

Significance
The ability of nucleic acids to catalyze reactions is important in the context of the origin of life and biomedical applications. However, the catalytic power of standard nucleic acids assembled from just four nucleotide building blocks is limited when compared with that of proteins. Using an artificially expanded genetic information system (AEGIS) carrying extra synthetic nucleotides, we show that DNA libraries with increased chemical diversity, higher information density, and larger searchable sequence spaces are at least one order of magnitude richer reservoirs of molecules able to catalyze the cleavage of RNA than DNA libraries built from a standard four-nucleotide alphabet. The AEGISzyme described here represents the first time that catalysts have been evolved from libraries built from expanded genetic alphabets.
Keywords: expanded genetic alphabets, RNA-cleaving DNAzymes, in vitro evolution, information density
Abstract
The ability of nucleic acids to catalyze reactions (as well as store and transmit information) is important for both basic and applied science, the first in the context of molecular evolution and the origin of life and the second for biomedical applications. However, the catalytic power of standard nucleic acids (NAs) assembled from just four nucleotide building blocks is limited when compared with that of proteins. Here, we assess the evolutionary potential of libraries of nucleic acids with six nucleotide building blocks as reservoirs for catalysis. We compare the outcomes of in vitro selection experiments toward RNA-cleavage activity of two nucleic acid libraries: one built from the standard four independently replicable nucleotides and the other from six, with the two added nucleotides coming from an artificially expanded genetic information system (AEGIS). Results from comparative experiments suggest that DNA libraries with increased chemical diversity, higher information density, and larger searchable sequence spaces are one order of magnitude richer reservoirs of molecules that catalyze the cleavage of a phosphodiester bond in RNA than DNA libraries built from a standard four-nucleotide alphabet. Evolved AEGISzymes with nitro-carrying nucleobase Z appear to exploit a general acid–base catalytic mechanism to cleave that bond, analogous to the mechanism of the ribonuclease A family of protein enzymes and heavily modified DNAzymes. The AEGISzyme described here represents a new type of catalysts evolved from libraries built from expanded genetic alphabets.
In vitro selection was conceived independently in 1990 by the laboratories of Larry Gold, Jack Szostak, and Gerald Joyce (1–3) as a way to obtain nucleic acid ligands and catalysts without the need to command chemical theory sufficient for direct design and without the directed trial and error that characterizes medicinal chemistry. When the process is seen to be “selection” without “evolution”, it presumes that a library of DNA or RNA (nucleic acid [NA]) molecules already contains one or more ligands for the target receptor or one or more catalyst for the target reaction. The experiment must only extract the desired ligands or catalysts from the library by exploiting their binding or catalytic powers. Then, PCR would make them in large amounts. If the presumption was incorrect, in vitro selection still could deliver, by allowing mutations during PCR to explore sequences not present in the original library.
Since it was introduced, the technology had successfully delivered aptamers for many targets. It has also been applied to evolve catalytic molecules (RNAzymes and DNAzymes), including RNA kinases, ligases, polymerases, and others (4–13).
Nevertheless, despite its success, as the concept developed, the question of initial library composition, both in the context of prebiotic molecular evolution and biotechnology, also arose. It was appreciated from the beginning that standard NAs contained fewer building blocks with less functional diversity than proteins. It was expected that this might mean that in vitro selection on standard NA platforms would not deliver performances comparable to those of contemporary proteins.
The last 20+ years of literature are rich with work attempting to understand requirements related to the composition of libraries built from four standard building blocks (for a comprehensive review, see ref. (14)). Studies of landscapes with standard RNA (15, 16) have revealed that, due to frustrated landscapes composed of largely disconnected islands of active sequences, chance emergence of an active RNA motif made of four building blocks would be more important in evolution than its optimization by natural selection, at least in the case of short sequences.
Depending on the specific function and other factors, the probability of finding a sequence that performs that function at an acceptable level in a pool of NA sequences with four building blocks ranges from 10−5 for mere binding to 10−30 for more-complex tasks, such as catalysis (14, 17).
These points prompt the question whether this platform is capable of supporting the “unfrustrated” evolution of catalysts for metabolic reactions matching protein enzymes or required for an RNA world.
Improvements to address the limited availability of building blocks in standard NAs compared with proteins were initially sought by adding functionalized side chains to one or more of the standard nucleotides used in the starting libraries (18, 19). This concept has been refined by the Mayer laboratory and elsewhere, showing good results with highly decorated aptamers (20, 21). Hirao showed that adding just one hydrophobic side chain can improve the affinities of aptamers to a picomolar level (22). Perrin and coworkers have had notable success obtaining catalytic DNAzymes using heavily functionalized libraries (23–25). Substituting polymerase-based amplification with ligation, the Liu laboratory was recently able to functionalize NA polymers with up to 32 variants of the building blocks (26). Following a different rationale, Gold’s group at SomaLogic obtained slow off-rate modified aptamers by appending hydrophobic side chains (benzyl, naphthyl, tryptamino, and isobutyl) to nucleobases (27, 28).
As an alternative approach, we and others have sought to improve the intrinsic functional value of NA libraries by increasing within them the number of independently replicating building blocks (29–32). This approach offers the possibility of adding functional groups more “lightly” to library components, as adding separate building blocks carrying new functional groups, rather than adding them to existing nucleobases, overcomes the issue of “overfunctionalization”, where the NA molecule no longer behaves like an NA molecule (33). Further, the information density of the library is increased by the number of building blocks, as reported in Table 1. This, in turn, may provide more control over folding, limit the possibility of inactive folds competing energetically with active folds (34), and allow for new kinds of folds (30, 35, 36).
Table 1.
Theoretical sequence space coverage for 1 or 2 nmol of starting libraries with different random region lengths and compositions (AEGIS = six, standard DNA = four building blocks), where numbers greater than 100% indicate the number of replicas per sequence that will be present in the library for that number of molecules
| 19 nt (1 nmol) | 25 nt (2 nmols) | 30 nt (2 nmols) | 40 nt (2 nmols) | 45 nt (2 nmols) | |
|---|---|---|---|---|---|
| AEGIS DNA | 100% | 4.3 × 10−3% | 5.5 × 10−7% | 9.2 × 10−15% | 1.2 × 10−18% |
| Standard DNA | 2.2 × 105% | 100% | 0.1% | 1 × 10−7% | 1 × 10−10% |
Many experiments from the Hirao, Benner, and Romesberg groups have shown that such addition is possible. The number of nucleotides in DNA and RNA is not constrained to four and can be increased to 6, 8, 10, and possibly 12 nucleotides (37–47). To date, however, expanded genetic alphabets have been examined only with respect to creating binders, avoiding the larger challenges presented by catalysis.
In this work, we assess, through in vitro selection, the evolutionary potential of libraries of NAs with different informational architectures as reservoirs for catalysis. Specifically, we compare in vitro selection experiments of two NA libraries carrying different information density, targeting the cleavage of RNA as a model reaction (48–50). This reaction has been widely explored with in vitro selection (for a review, see refs. (51–53)). The added information density and chemical diversity of the libraries come by adding two synthetic nucleotides from an artificially expanded genetic information system (AEGIS) developed in our laboratory.
Results
Standard/AEGIS In Vitro Selection of RNA-Cleaving DNAzymes/AEGISzymes.
Two parallel in vitro selection experiments to evolve RNA-cleaving DNAzymes were carried out with libraries containing either standard DNA or six-letter AEGIS DNA that included nucleobases Z (6-amino-3-(2′-deoxy)-D-ribofuranosyl)-5-nitro-1H-pyridin-2-one) and P (7-amino-9-(1′-beta-D-2′-deoxyribofuranosyl)-imidazo[1,2-c]pyrimidin-5(1H)-one). The starting libraries contained 25 nucleotides in their random regions. The standard library had a random region built from ACTG; the random region of the AEGIS library was built from ACTGZP (Fig. 1). The random regions were surrounded by standard DNA primer binding sites (PBSs).
Fig. 1.
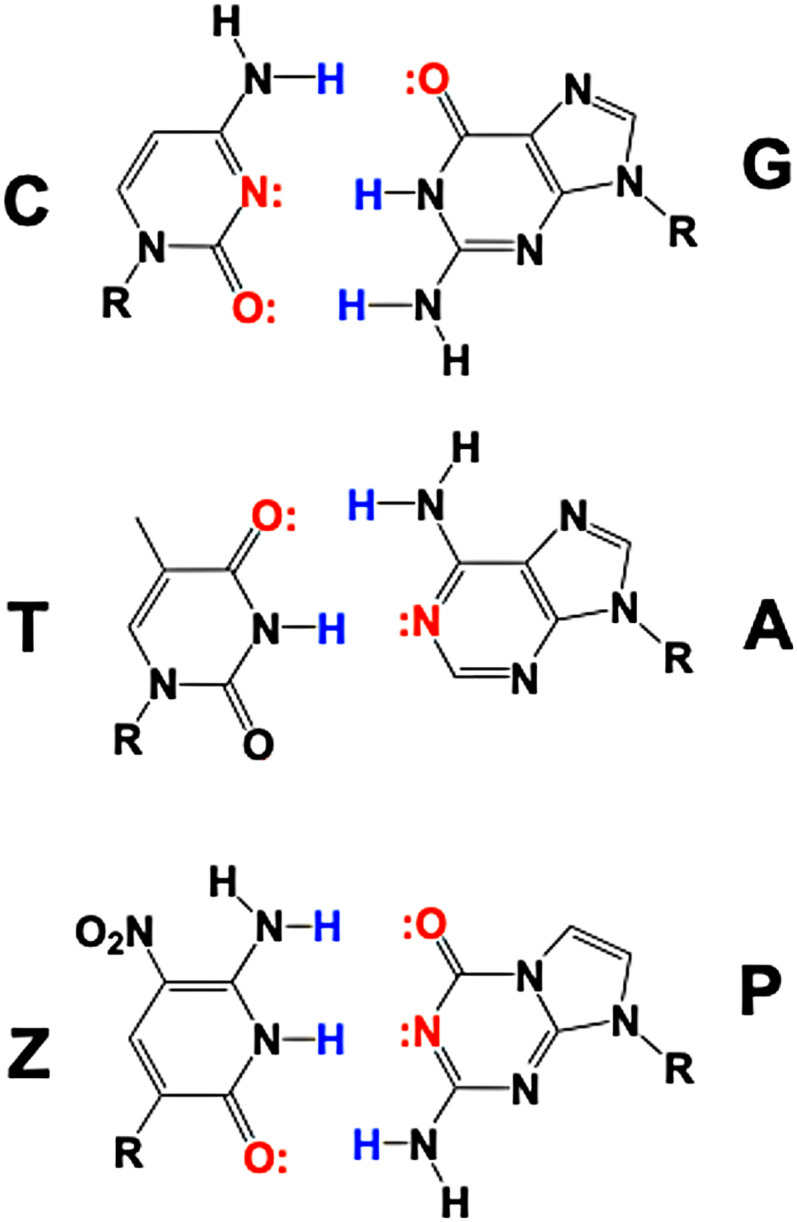
AEGIS and standard components of the libraries used in this study, showing hydrogen bond donors (in blue) and acceptors (in red).
The 25-nucleotide (nt) length for the random region was chosen for its being, for standard DNA, the largest library for which a nearly complete sequence space can be conveniently investigated with manageable scales of DNA material. Thus, a library with 25 random nucleotides built from standard nucleotides has 425, or 1015, possible sequences (Table 1). This sequence space can be, in principle, covered by ∼2 nmol of DNA; this amount would contain, on average, one exemplar of every possible sequence.
For six-letter AEGIS libraries, 25-nt-long random regions have 625 (2.8 × 1019) different sequences. This would require 47 μmol of library to similarly cover the sequence space. This scale is not conveniently attainable in the laboratory.
This mathematics captures the challenge of comparing “apples and oranges” outcomes from four-letter and six-letter evolution experiments. It shows that, with a four-letter, 25-nt library, the experiment actually seeks pre-existing catalysts already present in the library; no sequence evolution is possible. In the second case, only the initial rounds require that the library contain a selectable catalyst. Then, sequence evolution is possible and is a mechanism for obtaining receptors/catalysts that are improved over those originally in the library.
Exploiting this analysis, we chose to prepare 0.5 nmol of a standard DNA library as the starting point. This theoretically covered ∼25% of all possible sequences of this 25-nt length. We also prepared 0.5 nmol of the six-letter AEGIS library. This covered ∼0.0011% (0.000011 mol fraction) of the conceivable sequence space.
The laboratory selection/evolution strategy followed a protocol adapted from Santoro and Joyce (49), including AEGIS PCR protocols for the AEGIS in vitro evolution (40, 41, 43) (see SI Appendix, Fig. S1 for a schematic of the selection procedure). Either standard or six-letter AEGIS PCR was performed on the survivor pools after each selection cycle, using a 5′-biotinylated forward primer carrying a 12-nt-long RNA target. Here, we exploited an AEGIS-compatible polymerase able to read through RNA nucleotides. Pools for subsequent selections were made in single-stranded form using streptavidin magnetic beads capturing and treatment with base. Single-stranded DNA-RNA hybrids captured on magnetic beads through the RNA substrate were subjected to cleavage in buffer (2 mM MgCl2, 150 mM NaCl, and 50 mM Tris⋅HCl [pH 7.8]) for 30 min at 37 °C.
This gave components of the evolving pools an opportunity to fold over the target RNA sequence and cleave any one or more of 12 phosphodiester linkages, each resulting in the release of the active species into solution. These were collected and reamplified for the subsequent cycle. Selection cycles were repeated until substantial growth in catalysts was seen and for 16–18 cycles if no growth was seen.
Fig. 2 shows the progression of these selection experiments as percent of molecules released from magnetic beads versus the selection cycle No. Remarkably, the AEGIS pool was enriched with substantial apparent cleaving activity as early as cycle 9. Cycling was stopped after cycle 11, when a small decline in enrichment was seen.
Fig. 2.

AEGIS versus standard RNA-cleaving DNAzyme selection progression. Plot of percent of molecules released in solution (“% cleaved”) versus cycle No. Red dots, solid line: in vitro selection with AEGIS library; black dots, solid line: in vitro selection with standard library; gray triangles, dashed line: second in vitro selection with standard library with switched primers.
In contrast, sustained enrichment in activity in the standard DNA library was never seen, even though the selection was continued for 16 cycles. This implied that the initial library did not contain a single exemplar of a catalyst able to pass the survival challenge or one that was able to mutate by a site or two to cover the remaining sequence space not covered in the original library.
As the two different evolution results could have been due to the specific amplifying proficiency of the primer binding regions (which were different in the two starting libraries to avoid cross-contamination), a second attempt was made to extract DNAzymes from 25N standard libraries. Here, the standard library carried the PBSs originally used in the AEGIS library. Again, DNAzymes did not emerge, even after 16 cycles (gray dashed line in Fig. 2).
Deep Sequencing and Cluster Appearance.
Because the standard libraries did not produce catalysts, subsequent analyses were performed only on survivors of the AEGIS experiments. Here, pools from each cycle, including the starting library, were prepared and submitted for high-throughput sequencing (HTS) on an Illumina platform after the GACTZP sequences had been transliterated using two PCR conditions to replace Z and P with C and G in the first condition and to replace them with an approximately equivalent mixture of C+T and A+G in the second (54, 55). Sites that held Z and P in any surviving sequence were assigned based on being primarily C or G, respectively, in the results from the first PCR condition and C+T or A+G mixtures in those from the second PCR condition. This HTS analysis found that, out of ∼40 total clusters, at least seven of the most abundant clusters carried AEGIS nucleotides (Fig. 3A). The 10 most-abundant clusters were considered throughout the 10 selection cycles (C1 through 10 numbered approximately corresponding to relative abundance of reads in deep sequencing results and cycle appearance). Interestingly, all clusters except for C5 had at least two Zs; the best represented sequence (52% of reads) had four Zs and no Ps.
Fig. 3.

Sequences and appearance of clusters during AEGIS DNAzyme selection. (A) Schematic representation of cluster sequences. In blue: RNA target; underlined: primer binding regions. Locations of AEGIS P (green) and Z (red) are marked. (B) Plot of sequence abundance (expressed as No. of reads) versus selection cycle. (C) Table of cluster appearance color coded by type of cleavage. Blue: type 1, green: type 2, red: type 3 (see text for details), gray: no apparent cleavage.
Cluster 5 sequence (not shown in Fig. 3A) did not contain any AEGIS nucleotides. Its members did not show apparent cleavage activity in the selection conditions (SI Appendix, Fig. S3) and were not analyzed further.
Clusters 3 and 4 contained AEGIS nucleotides, but deep sequencing using our transliteration approach did not give a number of parallel reads sufficient to assign with confidence the location of the AEGIS nucleotides. Thus, these clusters were not analyzed further.
Deep sequencing after each selection cycle provided a panoramic view of sequence dynamics during the evolution experiment (Fig. 3B). Thus, some of the clusters (C1 and C2) were observed in cycle 1 and were evidently represented in the six-letter library. Others appeared later, in rounds 4, 6, 7, and 8. These might have been too infrequent in the starting pool for HTS to capture them or might have arisen by actual evolution.
Analysis of the cis-Cleaving Activity of Cluster Representatives.
Representative molecules from the 10 clusters selected from HTS data were resynthesized with a 5′-32P-label and tested for RNA-cleaving activity in cis, as species free in solution. As shown in Fig. 4 (and Fig. 3C), three classes of AEGISzymes were identified based on where they cleaved within the 12-nt RNA sequence:
Fig. 4.

Eighteen percent denaturing PAGE of cleavage reactions in cis for AEGISzymes cluster representatives, revealing three classes of AEGISzymes. “NR”: nonreacted; T-OH: partial alkaline hydrolysis of the RNA–DNA hybrid molecule used during selection and here as a ladder. RNA bases and sizes in nucleotides are indicated on the Right.
Type 1 AEGISzymes cleaved at G8 and included cluster 1, the most abundant survivor in the HTS data.
Type 2 AEGISzymes cleaved at A16 and included clusters C8 and C10, both appearing at cycle 7.
Type 3 AEGISzymes cleaved at A10 and included cluster C9, appearing at cycle 8.
Clusters 2, 6, and 7 did not appear to have substantial true self-cleavage activity when tested in free solution. However, they did show what we regarded as an increased susceptibility to hydrolysis at U9 and U13, which likely allowed them to survive the selection strategy (C6 not shown in Fig. 4). Interestingly, a selection experiment by Williams and coworkers also evolved DNAzymes seemingly cleaving at these two sites (both U*A) on a 12-nt-long RNA target with the same sequence as ours, using a library carrying imidazole and amino groups (56). As those authors noted, these sites are seen in the literature to be more favorable toward spontaneous cleavage, due to decreased base stacking and/or altered hydrogen bonding networks around the scissile linkage, depending on the specific sequence environment (57, 58).
The ability of these sequences to increase the rate of cleavage at those two RNA sites may be compared with other evolved sequences that do not have this effect on the same RNA target. This is especially interesting because we observe no obvious sequence similarity among clusters C2, C6, and C7 compared with sequences that show specific cleavage (but see below). This effect is likely due to the local structural environment enforced on these two ribonucleotides by the specific surrounding sequence; this might increase the time they spend sampling optimal in-line conformations for spontaneous nucleophilic attack of the phosphate by the adjacent 2’OH. These were not analyzed further in the present study.
It is noteworthy that the originally evolvable region in clusters C1 and C2 shared a 14-nt-long sequence containing the AEGIS nucleotides with 79% sequence identity (52% overall), where 11 out of 14 nt are identical, with two of the four Zs in C1 shared by C2 while the other two Zs are not (SI Appendix, Fig. S6). Since C1 has cleavage activity but C2 does not, this might indicate that the two internal Zs lacking in C2 but present in C1 are essential for catalysis. Clusters C8 and C10, both showing type 2 cleavage, also presented 56% overall sequence homology in the variable region, with 11 matching residues, including one Z. Moreover, when C8 and C10 sequences were run through the Dynalign web server (59) (https://rna.urmc.rochester.edu), which predicts shared secondary structures for two sequences, this calculated a stable (ΔG = −19.7 kcal/mol) stem-loop two dimensional (2D) conformation common to the two molecules (SI Appendix, Fig. S7).
The two clusters sharing highest overall sequence identity, 68% including two consecutive Zs, were C6 and C7. Neither of these showed specific cleavage within the RNA substrate. However, both presented, as mentioned, a level of increased hydrolysis at U6 and U13.
Interestingly, C1, which displayed a type 1 cleavage and had four Z nucleotides, was present in the initial library, persisted during all selection cycles, and was also the most-active cleaver when tested free in solution (∼27% cleavage after 4 h).
Assuming that the synthesis was random with respect to catalytic activity, these results offer prima facie evidence that the six-letter library was a richer reservoir for functional molecules than the standard four-letter library. Indeed, the density of catalysts meeting a certain threshold in the AEGIS library is here a positive number (∼10 in 1019), and that number is higher than that measured density in the standard library (0 in 1015 = exactly zero).
Enzymatic digestions were performed to assess a 2D conformation prediction produced with mFold (60) (Fig. 5A). Here, deoxyribonuclease I (DNase I), which nonspecifically cleaves DNA, and ribonuclease H (RNase H), which cleaves the RNA portion in RNA:DNA hybrid helices (61), were used in separate time courses (Fig. 6). As clearly shown by RNase H digestion results, part of the RNA substrate is entrapped in a 7-nt-long double-helix (with one G-U wobble), presenting the cleavable nucleotide to what would appear to be an unstructured region including the four Z (Fig. 5A). In the active 3D conformation, it is conceivable that this region is most likely not unstructured but folding over the cleavage site in an optimal configuration.
Fig. 5.

Cluster 1 AEGISzyme cleavage. (A) Two-dimensional rendering of AEGISzyme C1 generated with mfold (60) and RNA/DNA construct primary sequence. RNA target in blue, binding sites in green and underlined, Zs in red, and cleavage site in yellow. The RNA:DNA duplex portion of the molecule was confirmed by enzymatic digestion. (B) Denaturing 18% PAGE of cleavage reaction time course, with time points marked in minutes (‘) and hours (h). RNA bases and sizes in nucleotides are indicated on the Left. (C) Plot of percent cleavage versus time in minutes. Data were fitted to a double exponential equation. Data points represent the mean value of at least three replicates.
Fig. 6.

Enzymatic digestion of AEGISzyme C1 in native conditions. Eighteen percent, 7M urea PAGE. Left: time course of DNase I digestion. Right: time course of RNase H digestion. Time points are reported in minutes (‘). NR: nonreacted molecule. “DNA ladder”: 10-bp DNA ladder (New England Biolabs). “DNase I DNA ladder”: 10-bp DNA ladder digested with DNase I. Lengths in base pairs for the DNA ladders are in blue. RNA bases and sizes in nucleotides are indicated in black for ladders and DNA:RNA hybrid AEGISzyme C1. G8*: band resulting from the self-cleavage of C1 at its cleavage site during the digestion reaction, which is not a product of DNase I or RNase H digestion. U9**: hot spot for spontaneous hydrolysis of the RNA target, which is not a product of DNase I or RNase H digestion.
C8 and C10, showing type 2 cleavage, appeared together at the same cycle and showed high sequence similarity. They had somewhat different cleavage efficiencies, with ∼21% and 8% cleaved after 4 h incubation for C8 and C10. Type 3 cleaving cluster C9 appeared only at later cycles and gave ∼20% cleavage after a 4h incubation. These show that the AEGIS system actually was evolving during the process to deliver new catalysts that were not present in the original library.
We then performed a detailed kinetic analysis of AEGISzyme C1 self-cleavage under selection conditions. Experimental data observed a bimodal rate profile where the overall maximum fraction cleaved was ∼55% and the observed rates were kfast = 1.26 × 10−1 min−1 and kslow = 1.65 × 10−3 min−1. In contrast, clusters C8, C9, and C10 showed monophasic cleavage rates that fit a single exponential. The C8 and C10 representatives, cleaving at A16, had values for kobs of 0.76 × 10−3 and 0.34 × 10−3 min−1, respectively, with maximum cleavage of ∼25% (C8) and ∼31% (C10). The type 3 AEGISzyme from cluster C9 cleaved at A10 with kobs = 0.83 × 10−3 and maximum cleavage at ∼23%. The rate of 0.126 min−1 for C1 is fast for a primary NA enzyme lacking any postselection optimization or secondary reselection. Typically, first-generation self-cleaving DNAzymes arising directly from selection, before any optimization, present rates in the ∼10−3 range or lower (25, 56, 62). For example, C1’s kobs is at least one order of magnitude faster than the first-generation cleavers initially obtained by Perrin and coworkers when using a heavily modified 40N library carrying RNase A–mimicking modifications (25). Here, we obtained better rates with just one extra nucleotide in a 25N evolvable random region.
Standard versions of each AEGISzyme, where every Z was substituted with C and every P with G, were tested for activity in parallel with the original AEGISzymes (Fig. 7 and SI Appendix, Figs. S4 and S5). For every AEGISzyme tested, removal of the AEGIS components destroyed cleavage activity, indicating that AEGIS nucleotides were essential for activity.
Fig. 7.

(A) Self-cleavage of AEGISzymes C1, C8, C9, and C10. Data points were fitted to a double exponential equation and represent mean values of at least three replicates. (B) Eighteen percent denaturing PAGE showing an example of kinetic reactions for AEGISzyme C9 versus DNAzyme C9 – no ZPs, where AEGIS nucleotides were removed. T-OH: partial alkaline hydrolysis of the DNA/RNA ladder hybrid molecule used as forward primer during selection and here as a ladder.
Magnesium Dependence of Cleavage by AEGISzyme C1.
The dependence of self-cleavage activity of AEGISzyme C1 was tested at different magnesium concentrations, from 0 mM to 20 mM. Kinetic reactions were followed for 26 h. Results are shown in SI Appendix, Fig. S8. The highest catalytic rate was found when [Mg2+] = 5 mM. This suggests a requirement of at least one (but not two) bound Mg2+, either catalytic or, more likely, structural. Further, the bell-shaped curve in SI Appendix, Fig. 8B suggests that the AEGISzyme can bind with weaker affinity to additional Mg2+ ions to give complexes that are inactive or have lower activity or where the cation directly contributes to stabilizing the transition states of the spontaneous hydrolysis of any of the 12 nt in the target RNA.
pH Dependence of Cleavage by AEGISzyme C1.
The dependence on pH of self-cleavage activity of AEGISzyme C1 was tested over the range pH 6.0–9.0 (Fig. 8). Here, the log(kobs) values were seen to increase linearly in the range 6.0–7.0, with a slope of +0.93, decreasing in the range 7.0–9.0, with a negative slope of −0.233 (SI Appendix, Fig. S9). The bell-shaped curve is consistent with a kinetic model involving acid–base catalysis with at least two ionizable species, one acting as proton donor and the other acting as a proton acceptor. As the data did not fit well to a simple symmetrical Gaussian equation (SI Appendix, Fig. S9B), the ionizable species participating in the cleavage likely had two different pKas, perhaps in the range between 7 and 8 and influenced by local environments in the active site fold.
Fig. 8.

(A) Self-cleavage of AEGISzyme C1 at different pHs. Each data set was fitted with a biphasic, double-exponential equation. Data points are mean values of two to five replicates. (B) Plot of AEGISzyme C1 log(kfast) versus pH.
This profile is consistent with a SN2-type transesterification reaction typical of the well-known protein RNase A. It is noteworthy that the original selection experiment was performed at pH 7.8, which is the pKa of nucleobase Z free in solution. Here, it is conceivable that at least two of the Z nucleobases in AEGISzyme C1 are the ionizable participants in the reaction, with structurally induced perturbed pKas between ∼7 and 8. This would reflect an active site analogous to that of RNase A.
Discussion
This work compares libraries built from standard DNA nucleotides with libraries built from an Artificially expanded genetic Information System for their ability to deliver molecules that cleave a phosphodiester linkage within an RNA substrate 12 nt long (DNAzymes or AEGISzymes, respectively). Here, we chose to start with 25-nt random sequence libraries made of either four standard or six standard + AEGIS different building blocks.
Our working hypothesis premised that laboratory in vitro evolution applied to the second library would better deliver cleavers able to survive, be enriched, and evolve during selection than the first library, even though the second covered only 0.0011% (0.000011 mol fraction) of the available sequence space (Table 1).
Here, the results were clear. The standard library delivered no cleavers that survived to be enriched in our selection conditions. As the original pool covered 25% of the possible sequences, little evolution was possible in principle.
In contrast, the AEGIS library delivered several catalysts after only nine cycles of selection. At least one of these appears to have been present early and likely in the original library. Assuming that the library samples randomly with respect to cleavage activity, this implies that the total library would have contained ∼100,000 RNases with comparable activity. Since the standard library, which sampled a quarter of the sequence space, did not provide even a single RNase with this activity, we infer that the density of activity at this level is at least one order of magnitude higher in the AEGIS functionalized library than in the standard library.
The other AEGISzymes identified up to round 10 escaped detection by HTS in the original library, indicating that they were either present in very low numbers or not present at all at the beginning of the experiment. If the latter was the case, this would suggest that these experiments should not be seen as classical in vitro selection, where the only products are sequences already present in the original pool. Rather, this would suggest that there might have been actual in vitro evolution, where mutation delivered products whose sequences were not in the original library.
In this context, it should be noted that, while standard PCR is sufficiently faithful as to not likely permit much “search” of the space of standard sequences, AEGIS PCR is less faithful and can be compared with artificial steps of reselection and mutagenic PCR used after standard in vitro selection/evolution experiments. However, the mutation frequency is not uniform. Thus, the most frequent are transitions that interconvert Z and C and transitions that interconvert P and G. The PCR used here does not create many transitions that interconvert Z and T or T and C. Likewise, AEGIS-PCR does not create many transitions that interconvert P and A or A and G. Further, it does not likely create transversion mutations more frequently than standard PCR (54, 63).
The failure of the evolution experiment to find any RNase activity in standard libraries might appear to contradict literature. However, no previous literature, to our knowledge, reports selections of DNAzymes with libraries with such a short random region with no extra modifications and/or at these low salt concentrations. Moreover, the majority of recent literature uses a strategy where only one ribonucleotide is embedded in the target sequence, often with predesigned base-pairing arms to the sides of the random region (see ref. (50) for a description of RNA-cleaving DNAzyme strategies); a longer RNA target is seldom used (25, 56, 62, 64). This is because multiple targets in the same selection experiment decrease the overall reaction efficiency by diluting successful outcomes into many different catalysts. As a result, the few examples of this procedure generally have led to the isolation of first-generation DNAzymes with lower kobs (25, 56, 62) (here, we do not include Dz8–17 and 10–25 from Santoro and Joyce (49) and others who did not report intramolecular cleavage rates of first-generation selection survivors).
Thus, failure to evolve first-generation DNAzymes from standard libraries in our limiting conditions might be ascribed to the lack of enough “information” in natural DNA of this length, likely exacerbated by the lack of evolvability of comparably long PBSs required for in vitro evolution experiments (15).
Why might the GACTZP library be a richer reservoir of RNase activity than a standard GACT library? The pH-rate data and a general understanding of mechanism in protein RNase reactions and DNAzymes and RNAzymes reactions allow the suggestion that acid–base catalysis might be at play. In order to transfer a proton efficiently, the pKa of a species must be near the pH of the solution (when the protonated and deprotonated species are roughly equally represented). For canonical nucleobases, this requires that the pKa be shifted by ∼2, possible but requiring a large investment in perturbing the local environment of the active site (65–71). This investment may not be necessary when the nucleobase has a pKa near the solution’s pH.
With the pKa of the isolated heterocycle being measured at 7.8, Z provides DNA libraries a general acid–general base functionality near neutral pH that is absent in standard nucleobases; such general acid–base functionality is present in proteins, provided by histidine. In the well-studied protein RNase A from ox pancreas, two histidine residues act in concert in a “pull–push” general acid–general base mechanism. Here, one of the histidines must be protonated; the other must be deprotonated (Fig. 9).
Fig. 9.

General base–general acid type catalysis performed by RNase A (Top) and by Z nucleobase (Bottom).
We propose that the AEGISzyme effects RNA cleavage via an analogous mechanism but with the histidines replaced by Zs. Here, one of the Zs must be protonated; the other must be deprotonated. The pH-rate profile showing a maximum rate with a pH near the pKa of the Z heterocycle is consistent with this. With one Z protonated and one Z deprotonated around the phosphodiester linkage, general acid–base catalysis of RNA cleavage would follow. This hypothesis is also consistent with the observation that the selected RNases contain at least two Zs.
The apparent biphasic kinetics of AEGISzyme C1 fit well only with two exponentials, suggesting the presence of at least two conformations. Of these, one might represent an inactive conformation that slowly refolds into the active conformation with the help of magnesium ions. Similarly, the low kobs at higher magnesium concentrations might indicate that the AEGISzyme gets consistently trapped into magnesium-stabilized inactive conformations.
The efficient generation of cleavers from AEGIS 25N libraries and not with standard libraries supports our hypothesis that expanding the genetic alphabet enriches the sequence space as a reservoir of functional material. The rates of catalysis of AEGISzyme C1 are high, although not outstanding. This may represent an intrinsic limitation of the library itself. The fact that the PCR mutation profile allows only limited search of the sequence space may also be an important restraint. Because many transition mutations, and most transversion mutations, are not generated, the vast majority of the sequence space not represented in the original library remains unexplored.
Furthermore, these cis-acting molecules are long (74 nt, including the PBSs); much of this length is also not evolvable. This may allow the system to get “stuck” in kinetic conformational traps out of which they cannot evolve, a problem observed in other selections (34). Adding nucleotides, by increasing the information density of the oligonucleotides, allows for fewer conformational traps. This may explain in part why also four-letter libraries, even with extensive functionalization, including histamines, guanidinium, and amines, do not achieve outstanding rates (∼10−2–10−3 min−1) when coming “straight” from crude selection (25, 56, 62). Another consideration is that the selection strategy used here and in most other studies effectively prevents the selection of the best catalysts, as these are lost during selection steps at conditions that are optimal for RNA cleavage (however, as this is true for both selections, it does not affect our comparative analysis).
In at least one case in Perrin’s work on heavily modified RNase-like DNAzymes, reselection of 40N evolved molecules with 15% mutagenesis and error-prone PCR was needed to achieve the same rates of cleavage that we obtained from crude evolution experiments with just two extra nucleotides (from kobs = ∼10−2 min−1 to kobs fast = 0.21 min−1 for Dz7-45–28 (25)).
Granted, this postselection strategy on densely functionalized material also provided a DNAzyme that was metal free with a kobs fast = 4.9 min−1 (Dz7-38-32). Here, each molecule carries a modification on each of three nucleobases (dAim, dUga, and dCaa). Thus, the rate of the C1 AEGISzyme (kobs fast of 0.126 min−1) is especially noteworthy, since it requires no postselection and carries only four nitro-containing extra Zs in a 74-nt molecule. These comparisons also reinforce the point that AEGIS-PCR and AEGIS in vitro evolution carry embedded in themselves evolutionary features that standard selection do not present, needing artifices like mutagenesis and error-prone reselection to sample a larger set of sequence space that AEGIS in vitro evolution experiments have largely pre-embedded.
Materials and Methods
Materials.
Standard oligonucleotides and libraries were purchased from Integrated DNA Technologies. AEGIS nucleotide triphosphates were purchased from Firebird Bio. Radiolabeled [α-32P]ATP was from PerkinElmer. Dynal streptavidin-coated magnetic beads (M-270) were from Invitrogen.
Polynucleotide kinase, DNase I, RNase H, and relevant buffers were bought from New England Biolabs. Takara Taq HF polymerase was from Takara. Other general chemicals were from Sigma-Aldrich and Fisher Scientific.
Methods.
Oligonucleotide synthesis.
Oligonucleotides and libraries containing AEGIS nucleotides were prepared as previously reported (40, 41). The randomized sites in the library were prepared by coupling with a 1:1:1:1:1:1 mixture of the six (GACTZP) nucleoside phosphoramidites. The synthetic oligonucleotides and library were purified on denatured polyacrylamide gel electrophoresis (PAGE) (7 M urea) and then desalted using Sep-Pac Plus C18 cartridges (Waters).
In vitro selection (SI Appendix, Fig. S1).
-
(1)
The starting randomized libraries of interest (25N ACTG or ACTGZP, 2 nmol each starting material) were subjected to five cycles of PCR with a forward primer and a biotinylated reverse primer containing the 12-nt RNA target and a 7-nt unstructured DNA spacer between the RNA and a biotin tag. Specifically, the primer construct was composed of, in 5′ to 3′ orientation, (i) a biotin molecule; (ii) a 7-nt DNA spacer, with sequence d(GGAAAAA); (iii) the target RNA sequence, r(GUAACUAGAGAU); and (iv) a 15-nt-long DNA sequence complementary to the 3′ primer binding region of the starting library (reverse DNA/RNA hybrid primer). While sequences (i)–(iii) are the same as used in the original Santoro–Joyce selection (49), both 15-nt-long PBSs on the library differed in the two pools, to avoid cross-contaminations between selections. The primer pairs were switched in the second reiteration of the standard DNAzyme experiment. Primer extensions, and subsequently PCR amplifications for each cycle, were performed with Takara Taq HS DNA polymerase and a modified buffer, previously shown to be able to (a) read through an RNA/DNA template and (b) efficiently copy AEGIS DNA (40–43). The amplified products were gel purified in 10% native PAGE before subjecting them to treatment with streptavidin magnetic beads. For each starting library in cycle 1, 0.5 nmol of this material were used, corresponding to 25% and 0.0011% sequence space coverage for standard and AEGIS DNA, respectively.
-
(2)
Primer extended/amplified, internally 32P-labeled, double-stranded molecules were then bound to streptavidin-coated magnetic beads according to the manufacturer protocol and made single stranded with brief alkaline treatments to remove the nonbiotinylated strand. Five milligrams streptavidin beads (500 μL suspension) were used in cycle 1 for each library type, and 1.5 mg (150 μL suspension) were used in subsequent cycles. This resulted in the single-stranded construct 5′-biotin-DNA(7 nt)-RNA(12 nt)-DNA library (55 nt)-3′ being bound to the solid substrate via its 5′ end and able to be subjected to self-cleavage.
-
(3)
A self-cleavage reaction was started by resuspending the library/target-coated magnetic beads in a solution containing 2 mM MgCl2, 150 mM NaCl, and 50 mM Tris⋅HCl at pH 7.8 and incubating at 37 °C for 30 min.
-
(4)
After the prescribed incubation time, supernatants containing molecules released from the beads were collected, concentrated, and amplified with the same primers used as in step 1, at which point a selection cycle was completed.
Negative cycles for this selection were embedded in the cycles themselves: for RNA cleavage, a negative cycle would require the incubation of the RNA target–DNA library hybrid at conditions other than the ones set for cleavage and recovery of anything that did not cleave. This exactly was done at every cycle in this scheme when the single-stranded library construct coupled to the beads was washed with NaOH followed by three Tris⋅HCl, pH 7.8 washes before adding the cleavage buffer and incubating at 37 °C (between steps 2 and 3).
Transliteration and deep sequencing.
GACTZP libraries were sequenced using a transliteration method improved from the one described by Yang et al. (54, 55). Survivors from each cycle were PCR amplified under two separate conditions that transliterate each Z:P pair into C:G (condition 1) or a 1:1 mixture of C:G and T:A pairs (condition 2) using an error-prone polymerase methodology developed for this purpose. Tags were then added by 12 cycles of PCR, and primers included barcodes specific for the two conversion conditions. The amplicon mixture was purified by native PAGE, recovered by gel extraction, and submitted for deep sequencing. The full-length recovered sequences were clustered using a custom algorithm that considers only the variable nucleotide region by grouping those with a single base change between sequence reads, starting with the most common read and proceeding toward the least common, and iterating until all sequences were grouped. Clustered sequences were then separated into sets by barcode and variable sites compared between each set. Sites in the aligned sequences that are consistently G, A, C, and T in both conditions were assigned as G, A, C, and T, respectively. Conversely, we assigned Z residues to sites in the aligned sequences that showed ∼1:1 mixtures of C and T in condition 2 and P residues to sites in the aligned sequences that will show ∼1:1 mixtures of G and A.
Resynthesis of cluster representatives.
AEGIS DNA/RNA hybrid cluster representative constructs were prepared following either of two procedures: (i) by AEGIS PCR amplification followed by streptavidin beads–based strand separation using reverse-complement AEGIS DNA templates synthesized at Firebird Biomolecular Sciences, LLC, and primers as during selection but with biotin on the forward strand, with consequent recovery of the single-stranded DNA/RNA construct free in solution. These were then 5′-32P labeled and purified by gel extraction to remove any degradation product due to the hydrolysis of the RNA portion during manipulation; (ii) by primer extension with a 5′-32P-labeled DNA/RNA reverse primer, with subsequent strand separation and purification by denaturing PAGE (16%, 7M urea).
Enzymatic digestions.
Enzymatic digestions of AEGISzyme C1 in native conditions were performed as follows: 5′-[γ-32P]-labeled AEGISzyme C1 was denatured in water at 95 °C for 3′, after which the cleavage buffer was added to 1× final concentration and the tube passed on ice. Following this, enzyme-specific buffers and nucleases were added and a ∼10 s time point collected. The tubes were then passed at 37 °C. Five microliter time points were collected at 10 s, 30 s, 1 min, 2 min, 5 min, 8 min, 12 min, 16 min, and 20 min and quenched in 10 μL denaturing gel loading buffer (95% formamide, 10 mM ethylenediaminetetraacetic acid (EDTA), 0.025% bromophenol blue, and 0.025% xylene cyanol). Reaction conditions were as follows: DNase I digestion: 0.2 μM 5′-[γ-32P]-labeled AEGISzyme C1 in 1× cleavage buffer, 0.08 U/μL DNase I, 10 mM Tris⋅HCl, 2.5 mM MgCl2, 0.5 mM CaCl2, pH 7.6@25 °C and RNase H digestion: 0.2 μM 5′-[γ-32P]-labeled AEGISzyme C1 in 1× cleavage buffer, 0.2 U/μL RNase H, 50 mM Tris⋅HCl, 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, pH 8.3 @ 25 °C. Final reaction volumes were 50 μL.
Self-cleavage reactions.
Cleavage reactions in cis of resynthesized, 5′[32P]-labeled cluster representatives free in solution were performed as follows: 0.1–1 μM DNA/RNA hybrid construct (SI Appendix, Fig. S1) was denatured in water for 3 min at 95 °C, followed by slow renaturation to 37 °C at 0.1 °C/s. Reactions were started by the addition of cleavage buffer with final composition: 2 mM MgCl2, 150 mM NaCl, and 50 mM Tris⋅HCl (pH 7.8) and incubating at 37 °C for different amounts of time.
Single point reactions were taken at 4 h.
In kinetic reaction, aliquots were taken at 0 min, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 24 h, and 26 h and quenched in urea gel loading buffer (10 M urea, 15 mM EDTA, 0.025% bromophenol blue, and 0.025% xylene cyanol).
For AEGISzyme C1 Mg2+ cleavage dependence, the same reactions were performed at 0 mM, 1 mM, 2 mM, 5 mM, 10 mM, and 20 mM MgCl2.
For AEGISzyme C1 pH cleavage dependence, kinetic reactions were performed in 50 mM Tris⋅HCl at pH 6.5, 7, 7.5, 7.8, 8, 8.5, and 9. Kinetic reactions at pH 6.0 were performed in 50 mM 2-morpholinoethanesulfonic acid monohydrate.
Collected aliquots were run on 18%, 7 M urea denaturing PAGE; exposed to a phosphorimager screen; and radioactive signal read with a Personal Molecular Imager phosphorimager (Bio-Rad).
Gel images were quantified with the software Image Lab (Bio-Rad), and data were plotted with KaleidaGraph (Synergy Software). Data fitting was performed with either a single-exponential equation , where k is the cleavage rate and a is the maximum cleavage at the plateau, or a double-exponential equation + , where k1 and k2 are the fast and slow kobs and a and b are the fast and slow maximum cleavage at plateau, respectively.
AEGISzyme C1 pH profile was fitted with equation .
Supplementary Material
Acknowledgments
This material is based upon work supported by the NSF under grants CHE-2108028 to E.B. and MCB-1939086 to S.A.B.
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2208261119/-/DCSupplemental.
Data, Materials, and Software Availability
All data pertaining to this work will be made available to readers upon publication.
References
- 1.Tuerk C., Gold L., Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Robertson D. L., Joyce G. F., Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 344, 467–468 (1990). [DOI] [PubMed] [Google Scholar]
- 3.Ellington A. D., Szostak J. W., In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Bartel D. P., Szostak J. W., Isolation of new ribozymes from a large pool of random sequences [see comment]. Science 261, 1411–1418 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Biondi E., Maxwell A. W. R., Burke D. H., A small ribozyme with dual-site kinase activity. Nucleic Acids Res. 40, 7528–7540 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akoopie A., Arriola J. T., Magde D., Müller U. F., A GTP-synthesizing ribozyme selected by metabolic coupling to an RNA polymerase ribozyme. Sci. Adv. 7, eabj7487 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ekland E. H., Bartel D. P., RNA-catalysed RNA polymerization using nucleoside triphosphates. Nature 382, 373–376 (1996). [DOI] [PubMed] [Google Scholar]
- 8.Johnston W. K., Unrau P. J., Lawrence M. S., Glasner M. E., Bartel D. P., RNA-catalyzed RNA polymerization: Accurate and general RNA-templated primer extension. Science 292, 1319–1325 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Wochner A., Attwater J., Coulson A., Holliger P., Ribozyme-catalyzed transcription of an active ribozyme. Science 332, 209–212 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Ekland E. H., Szostak J. W., Bartel D. P., Structurally complex and highly active RNA ligases derived from random RNA sequences. Science 269, 364–370 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Attwater J., Raguram A., Morgunov A. S., Gianni E., Holliger P., Ribozyme-catalysed RNA synthesis using triplet building blocks. eLife 7, e35255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheitl C. P. M., Maghami M. G., Lenz A. K., Hobartner C., Site-specific RNA methylation by a methyltransferase ribozyme. Nature 587, 663–667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seelig B., Jäschke A., A small catalytic RNA motif with Diels-Alderase activity. Chem. Biol. 6, 167–176 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Pobanz K., Lupták A., Improving the odds: Influence of starting pools on in vitro selection outcomes. Methods 106, 14–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pressman A. D., et al. , Mapping a systematic ribozyme fitness landscape reveals a frustrated evolutionary network for self-aminoacylating RNA. J. Am. Chem. Soc. 141, 6213–6223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiménez J. I., Xulvi-Brunet R., Campbell G. W., Turk-MacLeod R., Chen I. A., Comprehensive experimental fitness landscape and evolutionary network for small RNA. Proc. Natl. Acad. Sci. U.S.A. 110, 14984–14989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorsch J. R., Szostak J. W., Chance and necessity in the selection of nucleic acid catalysts. Acc. Chem. Res. 29, 103–110 (1996). [DOI] [PubMed] [Google Scholar]
- 18.Battersby T. R., et al. , Quantitative analysis of receptors for adenosine nucleotides obtained via in vitro selection from a library incorporating a cationic nucleotide analog. J. Am. Chem. Soc. 121, 9781–9789 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Tarasow T. M., Eaton B. E., Dressed for success: Realizing the catalytic potential of RNA. Biopolymers 48, 29–37 (1998). [Google Scholar]
- 20.Tolle F., Mayer G., Dressed for success - applying chemistry to modulate aptamer functionality. Chem. Sci. (Camb.) 4, 60–67 (2013). [Google Scholar]
- 21.Pfeiffert F., Rosenthal M., Siegl J., Ewers J., Mayer G., Customised nucleic acid libraries for enhanced aptamer selection and performance. Curr. Opin. Biotechnol. 48, 111–118 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Kimoto M., Nakamura M., Hirao I., Post-ExSELEX stabilization of an unnatural-base DNA aptamer targeting VEGF165 toward pharmaceutical applications. Nucleic Acids Res. 44, 7487–7494 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paul S., Wong A. A. W. L., Liu L. T., Perrin D. M., Selection of M2+-independent RNA-cleaving DNAzymes with side-chains mimicking arginine and lysine. ChemBioChem 23, e202100600 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Hollenstein M., Hipolito C. J., Lam C. H., Perrin D. M., Toward the combinatorial selection of chemically modified DNAzyme RNase A mimics active against all-RNA substrates. ACS Comb. Sci. 15, 174–182 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Wang Y., Liu E., Lam C. H., Perrin D. M., A densely modified M2+-independent DNAzyme that cleaves RNA efficiently with multiple catalytic turnover. Chem. Sci. (Camb.) 9, 1813–1821 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Z., Lichtor P. A., Berliner A. P., Chen J. C., Liu D. R., Evolution of sequence-defined highly functionalized nucleic acid polymers. Nat. Chem. 10, 420–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gold L., et al. , Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 5, e15004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraemer S., et al. , From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS One 6, e26332 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piccirilli J. A., Krauch T., Moroney S. E., Benner S. A., Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature 343, 33–37 (1990). [DOI] [PubMed] [Google Scholar]
- 30.Switzer C., Moroney S. E., Benner S. A., Enzymatic Incorporation of a New Base Pair into DNA and Rna. J. Am. Chem. Soc. 111, 8322–8323 (1989). [Google Scholar]
- 31.Rich A., “On the problems of evolution and biochemical information transfer” in Horizons in Biochemistry, Kasha M., Pullmann B., Eds., (Academic press, New York, 1962), pp.103–126. [Google Scholar]
- 32.Zubay G., “A case for an additional RNA base pair in early evolution” in The Roots of Modern Biochemistry, H. Kleinkauf, H. von Döhren, L. Jaenicke Eds. (Walter de Gruiter and Co., Berlin, 1988), pp. 911–916. [Google Scholar]
- 33.Roychowdhury A., Illangkoon H., Hendrickson C. L., Benner S. A., 2′-deoxycytidines carrying amino and thiol functionality: Synthesis and incorporation by Vent (exo-) polymerase. Org. Lett. 6, 489–492 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Carrigan M. A., Ricardo A., Ang D. N., Benner S. A., Quantitative analysis of a RNA-cleaving DNA catalyst obtained via in vitro selection. Biochemistry 43, 11446–11459 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Hoshika S., et al. , “Skinny” and “Fat” DNA: Two new double helices. J. Am. Chem. Soc. 140, 11655–11660 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Matsuura M. F., Kim H. J., Takahashi D., Abboud K. A., Benner S. A., Crystal structures of deprotonated nucleobases from an expanded DNA alphabet. Acta Crystallogr. C Struct. Chem. 72, 952–959 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y., et al. , A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl. Acad. Sci. U.S.A. 114, 1317–1322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimoto M., Yamashige R., Matsunaga K., Yokoyama S., Hirao I., Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 31, 453–457 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Matsunaga K. I., Kimoto M., Hirao I., High-affinity DNA aptamer generation targeting von Willebrand factor A1-domain by genetic alphabet expansion for systematic evolution of ligands by exponential enrichment using two types of libraries composed of five different bases. J. Am. Chem. Soc. 139, 324–334 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Zhang L., et al. , Evolution of functional six-nucleotide DNA. J. Am. Chem. Soc. 137, 6734–6737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sefah K., et al. , In vitro selection with artificial expanded genetic information systems. Proc. Natl. Acad. Sci. U.S.A. 111, 1449–1454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L., et al. , Aptamers against cells overexpressing glypican 3 from expanded genetic systems combined with cell engineering and laboratory evolution. Angew. Chem. Int. Ed. Engl. 55, 12372–12375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biondi E., et al. , Laboratory evolution of artificially expanded DNA gives redesignable aptamers that target the toxic form of anthrax protective antigen. Nucleic Acids Res. 44, 9565–9577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biondi E., Benner S. A., Artificially Expanded Genetic Information Systems for New Aptamer Technologies. Biomedicines 6, 53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoshika S., et al. , Hachimoji DNA and RNA: A genetic system with eight building blocks. Science 363, 884–887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Futami K., Kimoto M., Lim Y. W. S., Hirao I., Genetic alphabet expansion provides versatile specificities and activities of unnatural-base DNA aptamers targeting cancer cells. Mol. Ther. Nucleic Acids 14, 158–170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dien V. T., et al. , Progress toward a semi-synthetic organism with an unrestricted expanded genetic alphabet. J. Am. Chem. Soc. 140, 16115–16123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breaker R. R., Joyce G. F., A DNA enzyme that cleaves RNA. Chem. Biol. 1, 223–229 (1994). [DOI] [PubMed] [Google Scholar]
- 49.Santoro S. W., Joyce G. F., A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. U.S.A. 94, 4262–4266 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silverman S. K., In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res. 33, 6151–6163 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou W., Liu J., Multi-metal-dependent nucleic acid enzymes. Metallomics 10, 30–48 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Huang P. J. J., Liu J., In vitro selection of chemically modified DNAzymes. ChemistryOpen 9, 1046–1059 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hollenstein M., DNA catalysis: The chemical repertoire of DNAzymes. Molecules 20, 20777–20804 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Z., Chen F., Alvarado J. B., Benner S. A., Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 133, 15105–15112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Z., et al. , Conversion strategy using an expanded genetic alphabet to assay nucleic acids. Anal. Chem. 85, 4705–4712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sidorov A. V., Grasby J. A., Williams D. M., Sequence-specific cleavage of RNA in the absence of divalent metal ions by a DNAzyme incorporating imidazolyl and amino functionalities. Nucleic Acids Res. 32, 1591–1601 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaukinen U., Lyytikäinen S., Mikkola S., Lönnberg H., The reactivity of phosphodiester bonds within linear single-stranded oligoribonucleotides is strongly dependent on the base sequence. Nucleic Acids Res. 30, 468–474 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bibillo A., Figlerowicz M., Ziomek K., Kierzek R., The nonenzymatic hydrolysis of oligoribonucleotides. VII. Structural elements affecting hydrolysis. Nucleosides Nucleotides Nucleic Acids 19, 977–994 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Fu Y., Sharma G., Mathews D. H., Dynalign II: Common secondary structure prediction for RNA homologs with domain insertions. Nucleic Acids Res. 42, 13939–13948 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zuker M., Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schultz S. J., Champoux J. J., RNase H activity: Structure, specificity, and function in reverse transcription. Virus Res. 134, 86–103 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taylor A. I., et al. , Catalysts from synthetic genetic polymers. Nature 518, 427–430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Z., Chen F., Chamberlin S. G., Benner S. A., Expanded genetic alphabets in the polymerase chain reaction. Angew. Chem. Int. Ed. Engl. 49, 177–180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santoro S. W., Joyce G. F., Sakthivel K., Gramatikova S., Barbas C. F. III, RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 122, 2433–2439 (2000). [DOI] [PubMed] [Google Scholar]
- 65.Mir A., Golden B. L., Two active site divalent ions in the crystal structure of the hammerhead ribozyme bound to a transition state analogue. Biochemistry 55, 633–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moody E. M., Lecomte J. T., Bevilacqua P. C., Linkage between proton binding and folding in RNA: A thermodynamic framework and its experimental application for investigating pKa shifting. RNA 11, 157–172 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gong B., et al. , Direct measurement of a pK(a) near neutrality for the catalytic cytosine in the genomic HDV ribozyme using Raman crystallography. J. Am. Chem. Soc. 129, 13335–13342 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Nakano S., Chadalavada D. M., Bevilacqua P. C., General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science 287, 1493–1497 (2000). [DOI] [PubMed] [Google Scholar]
- 69.Wilcox J. L., Ahluwalia A. K., Bevilacqua P. C., Charged nucleobases and their potential for RNA catalysis. Acc. Chem. Res. 44, 1270–1279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lupták A., Ferré-D’Amaré A. R., Zhou K., Zilm K. W., Doudna J. A., Direct pK(a) measurement of the active-site cytosine in a genomic hepatitis delta virus ribozyme. J. Am. Chem. Soc. 123, 8447–8452 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Kath-Schorr S., et al. , General acid-base catalysis mediated by nucleobases in the hairpin ribozyme. J. Am. Chem. Soc. 134, 16717–16724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data pertaining to this work will be made available to readers upon publication.