

Abstract
Sound—including music and noise—can relieve pain in humans, but the underlying neural mechanisms remain unknown. We discovered that analgesic effects of sound depended on a low (5-decibel) signal-to-noise ratio (SNR) relative to ambient noise in mice. Viral tracing, microendoscopic calcium imaging, and multitetrode recordings in freely moving mice showed that low-SNR sounds inhibited glutamatergic inputs from the auditory cortex (ACxGlu) to the thalamic posterior (PO) and ventral posterior (VP) nuclei. Optogenetic or chemogenetic inhibition of the ACxGlu→PO and ACxGlu→VP circuits mimicked the low-SNR sound-induced analgesia in inflamed hindpaws and forepaws, respectively. Artificial activation of these two circuits abolished the sound-induced analgesia. Our study reveals the corticothalamic circuits underlying sound-promoted analgesia by deciphering the role of the auditory system in pain processing.
As early as 1960, there were accounts from dental operations showing that music and noise can induce analgesic effects (1). Music delivered as an intervention can alleviate postoperative and procedural pain (2–4) and even refractory pain in the clinic (5, 6). Because diverse genres of music and even nature sounds can relieve pain to an equal extent (7), the inherent characteristics of music or contextual factors—that is, not only to music per se—have been hypothesized to drive these analgesic effects (8). However, it is still unknown how it works.
Functional magnetic resonance imaging (fMRI) studies have implicated changes in the activity of multiple brain areas mediating pain processing in humans exposed to music (9–12). To date, the neural substrates underpinning cross-modal audio-somatosensory interactions remain unclear. The thalamus relays multimodal sensory information (including auditory and somatosensory) (13–15) and reciprocally connects with pain-associated regions to process sensory discrimination and affective-motivation of pain through diverse subregions (16–19). Notably, the activity of the somatosensory thalamus can be affected by music and innocuous salient auditory stimuli (15, 20). Thus, the thalamus might function as a bridge for audio-somatosensory processing. However, the precise cell type-specific organization and the function(s) of the thalamic circuits mediating sound-induced analgesia remain largely unknown.
Sound-induced analgesia depends on a low signal-to-noise ratio (SNR) relative to ambient noise in mice
Inspired by findings in humans, we investigated whether consonant sound (e.g., pleasant music for humans) may elicit analgesic effects in mice with inflammatory pain induced by hindpaw injection of complete Freund’s adjuvant (CFA) (Fig. 1A and fig. S1A). Because loud noises (above ~75 dB) induce escape behavior in mice (21), we delivered consonant sound to CFA mice at a sound pressure level (SPL) of ~50 or ~60 dB through a closely positioned audio speaker (ambient noise at 45-dB SPL). Using von Frey filaments to assess mechanical sensitivity, we found that delivery of consonant sound at a SPL of ~50 dB, but not of ~60 dB, elevated the inflamed hindpaw nociceptive threshold compared with CFA mice exposed only to ambient noise (Fig. 1B). Notably, this effect lasted for at least 2 days after repeated exposure to ~50-dB SPL consonant sound for 3 days (20 min per day). Notably, no difference was detected in the nociceptive thresholds among CFA mice exposed to ~50-dB SPL consonant sound, dissonant sound, or white noise (Fig. 1, C and D, and fig. S1B).
Fig. 1. Low-intensity sound relative to ambient noise induces analgesia in mice.
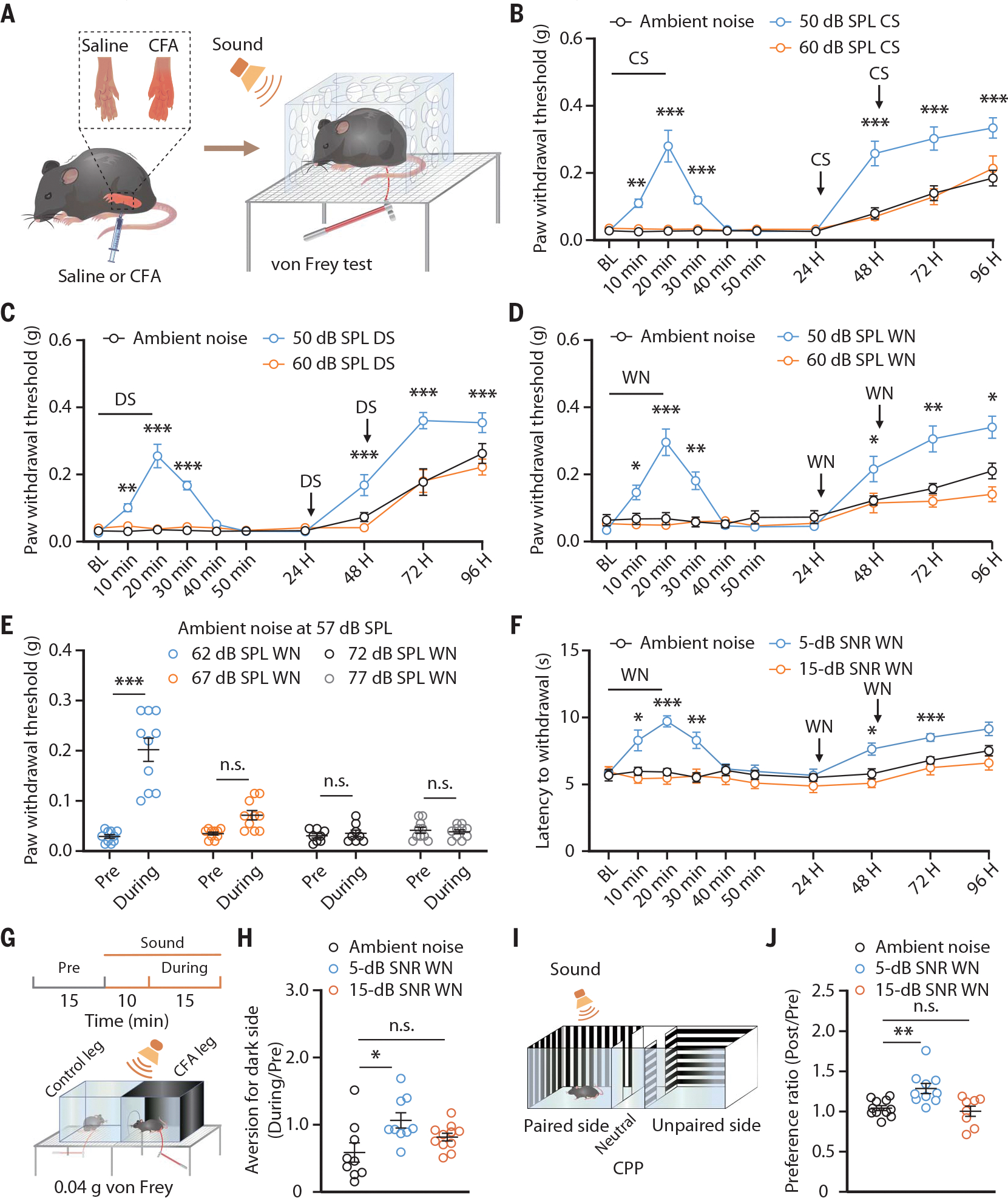
(A) Schematic for inducing inflammatory pain and the von Frey test to assess the mechanical nociceptive threshold. Sound refers to delivered consonant sound (CS), dissonant sound (DS), or white noise (WN) given at the indicated SPLs. (B to D) The mechanical nociceptive threshold in CFA mice treated with or without consonant sound (ambient noise, n = 10 mice; 50-dB SPL, n = 10 mice; 60-dB SPL, n = 8 mice; P < 0.0001) (B), dissonant sound (n = 10 mice each group; P < 0.0001) (C), and white noise (ambient noise, n = 10 mice; 50-dB SPL, n = 10 mice; 60-dB SPL, n = 8 mice; P < 0.0001) (D) in an environment with an ambient noise at 45-dB SPL. BL, baseline. (E) The mechanical nociceptive threshold of CFA mice exposed to white noise at different intensities in an environment with ambient noise at 57-dB SPL (62-dB SPL, n = 10 mice; 67-dB SPL, n = 10 mice; 72-dB SPL, n = 8 mice; 77-dB SPL, n = 10 mice; P < 0.0001). (F) The thermal nociceptive threshold assessed by the Hargreaves test in CFA mice exposed to different SNR white noise (n = 10 mice each group; P < 0.0001). (G) Schematic for the CPA test. (H) Summarized data for the von Frey filament stimulus-induced place aversion in CFA mice treated with or without white noise (ambient noise, n = 9 mice; 5-dB SNR, n = 9 mice; 15-dB SNR, n = 11 mice; P = 0.0165). (I) Schematic for the CPP test (J) Summarized data for preference ratio for the sound-delivery side in CFA mice from the indicated group (ambient noise, n = 11 mice; 5-dB SNR, n = 10 mice; 15-dB SNR, n = 8 mice; P = 0.0015). The data are expressed as the means ± SEMs. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. Details of the statistical analyses are presented in table S1.
Music-induced analgesia in humans may be attributable to the treatment environment (8). To test whether 50-dB SPL or the intensity difference relative to ambient noise (i.e., the SNR) increased the nociceptive threshold, we delivered 62-, 67-, 72-, or 77-dB SPL white noise to CFA mice under conditions of 57-dB SPL ambient noise. The mechanical nociceptive threshold was elevated only after exposure to 62-dB (5-dB SNR) SPL white noise (Fig. 1E). In addition, 35-dB (5-dB SNR) SPL white noise, but not 40-, 45-, or 50-dB SPL, produced similar effects in an environment with 30-dB SPL ambient noise (fig. S1C). We also found that 5-dB SNR sound elevated the thermal nociceptive threshold in the Hargreaves test (Fig. 1F and fig. S1D). Furthermore, the effects of 5-dB SNR sound on pain hypersensitivity were observed in mice with neuropathic pain induced by spared nerve injury (SNI) and tonic pain induced by capsaicin in both males and females (figs. S2 to S4). On the basis of these findings, we refer to 5-dB SNR as low SNR; henceforth, the ambient noise level used was 45-dB SPL, unless otherwise stated.
We next conducted conditioned place aversion (CPA) and conditioned place preference (CPP) tests to assay the potential effects of sound on the affective component of pain. Five-dB, but not 15-dB, SNR white noise abolished the subthreshold von Frey (0.04 g) stimuli-induced place aversion in the CPA test and evoked a preference for the sound-delivery side in the CPP test (Fig. 1, G to J, and fig. S5). Taken together, these findings support that low-SNR sound induces analgesia.
Pain perception is affected by emotion and stress (22,23). Behavioral tests, including open field, light-dark box, and elevated plus maze, showed that neither 5-dB nor 15-dB SNR white noise evoked anxiety-like behaviors in mice under acute pain conditions (CFA 3 days or SNI 7 days) (fig. S6, A to F), and neither sound reduced anxiety under chronic pain conditions (CFA 21 days or SNI 42 days) (fig. S6, G to L). Moreover, no stress effects were involved in the sound-induced analgesia: (i) The serum corticosterone level was unaffected by 5-dB SNR sound exposure (fig. S7), and (ii) intrathecal application of the μ opioid receptor antagonist naloxone had no effect on 5-dB SNR sound-induced increase in nociceptive thresholds (fig. S8).
Glutamatergic neurons in the auditory cortex (ACx) are functionally connected to the thalamic posterior (PO) and ventral posterior (VP) nuclei
We next investigated the neural circuit(s) underlying the observed low-SNR sound-induced analgesia. In the brain, sound signals are gated by the medial geniculate body (MGB) before reaching the ACx, which serves as the convergence point for detailed analysis of sound (24, 25). Because both of these nuclei are primarily composed of glutamatergic neurons (26), we examined the presynaptic outputs from the ACxGlu and MGBGlu neurons into the brain regions tightly linked to pain processing (27–29). Specifically, we injected an adeno-associated viral vector (AAV)-DIO-membrane-bound green fluorescent protein (mGFP)-Synaptophysin-mRuby into these nuclei in Ca2+/calmodulin-dependent protein kinase II (CaMKII)-Cre mice (fig. S9, A and B, and fig. S10, A and B).
mRuby+ terminals originating from ACxGlu neurons were observed in the insular cortex (ICx) but were very scarce in the medial prefrontal cortex, the anterior and posterior cingulate cortices, the primary and secondary somatosensory cortices, the nucleus accumbens, the ventrolateral periaqueductal gray, the dorsal raphe nucleus, the rostroventral medulla, the parabrachial nucleus, and the basolateral and central amygdala, and no signals were detected in the spinal cord (fig. S9). We detected abundant mRuby signals in thalamic nuclei, including the PO and VP, but not in the mediodorsal and central medial nuclei (fig. S9, M to P and R). Notably, we detected no mRuby signals originating from MGBGlu neurons in any of these nuclei (fig. S10). Because the PO and VP receive prominent inputs from the ACx and are known to relay nociceptive information (30–32), we subsequently focused on the role of ACx→PO and ACx→VP circuits in sound-induced analgesia.
In vivo multitetrode recordings in freely moving CFA mice showed that the spontaneous neuronal activity in the ACx was decreased in response to white noise at 5-dB, but not 15-dB, SNR (Fig. 2, A to C). Five-dB SNR white noise-induced neuronal inhibition was mimicked on the basis of (i) ACx infusion of Cre-dependent chemogenetic inhibitory hM4Di-mCherry viruses (AAV-DIO-hM4Di-mCherry) and (ii) intraperitoneal injection with the hM4Di-agent clozapine-N-oxide (CNO) in CaMKII-Cre mice. The nociceptive thresholds were elevated upon selective inactivation of ACxGlu neurons; conditioned aversion was abolished upon such inhibition (Fig. 2, D and E). Conversely, bilateral optical activation of ACxGlu neurons expressing AAV-DIO-ChR2-mCherry abolished the 5-dB SNR white noise-induced analgesia in CaMKII-Cre mice (fig. S11).
Fig. 2. ACxGlu neurons project to VPGlu and POGlu neurons.

(A) Schematic for multitetrode recording in freely moving mice. (B and C) Raster plots and voltage traces of the spontaneous firings recorded in the ACx (B) and summarized data (5-dB SNR, n = 25 cells from four mice; 15-dB SNR, n = 22 cells from four mice; P = 0.0053) (C). (D and E) Summarized data for the mechanical nociceptive threshold (mCherry, n = 10 mice; hM4Di-mCherry, n = 8 mice; BL, P = 0.3816; CNO, P < 0.0001) [(D), left], place aversion (n = 9 mice each group; P = 0.0006) [(D), right], and thermal nociceptive threshold (n = 10 mice each group; P < 0.0001) (E) in CFA mice upon chemogenetic inhibition of ACxGlu neurons. (F) Schematic for anterograde tracing and representative image of EGFP-expressing neurons in the PO and VP. Scale bar, 500 μm. LP, lateral posterior thalamic nucleus; st, stria terminalis. (G and H) Representative images showing the colocalization of EGFP-labeled neurons with glutamate (Glu) immunofluorescence (G) and summarized data (n = 4 slices) (H). Scale bars, 50 μm. DAPI, 4’,6-diamidino-2-phenylindole. (I) Schematic for retrograde tracing. (J) Representative images showing EGFP+ and tdTomato+ neurons in the ACx. Scale bars, 100 μm. (K and L) Representative images of the colocalization of EGFP-labeled PO- and VP-projecting ACx neurons with glutamate immunofluorescence (K) and summarized data (n = 4 slices) (L). Scale bars, 50 μm. (M) Schematic for viral injection and whole-cell recordings. R, recording. (N and O) Representative traces and summarized data for light-evoked postsynaptic currents recorded in PO neurons (n = 12 cells from four mice; P = 0.0002) (N) and VP neurons (n = 14 cells from four mice; P < 0.0001) (O). ACSF, artificial cerebrospinal fluid; DNQX, 6,7-dinitroquinoxaline-2,3-dione. (P) A model of ACxGlu→PO and ACxGlu→VP circuits. GluRs, glutamate receptors. The data are expressed as the means ± SEMs. ***P < 0.001; n.s., not significant. Details of the statistical analyses are presented in table S1.
We then characterized both ACx→PO and ACx→VP circuits in greater detail. We performed anterograde monosynaptic tracing by ACx injection with AAV1-Cre-enhanced green fluorescent protein (EGFP) virus along with ipsilateral PO and VP injection of Cre-dependent DIO-EGFP, which revealed numerous EGFP+ neurons in the PO and VP (Fig. 2F). These PO and VP EGFP+ neurons colocalized with glutamatergic neurons but not with γ-aminobutyric acid-releasing (GABAergic) neurons (Fig. 2, G and H, and fig. S12, A to C).
Retrograde tracing by PO injection with a retroAAV expressing tdTomato (retroAAV-tdTomato) and VP injection with a retroAAV expressing EGFP (retroAAV-EGFP) showed that retrogradely labeled tdTomato+ neurons were abundant in ACx layer 6 (L6), whereas EGFP+ neurons were abundant in ACx L5 (Fig. 2, I and J). Both the PO- and VP-projecting ACx neurons colocalized with glutamatergic rather than GABAergic neurons (Fig. 2, K and L, and fig. S12, D to F). Whole-cell recordings combined with optogenetics in brain slices showed that brief light stimulation of ChR2-containing ACxGlu terminals in the PO or VP reliably elicited glutamate-mediated excitatory postsynaptic currents (EPSCs) (Fig. 2, M to P).
Inhibition of the ACxGlu→PO circuit mediates sound-induced hindpaw analgesia
We conducted in vivo multitetrode and fiber photometry recordings in freely moving CFA mice. PO rather than VP neurons were rapidly activated by punctate mechanical stimulation of inflamed hindpaws (Fig. 3, A to C; fig. S13, A to D; and movies S1 and S2). PO neuronal activity was significantly enhanced in CFA mice compared with saline-treated controls (fig. S13, E and F), which was attenuated by 5-dB, but not 15-dB, SNR white noise (Fig. 3, D and E, and fig. S13G). Similar sound-induced inhibitory effects on PO neurons were observed after optical inhibition of the ACxGlu terminals in the PO of CaMKII-Cre mice treated with ACx infusion of a Cre-dependent AAV carrying eNpHR3.0-enhanced yellow fluorescent protein (EYFP) (AAV-DIO-eNpHR3.0-EYFP) (Fig. 3F and fig. S13, H and I).
Fig. 3. Low-SNR sound inhibits the ACxGlu→PO circuit to induce analgesia on hindpaws.

(A) Schematic for multitetrode recording in freely moving mice with punctate mechanical stimulation (von Frey filament, 0.04 g). (B and C) Raster plots, voltage traces, and summarized data for the spontaneous firings recorded in VP neurons (n = 19 cells from four mice; P = 0.5079) (B) and in PO neurons (n = 27 cells from five mice; P = 0.0003) (C) before and during punctate mechanical stimulation of CFA-injected hindpaws. FR BL, firing rate baseline. (D and E) Raster plots and voltage traces of the spontaneous firings recorded in PO neurons in CFA mice with or without 5-dB SNR white noise exposure (D) and summarized data (control, n = 24 cells from four mice; 5-dB SNR, n = 47 cells from eight mice; P < 0.0001) (E). (F) Raster plots of the spontaneous firings recorded in PO neurons before and during optical inhibition of the ACxGlu→PO circuit (left) and summarized data (n = 71 cells from seven mice; P < 0.0001) (right). (G) Schematic for optogenetic inhibition of the ACxGlu→PO circuit. (H to J) Summarized data for the thermal nociceptive threshold (EYFP, n = 10 mice; eNpHR3.0-EYFP, n = 9 mice; P < 0.0001) (H), mechanical nociceptive threshold (EYFP, n = 8 mice; eNpHR3.0-EYFP, n = 9 mice; P < 0.0001) (I), and place aversion (n = 10 mice each group; P = 0.0001) (J) after optical inhibition of the ACxGlu→PO circuit in CFA mice. (K) Schematic for vial injection and microendoscopic calcium imaging. (L) A typical image showing the GCaMP6m fluorescence and track of lens in the PO. Scale bar, 200 μm. rt, reticular thalamic nucleus. (M and N) Representative traces of spontaneous Ca2+ signal transient recorded in PO neurons receiving ACx projections (M) and summarized data (5-dB SNR, n = 20 cells from four mice; 15-dB SNR, n = 15 cells from four mice; P < 0.0001) (N). dF/F0, the change in fluorescence (dF) over the baseline fluorescence (F0) of calcium spikes. The data are expressed as the means ± SEMs. **P < 0.01; ***P < 0.001; n.s., not significant. Details of the statistical analyses are presented in table S1.
We then performed bilateral optical inhibition of the ACxGlu→PO circuit and conducted von Frey, Hargraves, CPA, and CPP tests. This selective inhibition mimicked the effects from the 5-dB SNR white noise in both male and female mice (Fig. 3, G to J; fig. S13J; and fig. S14). By contrast, optical activation of the ACxGlu→PO circuit abolished both neuronal inhibition in the PO and analgesia induced by 5-dB SNR sound (fig. S15).
To selectively monitor the response of PO-projecting ACx neurons to 5-dB SNR sound at single-neuron resolution, we infused a retroAAV-Cre virus into the PO and an AAV expressing Cre-dependent fluorescent Ca2+ indicator GCaMP6m (AAV-DIO-GCaMP6m) into the ipsilateral ACx, accompanied with the mounting of a microendoscopic gradient index (GRIN) lens at the top of the ACx (fig. S16A). The Ca2+ transient frequency of these ACx neurons was decreased after exposure to 5-dB, but not 15-dB, SNR white noise in freely moving CFA mice (fig. S16, B and C). The activity of PO neurons receiving ACx projections (POACx) was measured in mice with ACx infusion of AAV1-Cre virus and ipsilateral PO infusion of AAV-DIO-GCaMP6m (Fig. 3, K and L). The Ca2+ transient frequency of POACx neurons was rapidly increased by punctate mechanical stimuli (movies S3 and S4). The spontaneous Ca2+ transient frequency of these neurons was elevated in CFA mice compared with saline mice (fig. S16, D to F), and the increased frequency was attenuated during exposure to 5-dB, but not 15-dB, SNR white noise (Fig. 3, M and N).
The POACx neurons were then selectively manipulated to experimentally validate their participation in sound-induced analgesia. We injected AAV1-Cre virus into the ACx while concurrently injecting a viral vector expressing either Cre-dependent hM3Dq-mCherry or hM4Di-mCherry into the PO (fig. S16G). We found bilateral chemogenetic inhibition of POACx neurons recapitulated 5-dB SNR sound-induced analgesia (fig. S16H). By contrast, chemogenetic activation of these neurons blocked the 5-dB SNR sound-induced analgesia (fig. S16I).
Sound-induced forepaw analgesia mediated by inhibition of the ACxGlu→VP circuit
The VP mediates somatosensation of the upper limbs (33). Given our findings of a functional ACxGlu→VP connection, we further investigated whether this circuit participates in 5-dB SNR sound-induced analgesia. We observed a significant increase in nociceptive thresholds of the CFA-injected forepaw along with a reduction in place aversion upon 5-dB, but not 15-dB, SNR white noise (fig. S17, A to E). Both in vivo multitetrode and fiber photometry recordings in freely moving mice revealed that neuronal activity in the VP—but not the PO—was rapidly enhanced by punctate mechanical stimulation of inflamed forepaws (Fig. 4, A to C; fig. S17, F to H; and movies S5 and S6). Compared with saline mice, the VP neuronal activity was significantly increased in CFA mice (fig. S17, I and J), and this elevation in neuronal activity was attenuated by exposure to 5-dB SNR white noise, but not 15-dB SNR (Fig. 4, D and E, and fig. S17K).
Fig. 4. Inhibition of the ACxGlu→VP circuit mediates low-SNR sound-induced analgesia on forepaws.
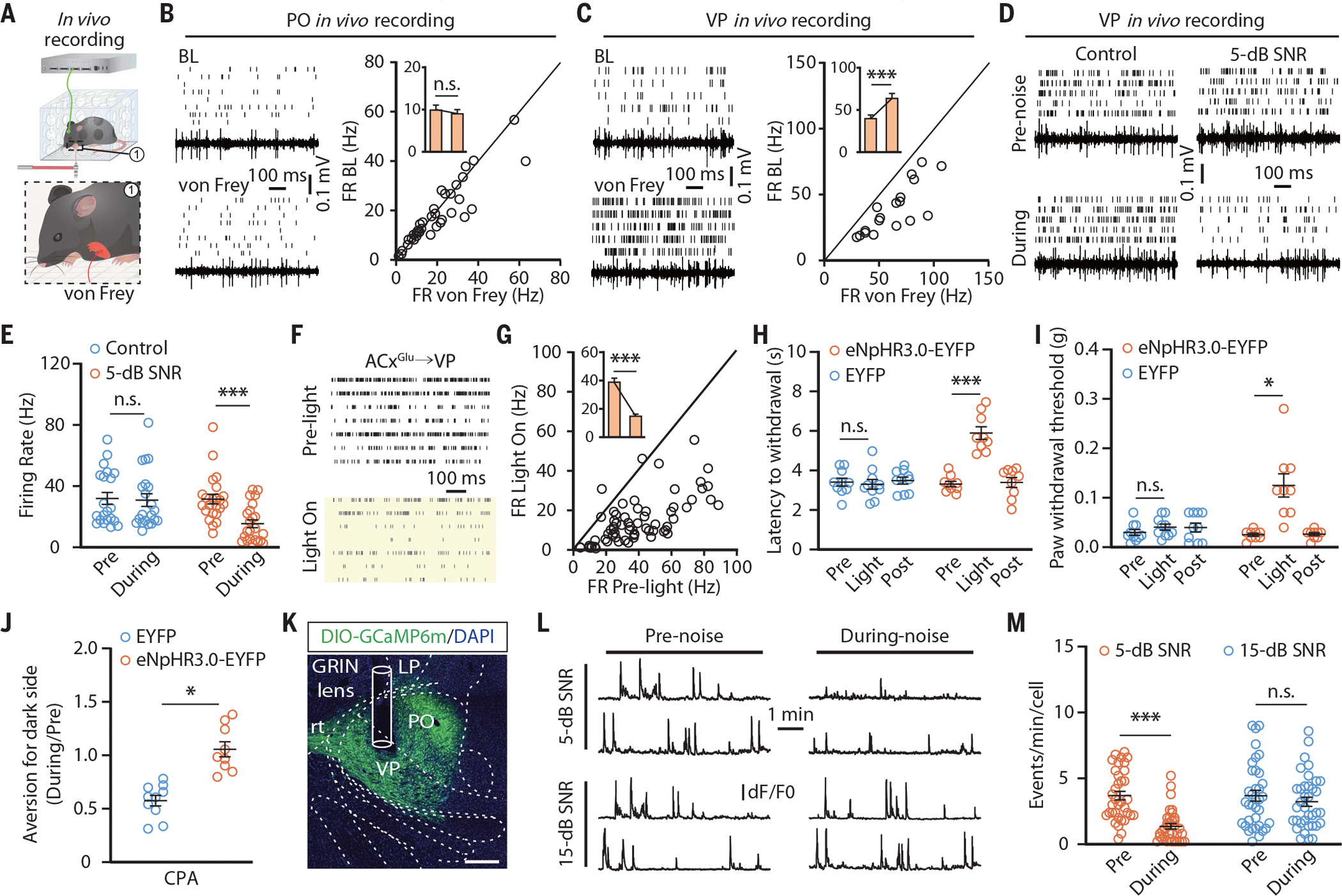
(A) Schematic for multitetrode recording in the VP or PO of freely moving mice. (B and C) Raster plots, voltage traces, and summarized data for the spontaneous firings recorded in PO neurons (n = 36 cells from four mice; t35 = 1.749; P = 0.089) (B) and VP neurons (n = 18 cells from four mice; t17 = 7.373; P < 0.0001) (C) before and during punctate mechanical stimulation (von Frey filament, 0.02 g) of inflamed forepaws. (D and E) Raster plots and voltage traces of the spontaneous firings recorded in VP neurons from CFA mice with or without 5-dB SNR white noise exposure (D) and summarized data (control, n = 21 cells from four mice; 5-dB SNR, n = 23 cells from four mice; F1,42 = 24.18; P < 0.0001) (E). (F and G) Raster plots of the spontaneous activity recorded in VP neurons before and during optical inhibition of the ACxGlu→VP circuit in CFA-treated mice (F) and summarized data (n = 67 cells from seven mice; t66 = 12.14; P < 0.0001) (G). (H to J) Summarized data for the thermal (EYFP, n = 10 mice; eNpHR3.0-EYFP, n = 9 mice; F2,34 = 20.98; P < 0.0001) (H) and mechanical (EYFP, n = 10 mice; eNpHR3.0-EYFP, n = 9 mice; F2,34 = 13.25; P < 0.0001) (I) nociceptive thresholds of CFA-injected forepaws and place aversion (EYFP, n = 10 mice; eNpHR3.0-EYFP, n = 9 mice; t17 = 5.648; P < 0.0001) (J) upon optical inhibition of the ACxGlu→VP circuit. (K) A typical image of GCaMP6m fluorescence and track of the lens in the VP. Scale bar, 200 μm. (L and M) Representative traces (L) of spontaneous Ca2+ signals recorded in VP neurons receiving ACx projections and summarized data (5-dB SNR, n = 35 cells from four mice; 15-dB SNR, n = 36 cells from four mice; F1,69 = 24.24; P < 0.0001) (M). The data are expressed as the means ± SEMs. *P < 0.05; ***P < 0.001; n.s., not significant. Details of the statistical analyses are presented in table S1.
In both male and female mice, bilateral optical inhibition of the eNpHR3.0-EYFP-containing ACxGlu terminals in the VP mimicked 5-dB SNR white noise-induced VP neuronal inhibition (Fig. 4, F and G) and analgesia (Fig. 4, H to J; fig. S17, L and M; and fig. S18). Optical activation of the ACxGlu→VP circuit abolished both elevation of nociceptive thresholds (fig. S19, A to C) and neuronal inhibition in the VP induced by 5-dB SNR sound (fig. S19, D to F).
Microendoscopic calcium imaging showed that the spontaneous Ca2+ transient frequency of VP-projecting ACx neurons was attenuated during exposure to 5-dB, but not 15-dB, SNR white noise (fig. S20, A to D). In contrast to that of POACx neurons, the Ca2+ transient frequency of VP neurons receiving ACx projections (VPACx) was rapidly increased by punctate mechanical stimulation of inflamed forepaws, but not hindpaws (movies S7 and S8). The spontaneous Ca2+ transient frequency of these neurons was enhanced in CFA mice compared with saline mice (fig. S20, E and F), which was attenuated by exposure to 5-dB, but not 15-dB, SNR white noise (Fig. 4, K to M).
The 5-dB SNR sound-induced elevation of the forepaw nociceptive threshold was mimicked upon chemogenetic inhibition of VPACx neurons and was blocked upon activation of these neurons (fig. S20, G to I). Notably, the hindpaw nociceptive threshold was not affected by optical activation or inhibition of the ACxGlu→VP circuit, and neither optical activation nor inhibition of the ACxGlu→PO circuit affected the forepaw nociceptive threshold (fig. S21). Comparable results were obtained upon chemogenetic manipulations of VPACx or POACx neurons (fig S22).
Given that the ICx, a higher-order cortex influenced by music in humans (9–11), receives ACx projections (fig. S9C), we examined the potential role of the ACxGlu→ICx circuit in sound-induced analgesia. Optical activation of ACxGlu terminals in the ICx did not affect 5-dB SNR sound-induced elevation of nociceptive thresholds of inflamed paws, whereas optical inhibition of this circuit in the absence of sound had no effects on pain hypersensitivity (fig. S23). Taken together, our results demonstrate that the observed analgesia from 5-dB SNR sound is mediated by the ACx→PO circuit for hindpaws and by the ACx→VP circuit for forepaws (fig. S24).
Discussion
The neural circuit responsible for processing sound-induced analgesia has long remained elusive. Here, we describe the analgesic effects of sound at low SNR through inhibition of audio-somatosensory corticothalamic circuits. Specifically, we reveal that the distinct roles of the ACxGlu→PO and ACxGlu→VP circuits in sound-induced analgesia depend on the physical location of the pain.
In mice, we found that sound-induced analgesia depended on its low SNR rather than harmony, which is supported by a previous hypothesis that music-induced analgesia is attributable to contextual factors of the treatment, not only to the music per se (8). It bears mention that the observed low-SNR sound-induced analgesia is unlikely to result from some reduction in anxiety or stress, and it probably does not directly involve attention-distraction in affecting pain perception (33), given that the analgesic effects persisted for at least 2 days after sound withdrawal.
The neural mechanisms underlying music-induced analgesia in humans are doubtlessly more complicated than those revealed in mice (34). In humans, multiple areas that are involved in pain processing, including the ICx, striatum, and midbrain dopamine system, do respond to music (35–38). Therefore, whether other brain regions receiving ACx projections (except the PO and VP) or how downstream projections of the PO and VP account for music-induced analgesia deserve further investigation.
This study suggests a complement for the classic pain sensation pathway that is implicated in the effect of sound on pain processing and could expedite the study of music-induced analgesia. In the future, these findings could spur the development of alternative interventions for treating pain.
Supplementary Material
ACKNOWLEDGMENTS
We thank Y. Yang for providing Ai9 (RCL-tdT) mice. We thank F. Xu and T. Xue for technical support, C. Weisz, and K. Suthakar for critical reading.
Funding:
This work was supported by the National Key Research and Development Program of China Brain Science and Brain-Like Intelligence Technology (2021ZD0203100), the National Natural Science Foundation of China (32025017, 32000717, and 82101301), the Science Fund for Creative Research Groups of the National Natural Science Foundation of China (32121002), the CAS Project for Young Scientists in Basic Research (YSBR-013), the Natural Science Foundation of Anhui Province (KJ2020A0138, 2008085QC115, and 2008085QC114), the University of Science and Technology of China Research Funds of the Double First-Class Initiative (YD3460002001), the NIH NIDCR intramural research program (ZIA DE000757), and the Innovative Research Team of High-level Local Universities in Shanghai.
Footnotes
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
science.org/doi/10.1126/science.abn4663
References (39, 40)
MDAR Reproducibility Checklist
View/request a protocol for this paper from Bio-protocol.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the supplementary materials.
REFERENCES AND NOTES
- 1.Gardner WJ, Licklider JC, Weisz AZ, Science 132, 32–33 (1960). [DOI] [PubMed] [Google Scholar]
- 2.Keenan A, Keithley JK, Oncol. Nurs. Forum 42, E368–E375 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Nguyen TN, Nilsson S, Hellström A-L, Bengtson A, J. Pediatr. Oncol. Nurs 27, 146–155 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Hartling L et al. , JAMA Pediatr. 167, 826–835 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Boyd-Brewer C, McCaffrey R, Holist. Nurs. Pract 18, 111–118 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Jensen MP, Hakimian S, Sherlin LH, Fregni F, J. Pain 9, 193–199 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Garza Villarreal EA, Brattico E, Vase L, Østergaard L, Vuust P, PLOS ONE 7, e29397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lunde SJ, Vuust P, Garza-Villarreal EA, Vase L, Pain 160, 989–993 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Dobek CE, Beynon ME, Bosma RL, Stroman PW, J. Pain 15, 1057–1068 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Sachs ME, Habibi A, Damasio A, Kaplan JT, Neuroimage 218, 116512 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Usui C et al. , Pain Med. 21, 1546–1552 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Bonetti L et al. , Neuroimage 245, 118735 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Jones EG, The Thalamus (Cambridge Univ. Press, ed. 2, 2007). [Google Scholar]
- 14.Sherman SM, Guillery RW, Phil. Trans. R. Soc. Lond. B 357, 1695–1708 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mouraux A, Diukova A, Lee MC, Wise RG, lannetti GD, Neuroimage 54, 2237–2249 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Vogt BA, Rosene DL, Pandya DN, Science 204, 205–207 (1979). [DOI] [PubMed] [Google Scholar]
- 17.Viaene AN, Petrof I, Sherman SM, Proc. Natl. Acad. Sci. U.S.A. 108, 18156–18161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meda KS et al. , Neuron 102, 944–959.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu X et al. , Nat Neurosci. 24, 542–553 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Wagner G, Koschke M, Leuf T, Schlösser R, Bär K-J, Neuropsychologia 47, 980–987 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Xiong XR et al. , Nat. Commun 6, 7224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilam G, Gross JJ, Wager TD, Keefe FJ, Mackey SC, Neuron 107, 17–21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grau JW, Hyson RL, Maier SF, Madden J 4th, Barchas JD, Science 213, 1409–1411 (1981). [DOI] [PubMed] [Google Scholar]
- 24.Koelsch S, Nat. Rev. Neurosci. 15, 170–180 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Read HL, Winer JA, Schreiner CE, Curr. Opin. Neurobiol. 12, 433–440 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Winer JA, in The Mammalian Auditory Pathway: Neuroanatomy, Webster DB, Popper AN, Fay RR, Eds. (Springer, 1992), pp. 222–409. [Google Scholar]
- 27.Salomons TV, lannetti GD, Liang M, Wood JN, JAMA Neurol. 73, 755–756 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK, Eur. J. Pain 9, 463–484 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Zhou W et al. , Nat. Neurosci. 22, 1649–1658 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Price DD, Mol. Interv. 2, 392–403 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Head H, Holmes G, Brain 34, 102–254 (1911). [Google Scholar]
- 32.Diamond ME, Armstrong-James M, Budway MJ, Ebner FF, J. Comp. Neurol. 319, 66–84 (1992). [DOI] [PubMed] [Google Scholar]
- 33.Wiech K, Ploner M, Tracey I, Trends Cogn. Sci. 12,306–313 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Leknes S, Tracey I, Nat. Rev. Neurosci. 9, 314–320 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Ren W et al. , Nat. Neurosci. 19, 220–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith ML, Asada N, Malenka RC, Science 371,153–159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salimpoor VN, Benovoy M, Larcher K, Dagher A, Zatorre RJ, Nat Neurosci. 14, 257–262 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Baliki MN et al. , Nat Neurosci. 15, 1117–1119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the supplementary materials.