

Abstract
Cell death is a crucial event underlying cardiac ischemic injury, pathological remodeling, and heart failure. Unlike apoptosis, necrosis had long been regarded as a passive and unregulated process. However, recent studies demonstrate that a significant subset of necrotic cell death is actively mediated through regulated pathways - a process known as “regulated necrosis”. As a form of regulated necrosis, necroptosis is mediated by death receptors and executed through the activation of receptor interacting protein kinase 3 (RIPK3) and its downstream substrate mixed lineage kinase-like domain (MLKL). Recent studies have provided compelling evidence that necroptosis plays an important role in myocardial homeostasis, ischemic injury, pathological remodeling, and heart failure. Moreover, it has been shown that genetic and pharmacological manipulations of the necroptosis signaling pathway elicit cardioprotective effects. Important progress has also been made regarding the molecular mechanisms that regulate necroptotic cell death in vitro and in vivo. In this review, we discuss molecular and cellular mechanisms of necroptosis, potential crosstalk between necroptosis and other cell death pathways, functional implications of necroptosis in heart disease, and new therapeutic strategies that target necroptosis signaling.
Keywords: Apoptosis, Necrosis, Necroptosis, Myocardial infarction, Heart failure
1. Introduction
Cell death has been implicated in the pathogenesis of multiple forms of heart disease such as myocardial infarction (MI), ischemia/reperfusion injury (I/R), congenital heart disease, and heart failure of diverse etiologies [1–4]. Cardiomyocyte death is the central event during MI that drives acute ischemic injury and adverse remodeling. Reperfusion therapy is effective in mitigating ischemic damage during MI, but it induces further myocyte death, a process referred to as I/R injury. During heart failure, which is a highly heterogeneous syndrome in terms of etiologies, the rate of cardiomyocyte death is relatively low compared to MI, but it is an important component in disease progression. Genetic perturbations of various cell death signaling pathways in mice have been shown to elicit cardioprotective effects.
Apoptosis is the first recognized form of regulated cell death, which is tightly regulated by death receptor- or mitochondria-mediated signaling pathways [1]. Genetic and pharmacological interventions that inhibit apoptosis signaling resulted in reductions in apoptosis markers in cardiomyocytes and myocardial ischemic injury [5–8]. However, more recent studies have questioned the role of apoptosis as a major contributor in cardiomyocyte loss during I/R injury [9]. Instead, it has been reported that the rates of cardiomyocyte death by necrotic means are greater than that of apoptosis during heart failure [10]. Unlike apoptosis, necrosis had long been regarded as a passive and unregulated process occurred in response to severe pathological stress, characterized by cell swelling, cell membrane and organelle disruption, and cell lyses [11]. However, emerging evidence has overturned this paradigm and revealed that a significant proportion of necrotic cell death is actively mediated through regulated pathways - a process known as “regulated necrosis”. Indeed, several forms of regulated necrosis have been reported, such as death receptor-mediated necrosis (termed “necroptosis”) [12–14], mitochondria-mediated necrosis [15,16], pyroptosis [17], parthanatos [18], and ferroptosis [19,20]. Of note, given some of these cell death modalities remain incompletely understood, it is unclear whether different modes of cell death represent overlapping entities or even the same cell death program that operates in different cellular contexts. Nonetheless, the discovery of regulated necrosis represents a breakthrough in targeting necrosis for the treatment of heart disease, which was deemed impossible in the past.
Multiple modes of cell death are involved in development, tissue homeostasis, and disease pathogenesis. These cell death programs are often activated by distinct extracellular or intracellular signals under different physiological or pathological settings. For example, necroptosis is activated when apoptosis is inhibited by certain viral proteins [e.g., cowpox cytokine response modifier A (CrmA)], which serves as a critical host response to limit viral load [1]. Moreover, necroptosis is induced by a specific cellular mechanism distinct from other forms of regulated necrosis such as ferroptosis and pyroptosis [1]. Targeting specific cell death mechanism(s) has important therapeutic implications in the treatment of human diseases. Recent studies have provided compelling evidence that necroptosis plays an important role in the pathogenesis of myocardial infarction and heart failure. New mechanistic insights have also been obtained regarding how necroptosis is regulated in the heart and its role in myocardial homeostasis and disease pathogenesis. Here we review current knowledge pertaining to regulatory mechanisms of necroptosis and the roles of this cell death modality in the pathogenesis of myocardial infarction and heart failure.
2. Necroptosis
Necroptosis is a caspase-independent, regulated necrotic cell death modality, which is mediated by death receptors and executed through the activation of receptor interacting protein kinase 3 (RIPK3), a serine/threonine kinase [12–14,21]. RIPK3 is activated through phosphorylation carried out by the homologous receptor interacting protein kinase 1 (RIPK1) or via other alternative mechanisms (discussed below). RIPK3 then phosphorylates and activates a pseudokinase termed mixed lineage kinase-like domain (MLKL), which oligomerizes and permeabilizes the plasma membrane to induce necroptotic cell death.
Necroptosis, like other forms of regulated necrosis, is characterized by cellular swelling and the loss of plasma membrane integrity, which is accompanied by release of damage-associated molecular patterns (DAMPs) and inflammation. Necroptosis is initiated by the binding of cell death ligands, which include, among others, TNFα, lymphotoxin-α (LTα), Fas ligand (FasL), CD40 ligand (CD40L), and TNF-related apoptosis-inducing ligand (TRAIL), to corresponding death receptors. Traditional death receptors include Fas (also termed CD95 or Apo-1), TNF receptor 1 (TNFR1), TNF-related apoptosis inducing ligand (TRAIL) receptor 1 (also termed DR4), and TRAIL receptor 2 (also termed DR5) [22–25]. Additionally, activation of toll-like receptors by LPS (via TLR4) and double-stranded RNA (via TLR3) can trigger necroptotic death via TIR domain-containing adapter-inducing interferon-β (TRIF), as can IFNγ receptor binding and viral activation of DNA-dependent activator of IFN-regulatory factors (DAI) [26–29].
2.1. Molecular mechanisms of necroptosis
Necroptosis is initiated by the binding of membrane-bound death receptors (DRs) with their ligands. DRs contain amino-terminal cysteine-rich domains (CRDs) which define their ligand specificity, and a death domain (DD) in their intracellular tail which mediates protein complex formation at the plasma membrane [30]. Two classes of protein complexes have been described: death inducing signaling complex (DISC) and complex I. The DISC consists of death receptor, FADD, and procaspase-8 or -10 [31], which has been best characterized in Fas, TRAIL-R1, and TRAIL-R2 signaling. Engagement of these receptors leads to the recruitment of the adaptor protein FADD through an interaction mediated by the DD [32]. FADD in turn, via its death effector domain (DED), mediates the recruitment and activation of procaspase-8. Caspase-8 then activates downstream effector procaspases to induce apoptosis. Ligation of Fas can also induce necroptosis under certain conditions such as depletion of cellular inhibitor of apoptosis proteins (cIAPs) [33]. Fas is found on most cells, whereas the basal expression of FasL is restricted to specific cell types [34]. However, FasL expression can be induced in response to pathological stress, such as cardiac I/R injury [7].
In contrast to the DISC, complex I can induce three divergent outcomes depending on cellular contexts: cell survival, apoptosis, or necroptosis [35]. Engagement of TNFR1 upon TNFα binding leads to the recruitment of another DD-containing adaptor protein TNFR1-associated death domain protein (TRADD), which then recruits RIPK1 through DD-DD interactions. The adaptor proteins TNF receptor-associated factors 2 and 5 (TRAF2 and TRAF5) and cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) are also recruited to TRADD and catalyze lysine 63 (K63)-linked ubiquitination of RIPK1 [36]. K63 ubiquitination of RIPK1 also facilitates the recruitment of the linear ubiquitination assembly complex (LUBAC), consisting of heme-oxidized IRP2 ubiquitin ligase 1 (HOIL-1), HOIL-1-interacting protein (HOIP), and shank-associated RH domain-interacting protein (SHARPIN) [37]. LUBAC catalyzes linear [methionine 1 (M1)-linked] ubiquitination of RIPK1. Also recruited into this complex are the deubiquitinase cylindromatosis (CYLD) and its adapter spermatogenesis-associated 2 (SPATA2), which remove K63-linked and linear ubiquitin chains from RIPK1 [38–40]; and A20, a ubiquitin-editing enzyme [41], which removes K63 ubiquitin chains from RIPK1 via its OUT domain and promotes K48 ubiquitination of RIPK1 via its ZnF4 motif. Together these enzymes determine the ubiquitination state of RIPK1 in complex I and downstream signaling [42].
The K63-linked ubiquitin chains on RIPK1 serve as a scaffold to facilitate the activation of transforming growth factor β-activated kinase-1 (TAK1, also known as MAPKKK7) by recruiting TAK1 binding protein 2 and 3 (TAB2 and TAB3) to complex I (Fig. 1). TAK1 promotes cell survival through the activation of nuclear factor-κB (NF-κB). TAK1-mediated activation of NF-κB is brought about through its phosphorylation of inhibitor of κB (IκB) kinase α and β (IKKα and -β) and subsequent phosphorylation and degradation of IκBα [43]. NF-κB mediates cell survival through transcriptional induction of prosurvival proteins such as B cell lymphoma-2 (BCL-2), BCL-2-like-1 long form (BCL-xL), cIAP1 and – 2, X-linked inhibitor of apoptosis (XIAP), and cellular-FADD like interleukin-1-converting enzyme-inhibitory protein (c-FLIP) [44–48].
Fig. 1.
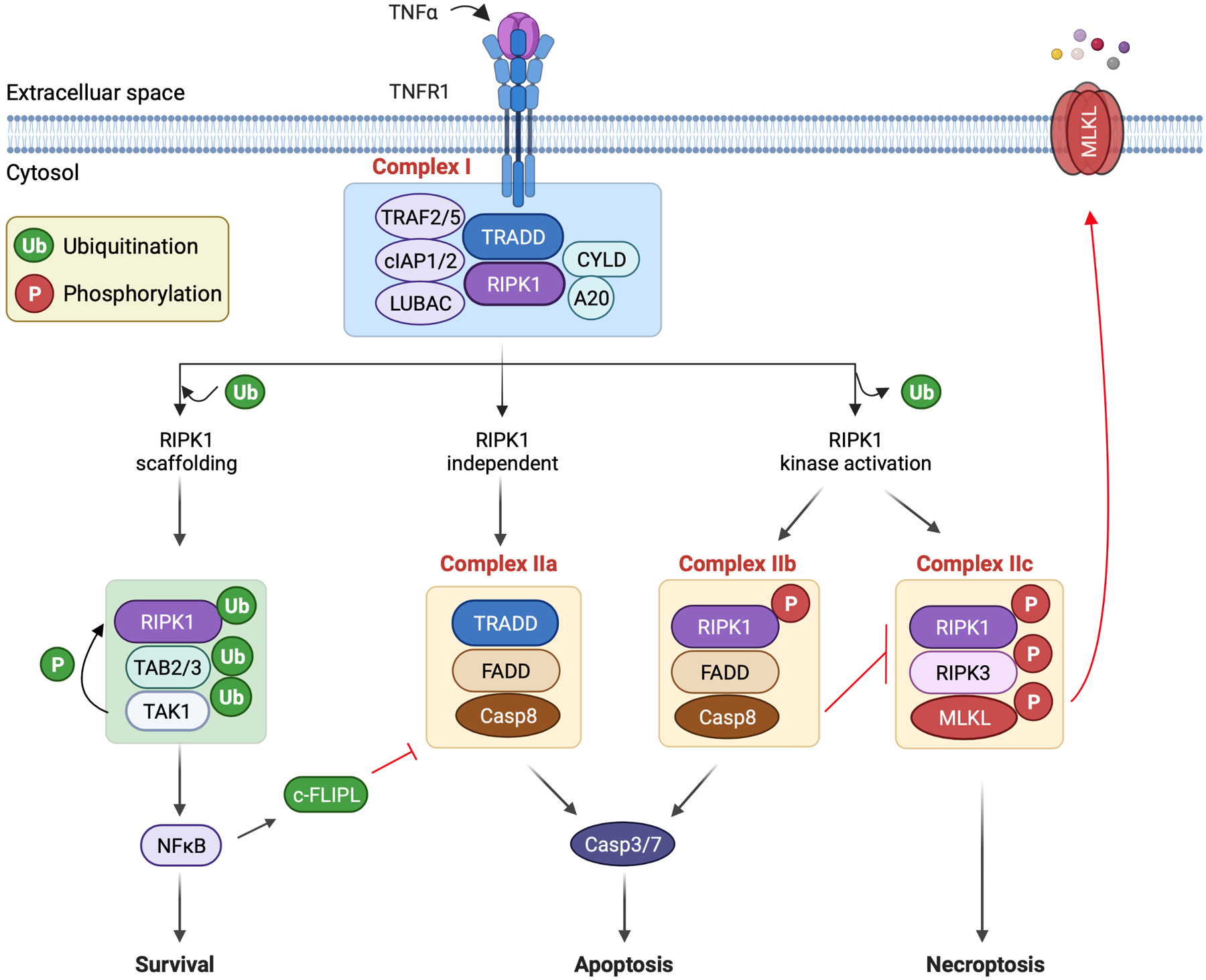
Mechanisms of necroptosis. Necroptosis is initiated by the binding of death ligands to membrane-bound death receptors. Shown here is the binding of tumor necrosis factor-α (TNFα) to TNF receptor 1 (TNFR1). This initiates assembly of complex I on the cytoplasmic tail of the receptor. Complex I assembly may induce divergent cellular outcomes including cell survival, apoptosis, or necroptosis, which are mediated by distinct downstream cytosolic signaling complexes. In the formation of complex I, TNFR1 recruits the adaptor protein TNFR1-associated death domain protein (TRADD), which then recruits receptor interacting protein kinase 1 (RIPK1). TRADD also recruits the adaptor proteins TNF receptor-associated factor 2 and 5 (TRAF2/5) and cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2), which catalyze lysine 63 (K63)-linked ubiquitination of RIPK1. The K63-linked ubiquitin chains also promote the recruitment of the linear ubiquitin chain assembly complex (LUBAC), which catalyzes linear [or methionine 1 (M1)-linked] ubiquitination of RIPK1. Also recruited into this complex are protein deubiquitinases cylindromatosis (CYLD) and A20, which mediate RIPK1 deubiquitination. The K63-linked ubiquitin chains on RIPK1 serve to recruit TAK1-binding protein 2 and 3 (TAB2 and TAB3) to facilitate the activation of transforming growth factor β-activated kinase-1 (TAK1). TAK1 promotes cell survival by activating nuclear factor-κB (NF-κB). TAK1 also prevents RIPK1 kinase activation through direct phosphorylation. Cell death initiation requires the transition from complex I to death-inducing cytosolic complexes. Complex IIa, which consists of TRADD-FADD-procaspase-8, signals RIPK1-independent apoptosis. In contrast, complex IIb, consisting of activated RIPK1-FADD-procaspase-8, mediates RIPK1-dependent apoptosis. Of note, caspase-8 inhibits necroptosis by cleaving RIPK1 and RIPK3. Moreover, in complex IIc (also termed “necrosome”), the binding of activated RIPK1 and RIPK3 leads to RIPK3 activation, which then phosphorylates and activates a pseudokinase called mixed lineage kinase-like domain (MLKL). MLKL undergoes oligomerization and translocates to and permeabilizes the plasma membrane to induce necroptosis.
Apoptosis and necroptosis are mediated by distinct cytosolic signaling complexes derived from complex I (Fig. 1). Complex IIa signals RIPK1-independent apoptosis, complex IIb mediates RIPK1-dependent apoptosis, and complex IIc directs the cell to undergo necroptosis. Intriguingly, a transient cytosolic complex, which lacks TNFR1 but includes TRADD, FADD and procaspases-8, subsequently forms in the cytoplasm after TNFα stimulation [49]. Long form of c-FLIP (c-FLIPL) is also detected in this complex under normal conditions, which represents a key mechanism to inhibit caspase-8 activation and apoptosis. However, under apoptosis-inducing conditions, such as upon NF-κB inhibition, little or no c-FLIPL is detected in this TRADD-FADD-procaspase-8 complex, termed complex IIa, where procaspase-8 is cleaved, leading to caspase-8 activation and apoptosis [49]. This apoptosis pathway is RIPK1 independent. Recent studies identified an additional RIPK1-dependent apoptosis pathway that is mediated by the complex consisting of RIPK1-FADD-procaspase-8, termed complex IIb. RIPK1 kinase activation is required for the assembly of the complex IIb. Therefore, the utilization of RIPK1-independent versus RIPK1-dependent apoptosis seems to be governed by c-FLIPL levels and RIPK1 activity [50]. Indeed, RIPK1-independent apoptosis is primarily inhibited by c-FLIPL, whereas RIPK1-dependent apoptosis is blocked by RIPK1 inactivation but not by c-FLIPL [50]. The common step in both RIPK1-independent and RIPK1-dependent modes of apoptosis is the recruitment of procaspase-8 to FADD, leading to caspase-8 activation through forced proximity and trans cleavage. Moreover, in addition to signaling apoptosis, caspase-8 activation inhibits necroptosis signaling by cleaving RIPK1 and RIPK3, key mediators of necroptosis [51,52].
Necroptosis is mediated by complex IIc, also known as the necrosome, which consists of RIPK1, RIPK3, and MLKL (Fig. 1). When apoptosis is inhibited, RIPK1 interacts with RIPK3 via RIP homotypic interaction motif (RHIM), leading to the assembly of the necrosome, an amyloid/microfilament-like aggregate [12–14,53]. Although RIPK1 can be bypassed (discussed below), RIPK1 kinase activity is often critical for RIPK3 activation and necrosome assembly. Within the necrosome, RIPK1 and RIPK3 engage in auto- and trans- phosphorylation to enhance their kinase activity, which is essential for the execution of necroptotic cell death [54,55]. It has been shown that K63-linked polyubiquitination of RIPK1 at K115 by pellino-1 is critical for the necrosome formation [56]. A20 removes K63-linked ubiquitin chains from RIPK3 and inhibits RIPK1-RIPK3 binding [57]. Linear ubiquitination of RIPK1 in the necrosome is mediated by LUBAC, which interferes with the binding of RIPK1 with RIPK3 to inhibit necrosome formation [58,59]. Moreover, K48-linked polyubiquitination of RIP3 by the E3 ligase CHIP is proposed to confer necrosome instability by targeting RIP3 for lysosomal degradation [60].
The kinase activity of RIPK3 is critical for the recruitment of the pseudokinase MLKL. Activation of MLKL by RIPK3-mediated phosphorylation marks an important execution step in necroptosis [61,62]. Upon phosphorylation, MLKL undergoes a defined conformational change and exposes a four helical bundle, which promotes its cysteine-dependent tetramerization, progression to amyloid-like filaments, and translocation to and permeabilization of the plasma membrane [63–65]. MLKL phosphorylation is antagonized by protein phosphatase 1b (Ppm1b) [66], a widely expressed enzyme that remains the only known natural inhibitor of necroptosis downstream of the necrosome. Necrosulfonamide, a MLKL inhibitor that forms a covalent adduct with Cys86 in MLKL, blocks MLKL polymerization and necroptosis. Conversely, forced MLKL polymerization is sufficient to trigger necroptosis even in the absence of RIPK3, suggesting that MLKL polymerization is indispensable for necroptosis [63,64]. The precise mechanisms by which MLKL executes necroptosis are incompletely understood. It has been reported that recombinant MLKL can bind phosphatidylinositol phosphates and cardiolipin and disrupt liposomes containing these phospholipids [64,67]. MLKL complexes can induce Na+ or Ca2+ influx and osmotic disruption of the plasma membrane [65,68]. Moreover, activated MLKL functions as cation channels or activates other cation channels such as TRPM7 to promote necroptosis [65,69]. MLKL also mediates membrane damage by antagonizing the function of the endosomal sorting complexes required for transport (ESCRT)-III machinery [70].
Although RIPK1 plays a critical role in the activation of canonical necroptosis signaling, RIPK1 or its kinase activity is dispensable for RIPK3 activation and necroptosis in other paradigms. For example, pathogen-associated molecular patterns (PAMPs), via Toll-like receptors (TLRs), can induce RIPK3 activation through Toll-interleukin-1 receptor (TIR) domain-containing adapter-inducing interferon (TRIF), which interacts with RIPK3 through its RHIM domain [26,71]. Moreover, cytomegalovirus infection can also induce necroptosis independently of RIPK1 by DNA-dependent activator of interferon regulatory factors/Z-DNA binding protein 1 (DAI/ZBP1), another RHIM-containing protein that interacts with RIPK3 [29]. Unlike RIPK1, TRIF or DAI does not possess protein kinase activity, which may induce RIPK3 activation through RHIM-mediated interactions, leading to a conformational change and autophosphorylation of RIPK3.
2.2. Scaffolding function versus kinase activity of RIPK1 in cell death signaling
The pro-survival role of RIPK1 is mediated by its “scaffolding” function that is independent of its kinase activity. Germline deletion of RIPK1 led to neonatal lethality accompanied by cell death in multiple tissue compartments [72]. Importantly, the detrimental effects of RIPK1 deletion were rescued by simultaneous inhibition of apoptosis and necroptosis but not by inactivation of either pathway alone [73–76]. These data indicate that the net effect of RIPK1 is to suppress apoptosis and necroptosis. In contrast to germline RIPK1 knockout model, kinase-dead RIPK1 knockin mice were normal at baseline, suggesting that the kinase-independent, scaffolding function of RIPK1 suppresses cell death in vivo [74,77,78].
In contrast to the scaffolding role of RIPK, which is critical required to suppress cell death, the kinase activity of RIPK1 is indispensable for its ability to mediate apoptosis and necroptosis in the death receptor pathway. RIPK1 kinase activity is tightly regulated by ubiquitination and phosphorylation. CYLD or SPATA2 deficiency promotes K63-linked ubiquitination of RIPK1, which inhibits RIPK1 activation and necroptosis [38–40]. Conversely, LUBAC deficiency reduces linear ubiquitination of RIPK1, promoting RIPK1 activation and necroptosis [59]. Moreover, ABIN-1 serves as a scaffold to recruit A20 which mediates the deubiquitination of RIPK1 [79], while optineurin promotes K48-linked ubiquitination and degradation of RIPK1 [80,81]. RIPK1 activity is also regulated through phosphorylation. IKKα/β and NF-κB essential modulator (NEMO) can mediate inhibitory phosphorylation on RIPK1 [82]. Similarly, MAPK-activated protein kinase 2 (MK2) phosphorylates RIPK1 at S321 and S336 to inhibit its kinase activity [83–85]. Moreover, TAK1 can phosphorylate RIPK1 on multiple sites of the intermediate domain (S321, S332 and S334), which negatively regulates RIPK1 kinase activity [86].
2.3. Role of mitochondria in necroptosis
RIPK1, RIPK3, and MLKL have all been shown to translocate to mitochondrial membranes during necroptosis in a variety of cell types [13,87,88]. Activated RIPK3 targets several metabolic enzymes, including glutamate dehydrogenase, glycogen phosphorylase, and glutamate-ammonia ligase, to promote mitochondrial ROS production. A recent study further showed that RIPK3 activates the pyruvate dehydrogenase complex in mitochondria to enhance aerobic respiration and ROS production [89]. Mitochondria derived ROS production has been implicated as a driving force in the execution of necroptosis. Mitochondrial ROS mediates oxidation of specific cysteines in RIPK1, which promotes RIPK1 autophosphorylation and the formation of functional necrosome [90,91]. However, the role of mitochondrial ROS in necroptosis seems to be cell type specific, as necroptosis in Jurkat cells and HT-29 cells seems to be ROS-independent [89,92]. Moreover, widespread mitochondria depletion with induced mitophagy still allows functional necroptosis in SVEC and 3 T3 cells [93]. It is also proposed that mitochondria permeability transition pore (mPTP) is linked to necroptosis. For example, the protective effect of RIPK1 inhibitor necrostatin-1 against myocardial I/R was not additive to that conferred by cyclophilin D ablation, suggesting these two components are part of the same genetic pathway [94]. Moreover, RIPK3 activation promotes mPTP opening through Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ) [95]. However, this model has been questioned by other studies. For example, caspase 8 deficiency-induced necroptosis was blocked by RIPK1 or RIPK3 deletion, but not by cyclophilin D inhibition [93,96,97]. Moreover, ablation of RIPK3 or cyclophilin D was protective in a kidney I/R model, but double knockout mice showed event greater protection, suggesting necroptosis and mPTP are distinct pathways [98]. The involvement of mitochondria in necroptosis is also demonstrated by the interaction between RIPK1/RIPK3/MLKL and the mitochondrial protein phosphatase PGAM5 in Hela cells [87]. This interaction induces phosphorylation and activation of PGAM5 in mitochondrial outermembranes, where it dephosphorylates the mitochondrial fission factor dynamin-related protein1 (Drp1) at Ser637 and activates its GTPase activity, leading to mitochondrial fragmentation, elevated ROS production, and cell death. However, subsequent studies showed that either PGAM5 or Drp1 is dispensable for necroptosis in several cell types [99,100]. Together, the role of mitochondria in necroptosis signaling seems to be dependent on cell types and cellular contexts, which warrants further investigation. Moreover, whether mitochondria play a role in necroptosis of cardiac myocytes has not been directly examined.
2.4. Crosstalk between necroptosis and other cell death pathways
A crosstalk between necroptosis and apoptosis has been revealed by genetic mouse models in vivo. Intriguingly, germline deletion of FADD or procaspase-8 caused embryonic lethality [101,102], which was rescued by simultaneous deletion of RIPK3 or knockin of RIPK3 kinase-dead mutants [55,96,103,104]. These observations indicate that inhibition of apoptosis promotes necroptosis activation. As a potential mechanism, it has been suggested that c-FLIPL-procaspase-8 heterodimers possess low, but detectable caspase-8 activity to suppress necroptosis [104]. Conversely, inhibition of necroptosis may also promote apoptosis activation. RIPK3 kinase-dead (D161N) knockin mice exhibited embryonic lethality due to excessive apoptosis in the yolk sac vasculature [55]. This was rescued by simultaneous deletion of procaspase-8 [55]. However, another RIPK3 kinase-dead (K51A) knockin mouse did not exhibit embryonic lethality or apoptosis induction [105]. Further work will be needed to determine the mechanisms underlying the differential effects of these two models on apoptosis activation. Necroptosis has been linked to mPTP-mediated necrosis through RIPK3-mediated phosphorylation and activation of CaMKII [95]. Moreover, pharmacological inhibition of CaMKII blocks mPTP opening and cardiomyocyte killing in response to RIPK3 overexpression [95]. Necroptosis may also promote pyroptosis through the release of DAMPs, which underlies the inflammatory response in post-ischemic heart failure [106,107]. Intriguingly, a crosstalk between necroptosis and ferroptosis has also been observed, where inhibition of necroptosis sensitized cells to ferroptotic cell death and vice versa, although the underlying mechanism is currently unknown [108].
3. Necroptosis in heart disease
Multiple cell death programs have been implicated in the pathogenesis of various forms of heart disease including MI, heart failure, cardiomyopathies, myocarditis, and others [1–4]. Recent studies have demonstrated that necroptosis plays an important role in major cardiac syndromes such as MI and heart failure. In this session, we review genetic and pharmacological manipulations of necroptosis and their effects on myocardial injury, pathological remodeling, and cardiac function in the setting of MI and heart failure (Table 1). We will also discuss how necroptosis is regulated in the heart in response to pathological stress and new therapeutic strategies that target necroptosis signaling (Fig. 2).
Table 1.
Genetic manipulations of necroptosis in cardiac disease models.
| Molecules | Cell type | Disease model | Outcomes | Reference |
|---|---|---|---|---|
| Ripk3 KO | Global | I/R | Reduced infarct size | [75,118] |
| Ripk3 KO | Global | MI, no reperfusion | Cardiac protection | [107] |
| Ripk3 KO | Global | Doxorubicin | Improved cardiac function | [75] |
| Pgam5 KO | Cardiomyocyte | I/R | Reduced infarct size | [121] |
| Pgam5 KO | Global | I/R | Reduced infarct size | [122] |
| miR-223 KO | Global | I/R | Increased infarct size | [117] |
| miR-223 TG | Cardiomyocyte | I/R | Reduced infarct size | [117] |
| miR-873 | I/R | Reduced infarct size | [123] | |
| miR-103/107 antagomir | I/R | Reduced infarct size | [124] | |
| Tab2 KO | Cardiomyocyte | Baseline | Dilated cardiomyopathy | [133] |
| Tab2 KO; Ripk1 K45A/K45A | Cardiomyocyte; Global | Baseline | Rescued pathological phenotype of Tab2 KO | [133] |
| Tak1 KO | Cardiomyocyte | Baseline | Dilated cardiomyopathy | [131] |
| Tak1 KO; Tnfr1 KO | Cardiomyocyte; Global | Baseline | Rescued pathological phenotype of Tak1 KO | [131] |
| Traf2 KO | Cardiomyocyte | Baseline | Dilated cardiomyopathy | [136] |
| Traf2 KO; Tnfr1 KO | Cardiomyocyte; Global | Baseline | Rescued pathological phenotype of Traf2 KO | [136] |
| Traf2 KO; Ripk3 KO | Cardiomyocyte; Global | Baseline | Rescued pathological phenotype of Traf2 KO | [136] |
| Tead1 KO | Cardiomyocyte | Baseline | Dilated cardiomyopathy | [142] |
| Cops8 KO | Cardiomyocyte | Baseline | Dilated cardiomyopathy | [143] |
| Cops8 KO; Ripk3 KO | Cardiomyocyte; Global | Baseline | Rescued pathological phenotype of Cops8 KO | [143] |
Fig. 2.
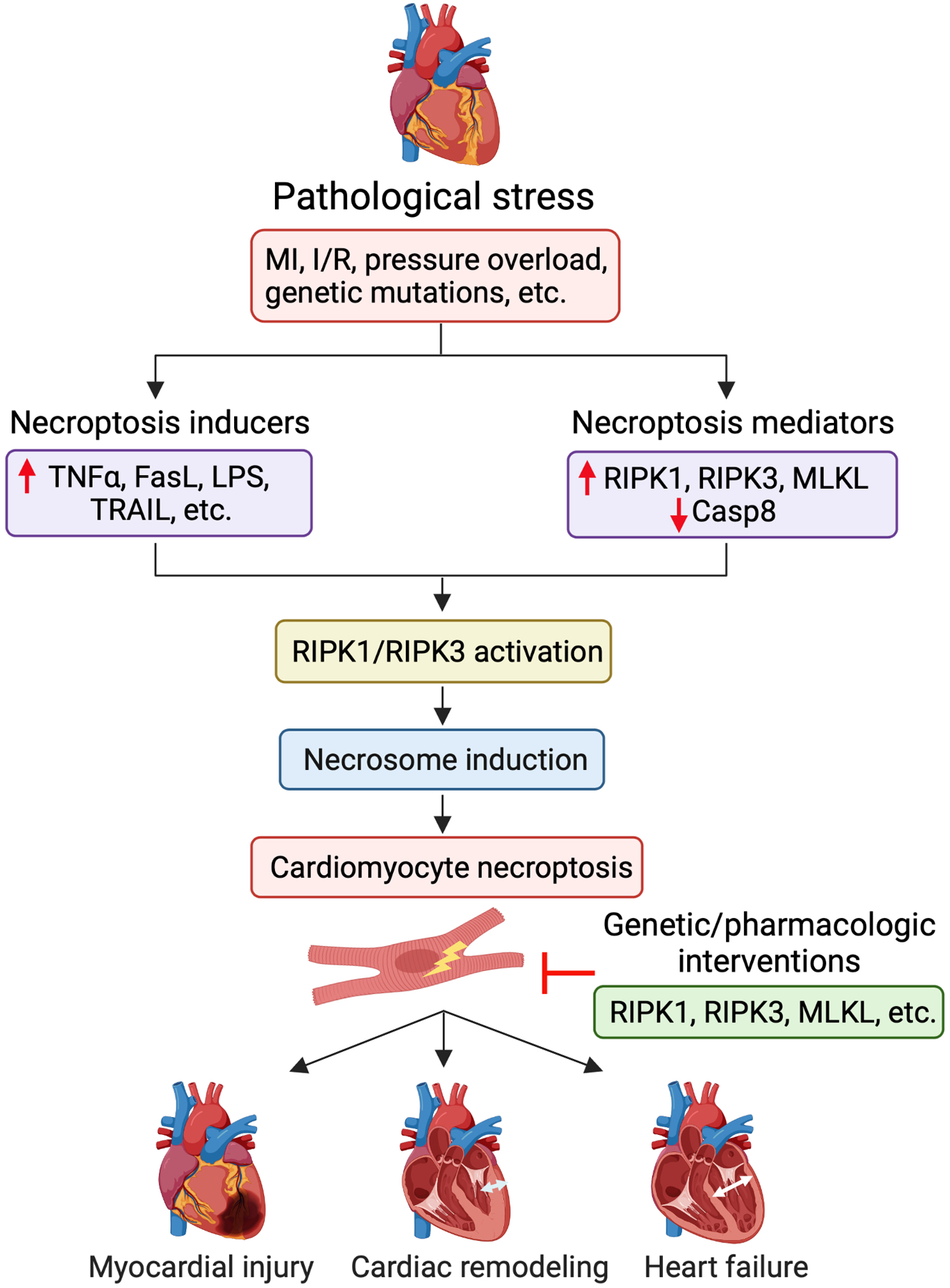
Necroptosis in the pathogenesis of heart disease. Cardiac necroptosis signaling is activated in response to pathological stress (e.g., MI, I/R, pressure overload, and genetic mutations), a process that involves the induction of key necroptosis inducers (e.g., TNF, FasL, LPS, and TRAIL) and mediators (e.g., upregulation of RIPK1, RIPK3, and MLKL; downregulation of caspase-8). Activation of RIPK1 and/or RIPK3 is critical for the assembly of the necroptosis signaling complex (termed “necrosome”), which induces cell death by activating MLKL and subsequent plasma membrane rupture. Cardiomyocytes death via necroptosis plays an important role in the pathogenesis of myocardial ischemic injury, pathological remodeling, cardiac dysfunction, and heart failure. Genetic or pharmacologic interventions targeting necroptosis signaling proteins (e.g., RIPK1, RIPK3, and MLKL) represent potential therapeutic strategies for heart disease.
3.1. Necroptosis in myocardial infarction and I/R injury
The role of necroptosis in cardiac I/R injury was first demonstrated using the small molecule inhibitor necrostatin-1 and its optimized analog necrostatin-1 s, which allosterically inhibit RIPK1 kinase activity [54,109]. Administration of necrostatin-1 reduced infarct size following I/R in mice, rats, guinea pigs, and pigs [110–113]. Reduced infarct was associated with decreased phosphorylation of RIPK1 and RIPK3, reduced necrosome formation, and attenuated necrotic cell death. Necrostain-1 has been shown to have off-target effects [114–116], such as inhibition of indoleamine 2,3-dioxygenase (IDO) and ferroptosis. However, a more specific RIPK1 inhibitor, necrostatin-1 s, also attenuated infarct size and myocardial necrosis after I/R [117], further implicating the role of necroptosis in I/R injury.
Protein levels of necroptotic mediators, such as RIPK1, RIPK3, and MLKL, were increased in the mouse heart following I/R [117]. A significant upregulation of RIPK3 was also detected in a nonreperfused MI model in mice [107]. Global deletion of RIPK3 attenuated infarct size following I/R [95,118]. Cardiac contractile function was also improved in RIPK3−/− mice 4 weeks after I/R. RIPK3 knockout also attenuated adverse cardiac remodeling and dysfunction at 30 days after MI injury induced by permanent coronary occlusion [107]. Adenoviral-mediated expression of RIPK3 in cardiomyocytes was sufficient to trigger necroptosis. Surprisingly, this effect was independent of RIPK1 or MLKL. Interestingly, Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ), a facilitator of mPTP-mediated necrosis [119,120], was identified as a novel target of RIPK3 [95]. Furthermore, pharmacological inhibition of CaMKII, blocks mPTP opening and cardiomyocyte death in response to RIPK3 overexpression [95]. As another downstream effector of RIPK3 [87], PGAM5 expression was elevated during cardiac I/R injury [121]. Cardiomyocyte-specific deletion of PGAM5 attenuated cardiac I/R injury by preventing Drp-1 dephosphorylation, mitochondrial fragmentation, ROS overproduction, and necroptosis [121]. However, another study using global PGAM5 knockout mice showed that ablation of PGAM5 exacerbated rather than reduced necroptosis in response to multiple in vitro and in vivo necroptotic stimuli, including I/R injury in the heart and brain [122]. Therefore, the role of PGAM5 in cardiac I/R injury and cardiomyocyte necroptosis warrants further investigation.
Several microRNAs (miRs) have been shown to regulate necroptosis in the setting of cardiac I/R. For example, miR-223 has been identified as a potential regulator of necroptosis in the heart and I/R injury [117]. miR-223 expression was upregulated in response I/R, and cardiacspecific overexpression of miR-223 reduced infarct size and markers of necrosis (but not apoptosis). Genetic deletion of miR-223 had the opposite effect. Mechanistically, miR-223 regulates multiple components of the death receptor pathway, including TNFR1, death receptor 6 (DR6), IKKα, NOD-like receptor family pyrin domain containing 3 (NLRP3), as well as RIPK1, RIPK3 and MLKL protein abundance. Similarly, miR-873 also inhibits necroptosis by decreasing the abundance of RIPK1 and RIPK3 in cardiomyocytes [123]. miR-873 was downregulated in the heart during I/R, and intracoronary delivery of miR-873 attenuated I/R-induced necrosis and infarct, leading to improved functional recovery. miR-873 downregulation is mediated by a long noncoding RNA named necrosis-related factor (NRF). NRF abundance was upregulated during I/R, possibly through a p53-mediated transcriptional mechanism [123]. In contrast to miR-223 and miR-873, which are negative regulators of necroptosis, miR-103/107 was found to promote this cell death program. miR-103/107 expression was upregulated in the ischemic region during I/R, and administration of specific anti-miRs attenuated necrosis, infarction, and improved cardiac function [124]. miR-103/107 was upregulated by oxidative stress and mediates necroptosis by decreasing FADD abundance [124]. Moreover, miR-103/107 is functionally inhibited by the lncRNA H19, which increases FADD abundance and promotes necroptosis resistance. These findings implicate specific miRs in I/R-induced necroptosis and suggest that targeting these miRs may serve as a potential strategy to ameliorate I/R injury.
3.2. Necroptosis in heart failure
Key mediators of necroptosis, including RIPK1, RIPK3, and MLKL, as well as their phosphorylated forms, were upregulated in the failing human heart compared with healthy controls [125]. In contrast, caspase-8 was significantly decreased, which may facilitate the induction of necroptosis [125]. A similar upregulation of necroptotic proteins was also observed in mice subjected to prolonged pressure overload with transverse aortic constriction (TAC) [126]. Intriguingly, the increase in RIPK1, RIPK3, and MLKL expression and phosphorylation levels after TAC was reversed by the administration of Hsp90 inhibitor 17-AAG [126]. Moreover, Hsp70 inhibition also attenuated TNFα-induced necroptosis in cardiomyocytes by suppressing RIPK1-RIPK3-MLKL interaction [126]. In addition to necroptosis regulatory proteins, TNFα was also upregulated in the myocardium after pressure/volume overload or myocardial infarction [127,128]. Upregulation of these necroptosis signaling proteins facilitates necroptosis activation in the heart following pathological stress.
The role of RIPK3 in heart failure was examined using a myocardial infarction model with permanent coronary occlusion [107]. Global RIPK3 knockout reduced cardiac dysfunction, hypertrophy, and inflammatory response 30 days after MI, suggesting that necroptosis may contribute to the pathogenesis of chronic pathological remodeling. RIPK3 has also been shown to play an important role in doxorubicin-induced cardiac injury [95]. Doxorubicin, a chemotherapeutic agent used to treat multiple solid tumors and leukemias, causes a dose-dependent cardiomyopathy in ~10% of patients. While doxorubicin-induced cardiomyopathy remains incompletely understood, multiple cell death programs have been implicated. RIPK3 was transcriptionally upregulated in cardiomyocytes following doxorubicin treatment [95]. Global deletion of RIPK3 attenuated doxorubicin-induced cardiac dysfunction, suggesting necroptosis is involved [95]. Moreover, global RIPK3 knockout also attenuated cardiac injury induced by catecholamine overload, an effect similar to RIPK1 inhibition with necrostatin-1 [129]. These results suggest that necroptosis is also involved in catecholamine overload-induced heart failure.
TAK1 has been identified as a key regulator of apoptosis and necroptosis through both NF-κB-dependent and -independent mechanisms [130,131]. The TAK1 signaling complex is recruited to the complex I upon TNFα stimulation via its adaptor proteins TAB2/3 which bind to K63-linked polyubiquitin chains [132]. Strikingly, cardiomyocyte-specific deletion of TAB2 in mice triggered dilated cardiomyopathy with massive apoptotic and necroptotic cell death [133]. Mutations in TAB2 gene have also been implicated in the pathogenesis of dilated cardiomyopathy and/or congenital heart disease in humans [134,135]. Myocardial TAB2 protein expression was markedly downregulated after pressure overload and MI [133]. Moreover, TAB2-deficient mice were predisposed to myocardial injury and adverse remodeling after pathological stress. Strikingly, genetic inactivation of RIPK1 with Ripk1-K45A knockin effectively rescued cardiac remodeling and dysfunction in Tab2-deficient mice. Mechanistically, TAB2, via TAK1, critically mediates RIPK1 phosphorylation at Ser321, preventing RIPK1 kinase activation and the formation of complex IIb (RIPK1-FADD-caspase-8) and complex IIc (RIPK1-RIPK3). Like TAB2 deficiency, cardiomyocyte-specific deletion of TAK1 in mice induced spontaneous apoptosis and necroptosis that led to adverse remodeling and heart failure, and these effects were abolished by ablation of TNFR1 or inhibition of RIPK1 with necrostatin-1 [131]. These results demonstrate that the TAB2-TAK1 signaling complex is a key regulator of myocardial homeostasis and remodeling by suppressing RIPK1-dependent apoptosis and necroptosis.
TRAF2 has been identified as a key suppressor of necroptosis. Indeed, cardiomyocyte-specific deletion of TRAF2 in mice, or expression of a TRAF2 mutant lacking the RING domain in neonatal cardiomyocytes, induced apoptosis and necroptosis [136]. The apoptosis component is RIPK1 dependent, which is sensitive to RIPK1 kinase inactivation by necrostatin-1. The necroptosis component is mitigated by genetic deletion of RIPK3 or MLKL. Although usually considered as an adaptor protein in complex I, TRAF2 also contains a RING domain with E3-ligase activity [137,138]. While the effects of the dominant negative TRAF2 mutant lacking RING domain suggest that its E3-ligase function is important in suppressing cell death, the precise mechanism remains to be defined. Potentially relevant TRAF2 substrates include RIPK1 and TAK1 [139,140], but their roles remain to be tested. Of note, TRAF2 appears to promote cell survival mainly through an NF-κB-independent mechanism, although TRAF2 overexpression can activates NF-κB [141]. The role of TRAF2 as a suppressor of RIPK1-dependent apoptosis and necroptosis is important in the pathogenesis of heart failure because TRAF2 was upregulated in response to pressure overload and post-MI heart failure and ablation of TRAF2 induced ventricular dilation and cardiac dysfunction [136].
The Hippo signaling effector TEAD1 has also been recognized as a potential regulator of necroptosis in the heart [142]. Cardiomyocyte-specific deletion of TEAD1 led to acute-onset dilated cardiomyopathy and heart failure. Notably, a marked increase in the abundance of key necroptotic mediators, including RIPK1, RIPK3, phospho-RIPK3, MLKL, and PGAM5, was detected in Tead1-deficient heart. Intriguingly, although full length caspase-8 was elevated, the cleaved form was decreased. Moreover, cardiomyocyte necrosis, but not apoptosis, was induced in Tead1-deficient heart, as assessed by Evans blue dye uptake and TUNEL staining, respectively. Administration of necrostatin-1 rescued Tead1 deletion-induced heart failure and attenuated cardiomyocyte necrosis. However, given the reported off-target effects of necrostatin-1 -such as inhibition of ferroptosis [114–116], the role of TEAD1 in necroptosis needs to be further validated by genetic manipulation of necroptosis signaling.
A recent study by Xiao et al. revealed a new role for the constitutive photomorphogenesis 9 (COP9) signalosome, a key regulator of protein ubiquitination, in suppressing cardiomyocyte necroptosis [143]. Cardiomyocyte-specific knockout of Cops8, an essential subunit of COP9, led to massive cardiomyocyte necrosis (but not apoptosis), dilated cardiomyopathy, and premature death [144]. Key necroptotic markers, including RIPK1, RIPK3, and MLKL, were significantly elevated in Cops8 knockout mice. Full-length caspase 8 was also increased but the cleaved form and the overall caspase-8 activity were decreased. Cardiomyocyte necrosis was evident in Cops8-deficient heart as assessed by Evans blue dye uptake, which was rescued by RIPK3 ablation or treatment with necrostatin-1, but not by cyclophilin D deficiency. These results suggest that cardiac Cops8/COP9 signalosome malfunction leads to cardiomyocyte necroptosis independently of the mitochondrial permeability transition pore. The molecular mechanism underlying cardiomyocyte necroptosis induced by Cops8 deficiency needs to be further determined.
4. Conclusions and perspectives
Studies presented above indicate that necroptosis contributes significantly to the pathogenesis of I/R injury, cardiac remodeling, and heart failure. During acute I/R injury, cardiomyocyte death is the central event that mediates acute loss of cardiomyocytes in the infarct zone and drives subsequent adverse remodeling in the remaining myocardium leading to heart failure. Genetic and pharmacologic inhibition of necroptosis has proven to be a promising strategy for I/R injury in animal studies. Unlike I/R injury, targeting cell death in chronic conditions, such as cardiomyopathies and heart failure, requires long-term inhibition of necroptosis, which may have undesirable systemic side effects. However, heart failure may still be a future candidate for antinecroptotic therapy, especially when the current treatments fail to prevent disease progression. The role of necroptosis in the pathogenesis of heart disease have also been demonstrated using pharmacological inhibitors that target necroptosis signaling components such as RIPK1, RIPK3, MLKL. These potential therapeutic reagents may provide beneficial effects under pathological conditions such as MI and heart failure by blocking necroptosis signaling and other downstream targets [1,118].
Besides necroptosis, other regulated cell death programs have also been implicated in I/R and heart failure. Indeed, multiple cell death programs, including apoptosis [5–8], mitochondria-mediated necrosis [132,145], necroptosis [95,118], ferroptosis [146–148], and pyroptosis [149,150], have been shown to mediate myocytes loss during I/R. Similarly, both apoptotic and necrotic cardiomyocyte death have been implicated in the pathogenesis of heart failure. Dissecting the relative contributions of these cell death programs under pathological conditions is challenging for several reasons. First, it is possible that multiple cell death processes may take place during I/R and heart failure, with some cells dying through one cell death modality and other cells through another. Second, crosstalk between different cell death mechanisms results in another cell death program taking over when a given program is inhibited. One such an example is the relationship between apoptosis and necroptosis discussed above. Third, distinguishing between different cell death programs is further complicated by the potential overlap among these mechanisms and the sharing of common signaling components. These issues need to be further determined through combined manipulations of distinct cell death pathways coupled with a comprehensive analysis of definitive markers for various cell death programs. An integrated approach is needed for careful delineation of necroptosis from other cell death programs in vivo, which includes the assessment of morphological, biochemical, and molecular features of necroptosis [151], especially the activation of key necroptosis molecules and the necrosome formation, combined with pharmacological and genetic approaches that target specific necroptosis signaling proteins. Furthermore, it has been shown that combined inhibition of apoptosis and necroptosis offered greater protection against cardiac I/R that apoptotic or necroptotic inhibitors alone [112]. Therefore, strategies that target multiple cell death programs may prove useful in the treatment of heart disease.
Acknowledgements
This work was supported by grants from the NIH (R01HL155035 and R01HL160767 to QL) and the American Heart Association (19TPA34850148 to QL). Figures were created with BioRender.
Footnotes
Disclosure
None.
References
- [1].Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN, Fundamental mechanisms of regulated cell death and implications for heart disease, Physiol. Rev 99 (2019) 1765–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kung G, Konstantinidis K, Kitsis RN, Programmed necrosis, not apoptosis, in the heart, Circ. Res 108 (2011) 1017–1036. [DOI] [PubMed] [Google Scholar]
- [3].Konstantinidis K, Whelan RS, Kitsis RN, Mechanisms of cell death in heart disease, Arterioscler. Thromb. Vasc. Biol 32 (2012) 1552–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whelan RS, Kaplinskiy V, Kitsis RN, Cell death in the pathogenesis of heart disease: mechanisms and significance, Annu. Rev. Physiol 72 (2010) 19–44. [DOI] [PubMed] [Google Scholar]
- [5].Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH, Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice, Am. J. Physiol. Heart Circ. Physiol 280 (2001) H2313–H2320. [DOI] [PubMed] [Google Scholar]
- [6].Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL, Reperfusion injury induces apoptosis in rabbit cardiomyocytes, J. Clin. Invest 94 (1994) 1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jeremias I, Kupatt C, Martin-Villalba A, Habazettl H, Schenkel J, Boekstegers P, et al. , Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia, Circulation 102 (2000) 915–920. [DOI] [PubMed] [Google Scholar]
- [8].Lee P, Sata M, Lefer DJ, Factor SM, Walsh K, Kitsis RN, Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo, Am. J. Physiol. Heart Circ. Physiol 284 (2003) H456–H463. [DOI] [PubMed] [Google Scholar]
- [9].Inserte J, Cardona M, Poncelas-Nozal M, Hernando V, Vilardosa Ú, Aluja D, et al. , Studies on the role of apoptosis after transient myocardial ischemia: genetic deletion of the executioner caspases-3 and -7 does not limit infarct size and ventricular remodeling, Basic Res. Cardiol 111 (2016) 18. [DOI] [PubMed] [Google Scholar]
- [10].Adameova A, Goncalvesova E, Szobi A, Dhalla NS, Necroptotic cell death in failing heart: relevance and proposed mechanisms, Heart Fail. Rev 21 (2016) 213–221. [DOI] [PubMed] [Google Scholar]
- [11].Edinger AL, Thompson CB, Death by design: apoptosis, necrosis and autophagy, Curr. Opin. Cell Biol 16 (2004) 663–669. [DOI] [PubMed] [Google Scholar]
- [12].Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. , Phosphorylation driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation, Cell 137 (2009) 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. , RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis, Science 325 (2009) 332–336. [DOI] [PubMed] [Google Scholar]
- [14].He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. , Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha, Cell 137 (2009) 1100–1111. [DOI] [PubMed] [Google Scholar]
- [15].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. , Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death, Nature 434 (2005) 658–662. [DOI] [PubMed] [Google Scholar]
- [16].Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, et al. , Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice, eLife 2 (2013), e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bergsbaken T, Fink SL, Cookson BT, Pyroptosis: host cell death and inflammation, Nat. Rev. Microbiol 7 (2009) 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].David KK, Andrabi SA, Dawson TM, Dawson VL, Parthanatos, a messenger of death, Front. Biosci 14 (2009) 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. , Ferroptosis: an iron dependent form of nonapoptotic cell death, Cell 149 (2012) 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. , Ferroptosis: a regulated cell death Nexus linking metabolism, redox biology, and disease, Cell 171 (2017) 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. , Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3, Mol. Cell 54 (2014) 133–146. [DOI] [PubMed] [Google Scholar]
- [22].Linkermann A, Green DR, Necroptosis, N. Engl. J. Med 370 (2014) 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Orlinick JR, Vaishnaw A, Elkon KB, Chao MV, Requirement of cysteine-rich repeats of the Fas receptor for binding by the Fas ligand, J. Biol. Chem 272 (1997) 28889–28894. [DOI] [PubMed] [Google Scholar]
- [24].Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, et al. , Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations, Science 288 (2000) 2354–2357. [DOI] [PubMed] [Google Scholar]
- [25].Von Karstedt S, Montinaro A, Walczak H, Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy, Nat. Rev. Cancer 17 (2017) 352–366. [DOI] [PubMed] [Google Scholar]
- [26].Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. , Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL, J. Biol. Chem 288 (2013) 31268–31279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, Saelens X, et al. , Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA, Cell Death Differ. 9 (2002) 981–994. [DOI] [PubMed] [Google Scholar]
- [28].Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. , DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB, EMBO Rep. 10 (2009) 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Upton JW, Kaiser WJ, Mocarski ES, DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA, Cell Host Microbe 11 (2012) 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H, The death domain superfamily in intracellular signaling of apoptosis and inflammation, Annu. Rev. Immunol 25 (2007) 561–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lavrik IN, Krammer PH, Regulation of CD95/Fas signaling at the DISC, Cell Death Differ. 19 (2012) 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME, Differential modulation of apoptosis sensitivity in CD95 type I and type II cells, J. Biol. Chem 274 (1999) 22532–22538. [DOI] [PubMed] [Google Scholar]
- [33].Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B, et al. , Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment, J. Cell Biol 187 (2009) 1037–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Green DR, Ferguson TA, The role of Fas ligand in immune privilege, Nat. Rev. Mol. Cell Biol 2 (2001) 917–924. [DOI] [PubMed] [Google Scholar]
- [35].Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P, Regulated necrosis: the expanding network of non-apoptotic cell death pathways, Nat. Rev. Mol. Cell Biol 15 (2014) 135–147. [DOI] [PubMed] [Google Scholar]
- [36].Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. , cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination, Mol. Cell 30 (2008) 689–700. [DOI] [PubMed] [Google Scholar]
- [37].Fujita H, Tokunaga A, Shimizu S, Whiting AL, Aguilar-Alonso F, Takagi K, et al. , Cooperative domain formation by homologous motifs in HOIL-1L and SHARPIN plays a crucial role in LUBAC stabilization, Cell Rep. 23 (2018) 1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, et al. , LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes, Cell Rep. 13 (2015) 2258–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G, The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination, Nature 424 (2003) 801–805. [DOI] [PubMed] [Google Scholar]
- [40].Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G, CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members, Nature 424 (2003) 793–796. [DOI] [PubMed] [Google Scholar]
- [41].Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. , Deubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling, Nature 430 (2004) 694–699. [DOI] [PubMed] [Google Scholar]
- [42].Vucic D, Dixit VM, Wertz IE, Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death, Nat. Rev. Mol. Cell Biol 12 (2011) 439–452. [DOI] [PubMed] [Google Scholar]
- [43].Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB, TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway, J. Mol. Biol 326 (2003) 105–115. [DOI] [PubMed] [Google Scholar]
- [44].Catz SD, Johnson JL, Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer, Oncogene 20 (2001) 7342–7351. [DOI] [PubMed] [Google Scholar]
- [45].Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R, Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1, J. Biol. Chem 275 (2000) 22064–22068. [DOI] [PubMed] [Google Scholar]
- [46].Kreuz S, Siegmund D, Scheurich P, Wajant H, NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling, Mol. Cell. Biol 21 (2001) 3964–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J, NF-kappaB signals induce the expression of c-FLIP, Mol. Cell. Biol 21 (2001) 5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, et al. , c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation, J. Biol. Chem 283 (2008) 24295–24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Micheau O, Tschopp J, Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes, Cell 114 (2003) 181–190. [DOI] [PubMed] [Google Scholar]
- [50].Wang L, Du F, Wang X, TNF-alpha induces two distinct caspase-8 activation pathways, Cell. 133 (2008) 693–703. [DOI] [PubMed] [Google Scholar]
- [51].Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M, Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain, Cell. Signal 19 (2007) 2056–2067. [DOI] [PubMed] [Google Scholar]
- [52].Lin Y, Devin A, Rodriguez Y, Liu ZG, Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis, Genes Dev. 13 (1999) 2514–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. , The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis, Cell. 150 (2012) 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. , Identification of RIP1 kinase as a specific cellular target of necrostatins, Nat. Chem. Biol 4 (2008) 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. , Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis, Science 343 (2014) 1357–1360. [DOI] [PubMed] [Google Scholar]
- [56].Wang H, Meng H, Li X, Zhu K, Dong K, Mookhtiar AK, et al. , PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP, Proc. Natl. Acad. Sci. U. S. A 114 (2017) 11944–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dziedzic SA, Su Z, Jean Barrett V, Najafov A, Mookhtiar AK, Amin P, et al. , ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC, Nat. Cell Biol 20 (2018) 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].de Almagro MC, Goncharov T, Newton K, Vucic D, Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination, Cell Death Dis. 6 (2015), e1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wei R, Xu LW, Liu J, Li Y, Zhang P, Shan B, et al. , SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination, Genes Dev. 31 (2017) 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Seo J, Lee EW, Sung H, Seong D, Dondelinger Y, Shin J, et al. , CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3, Nat. Cell Biol 18 (2016) 291–302. [DOI] [PubMed] [Google Scholar]
- [61].Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. , Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase, Cell 148 (2012) 213–227. [DOI] [PubMed] [Google Scholar]
- [62].Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. , Mixed lineage kinase domainlike is a key receptor interacting protein 3 downstream component of TNF-induced necrosis, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y, et al. , MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis, Proc. Natl. Acad. Sci. U. S. A 114 (2017) E7450–E7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. , Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3, Mol. Cell 54 (2014) 133–146. [DOI] [PubMed] [Google Scholar]
- [65].Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. , Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis, Nat. Cell Biol 16 (2014) 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen W, Wu J, Li L, Zhang Z, Ren J, Liang Y, et al. , Ppm1b negatively regulates necroptosis through dephosphorylating Rip3, Nat. Cell Biol 17 (2015) 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. , MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates, Cell Rep. 7 (2014) 971–981. [DOI] [PubMed] [Google Scholar]
- [68].Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. , Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death, Cell Res. 24 (2014) 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, et al. , MLKL forms cation channels, Cell Res. 26 (2016) 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. , ESCRT-III acts downstream of MLKL to regulate Necroptotic cell death and its consequences, Cell 169 (2017) 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].He S, Liang Y, Shao F, Wang X, Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 20054–20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P, The death domain kinase RIP mediates the TNF-induced NF-kappaB signal, Immunity 8 (1998) 297–303. [DOI] [PubMed] [Google Scholar]
- [73].Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. , RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3, Cell 157 (2014) 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, et al. , RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 7753–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. , RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis, Cell 157 (2014) 1175–1188. [DOI] [PubMed] [Google Scholar]
- [76].Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J, Functional complementation between FADD and RIP1 in embryos and lymphocytes, Nature 471 (2011) 373–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. , Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo, J. Immunol 193 (2014) 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Shutinoski B, Alturki NA, Rijal D, Bertin J, Gough PJ, Schlossmacher MG, et al. , K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo, Cell Death Differ. 23 (2016) 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, et al. , Polyubiquitin binding to ABIN1 is required to prevent autoimmunity, J. Exp. Med 208 (2011) 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. , RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS, Science. 353 (2016) 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, et al. , Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis, Nat. Commun 7 (2016) 12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. , NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and Necroptotic cell death during TNF signaling, Mol. Cell 60 (2015) 63–76. [DOI] [PubMed] [Google Scholar]
- [83].Dondelinger Y, Delanghe T, Rojas-Rivera D, Priem D, Delvaeye T, Bruggeman I, et al. , MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death, Nat. Cell Biol 19 (2017) 1237–1247. [DOI] [PubMed] [Google Scholar]
- [84].Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. , MK2 phosphorylates RIPK1 to prevent TNF-induced cell death, Mol. Cell 66 (2017) 698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Menon MB, Gropengießer J, Fischer J, Novikova L, Deuretzbacher A, Lafera J, et al. , p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection, Nat. Cell Biol 19 (2017) 1248–1259. [DOI] [PubMed] [Google Scholar]
- [86].Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. , Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis, Nat. Commun 8 (2017) 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang Z, Jiang H, Chen S, Du F, Wang X, The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways, Cell. 148 (2012) 228–243. [DOI] [PubMed] [Google Scholar]
- [88].Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, et al. , Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling, J. Biol. Chem 288 (2013) 16247–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, et al. , RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis, Nat. Cell Biol 20 (2018) 186–197. [DOI] [PubMed] [Google Scholar]
- [90].Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. , RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome, Nat. Commun 8 (2017) 14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Schenk B, Fulda S, Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death, Oncogene. 34 (2015) 5796–5806. [DOI] [PubMed] [Google Scholar]
- [92].Moquin D, Chan FK, The molecular regulation of programmed necrotic cell injury, Trends Biochem. Sci 35 (2010) 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, et al. , Widespread mitochondrial depletion via mitophagy does not compromise necroptosis, Cell Rep. 5 (2013) 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC, The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore, Cardiovasc. Drugs Ther 21 (2007) 467–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. , CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis, Nat. Med 22 (2016) 175–182. [DOI] [PubMed] [Google Scholar]
- [96].Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. , RIP3 mediates the embryonic lethality of caspase-8-deficient mice, Nature 471 (2011) 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM, Mechanisms of necroptosis in T cells, J. Exp. Med 208 (2011) 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. , Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 12024–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, et al. , The mitochondrial phosphatase PGAM5 is dispensable for Necroptosis but promotes Inflammasome activation in macrophages, J. Immunol 196 (2016) 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Moujalled DM, Cook WD, Murphy JM, Vaux DL, Necroptosis induced by RIPK3 requires MLKL but not Drp1, Cell Death Dis. 5 (2014), e1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, et al. , Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally, Immunity 9 (1998) 267–276. [DOI] [PubMed] [Google Scholar]
- [102].Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, et al. , FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis, Science 279 (1998) 1954–1958. [DOI] [PubMed] [Google Scholar]
- [103].Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, et al. , Survival function of the FADD-CASPASE-8-cFLIP(L) complex, Cell Rep. 1 (2012) 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. , Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis, Nature 471 (2011) 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. , RIP3 induces apoptosis independent of pronecrotic kinase activity, Mol. Cell 56 (2014) 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lichý M, Szobi A, Hrdlička J, Horváth C, Kormanová V, Rajtík T, et al. , Different signalling in infarcted and non-infarcted areas of rat failing hearts: a role of necroptosis and inflammation, J. Cell. Mol. Med 23 (2019) 6429–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, et al. , RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction, Cardiovasc. Res 103 (2014) 206–216. [DOI] [PubMed] [Google Scholar]
- [108].Müller T, Dewitz C, Schmitz J, Schröder AS, Bräsen JH, Stockwell BR, et al. , Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure, Cell. Mol. Life Sci 74 (2017) 3631–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. , Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury, Nat. Chem. Biol 1 (2005) 112–119. [DOI] [PubMed] [Google Scholar]
- [110].Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM, Necrostatin: a potentially novel cardioprotective agent? Cardiovasc. Drugs Ther 21 (2007) 227–233. [DOI] [PubMed] [Google Scholar]
- [111].Dmitriev YV, Minasian SM, Demchenko EA, Galagudza MM, Study of cardioprotective effects of necroptosis inhibitors on isolated rat heart subjected to global ischemia-reperfusion, Bull. Exp. Biol. Med 155 (2013) 245–248. [DOI] [PubMed] [Google Scholar]
- [112].Koshinuma S, Miyamae M, Kaneda K, Kotani J, Figueredo VM, Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury, J. Anesth 28 (2014) 235–241. [DOI] [PubMed] [Google Scholar]
- [113].Koudstaal S, Oerlemans MI, Van der Spoel TI, Janssen AW, Hoefer IE, Doevendans PA, et al. , Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs, Eur. J. Clin. Investig 45 (2015) 150–159. [DOI] [PubMed] [Google Scholar]
- [114].Cho Y, McQuade T, Zhang H, Zhang J, Chan FK, RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation, PLoS One 6 (2011), e23209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, et al. , Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models, Cell Death Dis. 3 (2012), e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. , Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice, Nat. Cell Biol 16 (2014) 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Qin D, Wang X, Li Y, Yang L, Wang R, Peng J, et al. , MicroRNA-223-5p and -3p cooperatively suppress necroptosis in ischemic/reperfused hearts, J. Biol. Chem 291 (2016) 20247–20259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. , RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury, Cell Death Differ. 23 (2016) 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, et al. , CaMKII determines mitochondrial stress responses in heart, Nature 491 (2012) 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP, Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis, J. Biol. Chem 282 (2007) 10833–10839. [DOI] [PubMed] [Google Scholar]
- [121].Zhu H, Tan Y, Du W, Li Y, Toan S, Mui D, et al. , Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control, Redox Biol. 38 (2021), 101777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Lu W, Sun J, Yoon JS, Zhang Y, Zheng L, Murphy E, et al. , Mitochondrial protein PGAM5 regulates mitophagic protection against cell necroptosis, PLoS One 11 (2016), e0147792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wang K, Liu F, Liu CY, An T, Zhang J, Zhou LY, et al. , The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873, Cell Death Differ. 23 (2016) 1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Wang JX, Zhang XJ, Li Q, Wang K, Wang Y, Jiao JQ, et al. , MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting FADD, Circ. Res 117 (2015) 352–363. [DOI] [PubMed] [Google Scholar]
- [125].Szobi A, Gonçalvesová E, Varga ZV, Leszek P, Kuśmierczyk M, Hulman M, et al. , Analysis of necroptotic proteins in failing human hearts, J. Transl. Med 15 (2017) 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Marunouchi T, Nishiumi C, Iinuma S, Yano E, Tanonaka K, Effects of Hsp90 inhibitor on the RIP1-RIP3-MLKL pathway during the development of heart failure in mice, Eur. J. Pharmacol 898 (2021), 173987. [DOI] [PubMed] [Google Scholar]
- [127].Kleinbongard P, Schulz R, Heusch G, TNFα in myocardial ischemia/reperfusion, remodeling and heart failure, Heart Fail. Rev 16 (2011) 49–69. [DOI] [PubMed] [Google Scholar]
- [128].Chen Y, Pat B, Zheng J, Cain L, Powell P, Shi K, et al. , Tumor necrosis factoralpha produced in cardiomyocytes mediates a predominant myocardial inflammatory response to stretch in early volume overload, J. Mol. Cell. Cardiol 49 (2010) 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Wu P, Cai M, Liu J, Wang X, Catecholamine surges cause Cardiomyocyte Necroptosis via a RIPK1-RIPK3-dependent pathway in mice, Front Cardiovasc Med. 8 (2021), 740839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Guo X, Yin H, Chen Y, Li L, Li J, Liu Q, TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints, Cell Death Dis. 7 (2016), e2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Li L, Chen Y, Doan J, Murray J, Molkentin JD, Liu Q, Transforming growth factor β-activated kinase 1 signaling pathway critically regulates myocardial survival and remodeling, Circulation. 130 (2014) 2162–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, et al. , TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway, Mol. Cell 5 (2000) 649–658. [DOI] [PubMed] [Google Scholar]
- [133].Yin H, Guo X, Chen Y, Zeng Y, Mo X, Hong S, et al. , TAB2 deficiency induces dilated cardiomyopathy by promoting RIPK1-dependent apoptosis and necroptosis, J. Clin. Invest 132 (2022), e152297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Cheng A, Dinulos MBP, Neufeld-Kaiser W, Rosenfeld J, Kyriss M, Madan Khetarpal S, et al. , 6q25.1 (TAB2) microdeletion syndrome: congenital heart defects and cardiomyopathy, Am. J. Med. Genet. A 173 (2017) 1848–1857. [DOI] [PubMed] [Google Scholar]
- [135].Engwerda A, Leenders EKSM, Frentz B, Terhal PA, Löhner K, de Vries BBA, et al. , TAB2 deletions and variants cause a highly recognisable syndrome with mitral valve disease, cardiomyopathy, short stature and hypermobility, Eur. J. Hum. Genet 29 (2021) 1669–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Guo X, Yin H, Li L, Chen Y, Li J, Doan J, et al. , Cardioprotective role of tumor necrosis Factor receptor-associated Factor 2 by suppressing apoptosis and Necroptosis, Circulation 136 (2017) 729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. , Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2, Nature 465 (2010) 1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lee TH, Shank J, Cusson N, Kelliher MA, The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2, J. Biol. Chem 279 (2004) 33185–33191. [DOI] [PubMed] [Google Scholar]
- [139].Fan Y, Yu Y, Shi Y, Sun W, Xie M, Ge N, et al. , Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1 beta-induced IKK/NFkappaB and JNK/AP-1 activation, J. Biol. Chem 285 (2010) 5347–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zhang L, Blackwell K, Shi Z, Habelhah H, The RING domain of TRAF2 plays an essential role in the inhibition of TNFalpha-induced cell death but not in the activation of NF-kappaB, J. Mol. Biol 396 (2010) 528–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Burchfield JS, Dong JW, Sakata Y, Gao F, Tzeng HP, Topkara VK, et al. , The cytoprotective effects of tumor necrosis factor are conveyed through tumor necrosis factor receptor-associated factor 2 in the heart, Circ. Heart Fail 3 (2010) 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Liu J, Wen T, Dong K, He X, Zhou H, Shen J, et al. , TEAD1 protects against necroptosis in postmitotic cardiomyocytes through regulation of nuclear DNA-encoded mitochondrial genes, Cell Death Differ. 28 (2021) 2045–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Xiao P, Wang C, Li J, Su H, Yang L, Wu P, et al. , COP9 signalosome suppresses RIPK1-RIPK3-mediated cardiomyocyte necroptosis in mice, Circ. Heart Fail 13 (2020), e006996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Su H, Li J, Menon S, Liu J, Kumarapeli AR, Wei N, et al. , Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice, Circ. Res 108 (2011) 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. , Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death, Nature 434 (2005) 652–658. [DOI] [PubMed] [Google Scholar]
- [146].Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. , Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes, Am. J. Physiol. Heart Circ. Physiol 314 (2018) H659–H668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. , Ferroptosis as a target for protection against cardiomyopathy, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Gao M, Monian P, Quadri N, Ramasamy R, Jiang X, Glutaminolysis and transferrin regulate Ferroptosis, Mol. Cell 59 (2015) 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. , Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury, Circulation 123 (2011) 594–604. [DOI] [PubMed] [Google Scholar]
- [150].Shi H, Gao Y, Dong Z, Yang J, Gao R, Li X, et al. , GSDMD-mediated cardiomyocyte pyroptosis promotes myocardial I/R injury, Circ. Res 129 (2021) 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Mishra PK, Adameova A, Hill JA, Baines CP, Kang PM, Downey JM, et al. , Guidelines for evaluating myocardial cell death, Am. J. Physiol. Heart Circ. Physiol 317 (2019) H891–H922. [DOI] [PMC free article] [PubMed] [Google Scholar]