevent TG2 inactivation via self-crosslinking. Data are representative of three or more experiments with equivalent results.
To validate the causative role of C230 in oxidative inactivation of extracellular TG2, we engineered two mammalian cell lines. First, we used CRISPR-Cas9 methodology to generate a homozygous, chromosomal C230S knock-in mutant of TG2 in the HT1080 human fibrosarcoma cell line (Fig. S2). HT1080 cells naturally deposit abundant quantities of TG2 in their extracellular matrix, most of which is inactive. We therefore reasoned that the C230S mutant line would show higher levels of extracellular TG2 activity under steady-state growth conditions. Second, we derived mouse embryonic fibroblasts (MEFs) from a TG2-knockout strain of C57Bl mice and transfected this cell line with a gene encoding wild-type mouse TG2 or its C230S mutant. Although MEFs do not naturally export significant quantities of TG2, we were able to increase steady-state TG2 levels in these transfected MEF lines upon treatment with betamethasone.15
Extracellular TG2 activity was measured via fluorescence microscopy, as described previously10, in cultures of HT1080 (Fig. 4A) or MEF derivatives (Fig. S3). Addition of 5BP to these cell cultures 2-4 h before fixation and microscopic evaluation results in biotinylation of matrix proteins. The absence of extracellular TG2 activity in HT1080 cultures expressing wild-type TG2 can be contrasted with the appearance of activity in the same system upon addition of TRX to the culture medium (Fig. 4B, 4C). In contrast, HT1080 cells expressing the C230S mutant exhibit TG2 activity in the extracellular matrix with or without addition of TRX (Fig. 4D). Similar results were observed with MEF transfectants (Fig. S3). Thus, together these findings highlight the pivotal role of disulfide bond formation as a counterbalance to Ca2+ mediated activation of TG2 in the extracellular matrix.
Figure 4.
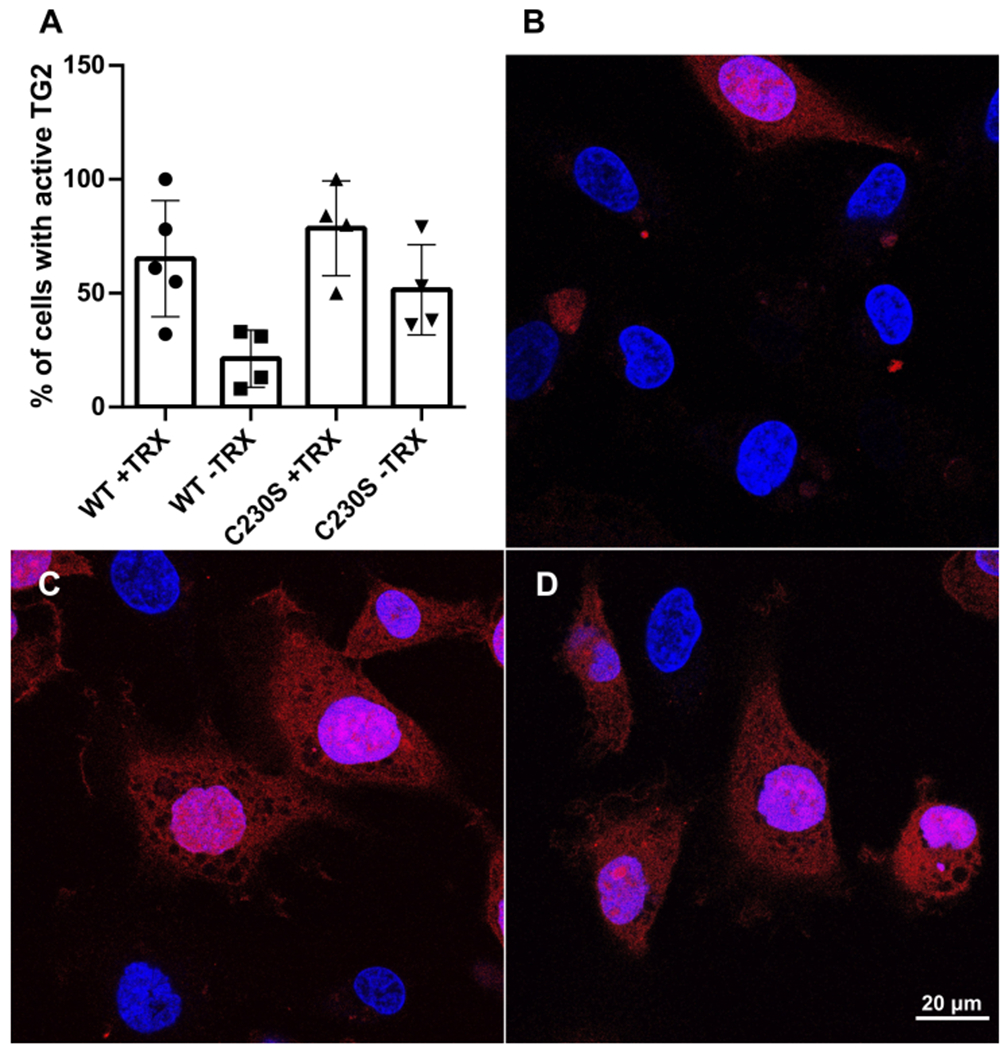
C230-promoted oxidative inactivation of extracellular TG2 in HT1080 cells. (A) 5BP incorporation in the extracellular matrices of wild-type or C230S HT1080 cells, as measured via fluorescence microscopy and image analysis. The incorporation of 5BP into extracellular matrix proteins is quantified by staining with fluorescent streptavidin followed by counting via ImageJ (see Supporting Information). Error bars depict standard deviation. The active groups were statistically different from the inactive reference (WT –TRX), as judged by Student’s t-test (p < 0.02). (B-D) Representative images of extracellular TG2 activity where 5BP incorporation is visualized in red and blue corresponds to a nuclear stain in (B) wild-type HT1080 cells with no pretreatment, (C) wild-type HT1080 pretreated with TRX, and (D) C230S HT1080 cells with no pretreatment.
In summary, our molecular and cellular studies demonstrate that extracellular TG2 activity is delicately balanced by two competitive mechanisms, calcium ions and redox agents that promote disulfide bond formation or cleavage, both of which act at the same site within the multifunctional protein. In other words, TG2 harbors an “OR” gate that can turn its enzymatic activity on (if high-affinity Ca2+ binding precedes disulfide bond formation) or off (if disulfide bond formation wins), as the protein is exported by cells.
An important corollary of our findings is that a missense mutant of TG2 at the C230 site would be expected to have a dominant phenotype that gives rise to constitutively elevated extracellular TG2 activity. Indeed, C230G and C230Y are rare germline mutants that have been discovered through whole-exome sequencing16, with all known individuals being heterozygotes. We expect these individuals to be more susceptible to CeD if they possess HLA-DQ2 or HLA-DQ8 alleles, given their constitutively active extracellular TG2.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Emma Chory, Dr. Brad A. Palanski, and Dr. Nick Plugis for the generation and transfection of MEF cell lines, and Dr. Lee Stunkard for assistance with PyMOL.
Funding Sources
This work was supported by National Institutes of Health (NIH) Grant R01 DK063158 (to C.K.).
ABBREVIATIONS
- 5BP
5-biotinamido pentylamine
- CeD
celiac disease
- ECM
extracellular matrix
- ERp57
endoplasmic reticulum-resident protein 57
- MEF
mouse endothelial fibroblast
- TG2
transglutaminase 2
- TRX
thioredoxin-1
- ZQG
Cbz-Gln-Gly
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org. Detailed description of methods, supplementary figures.
REFERENCES
- (1).Sollid LM The Roles of MHC Class II Genes and Post-Translational Modification in Celiac Disease. Immunogenetics 2017, 69 (8–9), 605–616. 10.1007/s00251-017-0985-7. [DOI] [PubMed] [Google Scholar]
- (2).Eckert RL; Kaartinen MT; Nurminskaya M; Belkin AM; Colak G; Johnson GVW; Mehta K Transglutaminase Regulation of Cell Function. Physiol. Rev 2014, 94 (2), 383–417. 10.1152/physrev.00019.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Király R; Cssz É; Kurtán T; Antus S; Szigeti K; Simon-Vecsei Z; Korponay-Szabó IR; Keresztessy Z; Fésüs L Functional Significance of Five Noncanonical Ca2+-Binding Sites of Human Transglutaminase 2 Characterized by Site-Directed Mutagenesis. FEBS J. 2009, 276 (23), 7083–7096. 10.1111/j.1742-4658.2009.07420.x. [DOI] [PubMed] [Google Scholar]
- (4).Palanski BA; Khosla C Cystamine and Disulfiram Inhibit Human Transglutaminase 2 via an Oxidative Mechanism. Biochemistry 2018, 57, 3359–3363. 10.1021/acs.biochem.8b00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bronner F Extracellular and Intracellular Regulation of Calcium Homeostasis. Sci. World 2001, 1, 919–925. 10.1100/tsw.2001.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pinkas DM; Strop P; Brunger AT; Khosla C Transglutaminase 2 Undergoes a Large Conformational Change upon Activation. PLoS Biol. 2007, 5 (12), 2788–2796. 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stamnaes J; Pinkas DM; Fleckenstein B; Khosla C; Sollid LM Redox Regulation of Transglutaminase 2 Activity. J. Biol. Chem 2010, 285 (33), 25402–25409. 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yi MC; Melkonian AV; Ousey JA; Khosla C Endoplasmic Reticulum-Resident Protein 57 (ERp57) Oxidatively Inactivates Human Transglutaminase 2. J. Biol. Chem 2018, 293 (8), 2640–2649. 10.1074/jbc.RA117.001382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Klöck C; Sollid LM; Stamnaes J; Khosla C; Jin X; DiRaimondo TR Activation of Extracellular Transglutaminase 2 by Thioredoxin. J. Biol. Chem 2011, 286 (43), 37866–37873. 10.1074/jbc.m111.287490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Plugis NM; Palanski BA; Weng C-H; Albertelli M; Khosla C Thioredoxin-1 Selectively Activates Transglutaminase 2 in the Extracellular Matrix of the Small Intestine. J. Biol. Chem 2017, 292 (5), 2000–2008. 10.1074/jbc.M116.767988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ahvazi B; Boeshans KM; Idler W; Baxa U; Steinert PM Roles of Calcium Ions in the Activation and Activity of the Transglutaminase 3 Enzyme. J. Biol. Chem 2003, 278 (26), 23834–23841. 10.1074/jbc.M301162200. [DOI] [PubMed] [Google Scholar]
- (12).Day N; Keillor JW A Continuous Spectrophotometric Linked Enzyme Assay for Transglutaminase Activity. Anal. Biochem 1999, 274 (1), 141–144. 10.1006/abio.1999.4255. [DOI] [PubMed] [Google Scholar]
- (13).Piper JL; Gray GM; Khosla C High Selectivity of Human Tissue Transglutaminase for Immunoactive Gliadin Peptides: Implications for Celiac Sprue. Biochemistry 2002, 41 (1), 386–393. 10.1021/bi011715x. [DOI] [PubMed] [Google Scholar]
- (14).Siegel M; Strnad P; Watts RE; Choi K; Jabri B; Omary MB; Khosla C Extracellular Transglutaminase 2 Is Catalytically Inactive , but Is Transiently Activated upon Tissue Injury. PLoS One 2008, 3 (3), 1–11. 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Seow BKL; McDougall ARA; Short KL; Wallace MJ; Hooper SB; Cole TJ Identification of Betamethasone-Regulated Target Genes and Cell Pathways in Fetal Rat Lung Mesenchymal Fibroblasts. Endocrinology 2019, 160 (8), 1868–1884. 10.1210/en.2018-01071. [DOI] [PubMed] [Google Scholar]
- (16).gnomAD browser Transglutaminase 2 (TGM2) https://gnomad.broadinstitute.org/gene/ENSG00000198959?dataset=gnomad_r2_1 (accessed Mar 1, 2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.