

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are considered one of the most commonly used medications globally. Seventeen isoxazole-containing compounds with various functional groups were evaluated in this work to identify which one was the most potent and which group was most selective toward COX-1 and COX-2 by using an in vitro COX inhibition assay kit. Their cytotoxicity was evaluated on the normal hepatic cell line (LX-2) utilizing the MTS assay. Moreover, these molecules' antibacterial and antifungal activities were evaluated using a microdilution assay against several bacterial and fungal species. In addition, molecular docking studies were conducted to identify the possible binding interactions between these compounds and their biological targets by using the X-ray crystal structure of the human COX enzyme and different proteins of bacterial and fungal strains. At the same time, the QiKProp module was used for ADME-T analysis. The results showed that all evaluated isoxazole derivatives showed moderate to potent activities against COX enzymes. The most potent compound against COX-1 and COX-2 enzymes was A13, with IC50 values of 64 and 13 nM, respectively, and a significant selectivity ratio of 4.63. It was clear that the 3,4-dimethoxy substitution on the first phenyl ring and the Cl atom on the other phenyl pushed the 5-methyl-isoxazole ring toward the secondary binding pocket and created the ideal binding interactions with the COX-2 enzyme in comparison with the other compounds. Compound A8 showed antibacterial and antifungal activities against Pseudomonas aeruginosa, Klebsiella pneumonia, and Candida albicans with MIC values of 2 mg/ml. In fact, this compound showed possible binding interactions with the elastase in P. aeruginosa and KPC-2 carbapenemase in K. pneumonia. Furthermore, for better understanding, molecular dynamics simulations were undertaken to study the change in dynamicity of the protein backbone and ligand after the ligand binds to the protein and to ensure the stability of ligand–protein complexes.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-022-03408-8.
Keywords: Isoxazole, COX, LX-2, Bacterial, Molecular docking
Introduction
The biosynthesis of prostaglandin H2 from arachidonic acid is catalyzed by the cyclooxygenase (COX) enzyme, resulting in mediating various pathogenic mechanisms (Park et al. 2006; Smith et al. 2011). Prostaglandin H2 is the first step in making different prostaglandins, prostacyclin, and thromboxane, which play important roles in many critical physiological and pathological processes via their reversible interaction with G-protein coupled membrane receptors (Burdan et al. 2006). The inhibition of COX confers relief from inflammatory, thrombotic, neurodegenerative, pyretic, and oncological disorders. COX enzymes are membrane-bounded enzymes with two main isoforms: COX-1 and COX-2 (Paragi-Vedanthi and Doble 2010). The COX-1 enzyme is involved in the production of prostaglandins, which are essential to maintain the function of cardiovascular physiology through their vasodilatory, antiadhesive, antiplatelet, and antiproliferative properties.
Additionally, the COX-1 enzyme maintains the gastrointestinal system by protecting the stomach from the digestive juices and maintaining the normal lining of the stomach and intestines (Vane and Botting 2003; FitzGerald and Patrono 2001; Linton and Fazio 2008). Also, the COX-1 isozyme stimulates neovascularization and tumor development in the ovary. Therefore, COX-1 inhibitors could be helpful as adjuvant therapy to control cancer pathogens such as ovarian cancer. However, since the primary role of COX-1 is to protect the gastrointestinal tract and mediate blood clotting, cyclooxygenase inhibitors can lead to unwanted side effects. The COX-2 enzyme is overexpressed in different pathophysiological conditions like cancer and inflammation (Zidar et al. 2009). While COX-1 isozymes are mainly found in the stomach, kidney, platelets, and intestinal endothelium, the COX-2 isozymes localize mainly in leukocytes, macrophages, fibroblasts, and endothelial cells. Thus, the variant functions of both enzymes could be estimated (Seibert et al. 1997).
The structures of both COX enzymes are identical in 67% of their amino acid chains. The major difference between them is the presence of isoleucine (Ile523) in COX-1 instead of valine (Val523) in COX-2, which makes a greater available region for binding in COX-2 in comparison to the COX-1 enzyme (Vane and Botting 1998). Nowadays, the most prescribed medications for pain relief and inflammation are non-steroidal anti-inflammatory drugs. Their mechanism of action is to inhibit the COX enzyme, which will decrease prostaglandin production (Bacchi et al. 2012). Because they inhibit the COX-1 enzyme and the COX-2 enzyme, their long-term use will lead to undesirable side effects such as GIT ulcers and damage to the liver and kidney (Kumar et al. 2018). So, selective non-steroidal anti-inflammatory drugs will play an important role in overcoming the side effects of non-selective NSAIDs. Valdecoxib, celecoxib, and rofecoxib are marketed as selective NSAIDs (Mahboubi Rabbani and Zarghi 2019). These agents treat various inflammatory conditions, including osteoarthritis and rheumatoid arthritis, with fewer side effects than non-selective NSAIDs (Rao and Knaus 2008).
On the other hand, chronic use of these selective COX-2 inhibitors causes a decrease in the production of prostaglandin I2, which leads to cardiovascular side effects (Hermann and Ruschitzka 2006). Many of these agents were withdrawn from the market due to their cardiotoxicity. So there is a need for more safe and selective inhibitors. Studies showed that the ketoprofen analogs and tricyclic compounds had better COX-2/COX-1 ratios compared to ketoprofen (Zarghi and Ghodsi 2010). As many commonly used drugs containing heterocycles, such as rofecoxib and celecoxib, demonstrate COX inhibitory activity, we recently synthesized chemical structures of pyrazole or tetrazole derivatives as new promising COX inhibitors (Assali et al. 2020).
As various heterocycle scaffolds like benzodioxole (Khalil et al. 2021), benzoxazole (Nacak et al. 2005), or phenyl-isoxazole (Panda et al. 2009; Eccles et al. 2008; Zhang et al. 2010; Santos et al. 2010; Mao et al. 2009) were found to play a decisive role in boosting the antimicrobial, anticancer, anti-tuberculosis activity besides the recently reported tertiary action of COX-2 inhibitors (e.g., Celecoxib) as anti-inflammatory, antibacterial, and antifungal agents, a new and appealing field of drug research has been sparked that aimed to interfere with and effectively manage the inflammatory-associated infections. Celecoxib's and ibuprofen's synergistic antimicrobial and antifungal actions were demonstrated to be the result of blocking multi-drug resistance (MDR) transporters within the bacterial and fungal cell membranes, respectively. The accumulation of antibiotics inside the invasion organisms led to enhanced sensitivity of antibiotics, besides reversing bacterial and fungal resistance via inhibiting the pumping of antibiotics out of the cell (Kalle and Rizvi 2011; Thangamani et al. 2015; Ricardo et al. 2009).
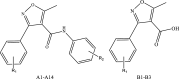
Previously, three series of isoxazole-carboxamide derivatives, phenyl-isoxazole-carboxamide (A1-A5), fluoro-phenyl-isoxazole-carboxamide (A6-A11), and chloro-phenyl-isoxazole-carboxamide (A12-A14) (Fig. 1) were synthesized by our research team and were evaluated against a panel of cancer and normal cell lines. Almost all of these compounds showed very weak negligible toxicity on the normal cell lines (Hek293t) with IC50 values > 112 μM, and some of these compounds showed moderate to potent cytotoxic activities on some cancer cell lines with varying IC50 values (Eid et al. 2021; Hawash et al. 2021a, 2021b).
Fig. 1.

Phenyl-isoxazole-carboxamide series’ main structures where R groups were H, methoxy substituent/s or t-butyl
Depending on the previous data, the current work aims to evaluate previously synthesized phenyl-isoxazole-carboxamide derivatives (A1-A14; Fig. 1) by our team on COX enzymes and their cytotoxicity on normal hepatic cell line (LX-2) as well as their antimicrobial activities against several bacterial and fungal species. However, the commercially available phenyl-isoxazole-carboxylic acid derivatives were evaluated on COX enzymes to create the structure–activity relationship (SAR) and find the pharmacophore group, whether COOH or CONHR. Finally, molecular docking studies were performed to justify the possible binding interactions between the COX enzymes and our compounds. Thus, developing and discovering new COX inhibitors that exhibit both antimicrobial and antifungal activity is warranted. The efforts here in our study to investigate the binding patterns of native and newly discovered agents within the binding domain of both COX-1 and COX-2 isozymes. Additionally, the reported antimicrobial and antifungal activities have been supported by estimating the interaction behavior between bacterial administrative enzymes (P. aeruginosa elastase B and K. pneumonia KPC-2 carbapenemase) and cytochrome P450 14-α-sterol demethylase (Cyp51) which is a critical protein in the fungal type (ex. C. albicans). This newly achieved research here, along with previous studies, could finally pull off that supreme goal.
Methods
Chemistry
The phenyl-isoxazole-carboxamide series (A1-A14) were synthesized as outlined in Scheme 1. The coupling reaction to form isoxazole-carboxamide compounds was afforded by EDCI and DMAP as activating agents and covalent nucleophilic catalysts, respectively (Hawash et al. 2021c, 2021d). These compounds had already been studied with IR, HRMS, and NMR spectrometers, and the results of these studies are listed in the supplementary file Table S1 (Eid et al. 2021; Hawash et al. 2021a, 2021b).
Scheme 1.

Acid + appropriate aniline derivatives, stirred in 15 ml DCM, then DMAP and EDC were added under inert nitrogen gas and stirred for 24–72 h (R1 = H, tri-OCH3, di-OCH3, Cl + di-OCH3, and t-butyl)
Biological COX assay method
The COX-1 and COX-2 inhibitory activities were evaluated on the COX (human) Inhibitor Screening Assay Kit (supplied by Cayman chemicals, Ann Arbor, MI, USA). The preparation of the reagents and the testing procedure were performed according to the manufacturer’s recommendations. In brief, various concentrations of the compounds and ketoprofen (concentration range of 100 μM–1 μM) dissolved in a minimum quantity of dimethylsulfoxide (DMSO) were incubated with a mixture of COX-1 or COX-2 enzyme. The reaction was initiated by adding 50 μl of arachidonic acid, followed by incubation at 37 °C for exactly 30 s. Then, the reaction was stopped by adding 30 μl of stannous chloride solution to each reaction tube, followed by incubation for 5 min at room temperature. The produced PGF2a in the samples by COX reactions was quantified via enzyme-linked immunosorbent assay (ELISA). The 96-well plate was covered with plastic film and incubated for 18 h at room temperature on an orbital shaker. After incubation, the plate was rinsed five times with the washed buffer followed by the addition of Ellman’s reagent (200 μl) and incubated for about 60–90 min at room temperature until the absorbance of the Bo well was in the range of 0.3–0.8 at 405 nm. The plate was then read by an ELISA plate reader Unilab microplate reader 6000. The inhibitory percentage was measured for the different tested concentrations against the control. The IC50 was calculated from the concentration inhibition response curve, and the selectivity index (SI) was calculated by dividing the IC50 values of COX-1 by the IC50 values of COX-2. Ketoprofen and celecoxib were used as positive control drugs in this study (Assali et al. 2020; Hawash et al. 2020b).
Cell culture cytotoxicity assay
Human hepatic stellate (LX-2), cells (ATCC, Rockville, MD, USA) were cultured in RPMI-1640 media and supplemented with a 1% mixture of streptomycin, penicillin, 1% l-glutamine, and 10% fetal bovine serum. Cells were grown at 37 °C in a humidified atmosphere with 5% CO2. Cells were seeded at 2.6 × 104 cells/well in a 96-well plate. After 48 h, cells were incubated with various concentrations (300, 100, 50, 10, and 1 µM) of each compound for 24 h. At the same concentration, doxorubicin (DOX) was used as a positive control. Cell viability was assessed by the CellTilter 96® Aqueous One Solution Cell Proliferation (MTS) Assay according to the manufacturer’s instructions (Promega Corporation, Madison, WI). In brief, at the end of the treatment, 20 μL of MTS solution per 100 μL of media was added to each well and incubated at 37 °C for 2 h. Absorbance was measured at 490 nm (Jaradat et al. 2018; Hawash et al. 2020a).
Antimicrobial assay
Microorganisms
The organisms used for the bacterial evaluation were Type Culture Collection (ATCC); Klebsiella pneumonia (ATCC13883), Staphylococcus aureus (ATCC 25,923), Pseudomonas aeruginosa (ATCC 9027), Proteus vulgaris (ATCC 8427), and Escherichia coli (ATCC 25,922), in addition to the clinical isolates of MRSA (Methicillin-Resistant Staphylococcus aureus). Moreover, Candida albicans (ATCC 90,028) was used for the antifungal evaluation.
Culture media
Five milligrams of each compound was fully dispersed in 5 ml of dimethyl sulfoxide (DMSO) to establish a final 1 mg/ml concentration. Filter sterilization was carried out on the resulting solution by serial microdilution of two- and sixfolds in sterilized nutrient broth. The dilution processes were carried out using aseptic conditions in the available 96-well dishes. The media were prepared by combining the components and then heating them with restlessness until they boiled accordingly. After that, the medium was then autoclaved at 121 °C for about 20 min. Following cooling, the agar was poured onto a flat surface in sterile Petri dishes, and held at a depth of 4 °C. Sabouraud dextrose agar containing 40.0 g of dextrose, 10.0 g of peptone, and 20.0 g of agar/liter of distilled water was the medium used for fungal production. As mentioned, the media were prepared. McFarland turbidity standards were used to standardize the inoculums. McFarland 0.50 standard was used for turbidity comparison, which offers turbidity similar to that of a bacterial suspension containing 1.5 × 108 CFU/mL (Jaradat et al. 2021). For all tested bacteria, we had four controls: (1) positive control, which contains media and bacteria; (2) negative control, which only contains media; (3) compound control (compound + media) to be sure that there is no contamination and turbidity and that the changes are not due to the compound itself (so the compounds were serially diluted in this control), and (4) DMSO, which was tested for every microbe separately to check the effect on each one, and the antimicrobial activity of DMSO was also considered (Khalil et al. 2021; Hawash et al. 2022).
Molecular docking studies
To identify the predicted protein–ligand interactions of the synthetic compounds within the binding site, molecular docking studies were carried out using glide standard-precision mode (Glide score SP) Maestro 12.1 (Schrödinger, New York, NY, USA) running on a Windows 10 operating system (Kumar et al. 2021). Docking simulations were also utilized to realize the possible best binding poses of the docked molecules, so they could be used to explain the reported results and to distinguish the promising leads. Molecular docking studies usually follow a general procedure that starts with the selection and preparation of an appropriate protein, grid generation, and ligand preparation, followed by analyzing docking outcomes and investigating the ligand–receptor interaction patterns. The X-ray crystal-structure data applied for docking studies were taken from the Brookhaven protein data bank (PDB; http://www.rcsb.org/pdb. The human COX-1 (PDB ID 3KK6) and human COX-2 (PDB ID 5KIR) were selected based on their relevant binding orientations and high resolution (e.g., resolutions were 2.75 A˚ and 2.70 A˚, respectively) for molecular docking studies regarding cyclooxygenase enzymes, which were previously applied in our previous work (Assali et al. 2020).
Also, the biological assay of antimicrobial activity of molecules was supported by conducting molecular docking studies within critical bacterial target enzymes present in Gram-negative bacteria like P. aeruginosa elastase B protein (PDB ID 1U4G) and K. pneumonia KPC-2 carbapenemase protein (PDB ID 2OV5) (Magpantay et al. 2021). P. aeruginosa elastase B (pseudolysin), the main peptidase found in pseudomonal secretions, plays multi-functional roles in different aspects of the pathogen–host interaction starting from the invasiveness stage through breaking the basolateral intercellular junctions present in the host tissues to the post-invasiveness stage via hydrolyzing of many immunologically relevant molecules like complement components and antibodies (Galdino et al. 2019; Bommagani et al. 2021). K. pneumonia carbapenemases (KPC) are hydrolysis enzymes that are suggested to be the main reason for inactivation B -lactam antibiotics (including Carbapenems) and imipenem results in drug-resistance generating (Malathi et al. 2019; Sharma et al. 2021). Thus, P. aeruginosa elastase B and K. pneumoniae carbapenemase become highly attractive targets to defeat antibiotic-refractory infections as a recently urgent issue caused by P. aeruginosa and K. pneumonia.
Additionally, molecular docking studies were performed within the administrative fungal enzyme cytochrome P450 14-α-sterol demethylase (Cyp51), which is found in a complex with fluconazole (PDB1 ID 1EA1), and it is a representative critical protein in C. albicans (Ghabbour et al. 2014). The final score was presented as a Glide score based on the energy-minimized poses. The lowest Glide score value related to the best-docked pose for each ligand was recorded (Friesner et al. 2004). Using the Graphical User Interface of Maestro 12.1 (Zervou et al. 2021), the ligands were drawn, followed by ligand preparation, including parameterizing the atom types and protonation states at target pH (7.0 ± 2.0) with the OPLS2005 force field using LigPrep (Assali et al. 2020; Panwar and Singh 2021). The crystal structures were prepared using the Protein Preparation Wizard (Schrödinger 2016), which started with preprocessing that involved: filling in missing side chains using prime, creating zero-order bonds to metals, adding hydrogens, creating disulfide bonds, deleting water molecules beyond 5 Å from the hit group, and removing waters with less than 3 H-bonds to non-waters. The preprocessing step was followed by optimizing the protein structure’s hydrogen bonds, followed by minimizing to a root-mean-square deviation of 0.3 Å. The receptor grids were generated using the prepared proteins; the default setup was conserved. The PLIP server was applied to evaluate the binding modes (Adasme et al. 2021). The final results were visualized with PyMOL 2.5.2 (Elokely and Doerksen 2013). In order to validate the docking method concerning each prepared protein, the native ligand was drawn, prepared, and re-docked into the binding pockets. Then, the native ligand was superimposed with the best pose of the re-docked ligand, followed by calculating the root mean square deviation (RMSD). Getting an RMSD value less than 2.0 Å indicates that the protein crystal structures and ligands were appropriately prepared and the docking method precisely works (Bell and Zhang 2019).
Molecular dynamics (MD) simulations
The molecular dynamics simulations were performed utilizing the online WEBGRO Macromolecular Simulations server (https://simlab.uams.edu/ProteinWithLigand/index.html), available by the University of Arkansas for Medical Sciences (UAMS) as a public service. It is a GRACE High-Performance Computing Facility and is based on GROMAC (de Groot and Nadrchal 1993). After creating the ligand topology using the PRODRG 2.3 online server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg/submit.html) (Van Aalten et al. 1996) and choosing the GROMOS96 54a7 force field, the ligand–protein complex structures and apo structures were subjected to the simulation at 300 K in an explicit solvent. Then each system was subjected to the triclinic box and SPC water models for salvation. The Na + and/or Cl− ions were added as salt types to neutralize the system. In the presence of solvent, the proteins were energetically minimized using the integrator steepest descent to a max of 5000 steps. After that, the equilibration process was performed by applying the NVT and NPT ensemble at 300 k temperature. Finally, for each system, the molecular dynamics were performed for a simulation of time 100 ns and the frames per MD simulation were fixed to 5000. The MD simulations using the WEBGRO server output various trajectories such as the root mean-square deviation (RMSD) and the radius of gyration (Rg) to understand more the formation and stability of complexes (Lindorff‐Larsen et al. 2010; Abraham et al. 2015).
ADME-T calculations
In order to examine the future drug properties of our new tested molecules, a set of ADME-T calculations (molecular weight, dipole moment, octanol/gas partition coefficient, octanol/water partition coefficient, aqueous solubility, brain/blood partition coefficient, binding to human serum albumin, oral absorption, polar surface area, number of violations of Lipinski's rule of 5, and number of violations of Jorgensen’s rule of 5) were performed. The QiKProp module (Schrödinger 12.1, LLC, NY) was used for ADME-T analysis (Bell and Zhang 2019).
Statistical analysis
The COX, antibacterial, antifungal, and cell culture assays were conducted in triplicates. The results were expressed as means of the standard deviation (± SD), while the results were considered significant when the p value was < 0.05.
Result and discussion
Chemistry
About 19 compounds were evaluated against COX enzymes, and these compounds were classified into five groups: the first group includes five compounds (A1-A5) which belong to phenyl-isoxazole-carboxamide, and the second group consists of six compounds (A6-A11) which belong to fluoro-phenyl-isoxazole-carboxamide; the third group belongs to chloro-phenyl-isoxazole-carboxamide which includes three compounds (A12-A14); the fourth group belongs to phenyl-isoxazole-carboxylic acid that related to the three compounds B1-B3, and the final group as positive controls (ketoprofen and celecoxib). The first three groups were synthesized previously, as mentioned in Methods section, and the other groups were ordered from Sigma-Aldrich. The structures of all evaluated compounds are presented in Table 1.
Table 1.
The structures of isoxazole derivatives and the IC50 values against COX enzymes
| |||||
|---|---|---|---|---|---|
| Name | R1 | R2 | IC50 COX-1 (µM) | IC50 COX-2 (µM) | Selectivity index |
| A1 | H | H | 0.839 ± 0.04 | 1.214 ± 0.78 | 0.69 |
| A2 | H | 3-OCH3, 4-OCH3, 5-OCH3 | 0.269 ± 0.17 | 3.949 ± 1.02 | 0.07 |
| A3 | H | 3- OCH3, 5-OCH3 | 0.122 ± 0.11 | 0.310 ± 0.25 | 0.39 |
| A4 | H | 3-OCH3, 4-OCH3 | 0.076 ± 0.06 | 0.258 ± 0.28 | 0.29 |
| A5 | H | 4-t-butyl | 0.312 ± 0.16 | 0.486 ± 0.01 | 0.65 |
| A6 | 4-F | H | 0.140 ± 0.05 | 0.218 ± 0.11 | 0.6418 |
| A7 | 4-F | 3-OCH3, 4-OCH3, 5-OCH3 | 0.350 ± 0.05 | 0.346 ± 0.24 | 1.0113 |
| A8 | 4-F | 3-OCH3,5-OCH3 | 0.317 ± 0.02 | 0.878 ± 0.47 | 0.3618 |
| A9 | 4-F | 3-OCH3, 4-OCH3 | 0.335 ± 0.07 | 0.829 ± 0.15 | 0.4047 |
| A10 | 4-F | 4-t-butyl | 0.126 ± 0.10 | 0.370 ± 0.08 | 0.3403 |
| A11 | 4-F | 2-OCH3, 4-Cl, 5-OCH3 | 0.478 ± 0.21 | 0.334 ± 0.03 | 0.5604 |
| A12 | 2-Cl | 3-OCH3,5-OCH3 | 0.413 ± 0.19 | 0.293 ± 0.11 | 1.4117 |
| A13 | 2-Cl | 3-OCH3, 4-OCH3 | 0.064 ± 0.02 | 0.013 ± 0.01 | 4.6341 |
| A14 | 2-Cl | 2-OCH3, 5-OCH3 | 0.077 ± 0.01 | 0.376 ± 0.21 | 0.2066 |
| B1 | 2-Cl | – | 1.210 ± 0.55 | 0.856 ± 0.15 | 1.414454 |
| B2 | 4-F | – | 0.702 ± 0.28 | 0.033 ± 0.07 | 20.72017 |
| B3 | H | – | 0.668 ± 0.08 | 1.040 ± 0.85 | 0.642553 |
| Ketoprofen | – | – | 0.043 ± 0.02 | 0.066 ± 0.05 | 0.643249 |
| Celecoxib | – | – | 1.479 ± 0.47 | 0.004 ± 0.003 | 369.75 |
In vitro COX-1 and COX-2 inhibition assay
All compounds were evaluated for inhibition assay on COX-1 and COX-2 enzymes using the COX-1 (human) Inhibitor Screening Assay Kit and COX-2 (human) Inhibitor Screening Assay Kit (Cayman Chemical Company, Ann Arbor, MI, USA). The first three groups of evaluated compounds (PICA, FPICA, and CPICA) have a tricyclic structure similar to celecoxib NSAID so that they could display some selectivity toward the COX-2 enzyme. Celecoxib, a selective COX compound, and ketoprofen, a non-selective compound, were used as positive controls to compare the biological activity and selectivity found in our newly evaluated series. The results of the COX inhibitory assay are presented in Table 1.
All evaluated isoxazole derivatives showed moderate to potent activities, as presented in Table 1, with IC50 values ranging < 4 µM against both COX enzymes. However, the most potent compound was A13 against COX-1 and COX-2 with IC50 equal to 64 and 13 nM, respectively, which was more active on COX-2 than Ketoprofen drug with a selectivity ratio of 4.63, as well as the most selective compound was B2 with a selectivity ratio value of 20.7.
Cytotoxic evaluation on LX-2
MTS assay was used to determine the cytotoxicity effect of phenyl-Isoxazole-carboxamide derivatives on LX-2 (normal hepatic cells). The percentage of cell viability on LX-2 cell line of isoxazole-carboxamide derivatives, positive control (5-Fu), and negative control (DMSO) at a concentration of 100 µM is shown in Fig. 2, as well as Fig. S1 (supplementary file), which showed the results of cell viability of the most cytotoxic compound (A6) and the least cytotoxic compound (A13), in comparison with 5-Fu at all used concentrations. The percentage was in the range of 55.12–90.47% for all carboxamide derivatives, in comparison with the positive control anticancer drug percentage value of 7.87%, which means that all of these compounds have very weak or negligible cytotoxic activities on the normal liver cell line, and these compounds seem safe at the effective dose.
Fig. 2.

The cell viability percentage at a concentration of 100 µM of isoxazole-carboxamide derivatives, positive control (5-Fu), and negative control (DMSO) on the LX-2 normal cell line
Antimicrobial activity
The isoxazole-carboxamide derivatives were further evaluated to observe their antibacterial activities. The antibacterial activities were very weak or negligible, and all bacterial strains showed resistance to the evaluated compounds (Table 2) except compounds A8 and A9. These two compounds showed significant antibacterial activities against P. aeruginosa bacterial strain and C. albicans fungal strain at MIC 2.00 mg/ml, as well as compound A8, which showed antibacterial activities against K. pneumonia bacterial strain at the same concentration. Two positive controls were used to make a comparison, fluconazole as an antifungal agent and ciprofloxacin as an antibacterial agent.
Table 2.
Microbial and fungal growth inhibition MIC values (mg/ml) of the synthesized compounds and positive controls
| Microbial strains | S. aureus | E. coli | K. pneumonia | P. vulgaris | P. aeruginosa | MRSA | C. albicans |
|---|---|---|---|---|---|---|---|
| A1 | R | R | R | R | R | R | R |
| A2 | R | R | R | R | R | R | R |
| A3 | R | R | R | R | R | R | R |
| A4 | R | R | R | R | R | R | R |
| A5 | R | R | R | R | R | R | R |
| A6 | R | R | R | R | R | R | R |
| A7 | R | R | R | R | R | R | R |
| A8 | R | R | 2 | R | 2 | R | 2 |
| A9 | R | R | R | R | 2 | R | 2 |
| A10 | R | R | R | R | R | R | R |
| A11 | R | R | R | R | R | R | R |
| A12 | R | R | R | R | R | R | R |
| A13 | R | R | R | R | R | R | R |
| A14 | R | R | R | R | R | R | R |
| Ciprofloxacin | 0.78 | 1.56 | 0.125 | 3.12 | 0.015 | 0.125 | R |
| Fluconazole | R | R | R | R | R | R | 1.56 |
Molecular docking study
The crystallized poses of native ligands were superimposed with reduced poses in order to confirm the authenticity of the molecular docking process used in this study for each prepared protein (Fig. 3). The RMSD values were calculated to be 0.40 for celecoxib docked to COX-1 (PDB ID 3KK6), 0.01 for rofecoxib docked to COX-2 (PDB ID 5KIR), 0.90 for HPI docked to Elastase of P. aeruginosa (PDB ID 1U4G), 1.40 for BCN docked to K. pneumonia KPC-2 carbapenemase (PDB ID 2OV5), and 1.43 for fluconazole docked to cytochrome P450 14 α-sterol demethylase (PDB ID 1EA1). As all RMSD values were located much lower than the acceptable limits of 2.0 Ao, the docking protocol was well optimized and could be confirmed.
Fig. 3.

Superimposition of crystal form and docking result of (A) celecoxib for human COX 1(PDB ID 3KK6), (B) rofecoxib for human COX 2 (PDB ID 5KIR), (C) HPI for Elastase of Pseudomonas aeruginosa (PDB ID 1U4G), (D) BCN for Klebsiella pneumonia KPC-2 carbapenemase (PDB ID 2OV5), and (E) fluconazole for Cytochrome P450 14 alpha-sterol demethylase (PDB ID 1EA1)
Both COX-1 and COX-2 isoforms are highly related to each other, with amino acid sequence identity equaling 67%. The selectivity of NSAIDs toward the COX-2 isozyme mainly stemmed from optimally occupying the additional secondary binding pocket that does exist only in the COX-2 isoform. The conformational change presented as a result of replacing ILE-532 amino acid in COX-1 with VAL-532 in COX-2 led to unlocking this secondary binding pocket and unveiling the polar amino acid ARG-513 which is considered the ameliorating COX-2 selectivity (Badrey et al. 2015). The molecular docking studies here aimed to scrutinize the expecting binding profile of newly synthesized molecules besides the well-known controls within the binding site of crystallized human COX-1 (PDB code 3KK6) and COX-2 (PDB code 5KIR). Thus, understanding more about the structure–activity relationship (SAR) alongside finding differences and similarities is a fundamental strategy to enhance the drug profile, including activity alongside selectivity. The results of docking analysis scores of all tested docked ligands are summarized in Table S2 (in supplementary data), and the docking figures for all evaluated compounds are shown in Figs S2 and S3. In Fig. 4, the crystal binding modes of celecoxib, ketoprofen, and the most potent compound A13 are shown, it was clear that the presence of Cl atom at the ortho position of the first phenyl ring and 3,4-dimethoxy on the other phenyl ring create ideal binding interaction between A13 compound and the crystal of COX enzymes, especially COX2, in comparison with the other compound. Regarding the docking studies within the COX-1 isozyme, the compounds A13, A14, and A4 revealed significant binding affinity toward the binding domain of COX-1 which was comparable to the binding affinity of ketoprofen (located within the range 0.064–0.076 μM) (Figure S3; supplementary data). The three compounds showed a low selectivity index (below 0.3) which means that have a higher affinity toward COX-1 than COX-2 except for compound A13 which exhibited a reverse affinity with a selectivity index equaled to 4.6.
Fig. 4.

Crystal binding mode and predicted binding orientations of (A) celecoxib, (B) ketoprofen, (C) A13 visualized in COX-2 active site (PDB code 5KIR); ligands are shown in orange and amino acids are shown in blue. Only the interacting residues and their interaction patterns are shown
The aforementioned precious anti-inflammatory activity of A8 compound against COX-1 and COX-2 isozymes (0.31 and 0.87 μM) was boosted via further reporting antimicrobial activity against two serious strains of gram-negative bacteria (P. aeruginosa and K. pneumonia) and antifungal activity against C. albicans within acceptable ranges. These observed activities are continued to be examined within selected decisive proteins related to gram-negative bacteria, Elastase of P. aeruginosa and KPC-2 carbapenemase of K. pneumonia utilizing the crystallized enzymes of PDB ID 1U4G and 2OV5, respectively. Additionally, the interaction profile of the A8-molecule was compared to crystallized fluconazole within the Cytochrome P450 14 alpha-sterol demethylase (PDB ID 1AE1) in order to state the antifungal activity observed. All the docking data are shown in Fig. 5, various kinds of binding interactions were predicted between this compound and possible targets of strains.
Fig. 5.

The predicted binding orientations of ciprofloxacin and A8, visualized in a the Elastase of P. aeruginosa (PDB ID 1U4G). and b K. pneumonia KPC-2 carbapenemase (PDB ID 2OV5). The crystal binding mode of c fluconazole and predicted binding orientations of A8, visualized in cytochrome P450 14 α-sterol demethylase (PDB ID 1EA1); ligands are shown in orange and amino acids are shown in blue. Only the interacting residues and their interaction patterns are shown
In return to Fig. 4 and supplementary materials (Figs S2 and S3), discussing the molecular docking studies in more detail reveal that compound B2 and A13 exhibited the best COX-2 selectivity index (IC50COX-1/ IC50COX-2) that equals 20.7 and 4.6, respectively. Compared to the tested FDA-approved drugs (celecoxib and ketoprofen) as controls, B2 and A13 compounds revealed a greater COX-2 inhibition activity than ketoprofen but with a lower selectivity index than celecoxib. Starting from investigating the interaction pattern of celecoxib as an ideal selective COX-2 ligand, the binding site is optimally fitted, including the secondary binding domain, and forms highly favorable five hydrogen bonds (bond lengths about 2 Ao) with ARG-513 (ameliorated COX-2 residue), HIS-90, SER-353, ILE-517, and PHE-518. Additionally, celecoxib ordinary set within the binding site via forming favorable hydrophobic interactions with eight residues that mainly participate in boosting affinity. Regarding the predicted interaction profile of ketoprofen, the inability to associate deeply with the secondary polar domain at the right side led to the loss of critical interactions with ARG-513 and HIS-90, resulting in loss of selectivity and decreased affinity toward the COX-2 enzyme. The moderate activity of ketoprofen is mainly the result of forming a lot of hydrophobic interactions at the left side (with at least 8 residues) and two hydrogen bonds with TYR-385 and SER-530. The interaction patterns of both celecoxib and ketoprofen within the binding domain of the COX-2 enzyme were identical to the previously reported studies (Plount Price and Jorgensen 2000; Orlando et al. 2015).
Substituting the phenyl-amide ring with 3,4-dimethoxy in A13-compound led to pushing in the 5-methyl-isoxazole ring toward the secondary binding pocket and thus could occupy that domain partially and forming a hydrogen bond with ARG-513. The favorable conformation of A13 that exists within the binding site results in forming additional three hydrogen bonds with TYR-355, TYR-385, and SER-530, besides many hydrophobic interactions. The presence of Cl at position-2 at the phenyl-isoxazole ring participated mainly in optimizing the ligand conformation to fit the binding domain more. A13 molecule showed better affinity than A12 or A14 when the 3,4-dimethoxy changed to 2,5 or 3,5 dimethoxy, respectively. Docking studies here showed that the steric clashes of 2,5 or 3,5 dimethoxy pushed the isoxazole ring out of the secondary binding site leading to the loss of the ameliorating interactions with ARG-513. Replacing 2-Cl with 4-F in A9-compound led to a decrease in activity by fivefold due to the generated steric clashes with the surrounded residues. Removing the 2-Cl substituent at A4-compound resulted in a few decreases in potency but kept it in an arrange of 0.2 μM due to the favorable conformation of the 3,4-dimethoxy phenyl ring. Regarding the other non-substituted phenyl-isoxazole ring, shifting the 4-methoxy in A4 to 5-methoxy in A3 resulted in changing some residues directions that led to a decrease in the number of hydrophobic interactions from eleven to just seven with the surrounding residues.
The addition of t-butyl at para-position at compound A5 led to a further decrease in affinity due to flipping the molecule and the t-butyl exposed to the second binding domain so losing more favorable interactions. The free substituted phenyl-amide ring in compound-A1 showed better affinity than 3,4,5 trimethoxy phenyl-amide in compound-A2 by threefold but lower than the dimethoxy substituted compounds. However, A2 molecules formed three favorable hydrogen bonds with the surrounding residues, including ARG-513. Just one methoxy formed a hydrogen bond interaction, and the other two methoxy groups paid desolvation and conformation penalties that resulted in a remarkable decrease in affinity. Compound A1 formed at least 13 hydrophobic interactions that participated mainly in the binding affinity. As noticed in general, in all carboxamide derivatives, substituting the 4-position of the phenyl-amide ring with F (compounds—A6, A7, A8, A10, and A11) resulted in more conformation penalties and steric clashes inside the COX-2 binding site resulted in decreasing the binding affinity, especially if the phenyl-amide ring is substituted with 3,4 or 3,5 dimethoxy groups in A9 and A8, respectively. Concerning the carboxylic acid derivatives, the existence of fluor at para-position (Compound-B2) led to increasing the lipophilicity of the phenyl ring connected to the isoxazole ring as a result of blocking the π-resonance thus, creating sturdy hydrophobic interactions within the binding site and robust the affinity to about 0.033 μM (Meanwell 2018; Griffen et al. 2018).
Squandering these advantageous features by replacing F with Cl (larger size and could activate the resonance of π-electrons) at position-2 or removing the F atom in compounds B1 and B3, respectively, resulted in highly diminished affinity. Compared to the other 4-fluoro carboxamide derivatives, B2 had a higher affinity. This is because it was able to get more favorable interactions and had the least number of conformations and steric penalties.
Regarding the docking studies within the COX-1 isozyme, the compounds A13, A14, and A4 revealed significant binding affinity toward the binding domain of COX-1, which was comparable to the binding affinity of ketoprofen (located within the range 0.064–0.076 μM). The three compounds showed a low selectivity index (below 0.3) which means that they have a higher affinity toward COX-1 than COX-2 except for compound A13, which exhibited a reverse affinity with a selectivity index equaled to 4.6.
The high affinity of ketoprofen toward the COX-1 binding site is retrieved via forming an extremely favorable salt bridge between the positively charged guanidine group of ARG-120 residue with the negatively charged carboxylate group of ketoprofen which greatly contributed to the further deepening of the molecule within the binding site. Additionally, the biphenyl ketone fitted optimally within the binding residues and succeeded in forming hydrophobic interactions with VAL-116, VAL-349, LEU-352, LEU-359, TYR-385, TRP-387, PHE-518, and ALA-527. However, celecoxib within the COX-1 binding site can form many hydrogen bonds and hydrophobic interactions, but the presence of ILE-523 in COX-1 instead of VAL-523 in COX-2 isozyme caused to diminish the cavity space (by 25%) available to catch celecoxib optimally. This led to restricting the entrance of the sulfonamide group to the relatively polar side pocket beside the steric clashes exposed to the trifluoromethyl group (Plount Price and Jorgensen 2000).
Analyzing the docking data showed that all the carboxamide derivatives (except A1) exhibited a binding affinity over the carboxylic acid derivatives and celecoxib. The incorporation of phenyl amide provided more favorable conformations and fit the binding site of COX-1more properly. The existence of Cl at position 2 of phenyl isoxazole ring coupled to 3,4 dimethoxy-phenyl amide caused to set A13-compound optimally at ideal binding distances. The 4-methoxy group entered perfectly into the relatively polar side pocket-forming hydrogen bonds with HIS-90, GLN-192, and SER-516. The 3-methoxy group and carbonyl formed hydrogen bonds with ILE-517 and TYR-355, respectively. Comparing the binding pattern of other synthesized compounds to A13 as a reference, eliminating the Cl group or replacing the 3,4 dimethoxy with 2,5 dimethoxy in compounds A4 and A14, respectively, led to a slight deviation in the conformation besides desolvation penalties that finally caused a minimal decrease in the binding affinity. Keeping the 2-Cl group in compound A13, but replacing 3,4 dimethoxy with 3,5 dimethoxy resulted in flipping the conformation of the A12 compound and creating high steric clashes exposed to the dimethoxy groups which led to decreased potency by sevenfold. Substituting position 4 of carboxamide derivatives (compounds A6-A10) instead of 2-Cl led to a decrease in the potency located within a narrow range (0.12–0.4 μM). It is expected that the large size and the preferred position 2 of the Cl atom could play an essential role in directing the molecules to occupy and fit the binding site more optimally than F at position 4.
Non-substituting, the phenyl isoxazole ring of carboxamide derivatives (compounds A1-A5) revealed various binding affinities that ranged from 0.076 μM in compound A4 to 0.839 μM in compound A1. The affinity is differentiated according to the position and type of substituents at the phenyl-amide ring. The 3,4 dimethoxy showed the best binding pattern within the non-substituting phenyl isoxazole group and could partially enter the relatively polar side pocket-forming many favorable interactions. Replacing it with 3,5 dimethoxy or 3,4,5-trimethoxy in compounds A3 and A2, respectively, resulted in pushing it away from the relative polar side and flipping the methoxy group to the lipophilic part, which led to losing the ability to form hydrogen bonds. Eliminating the halogen at the phenyl isoxazole ring and methoxy groups at the phenyl amide ring in compound-A1 resulted in a further decrease in potency due to losing advantageous filling and conformation character caused by substituents.
The carboxylic acid derivatives (B1-B3) exhibited weaker affinity than carboxamide derivatives. Docking these compounds to the COX-1 enzyme showed lower interacting residues without any propitious interactions besides paying desolvation penalties due to the non-bonding carboxylic acid group. This non-preferable interaction profile observed low inhibition activity when tested against COX-1 isozyme. The binding profiles of celecoxib and ketoprofen were similar to previously recorded data (Plount Price and Jorgensen 2000; Viegas et al. 2011).
Concerning both gram-negative strains, the ciprofloxacin was used as a reference drug which is FDA-approved potent antimicrobial agent. Docking of ciprofloxacin against the Elastase of P. aeruginosa (PDB ID 1U4G) showed very favorably positively contributing binding interactions within the binding site including two salt bridges with HIS-140 and HIS-144 beside three optimally oriented and distanced hydrogen bonds with GLU-141, HIS-223, and ALA-113, and ASP-221 with the carboxylic acid group. The high basicity of terminal the nitrogen of the piperazine ring is involved in fitting the molecule's conformation optimally inside the binding domain via forming a hydrogen bond with the carboxylate group of ASP-221. For the A8 molecule, a valuable interaction profile was observed within the binding site of the Elastase enzyme. The four generated hydrogen bonds optimized the conformation, matching the space available for binding.
Regarding the docking studies within KPC-2 carbapenemase of K. pneumonia (PDB ID 2OV5), ciprofloxacin settled ordinary inside the binding domain via constructing highly favorable binding interaction including two salt bridges with LYS-73 and LYS-235, five hydrogen bonds with SER-130, THR-237, THR-235, and GLU-276, supported with hydrophobic interaction with many residues. Otherwise, docking the A8 molecule did not show hydrogen bonds but was able to form a water bridge between the isoxazole ring and THR-216 residue. Therefore, the activity of the A8 molecule is expected to be retrieved from the hydrophobic interactions with the surrounding residues.
Docking of compound A8 to both enzymes (Elastase and KPC-2 carbapenemase) cleared that the methoxy substituents played a negative role through generating steric clashes with the exposed residues beside desolvation penalties that led to decreased affinity compared to ciprofloxacin. It’s supposed that replacing the methoxy group with a smaller lipophilic group is supposed to enhance the antimicrobial activity of compound A8 against both gram-negative bacterial strains.
The binding mode of crystallized fluconazole within the binding site of Cytochrome P450-14-alpha-sterol demethylase (PDB ID 1AE1) was investigated and taken as a control to explain the antifungal activity of the A8-compound. Concerning fluconazole, the triazole ring coordinated to heme iron (with a distance equal to 3.8 Ao) resulted in positioning fluconazole almost perpendicular to the porphyrin plan (Fe+2-cofactor). Other polar interactions were observed, including three salt bridges and hydrogen bonds with at least seven residues. Besides the ability to coordinate to heme cofactor, the capability to generate hydrophobic interactions is also crucial to match the binding site of Cytochrome P450-14-alpha-sterol demethylase (Hargrove et al. 2017), which could be observed here regarding fluconazole. This unique interaction profile succeeded in settling the molecule optimally inside the binding domain resulting in high antifungal activity. Docking the A8 molecule showed the proper accommodation over the porphyrin plan but with a longer distance (measured to 4.9 Ao), resulting in minimizing the participation of that critical bond in the binding energy. Ability to form sufficient hydrophobic interaction with surrounding lipophilic residues (PHE-78, PHE-83, and LEU-100 relatively boosted the binding affinity. A8 showed prorated fitting compared to fluconazole, concluded to slightly decrease in antifungal activity but still considered a promising ligand as an antifungal agent.
All the examined binding patterns of reference used drugs utilized in the cyclooxygenase COX-1 and COX-2, antibacterial, and antifungal assays besides the newly synthesized compounds are shown in Figs. 4, S2, and S3.
Molecular dynamics simulations
Molecular dynamic simulation is defined as a technique applied to investigate the conformation changes through simulating the whole protein–ligand within a course of a certain time (Lindorff‐Larsen et al. 2010). For a given period time of simulation, various trajectories are generated through applying Newton’s law of motion to the macromolecular system. As a result of analyzing the coordinates of trajectories points, certain parameters such as the root mean square deviation (RMSD) could be obtained and analyzed to realize the stability of protein–ligand complex (Meena et al. 2022; Kumar et al. 2019, 2020; Vishvakarma et al. 2015).
The RMSD plot in MD simulation is used to investigate the compactness of protein after the ligand-induced fit into.
it (Abraham et al. 2015). It is calculated by rooting the average value of the square of atomic coordinates of backbone atoms. The less fluctuation in the RMSD values (within 3 angstroms) of the ligand–protein complex indicated greater stability, thus successful fit or docking.
The conformational stability of the A13-COX-2 (PDB ID 5KIR), A8-Klebsiella P. KPC-2 carbapenemase (PDB ID 2OV5), A8-Elastase of P. aeruginosa (PDB ID 1U4G), and A8-cytochrome P450 14 α-sterol demethylase (PDB ID 1EA1) complexes along with the molecular dynamics trajectories was evaluated from the backbone and ligand RMSD plots. The RMSD plots of the fitted ligands and proteins backbones were extracted, and the time series of RMSD values within 100 ns are shown in Fig. 6A–C, respectively. All the proteins’ backbones showed trivial fluctuations (within 0.15 nm) as shown in RMSD plots that reached the plateau of equilibrium at approximately 20 ns of simulation trajectory. The A13-COX-2 complex presented an ideal ligand and backbone RMSD plots which showed minimal RMSD fluctuation (0.2 nm) at the first 20 ns of molecular dynamic simulations and then retained a stable state. With respect to the A8- KPC-2 carbapenemase complex, the A8 structure revealed strong fluctuation at the initial simulation trajectory and the plateau remained stable directly after that until 80 ns of simulation trajectory then showed high dynamicity (RMSD ranged within 1.5 nm) for a few nanoseconds and tried to return to the stable state within the last time of trajectory simulation. The A8 structure upon fitting to the binding site of elastase protein of P. aeruginosa presented high dynamicity (RMSD = 1.6 nm), especially at the primary simulation time and after 50 ns showed a weak plateau and all fluctuations were within the normal range ( less than 2 nm) which indicate the stability state of that complex. The A8-ligand revealed lighter dynamicity (RMSD = 0.3 nm) within the cytochrome P450 14 α-sterol demethylase protein and retained a plateau state after the first 5 ns of simulation trajectory.
Fig. 6.

The time-series of RMSD values of MD simulation trajectory. The RMSD plot obtained for A A13—COX2 complex, B A8—K. Pneumonia KPC-2 carbapenemase, (C) A8—Elastase of Pseudomonas aeruginosa, and (D) A8—cytochrome P450 14 α-sterol demethylase for 100 ns of simulation trajectory at 300 K temperature. The figures present the RMSD plots of proteins’ backbones (red) and ligands (blue). The RMSD values were calculated with respect to the initial structures
Another parameter obtained to measure the strength of the ligand–protein complexes and macromolecular systems is the radius of gyration (Rg). Considering the trajectories points obtained from MD simulations, it calculates the distance between the center of mass and the axis of rotation. In term of Rg, less the average value of Rg indicates a more compact macromolecular system or ligand–protein complex, while less fluctuations in Rg values means more conformational stability of the system.
As shown in Fig. 7, the complexes A13-COX-2 and A8-cytochrome P450 14 α-sterol demethylase present an Rg average value within 2–2.5 nm, while the A8-Klebsiella P. KPC-2 carbapenemase and A8-Elastase of P. aeruginosa complexes showed Rg average values within 1.5 – 2 nm. All complexes showed less fluctuation in the Rg values and did not exceed 0.5 nm which indicates the high conformational stability and system compactness for a simulation trajectory of 100 ns and 300 K temperature..
Fig. 7.

The radius of gyration plots for A13-COX2 (blue), A8—K.Pneumonia KPC-2 carbapenemase (red), A8—Elastase of Pseudomonas aeruginosa (green), A8—cytochrome P450 14 α-sterol demethylase (yellow) for 100 ns of simulation trajectory at 300 K temperature
ADME-T analysis
The predictive ADME module (Qikprop) within the Maestro-schrödinger software was applied to predict virtually a lot of physicochemical properties (ADME-T parameters) of our new molecules to check if they could proceed to the next stages of study potentially. As shown in supplementary data Table S3, all our new tested molecules (without any exceptions) demonstrated outstanding and optimum ADME-T values within the recommended doses. These data, alongside the previously reported biological results, give a hopeful conception of these newly designed ligands.
Conclusion
To sum up, this study demonstrates the discovery of a series of COX-1 and COX-2 inhibitors that was recorded with a moderate selectivity index (SI) toward the COX-2 isozyme like B2- and A13-compounds, if compared to the FDA-approved COX-2 selective drug (celecoxib). But if compared to ketoprofen, as a non-selective COX inhibitor, the compounds A13, A-14, and A4 showed comparable inhibition activity against the COX-1 isozyme and selectivity toward COX-2. Indeed, a feature that could be figured in this study is the discovery of new drugs with tertiary anti-inflammatory, antibacterial, and antifungal actions. Compound A8 could crucify two gram-negative invasion strains alongside the antifungal and anti-inflammatory activities toward C. albicans and COX, respectively. Studying the safety and anti-proliferation of our newly discovered ligands against LX-2 cell cultures (normal hepatic cell line) did not show any cytotoxic effect. The results of the in vitro assays applied here were supported by investigating the ligand-receptor interaction profile and stability within the binding domain via conducting molecular docking and molecular dynamic simulations studies. All the complexes studied showed favorable interactions patterns within the binding pocket that explicated that distinctive results observed and showed high stability for 100 ns of molecular dynamic simulation trajectory. Additionally, analyzing the chemical structures utilizing the QiKProp module to check the future drug properties via calculating the ADME-T parameters showed the existence of all ligands within the ideal and optimal ranges, which supports the outcomes of the in vitro assay obtained here to push ahead to further investigation assays, including clinical studies.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank An-Najah National University's Faculty of Medicine and Health Sciences for their support.
Funding
No funds, grants, or other support was received.
Data availability
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Adasme MF, Linnemann KL, Bolz SN, Kaiser F, Salentin S, Haupt VJ, Schroeder M. PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021;49(W1):W530–W534. doi: 10.1093/nar/gkab294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assali M, Abualhasan M, Sawaftah H, Hawash M, Mousa A. Synthesis, biological activity, and molecular modeling studies of pyrazole and triazole derivatives as selective COX-2 inhibitors. J Chem. 2020 doi: 10.1155/2020/6393428. [DOI] [Google Scholar]
- Bacchi S, Palumbo P, Sponta A, Coppolino MF. Clinical pharmacology of non-steroidal anti-inflammatory drugs: a review. Antiinflamm Antiallergy Agents Med Chem. 2012;11(1):52–64. doi: 10.2174/187152312803476255. [DOI] [PubMed] [Google Scholar]
- Badrey MG, Abdel-Aziz HM, Gomha SM, Abdalla MM, Mayhoub AS. Design and synthesis of imidazopyrazolopyridines as novel selective COX-2 inhibitors. Molecules. 2015;20(8):15287–15303. doi: 10.3390/molecules200815287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EW, Zhang Y. DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J Cheminformatics. 2019;11(1):1–9. doi: 10.1186/s13321-019-0362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommagani MB, Yerrabelly JR, Chitneni M, Thalari G, Vadiyala NR, Boda SK, Chitneni PR. Synthesis and antibacterial activity of novel cinnoline-isoxazole derivatives. Chem Data Collect. 2021;31:100629. doi: 10.1016/j.cdc.2020.100629. [DOI] [Google Scholar]
- Burdan F, Chałas A, Szumiło J. Cyklooksygenaza i prostanoidy–znaczenie biologiczne* cyclooxygenase and prostanoids–biological implications. Postepy Hig Med Dosw. 2006;60:129–141. [PubMed] [Google Scholar]
- de Groot RA, Nadrchal J (1993) Physics Computing'92: Proceedings of the 4th International Conference. In: Physics Computing'92: Proceedings of the 4th International Conference. Edited by NADRCHAL J ET AL. Published by World Scientific Publishing Co. Pte. Ltd
- Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven BA, Gowan S, Boxall F. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Can Res. 2008;68(8):2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- Eid AM, Hawash M, Amer J, Jarrar A, Qadri S, Alnimer I, Sharaf A, Zalmoot R, Hammoudie O, Hameedi S. Synthesis and biological evaluation of novel isoxazole-amide analogues as anticancer and antioxidant agents. BioMed Res Int. 2021 doi: 10.1155/2021/6633297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elokely KM, Doerksen RJ. Docking challenge: protein sampling and molecular docking performance. J Chem Inf Model. 2013;53(8):1934–1945. doi: 10.1021/ci400040d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345(6):433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Galdino ACM, de Oliveira MP, Ramalho TC, de Castro AA, Branquinha MH, Santos AL. Anti-virulence strategy against the multidrug-resistant bacterial pathogen Pseudomonas aeruginosa: pseudolysin (elastase B) as a potential druggable target. Curr Protein Pept Sci. 2019;20(5):471–487. doi: 10.2174/1389203720666190207100415. [DOI] [PubMed] [Google Scholar]
- Ghabbour HA, Qabeel MM, Eldehna WM, Al-Dhfyan A, Abdel-Aziz HA. Design, synthesis, and molecular docking of 1-(1-(4-chlorophenyl)-2-(phenylsulfonyl) ethylidene)-2-phenylhydrazine as potent nonazole anticandidal agent. J Chem. 2014 doi: 10.1155/2014/154357. [DOI] [Google Scholar]
- Griffen EJ, Dossetter AG, Leach AG, Montague S. Can we accelerate medicinal chemistry by augmenting the chemist with big data and artificial intelligence? Drug Discov Today. 2018 doi: 10.1016/j.drudis.2018.03.011. [DOI] [PubMed] [Google Scholar]
- Hargrove TY, Friggeri L, Wawrzak Z, Qi A, Hoekstra WJ, Schotzinger RJ, York JD, Guengerich FP, Lepesheva GI. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J Biol Chem. 2017;292(16):6728–6743. doi: 10.1074/jbc.M117.778308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawash M, Eid AM, Jaradat N, Abualhasan M, Amer J, Naser Zaid A, Draghmeh S, Daraghmeh D, Daraghmeh H, Shtayeh T, Sawaftah H, Mousa A. Synthesis and biological evaluation of benzodioxole derivatives as potential anticancer and antioxidant agents. Heterocycl Commun. 2020;26(1):157–167. doi: 10.1515/hc-2020-0105. [DOI] [Google Scholar]
- Hawash M, Jaradat N, Hameedi S, Mousa A. Design, synthesis and biological evaluation of novel benzodioxole derivatives as COX inhibitors and cytotoxic agents. BMC Chem. 2020;14(1):1–9. doi: 10.1186/s13065-020-00706-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawash M, Jaradat N, Abualhasan M, Amer J, Levent S, Issa S, Ibrahim S, Ayaseh A, Shtayeh T, Mousa A. Synthesis, chemo-informatics, and anticancer evaluation of fluorophenyl-isoxazole derivatives. Open Chem. 2021;19(1):855–863. doi: 10.1515/chem-2021-0078. [DOI] [Google Scholar]
- Hawash M, Jaradat N, Bawwab N, Salem K, Arafat H, Hajyousef Y, Shtayeh T, Sobuh S. Design, synthesis, and biological evaluation of phenyl-isoxazole-carboxamide derivatives as anticancer agents. Heterocycl Commun. 2021;27(1):133–141. doi: 10.1515/hc-2020-0134. [DOI] [Google Scholar]
- Hawash M, Kahraman DC, Cetin-Atalay R, Baytas SN. Induction of apoptosis in hepatocellular carcinoma cell lines by novel indolylacrylamide derivatives: synthesis and biological evaluation. Chem Biodivers. 2021;18(5):e2001037. doi: 10.1002/cbdv.202001037. [DOI] [PubMed] [Google Scholar]
- Hawash M, Kahraman DC, Ergun SG, Cetin-Atalay R, Baytas SN. Synthesis of novel indole-isoxazole hybrids and evaluation of their cytotoxic activities on hepatocellular carcinoma cell lines. BMC Chem. 2021;15(1):1–14. doi: 10.1186/s13065-021-00793-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawash M, Jaradat N, Abualhasan M, Qneibi M, Rifai H, Saqfelhait T, Shqirat Y, Nazal A, Omarya S, Ibrahim T, Sobuh S, Zarour A, Mousa A. Evaluation of cytotoxic, COX inhibitory, and antimicrobial activities of novel isoxazole-carboxamide derivatives. Lett Drug Des Discov. 2022;19:1–1. doi: 10.2174/1570180819666220819151002. [DOI] [Google Scholar]
- Hermann M, Ruschitzka F. Coxibs, non-steroidal anti-inflammatory drugs and cardiovascular risk. Intern Med J. 2006;36(5):308–319. doi: 10.1111/j.1445-5994.2006.01056.x. [DOI] [PubMed] [Google Scholar]
- Schrödinger L. Schrödinger suite. LLC, New York, NY: Schrödinger; 2016. [Google Scholar]
- Jaradat N, Al-lahham S, Abualhasan MN, Bakri A, Zaide H, Hammad J, Hussein F, Issa L, Mousa A, Speih R. Chemical constituents, antioxidant, cyclooxygenase inhibitor, and cytotoxic activities of teucrium pruinosum boiss. Essent Oil Biomed Res Int. 2018;18:1–9. doi: 10.1155/2018/4034689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaradat N, Khasati A, Hawi M, Qadi M, Amer J, Hawash M. In vitro antitumor, antibacterial, and antifungal activities of phenylthio-ethyl benzoate derivatives. Arab J Sci Eng. 2021;46(6):5339–5344. doi: 10.1007/s13369-020-05114-0. [DOI] [Google Scholar]
- Kalle AM, Rizvi A. Inhibition of bacterial multidrug resistance by celecoxib, a cyclooxygenase-2 inhibitor. Antimicrob Agents Chemother. 2011;55(1):439–442. doi: 10.1128/AAC.00735-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil A, Jaradat N, Hawash M, Issa L. In vitro biological evaluation of benzodioxol derivatives as antimicrobial and antioxidant agents. Arab J Sci Eng. 2021;46(6):5447–5453. doi: 10.1007/s13369-021-05332-0. [DOI] [Google Scholar]
- Kumar JD, Zanderigo F, Prabhakaran J, Rubin-Falcone H, Parsey RV, Mann JJ. In vivo evaluation of [11C] TMI, a COX-2 selective PET tracer, in baboons. Bioorg Med Chem Lett. 2018;28(23–24):3592–3595. doi: 10.1016/j.bmcl.2018.10.049. [DOI] [PubMed] [Google Scholar]
- Kumar D, Singh P, Jayaraj A, Kumar V, Kumari K, Patel R. A theoretical model to study the interaction of erythro-noscapines with nsP3 protease of chikungunya virus. ChemistrySelect. 2019;4(17):4892–4900. doi: 10.1002/slct.201803360. [DOI] [Google Scholar]
- Kumar D, Singh P, Jayaraj A, Kumar V, Kumari K, Chandra R, Ramappa VK. Selective docking of pyranooxazoles against nsP2 of CHIKV eluted through isothermally and non-isothermally MD simulations. ChemistrySelect. 2020;5(14):4210–4220. doi: 10.1002/slct.202000768. [DOI] [Google Scholar]
- Kumar S, Gupta Y, Zak SE, Upadhyay C, Sharma N, Herbert AS, Durvasula R, Potemkin V, Dye JM, Kempaiah P. A novel compound active against SARS-CoV-2 targeting uridylate-specific endoribonuclease (NendoU/NSP15): in silico and in vitro investigations. RSC Medicinal Chemistry. 2021;12(10):1757–1764. doi: 10.1039/D1MD00202C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the amber ff99SB protein force field. Proteins Struct Funct Bioinform. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton MF, Fazio S. Cyclooxygenase products and atherosclerosis. Drug Discov Today: Ther Strateg. 2008;5(1):25–36. doi: 10.1016/j.ddstr.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magpantay HD, Malaluan IN, Manzano JAH, Quimque MT, Pueblos KR, Moor N, Budde S, Bangcaya PS, Lim-Valle D, Dahse H-M. Antibacterial and COX-2 inhibitory tetrahydrobisbenzylisoquinoline alkaloids from the Philippine medicinal plant Phaeanthus ophthalmicus. Plants. 2021;10(3):462. doi: 10.3390/plants10030462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahboubi Rabbani SMI, Zarghi A. Selective COX-2 inhibitors as anticancer agents: a patent review (2014–2018) Expert Opin Ther Pat. 2019;29(6):407–427. doi: 10.1080/13543776.2019.1623880. [DOI] [PubMed] [Google Scholar]
- Malathi K, Anbarasu A, Ramaiah S. Identification of potential inhibitors for Klebsiella pneumoniae carbapenemase-3: a molecular docking and dynamics study. J Biomol Struct Dyn. 2019;37(17):4601–4613. doi: 10.1080/07391102.2018.1556737. [DOI] [PubMed] [Google Scholar]
- Mao J, Yuan H, Wang Y, Wan B, Pieroni M, Huang Q, Van Breemen RB, Kozikowski AP, Franzblau SG. From serendipity to rational antituberculosis drug discovery of mefloquine-isoxazole carboxylic acid esters. J Med Chem. 2009;52(22):6966–6978. doi: 10.1021/jm900340a. [DOI] [PubMed] [Google Scholar]
- Meanwell NA. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J Med Chem. 2018;61(14):5822–5880. doi: 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Meena MK, Kumar D, Kumari K, Kaushik NK, Kumar RV, Bahadur I, Vodwal L, Singh P. Promising inhibitors of nsp2 of CHIKV using molecular docking and temperature-dependent molecular dynamics simulations. J Biomol Struct Dyn. 2022;40(13):5827–5835. doi: 10.1080/07391102.2021.1873863. [DOI] [PubMed] [Google Scholar]
- Nacak S, Ökçelik B, Ünlü S, Şahin MF, Özkan S, Abbasoğlu U. Synthesis and antimicrobial activity of some new mannich bases of 7-acyl-5-chloro-2-oxo-3H-benzoxazole derivatives. Turk J Pharm Sci. 2005;2(1):25–33. [Google Scholar]
- Orlando BJ, Lucido MJ, Malkowski MG. The structure of ibuprofen bound to cyclooxygenase-2. J Struct Biol. 2015;189(1):62–66. doi: 10.1016/j.jsb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Chowdary PR, Jayashree B. Synthesis, antiinflammatory and antibacterial activity of novel indolyl-isoxazoles. Indian J Pharm Sci. 2009;71(6):684. doi: 10.4103/0250-474X.59554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panwar U, Singh SK. Atom-based 3D-QSAR, molecular docking, DFT, and simulation studies of acylhydrazone, hydrazine, and diazene derivatives as IN-LEDGF/p75 inhibitors. Struct Chem. 2021;32(1):337–352. doi: 10.1007/s11224-020-01628-3. [DOI] [Google Scholar]
- Paragi-Vedanthi P, Doble M. Comparison of PGH2 binding site in prostaglandin synthases. BMC Bioinformatics. 2010;11(1):1–8. doi: 10.1186/1471-2105-11-S1-S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol. 2006;119(3):229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Plount Price ML, Jorgensen WL. Analysis of binding affinities for celecoxib analogues with COX-1 and COX-2 from combined docking and Monte Carlo simulations and insight into the COX-2/COX-1 selectivity. J Am Chem Soc. 2000;122(39):9455–9466. doi: 10.1021/ja001018c. [DOI] [Google Scholar]
- Rao P, Knaus EE. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci. 2008;11(2):81s–110s. doi: 10.18433/j3t886. [DOI] [PubMed] [Google Scholar]
- Ricardo E, Costa-de-Oliveira S, Silva Dias A, Guerra J, Rodrigues AG, Pina-Vaz C. Ibuprofen reverts antifungal resistance on Candida albicans showing overexpression of CDR genes. FEMS Yeast Res. 2009;9(4):618–625. doi: 10.1111/j.1567-1364.2009.00504.x. [DOI] [PubMed] [Google Scholar]
- Santos MM, Faria N, Iley J, Coles SJ, Hursthouse MB, Martins ML, Moreira R. Reaction of naphthoquinones with substituted nitromethanes. Facile synthesis and antifungal activity of naphtho [2, 3-d] isoxazole-4, 9-diones. Bioorganic Med Chem Lett. 2010;20(1):193–195. doi: 10.1016/j.bmcl.2009.10.137. [DOI] [PubMed] [Google Scholar]
- Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Isakson P. Eicosanoids and other bioactive lipids in cancer, inflammation, and radiation injury 2. Springer; 1997. Distribution of COX-1 and COX-2 in normal and inflamed tissues; pp. 167–170. [DOI] [PubMed] [Google Scholar]
- Sharma S, Kumar-M P, Singh S, Sohal A, Yadav R, Gupta N. Discovery of dual inhibitors of KPC-3 and KPC-15 of Klebsiella pneumoniae–an in-silico molecular docking and dynamics study. bioRxiv. 2021 doi: 10.1101/2019.12.23.886671. [DOI] [Google Scholar]
- Smith WL, Urade Y, Jakobsson P-J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem Rev. 2011;111(10):5821–5865. doi: 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangamani S, Younis W, Seleem MN. Repurposing celecoxib as a topical antimicrobial agent. Front Microbiol. 2015;6:750. doi: 10.3389/fmicb.2015.00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aalten DM, Bywater R, Findlay JB, Hendlich M, Hooft RW, Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J Comput Aided Mol Des. 1996;10(3):255–262. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104(3S1):2S–8S. doi: 10.1016/S0002-9343(97)00203-9. [DOI] [PubMed] [Google Scholar]
- Vane J, Botting R. The mechanism of action of aspirin. Thromb Res. 2003;110(5–6):255–258. doi: 10.1016/S0049-3848(03)00379-7. [DOI] [PubMed] [Google Scholar]
- Viegas A, Manso J, Corvo MC, Marques MMB, Cabrita EJ. Binding of ibuprofen, ketorolac, and diclofenac to COX-1 and COX-2 studied by saturation transfer difference NMR. J Med Chem. 2011;54(24):8555–8562. doi: 10.1021/jm201090k. [DOI] [PubMed] [Google Scholar]
- Vishvakarma VK, Kumari K, Patel R, Dixit V, Singh P, Mehrotra GK, Chandra R, Chakrawarty AK. Theoretical model to investigate the alkyl chain and anion dependent interactions of gemini surfactant with bovine serum albumin. Spectrochim Acta Part A Mol Biomol Spectrosc. 2015;143:319–323. doi: 10.1016/j.saa.2015.01.068. [DOI] [PubMed] [Google Scholar]
- Zarghi A, Ghodsi R. Design, synthesis, and biological evaluation of ketoprofen analogs as potent cyclooxygenase-2 inhibitors. Bioorg Med Chem. 2010;18(16):5855–5860. doi: 10.1016/j.bmc.2010.06.094. [DOI] [PubMed] [Google Scholar]
- Zervou M, Andreou A, Goulielmos G, Eliopoulos E. AB0004 the association of the rare RS35667974 IFIH1 gene polymorphism with six autoimmune diseases: structural biological insights. BMJ Publishing Group Ltd; 2021. [Google Scholar]
- Zhang D, Jia J, Meng L, Xu W, Tang L, Wang J. Synthesis and preliminary antibacterial evaluation of 2-butyl succinate-based hydroxamate derivatives containing isoxazole rings. Arch Pharmacal Res. 2010;33(6):831–842. doi: 10.1007/s12272-010-0605-7. [DOI] [PubMed] [Google Scholar]
- Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. Cyclooxygenase in normal human tissues–is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med. 2009;13(9b):3753–3763. doi: 10.1111/j.1582-4934.2008.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.