

Abstract
Aquaporin-4 (AQP4) is characterized by the formation of orthogonal arrays of particles (OAPs) comprising its M1 and M23 isoforms in the plasma membrane. However, the biological importance of OAP formation is obscure. Here, we developed an OAP depolymerization male mouse model by transgenic knock-in of an AQP4-A25Q mutation. Analyses of the mutant brain tissue using blue native polyacrylamide gel electrophoresis, super-resolution imaging, and immunogold electron microscopy revealed remarkably reduced OAP structures and glial endfeet localization of the AQP4-A25Q mutant protein without effects on its overall mRNA and protein expression. AQP4A25Q/A25Q mice showed better survival and neurologic deficit scores when cerebral edema was induced by water intoxication or middle cerebral artery occlusion/reperfusion. The brain water content and swelling of pericapillary astrocytic endfeet processes in AQP4A25Q/A25Q mice were significantly reduced, functionally supporting decreased AQP4 protein expression at the blood–brain barrier. The infarct volume and neuronal damage were also reduced in AQP4A25Q/A25Q mice in the middle cerebral artery occlusion/reperfusion model. Astrocyte activation in the brain was alleviated in AQP4A25Q/A25Q mice, which may be associated with decreased cell swelling. We conclude that the OAP structure of AQP4 plays a key role in its polarized expression in astrocytic endfeet processes at the blood–brain barrier. Therefore, our study provided new insights into intervention of cerebral cellular edema caused by stroke and traumatic brain injury through regulating AQP4 OAP formation.
SIGNIFICANCE STATEMENT Aquaporin-4 (AQP4) is characterized by orthogonal arrays of particles (OAPs) comprising the M1 and M23 isoforms in the membrane. Here, an OAP depolymerization male mouse model induced by AQP4-A25Q mutation was first established, and the functions of OAP depolymerization in cerebral edema have been studied. The results revealed that AQP4 lost its OAP structure without affecting AQP4 mRNA and protein levels in AQP4-A25Q mice. AQP4-A25Q mutation mice has neuroprotective effects on cerebral edema induced by water intoxication and middle cerebral artery occlusion/reperfusion through relieving the activation of astrocytes and suppressed microglia-mediated neuroinflammation. We concluded that the OAP structure of AQP4 plays a key role in its polarized expression in astrocytic endfeet processes at the blood–brain barrier. Therefore, our study provided new insights into intervention of cerebral cellular edema caused by stroke and traumatic brain injury through regulating AQP4 OAP formation.
Keywords: astrocyte, BBB, brain edema, MCAO/R, neuronal injury, water intoxication
Introduction
Aquaporin-4 (AQP4) is a water-selective membrane channel expressed in various epithelial type cells as well as astroglial cells in the CNS (Papadopoulos and Verkman, 2012; Nagelhus and Ottersen, 2013). In the CNS, AQP4 is strongly expressed in astrocytic endfeet membrane facing microvessels at the blood–brain barrier (BBB) and plays a key role in brain fluid and ion homeostasis, brain edema formation and resolution, astrocyte migration, glial scar formation, and proinflammatory factor secretion (Papadopoulos and Verkman, 2012, 2013; Kim et al., 2017). AQP4 is also the target of the autoimmune IgG antibody of neuromyelitis optica (NMO) (Papadopoulos et al., 2014; Guo et al., 2017). AQP4 is distinguished from other aquaporins in the AQP family by its formation of well-ordered square structures called orthogonal arrays of particles (OAPs) in the membrane. The formation of OAPs primarily involves two major polypeptides of AQP4: M1 and M23 (Crane and Verkman, 2009; Papadopoulos and Verkman, 2012). The full-length “long” (Ml) form of AQP4 is organized into tetramers or very small OAPs, whereas the “short” (M23) form of AQP4 constitutes large OAPs. Coexpression of these two isoforms leads to the formation of OAPs of different sizes based on the isoform ratio (Smith and Verkman, 2015). Astrocytes can regulate the OAP size by changing the ratio of M1 and M23 according to functional requirements (migration or cell adhesion/polarization) (Smith et al., 2014). However, the biological significance of AQP4 OAP structures in the cell membrane remains unclear.
AQP4 deficiency appears to protect ischemic and water intoxication induced brain injury by reducing fluid accumulation in astrocytic endfeet at the BBB (Manley et al., 2000). AQP4 has a highly polarized distribution in brain. It is greatly enriched in the perivascular plasma membrane of astrocytic endfeet (Nielsen et al., 1997). It was proposed that the OAP structure may be important for polarized expression of AQP4 protein at the glial endfeet microdomains to support AQP4 interaction with the extracellular matrix (Hiroaki et al., 2006) and high water permeability of the plasma membrane (Lisjak et al., 2017). Previous mutagenesis studies found that glutamine substitution of alanine 25 (M23-A25Q) reduced the OAP content by ∼80% on average, which was similar to the result obtained for the M1 isoform alone in transfected cell models (Crane and Verkman, 2009). No further in vivo studies of this mutation have been reported. Recently, de Bellis et al. (2021) reported an “OAP-null” mouse model that selectively lacks the M23-AQP4 isoform because of targeted removal of its start codon from the AQP4 genomic sequence. They found a massive reduction in overall, membrane and intracellular expression of the AQP4 protein but relatively increased M1-AQP4 tetramers in astrocytes. They concluded that OAP aggregation is fundamental to ensuring a normal level of AQP4 expression in the plasma membrane of astrocytes; therefore, AQP4 tetramers cannot be used by astrocytes as an alternative to OAPs without affecting AQP4 expression levels. However, the study did not answer the question whether the AQP4 OAP structure determines its polarized expression in astrocytic endfeet.
In the present study, we generated a transgenic knock-in mouse model with the A25Q mutation in AQP4. We found diminished OAP structures and dramatically decreased AQP4 localization in astrocytic endfeet at the BBB without significant reduction in overall mRNA and protein expression in the brains of the AQP4A25Q/A25Q mice. AQP4-A25Q significantly reduced cerebral edema induced by water intoxication and middle cerebral artery occlusion/reperfusion (MCAO/R). Our study provided the first evidence that AQP4-OAP formation plays a key role in its polarized expression in astrocytic endfeet at the BBB.
Materials and Methods
Generation of AQP4-A25Q mice
The AQP4-A25Q transgenic knockin mouse model was generated using a gene replacement strategy in embryonic stem cells as shown in Figure 1A. A targeting vector was constructed based on the gene structure of mouse AQP4 (Ma et al., 1996) with an A25Q point mutation in the long arm. The targeting vector uses PolII-neo-bpA cassette for positive selection, and the PGK-tk cassette for negative selection after transfection into E14 embryonic stem cells as described previously (Ma et al., 1997). Properly targeted embryonic stem cell clones were identified by PCR amplification of a 1.4 kb fragment using a sense primer (5′-CTCTATGGCTTCTGAGGCGGAAAG-3′) located in the neogene and an antisense primer (5′-GGTGAGGTTTCCATGAACCTAG-3′) located downstream from the short arm. Embryonic stem cells carrying the AQP4-A25Q mutation were injected into postcoital 3.5 d 8-cell morula stage C57BL/6J zygotes, cultured overnight into blastocysts, and transferred into pseudopregnant Kunming female mice. Offspring were genotyped by PCR using the following primers: a-sense, 5′-GCATTGTCTGAGTAGGTGTC-3′, located at the 3′-end of the neogene; b-sense, 5′-CATGTGCTTGCAAACTCTGGTG-3′, located at the junction of the long arm and short arm; and c-antisense, 5′-CCTGCCTAAACTACTCAAGTGC-3′, located at the 5'-end of the short arm. Southern blot analysis was performed to confirm correct gene targeting. Heterozygous AQP4A25Q-neo/+ mice were then mated with EIIa-cre mice (purchased from Model Animal Research Center of Nanjing University) to excise the neomycin-resistance (neo) cassette from the intron. The resulting heterozygous AQP4A25Q/+ mice were mated to generate AQP4A25Q/A25Q mice.
Figure 1.
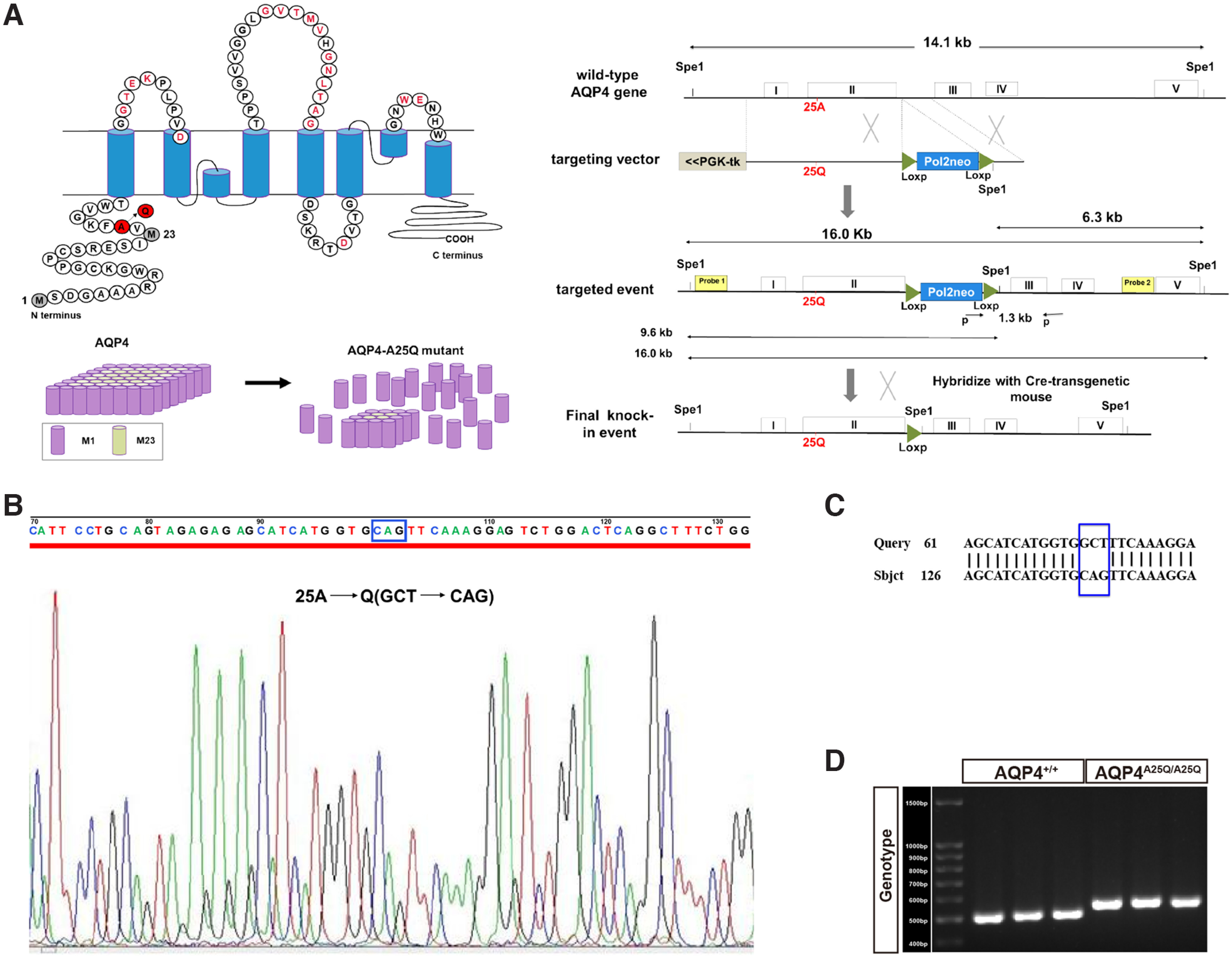
Generation of the AQP4-A25Q gene knocked-in mouse model. A, Schematic depicting the primary amino acid sequence of AQP4 and A25Q point mutation (left) and flowchart of the gene targeting strategy (right). B, Sequence analysis of the mutant alleles in AQP4A25Q/A25Q mice. The desired mutation “GCT” to “CAG” in the AQP4 gene was confirmed in knock-in mice. C, DNA sequence alignment of AQP4+/+ and AQP4A25Q/A25Q mice. D, Genotyping of AQP4+/+ and AQP4A25Q/A25Q mice. The size of the AQP4+/+ amplicon is 500 bp, and the size of the AQP4A25Q/A25Q amplicon is 540 bp. Primers used are provided in Extended Data Table 1-1.
The mice were maintained at the Specific Pathogen Free Model Animal Center of Dalian Medical University, China. Age-matched, male AQP4+/+ and AQP4A25Q/A25Q mice (10-12 weeks old) were used for water intoxication and MCAO/R. All animal experiments were conducted according to the Institutional Animal Care and Use Committee guidelines of the National Institutes of Health (National Institutes of Health publication no. 86-23, revised 1987), and approved by the Institutional Ethics Committee of Dalian Medical University (no. AEE19105).
Genotyping of AQP4A25Q/A25Q mice
The genotype of AQP4A25Q/A25Q mice was confirmed before the animals were used for the subsequent experiments. The primers used for genotyping (Invitrogen, Thermo Fisher Scientific) are listed in Extended Data Table 1-1.
The PCR conditions were set at 94°C (5 min) for enzyme activation, with 30 cycles of denaturation at 94°C for 30 s; annealing at 56.5°C for 30 s; and extension at 72°C for 60 s, 72°C for 10 min, and, finally, 10°C infinitely. The amplified DNA was separated on a 2% agarose gel by electrophoresis using a current of 120 mA for 60 min. AQP4+/+ and AQP4A25Q/A25Q bands were detected at 500 and 540 bp, respectively, compared with the standard DNA ladder.
qRT-PCR
qRT-PCR was conducted as reported previously (Ntim et al., 2020). Total RNA from brain tissues was extracted with TRIzol reagent according to the manufacturer's instructions. One microgram of total RNA was reverse-transcribed into cDNA using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech). The primers used for AQP4 and the housekeeping gene GAPDH (Invitrogen) are listed in Extended Data Table 1-1. The expression levels of targeted genes were determined using SYBR Green qPCR SuperMix (TransGen Biotech) on a CFX96 real-time system (Bio-Rad). Cycle threshold (CT) values for the genes were calculated, and changes were assessed using the formula 2−△△CT, where △CT represents the difference in CT values between the target and housekeeping gene (GAPDH) mRNA.
Membrane protein extraction
Membrane protein extraction was conducted following the instructions of a Mem-PERTM Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific, 89842).
SDS-PAGE and Western blot analysis
SDS-PAGE was conducted as previously reported with modifications (Jiang et al., 2020). Brain samples were homogenized in RIPA buffer and protease inhibitor cocktail. Equal amounts of proteins (20-150 μg) were size-fractionated by 8%-12% SDS-PAGE. The primary antibodies used included AQP4 (rabbit, 1:1000, Abcam ab46182) and anti-β actin (mouse, 1:3000, Zen Bioscience, 200068-8F10), and the sample-loaded membranes were incubated with the primary antibodies at 4°C overnight. The membranes were then incubated with the secondary antibodies goat anti-rabbit (1:5000, Thermo Fisher Scientific, A16104) and goat anti-mouse IgG (1:5000, Thermo Fisher Scientific, 31430), along with electrochemiluminescence (Millipore) reagent. Band signals were detected using Bio-Rad gel analysis software.
Blue native PAGE (BN–PAGE)
BN–PAGE was performed as previously described (Rossi et al., 2012). Briefly, brain tissue samples were suspended in blue native lysis buffer (NativePAGE Sample Prep Kit, Invitrogen, BN2008) and incubated for 30 min. The lysate was centrifuged at 20,000 × g for 30 min at 4°C. Aliquots of the supernatant were placed into sterile microcentrifuge tubes and stored at −80°C until use. NativePAGE 5% G-250 solution was added to the detergent containing 900 μg of protein sample to reach a final G-250 detergent concentration ratio of 1:4 before native gel electrophoresis. A 20 μg protein sample and the molecular mass standard (NativeMark Unstained Protein Standard, Invitrogen, LC0725) were loaded onto polyacrylamide native gradient gels (3%-12%) and electrophoresed with NativePAGE running buffers according to the manufacturer's protocol (Invitrogen, BN2007). The proteins were then transferred onto PVDF membranes (Millipore) by blotting at 240 mA for 1.5 h using NuPAGE transfer buffer (Invitrogen, NP0006). Subsequently, the Western blot procedure was performed.
Cell line culture and treatment
The Chinese hamster lung fibroblast cell line (V79; a gift from Bari University, Italy) was maintained in Dulbecco's high-glucose medium (DMEM, D7777; Sigma Aldrich) supplemented with 10% FBS (Tissue Culture Biologicals) and supplemented with 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. V79 cells were cultured at 37°C in a humidified incubator with an atmosphere of 5% CO2. The V79 cell line was stably transfected with the plasmid encoding human M23-AQP4, amplified by PCR from brain cDNA, and cloned into a mammalian expression vector pmCherry-N1 (Clontech Laboratories) according to the instructions of the manufacturer (Sun et al., 2016). Transfections were performed using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions for stable transfection.
Human-derived serum collection
Sera from patients with NMO and healthy controls were used for this study. All NMO patients were selected as consecutively diagnosed patients according to Wingercuck's criteria (Wingerchuk et al., 2006), and the institutional Ethics Committee (Dalian Medical University, Dalian, China) approved the study. Serum was prepared from blood samples and working aliquots were stored at −80°C.
Immunofluorescence
Immunofluorescent staining was conducted as previously reported (Jiang et al., 2020). Briefly, brain samples were fixed overnight in 4% PFA and then cryosectioned at 10 μm depth and subjected to immunofluorescence staining. V79-AQP4-M23-mCherry cells and V79-WT cells without AQP4-M23 expression were incubated with human serum diluted in culture medium for 1 h on ice to bind surface antigens onto living cells. After washing, cells were fixed in 4% PFA, permeabilized in 0.3% Triton X-100, and blocked in 2% BSA. The brain samples were incubated with primary antibodies listed in Extended Data Table 2-1, at 4°C overnight. The cells were then incubated with the secondary antibodies listed in Extended Data Table 2-1 and DAPI stain were incubated for 1 h at room temperature. All images were collected with a Pannoramic MIDI scanner (3D Histech) equipped with a GS3-U3-51S5 M-C camera (FLIR), Lumencor SOLA light engine, and Semrock filters and confocal microscope (Leica, TCS SP8 3×). The mean fluorescence intensities were determined blindly using ImageJ software (National Institutes of Health) and Image-Pro Plus 6.0.
Time-gated stimulated emission depletion (g-STED) imaging
Tissue sections were analyzed by g-STED as previously described (Rosito et al., 2018). g-STED images were acquired with a Leica TCS SP8 3× STED 3× fully automated epifluorescence confocal microscope equipped with an HC PL APO CS2 100 × 1.40 NA oil-immersion objective and operated with LAS-X imaging software. Excitation was performed with a tunable white light laser, and emissions were detected by HyD4 standard mode detectors. In g-STED mode, the excitation of AlexaFluor-594 and -488 dye was sequentially performed with a tunable white light laser at 594 and 660 nm, respectively. A 660 nm depletion beam was used as the STED beam. All g-STED images were acquired in the XY plane over an XY area of 22.97 μm × 22.97 μm with a fixed Z plane.
The spatial resolution of the STED-system was 30 nm. The image processing package Fiji was used to analyze the size of the OAPs in images as follows. The astrocyte endfeet outline was traced with the polygon selection tool, and the area outside the astrocyte endfeet was cropped to exclude extracellular noise. Images were converted from 16-bit to 8-bit grayscale and transformed into binary images by threshold adjustment. The particle analyzer plug-in was then used to determine the size (area) of connected pixels by fitting to ellipses. Elongation ratios were determined as the long-to-short diameter of fitted ellipses using the FracLac plug-in for ImageJ. Representative single OAPs have been encircled in Figure 3A.
Figure 3.
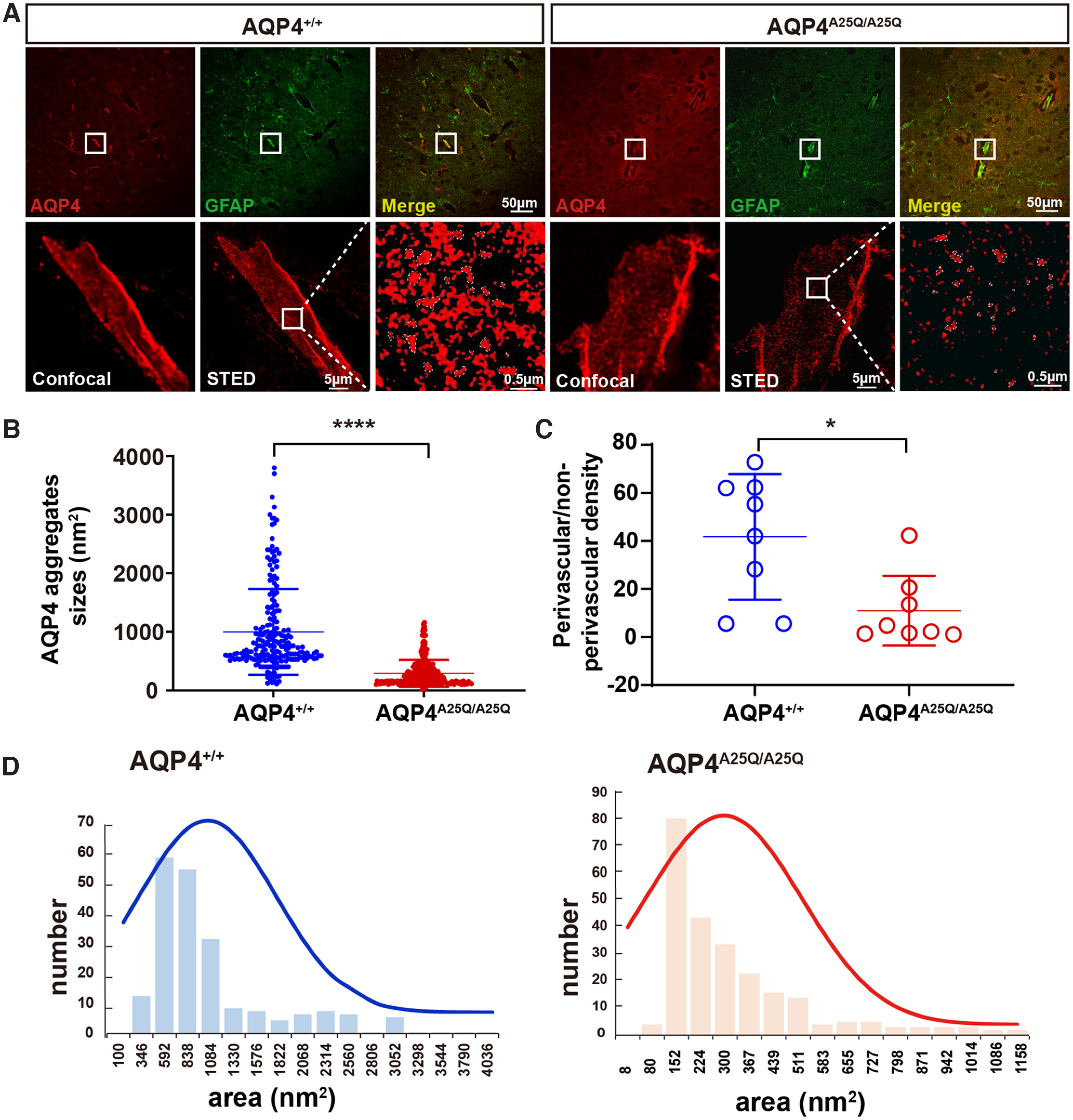
AQP4-A25Q mutation modulates supramolecular clustering of AQP4. A, Brain blood vessel stained with commercial anti-AQP4 and anti-GFAP antibodies and imaged using confocal microscopy and g-STED. Top, The typical staining of AQP4 astrocyte processes surrounding brain cortex capillaries is shown in the white box. Scale bar: 50 μm. Bottom, a comparison of confocal and g-STED imaging, with magnification of the white-boxed region indicated by g-STED. Scale bars: 5 μm, 0.5 μm. B, Analysis of AQP4 clusters was performed in AQP4+/+ and AQP4A25Q/A25Q mice. Scatter plot of AQP4 cluster sizes measured from the intensity profiles by the FWHM intensity. n = 214 clusters for the AQP4+/+ group; n = 247 clusters for the AQP4A25Q/A25Q group. C, Quantification of the ratio of AQP4 staining density in GFAP-labeled processes adjacent to blood vessels versus nonadjacent processes was performed in AQP4+/+ and AQP4A25Q/A25Q mice. n = 8 mice for each group. D, The size distributions of OAPs were also different, measuring 291.7 ± 14.6 nm2 in AQP4A25Q/A25Q mice and 998.7 ± 50.0 nm2 in AQP4+/+ mice on average. B, C, Data are median ± SD. *p < 0.05; ****p < 0.0001; Mann–Whitney test.
Immunogold electron microscopy (IEM)
Brains were collected and kept in 4% PFA after perfusion. The tissues were washed with 50%, 70%, 80%, 90%, 95%, and 100% ethanol at 4°C for 20 min, placed into solutions containing different ratios of resin to ethanol (1:2, 1:1, and 2:1) for 1 h, and fixed in resin overnight. The brains were then placed under ultraviolet light for photopolymerization for 72 h at −20°C. Sixty-nanometer sections were cut using a Leica UC7 ultramicrotome, and a nickel net was used to dredge the sections. On-grid immunogold labeling for AQP4 was conducted on cortices from AQP4+/+ and AQP4A25Q/A25Q mice. The immunogold procedure included the following steps: sample incubation with 1% BSA in Dulbecco's PBS for 20 min, followed by incubation with the first antibody overnight at room temperature, and, finally, incubation with 10 nm of the second antibody for 3 h. The sections were then counterstained with uranyl acetate followed by lead citrate before examination under a transmission electron microscope (Tecnai Spirit Biotiwm, FEI, accelerating voltage 120 V). The linear density of AQP4 toward the endothelium was subsequently analyzed using a MATLAB-based toolbox (Hasan-Olive et al., 2019) designed for the automatic evaluation of immunogold-labeled membranes.
Establishment of the water intoxication model
AQP4+/+ and AQP4A25Q/A25Q mice received intraperitoneal injections of distilled water equal to 20% of their body weights with desmopressin/1-deamino-8-D-arginine vasopressin (0.4 g/kg). For morbidity analysis, the mice were observed continuously and evaluated using a 100 point neurologic deficit scale, with a score of 0 indicating a normal status and a score of 100 representing brain death 30 min after acute water intoxication (Manley et al., 2000). The scale assessed general behavioral deficits (40 points: attempt to explore (0), no attempt to explore (20), normal respiration (0), and abnormal respiration (20)); cranial nerve reflexes (20 points: olfactory sensation present (0) or absent (4), vision present (0) or absent (4), corneal response present (0) or absent (4), whisker movement present (0) or absent (4), and hearing present (0) or absent (4)); motor deficits (20 points: leg-tail movement normal (0), stiff (5), or paralyzed (10)); sensory deficits (10 points: withdrawal response to pain present (0) or absent (10)); and coordination deficits (20 points: travel along a ledge present (0) or absent (5), placement testing present (0) or absent (5), righting reflex present (0) or absent (5), and stopping at the edge of the table present (0) or absent (5)). The time of death was recorded for log-rank (Mantel–Cox) survival analysis.
Establishment of the MCAO/R model
Eight- to 12-week-old male AQP4+/+ and AQP4A25Q/A25Q C57BL/6 mice were acutely anesthetized with volatilized isoflurane to ensure survival during surgery. A total number of 135 mice were used in MCAO/R model with an overall mortality rate of ∼30%. The MCAO/R model was established according to a published method (Wang et al., 2020a) with minor modifications. Mice were positioned on a heated operating table in the supine position. The skin was disinfected with iodine and 75% ethanol. A midline neck incision was made, and then the right common carotid artery (CA), internal carotid artery (ICA), and external carotid artery (ECA) were surgically exposed. The ECA was then separated and occluded with two knots. Next, the ICA was isolated, and the CA and ICA were clipped with a slipknot. A small hole was cut in the ECA between a knot and the bifurcation point. An MCAO suture (Southern Medical University, China) was introduced into the ICA and inserted until it stopped at the clip. The clipped arteries were opened while the suture was inserted to occlude the vessel. A third knot was closed on the ICA to hold the suture in position. Each mouse was kept in a heated cage for 2 h after occlusion, and the wound was closed with a saline cotton ball. The third knot was loosened, and the filament was withdrawn for reperfusion. Finally, the skin was closed, and the mouse was returned to an individual cage (Dong et al., 2019). The body temperature was carefully maintained at 37°C by a heating pad until the animal completely recovered from anesthesia. The same surgical procedure was performed on sham-operated animals, excluding occlusion of the artery. We used doppler flow imaging system (Vevo 3100 Preclinical Imaging system, FUJIFILM, Visual Sonics) to monitor the perfusion of the ipsilateral hemisphere in the circle of Willis and laser speckle contrast imaging (RWD Life Science) to monitor regional cerebral blood flow (rCBF) between sham and MCAO group. Animals that died or failed to show 70% rCBF reduction of the preischemia levels were excluded from further analyses. Mouse neurologic functions were measured using neurologic severity scores established by Clark et al. (1997) after 2 h of MCAO and 24 h of blood flow reperfusion. The neurologic deficit scores (NDSs) were recorded blindly by two investigators.
Brain water content
After 30 m of acute water intoxication or 2 h of MCAO and 24 h of blood flow reperfusion, the mice were killed after anesthetization with volatilized isoflurane, and the cerebrum was collected quickly and placed on an ice plate with physiological saline infiltration to determine wet brain weight (W). The dry brain weight (D) was measured after drying at 120°C overnight. The brain water content (%) = (wet weight – dry weight)/wet weight × 100%.
Triphenyl tetrazolium chloride (TTC) staining
Measurements of infarct volumes were performed as described previously (P. Li et al., 2013; Yang et al., 2017). The brain was sectioned into three slices, starting 3 mm from the anterior tip of the frontal lobe in the coronal plane. Slices were placed in 2% TTC and incubated at 37°C for 15 min. The cerebral infarct volumes were measured with Pro Plus Image. Infarct volumes were corrected for brain edema by reporting the volume of the contralateral hemisphere minus the noninfarcted volume of the ipsilateral hemisphere.
Nissl staining
Nissl staining was conducted as previously reported (Wang et al., 2020a). Brain samples were perfused with sterile saline, fixed overnight in 4% phosphate-buffered formaldehyde, successively transferred to 15% and 30% sucrose for 24 h, sectioned at 10 µm depth for staining (Leica CM 1850, Leica Microsystems), stained with 0.1% cresyl violet for 10 min, and subjected to gradient elution with 75%, 95%, and 100% ethanol. Finally, the sections were immersed in dimethyl benzene for 3 min twice and sealed with neutral balsam. The proportion of intact neurons in the MicroVision field was recorded. Three views of each group were selected randomly.
Electron microscopy
Electron microscopy was conducted as previously reported (Wang et al., 2019). The brain ultrastructure was evaluated 30 min after water intoxication, after 2 h of MCAO, and after 24 h of blood flow reperfusion. The brains were collected and maintained in 2.0% PFA and 2.0% glutaraldehyde for 24 h at 4°C. Samples were postfixed in osmium tetroxide, washed in buffer, and then embedded in epoxy resin. One-micron-thick sections were stained with toluidine blue and examined with light microscopy. Ultrathin sections (60 nm) were stained with 2.0% uranyl acetate and lead citrate and examined under a JEOL electron microscope (JEM-2000EX). The astrocytic foot process cross-sectional area was determined using randomly selected transmission electron micrographs containing astrocytic foot processes adjacent to brain capillaries by an investigator blinded to the genotypes of the mice.
Experimental design and statistical analysis
All quantitative analyses were performed with the researcher blinded to the condition. All data are shown as mean ± SD. Prism 8.0.2 (GraphPad Software) was used to produce all graphs. First, a data normality test was performed by Shapiro–Wilk normality test, and then statistical significance was evaluated by performing unpaired Student's t tests for normally distributed data and Mann–Whitney U tests for non-normally distributed data. For two independent variables, two-way ANOVA was performed, followed by Tukey's post hoc test. Significance in the cumulative survival studies was determined with the log-rank (Mantel–Cox) test. A value of p < 0.05 was considered statistically significant.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article.
Ethics statement
Human-derived serum was obtained with consent. Obtaining and using human serum were approved by the Ethics Committee of our institution (Dalian Medical University, Dalian, China). All animal experiments were conducted according to the Institutional Animal Care and Use Committee guidelines of the National Institutes of Health (Publication no. 86-23, revised 1987), and approved by the Institutional Ethics Committee of Dalian Medical University (no. AEE19105).
All experiments were designed to minimize the number of animals used and animal suffering. Mice were maintained under a 12 h dark to light cycle at constant room temperature and humidity, with food and water provided ad libitum, and supplied with environmental enrichment materials, such as toys and shelters.
Results
Generation of the AQP4-A25Q knock-in mouse model
The AQP4 gene is composed of five exons, and the alanine residue at position 25 is encoded by the second exon (Fig. 1A). An AQP4-A25Q knock-in model was generated by introducing a GCT→CAG point mutation into exon 2, followed by a homologous recombination strategy and cleaving of the neo cassette with the Cre-LoxP system (Fig. 1A). The mutation was confirmed by DNA sequencing of the PCR product of AQP4-exon 2 from the brains of AQP4+/+ and AQP4A25Q/A25Q mice (Fig. 1B,C). Figure 1D shows the PCR genotyping results of AQP4A25Q/A25Q mice.
Effects of the A25Q mutation on AQP4 expression and OAP formation
We first analyzed the expression of AQP4 by comparing brain mRNA and protein levels in the AQP4A25Q/A25Q versus AQP4+/+ mice. We detected the AQP4 mRNA level of mouse brain tissue using primers flanking exon 0 (GenBank: AF219992.2) (Alikina et al., 2012) by SYBR Green qRT-PCR. The results indicated no significant difference between AQP4A25Q/A25Q and AQP4+/+ mice. The mRNA levels of different AQP4 isoforms (e.g., M1, M23) were also detected using sense primers 58F (5′-catcgccaaactgcaagac-3′) for M23 (exon-1), 135F (5′-acagagctgcggcaagg-3′) (Alikina et al., 2012) for M1 (exon-0) and a common antisense primer from exon-2 1078B (5′-aaagtgattattaactccaccagg-3′) (Alikina et al., 2012) for all isoforms. There were no significant differences of AQP4-M1 mRNA level and the AQP4-M23 mRNA level in AQP4+/+ and AQP4A25Q/A25Q mouse brain (p>0.05, Mann–Whitney test) (Fig. 2A).
Figure 2.
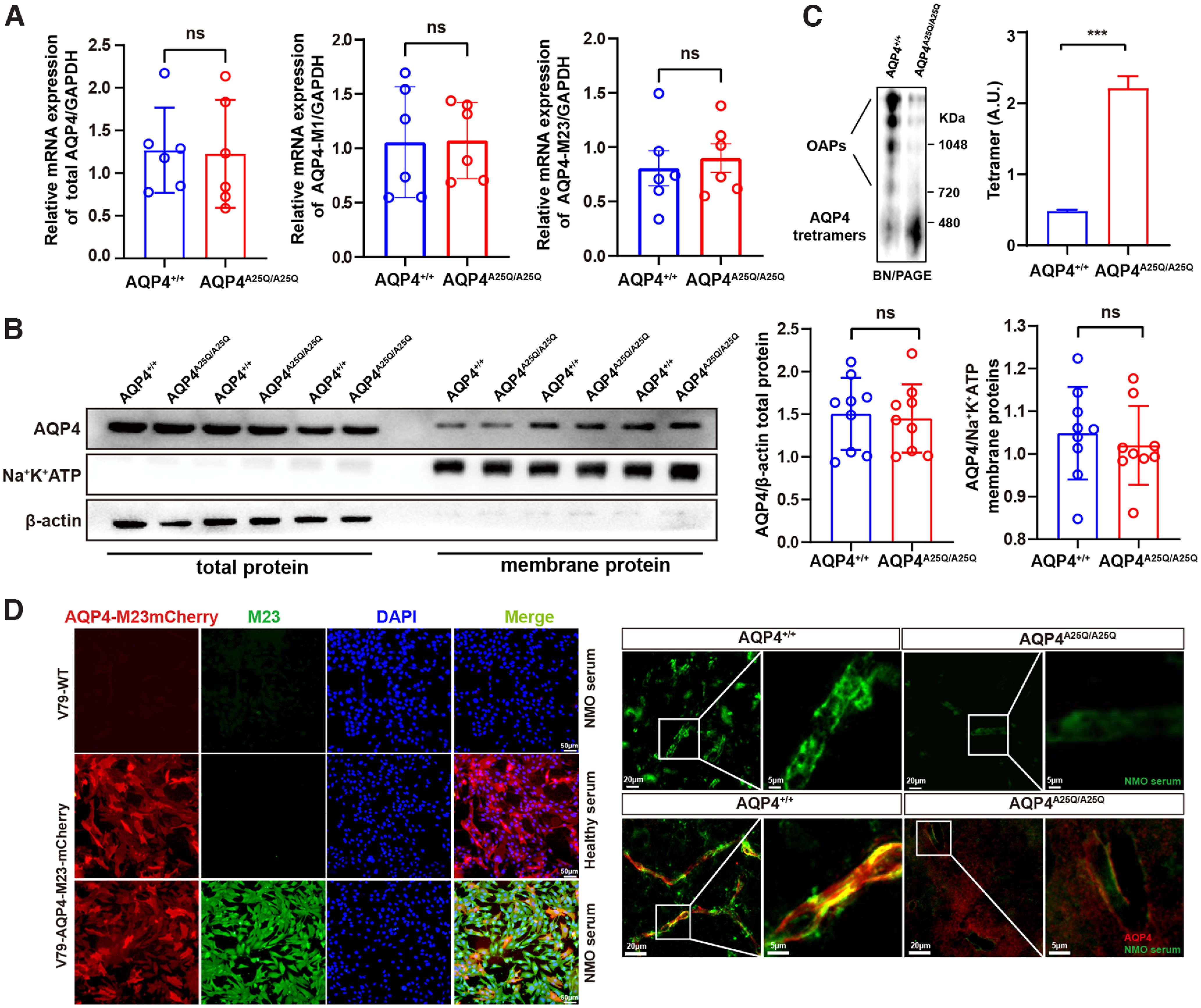
Effects of A25Q mutation on AQP4 expression and OAP formation in the brains of AQP4A25Q/A25Q mice. A, Densitometry analysis of the mRNA levels of total AQP4, AQP4-M1, and AQP4-M23 in AQP4+/+ and AQP4A25Q/A25Q mouse brains. n = 6 mice for each group. Primers used are provided in Extended Data Table 1-1. B, Representative bands and densitometry analysis of the AQP4 total protein and the AQP4 membrane protein expression level in AQP4+/+ and AQP4A25Q/A25Q mouse brains. n = 9 mice for each group. C, AQP4 immunoblot of brain tissues following BN–PAGE, and bar graph represents the results of densitometry analysis of AQP4 tetramers, which was shown in arbitrary units (A.U.). n = 3 mice for each group. D, Left, Live V79-AQP4-M23-mCherry cells and V79-WT cells (without AQP4-M23 expression) exposed to NMO patient serum. Scale bar, 50 μm. Right, immunofluorescence pattern of bound NMO serum and AQP4 antibody in the mouse CNS. Scale bars: top, 20 μm, 5 μm; bottom, 20 μm, 5 μm. ***p < 0.001 (Mann–Whitney test and unpaired Student's t test). Bar graphs indicate mean ± SD. Antibodies used are provided in Extended Data Table 2-1.
Western blot analysis of total protein extracts also revealed similar overall AQP4 protein expression in AQP4A25Q/A25Q and AQP4+/+ mouse brains (Fig. 2B). We further quantified the expression of AQP4 protein in the plasma membrane fraction of the mouse brain (purity was indicated by minimal β-actin detection). As shown in Figure 2B, there were no significant differences in AQP4+/+ and AQP4A25Q/A25Q protein expression. However, the OAPs in the AQP4A25Q/A25Q mouse brain were largely depolymerized into tetrameric forms as determined by BN–PAGE, which indicated the presence of nearly 5 times more tetramers (p<0.001, Mann–Whitney test) in the AQP4A25Q/A25Q versus AQP4+/+ mouse brains (Fig. 2C).
Previous reports have shown that the serum biomarker NMO-IgG in patients can specifically bind to OAPs formed by the AQP4-M23 isoform (Hinson et al., 2012). We first verified the serum from NMO patients with cultured live V79 cells stably expressing AQP4-M23-mCherry and without AQP4-M23 expression to identify the specific NMO (Fig. 2D). Then, we performed immunofluorescence with a high-titer NMO serum (1:4000) on cryosections of AQP4+/+ and AQP4A25Q/A25Q mouse brains. The AQP4A25Q/A25Q mouse brains showed much less staining of AQP4-M23 aggregates (OAPs) in endfeet adjacent to microvessels compared with the AQP4+/+ mice (Fig. 2D). Furthermore, we also performed NMO serum (for M23) and pan-AQP4 double staining in mouse brain tissue. The AQP4A25Q/A25Q mouse brains showed much less staining of AQP4 and AQP4-M23 aggregates (OAPs) at the endfeet adjacent to microvessels and a stronger immunoreactivity of AQP4 at the nonperivascular parts of astrocytes than the AQP4+/+ mice (Fig. 2D).
Collectively, these results suggest that the AQP4-A25Q mutation did not affect overall AQP4 mRNA and protein expression but decreased OAP formation and polarized AQP4 expression in astrocytic endfeet at BBB in the mouse brain.
AQP4-A25Q mutation modulates the supramolecular clustering of AQP4
We evaluated the AQP4 supramolecular structures in cerebral perivascular astrocyte endfeet using g-STED microscopy. This technique also allowed the determination of the size and distribution of individual AQP4 clusters (OAPs) at nanoscale resolution (Rosito et al., 2018). Confocal images showed much fewer AQP4 OAPs in tangential perivascular glial endfeet processes in the AQP4A25Q/A25Q versus AQP4+/+ mice (Fig. 3A). Quantitative analysis (Fig. 3B) indicated an ∼3.5 times smaller average OAP size in the AQP4A25Q/A25Q mice than in the AQP4+/+ mice (p<0.0001, Mann–Whitney test). Calculation of the relative density of AQP4 staining in the perivascular and nonperivascular GFAP-labeled structures showed significantly lower AQP4 expression (p = 0.0104, Mann–Whitney test) in astrocyte endfeet at the BBB in AQP4A25Q/A25Q versus AQP4+/+ mice (Fig. 3C). The size distributions of OAPs were also different, measuring 291.7 ± 14.6 nm2 in AQP4A25Q/A25Q mice and 998.7 ± 50.0 nm2 in AQP4+/+ mice on average (Fig. 3D). The OAP size distribution in AQP4+/+ mice was consistent with previously reported data in the mouse brain (Rossi et al., 2012).
AQP4-A25Q mutation decreases polarized expression of AQP4 in glial endfeet
AQP4 is highly enriched in astrocytic endfeet, where it is anchored to the basement membrane by interactions with adhesion complexes (Steiner et al., 2012). AQP4 OAPs play important roles in this process, as clustered AQP4 may form more stable interactions with adhesion complexes than individual AQP4 tetramers in the endfeet membrane (Smith et al., 2014). To determine the localization pattern of AQP4 in the AQP4A25Q/A25Q mouse brain, we performed immunofluorescence staining of cryosections for AQP4 and GFAP. Strong AQP4 staining was mainly seen around microvessels in the AQP4+/+ brain, whereas diffuse AQP4 labeling was seen throughout the brains of AQP4A25Q/A25Q mice (Fig. 4A). A comparative 3D fractal analysis also indicated less AQP4 aggregation in the perivascular region of AQP4A25Q/A25Q versus AQP4+/+ mouse brain (Fig. 4A). Furthermore, using IEM, specific AQP4 aggregation patterns of gold particles along the perivesicular astrocytic endfeet membranes were detected in the AQP4+/+ but not AQP4A25Q/A25Q mice (Fig. 4B). The quantification of gold particles demonstrated remarkably reduced AQP4 labeling (23.5 gold particles/μm in AQP4+/+ mice and 8.5 gold particles/μm in AQP4A25Q/A25Q mice, p = 0.0022, Mann–Whitney test) in the glial endfeet membranes at the BBB in AQP4A25Q/A25Q mouse brain (Fig. 4B). In addition, quantification of immune-gold particles at other parts of astrocytes demonstrated remarkably increased density in AQP4A25Q/A25Q versus AQP4+/+ mouse brains (Fig. 4B). These results revealed substantially decreased expression of AQP4 in astrocytic endfeet at the BBB.
Figure 4.
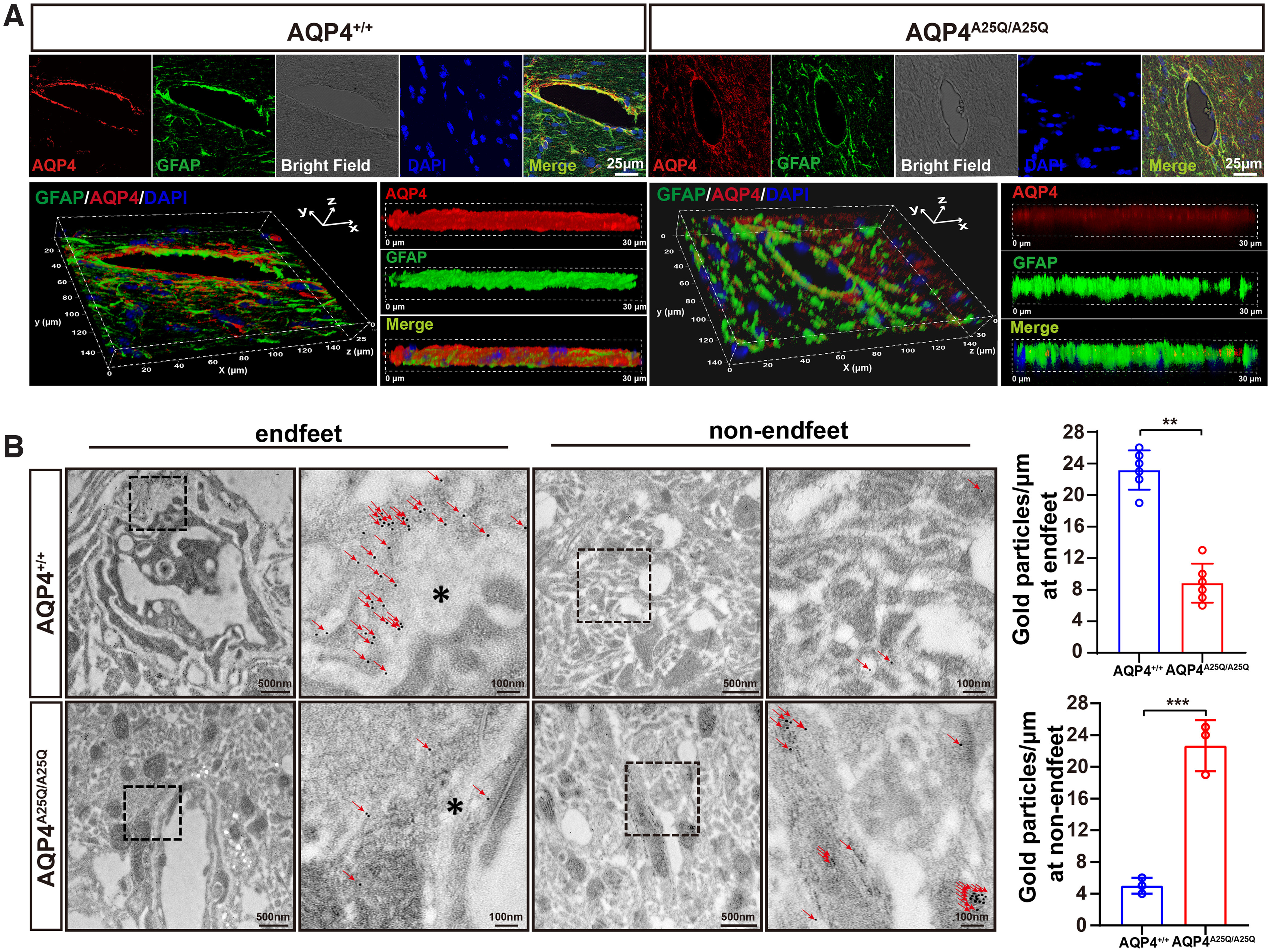
AQP4-A25Q mutation decreases polarized AQP4 expression in glial endfeet. A, Distribution of AQP4 in astrocytes of AQP4+/+ and AQP4A25Q/A25Q mice. Coimmunostaining with anti-AQP4 (red) and anti-GFAP (green) antibodies with DAPI staining for cell nucleus was performed. Bright field represents the microvasculature. Scale bar: 25 μm. 3D reconstruction of a confocal z stack of AQP4+/+ and AQP4A25Q/A25Q mouse brain slices. Scale bar: 30 μm. B, Left, Representative locations of AQP4 in AQP4+/+ and AQP4A25Q/A25Q mice indicated by IEM staining. In AQP4+/+ mice, gold particles (red arrowheads) were concentrated in astrocyte endfeet (asterisks), whereas in AQP4A25Q/A25Q mice, AQP4 labeling was scattered. Scale bars: 100 nm, 500 nm. Right, Quantitative analysis of AQP4 immunogold particles labeling in AQP4+/+ and AQP4A25Q/A25Q mice. **p<0.01; ***p<0.001; Mann–Whitney test. n = 6 mice for gold particles at endfeet. n = 3 mice for gold particles at non-endfeet region. Bar graphs represent the mean ± SD. Antibodies used are provided in Extended Data Table 2-1.
AQP4A25Q/A25Q mice exhibited improved brain edema, neurologic outcomes, and survival after water intoxication and MCAO/R
To evaluate the effect of the AQP4-A25Q mutation on the development of cerebral edema, we first used acute water intoxication to induce cytotoxic brain edema, which is a well-established cerebral edema model (Manley et al., 2000). Water intoxication was induced in age- and weight-matched AQP4+/+ and AQP4A25Q/A25Q mice by intraperitoneal infusion of distilled water and desmopressin acetate (desmopressin/1-deamino-8-D-arginine vasopressin). At 150 min after water intoxication, only 14.3% of the AQP4+/+ mice survived (Fig. 5A), whereas 42.9% of the AQP4A25Q/A25Q mice were alive. The NDSs of the AQP4A25Q/A25Q mice were significantly lower than those of the AQP4+/+ mice (NDS = 49.29 ± 4.989 in AQP4A25Q/A25Q mice, NDS = 74.57 ± 3.847 in AQP4+/+ mice, p < 0.001) (Fig. 5B). The brain water content measured by the wet-to-dry weight ratio of the mouse brain was significantly lower in AQP4A25Q/A25Q mice than in AQP4+/+ mice (Fig. 5C). Astrocytes are the most abundant cells in the CNS, and astrocyte edema is the major component of cerebral edema. The area of swollen astrocytic foot processes in the AQP4A25Q/A25Q mice was significantly smaller (p = 0.0146, two-way ANOVA followed by Tukey's post hoc test) than that in the AQP4+/+ mice following water intoxication (Fig. 5D). Astrocytes were labeled with GFAP to determine astrocyte swelling. Furthermore, the intensity of GFAP fluorescence was remarkably lower in AQP4A25Q/A25Q mice than in AQP4+/+ mice with water intoxication. Similarly, the GFAP-positive area and the total number of GFAP-positive cells in the AQP4A25Q/A25Q mice were also significantly lower than that in the AQP4+/+ mice with water intoxication (Fig. 5E; Extended Data Fig. 5-1), indicating less swelling and activating of astrocytes in AQP4A25Q/A25Q mice in the water intoxication model. These results demonstrated that polarized AQP4 expression in astrocytic endfeet membranes plays a key role in cellular edema caused by water intoxication.
Figure 5.
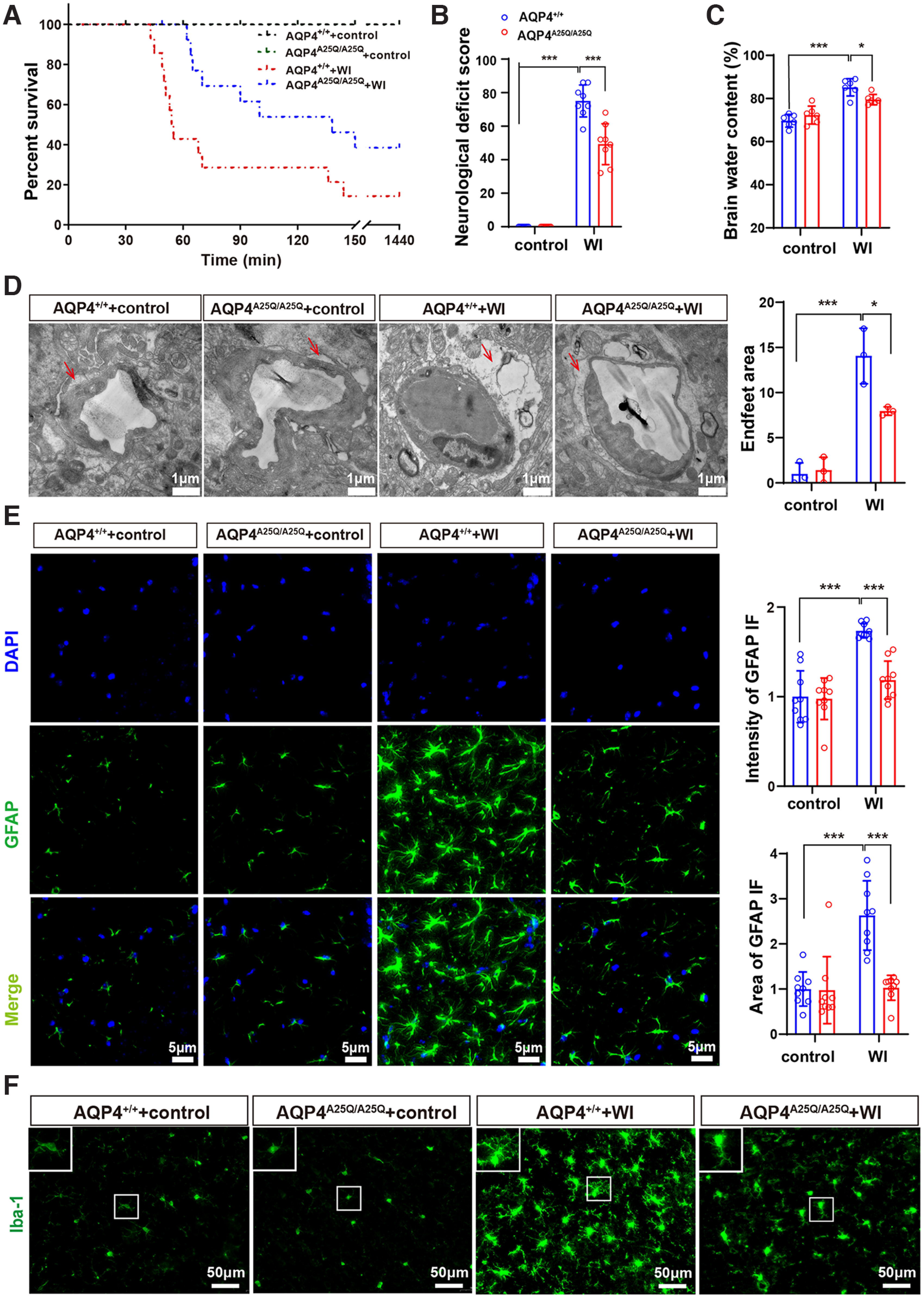
AQP4A25Q/A25Q mice exhibited improved brain edema, neurologic outcomes, and survival after water intoxication (WI). A, Cumulative survival of AQP4+/+ and AQP4A25Q/A25Q mice with and without WI; n = 14 mice. B, Neurologic scores of AQP4+/+ and AQP4A25Q/A25Q mice with and without water intoxication; n = 8 mice. C, Brain water contents of AQP4+/+ and AQP4A25Q/A25Q mice with and without water intoxication; n = 6 mice. D, Left, Transmission electron micrograph represents an edematous cerebral cortex at 30 min after water intoxication, the red arrow indicates astrocyte endfeet process. Scale bar, 1 μm. Right, Quantification of the pericapillary astrocyte endfeet process area was performed with 3 fields/mouse; n = 3 mice for each group. E, Left, Representative photographs of GFAP staining in the astrocytes of AQP4+/+ and AQP4A25Q/A25Q mice with and without water intoxication. Scale bar, 5 μm. Right, The intensity and area of GFAP in the brain were divided by the number of cells counterstained with DAPI; n = 9 mice for each group. Total number of GFAP-positive cells in control and mutant mice with water intoxication is provided in Extended Data Figure 5-1. F, Immunofluorescence staining of Iba-1 in AQP4+/+ and AQP4A25Q/A25Q mice with and without water intoxication. Scale bar, 50 μm. Expression of AQP4 in a different group of mice is provided in Extended Data Figure 5-2. A, Log-rank (Mantel–Cox) test was used. B-E, Data are mean ± SD. *p < 0.05; ***p<0.001; two-way ANOVA with Tukey's post hoc test.
We also performed the experiment to show the expression of AQP4 in control and mutant mice in both water intoxication and MCAO/R models (Extended Data Fig. 5-2). Water intoxication and MCAO/R significantly upregulated AQP4 protein expression, which were consistent with previously reported data (Karmacharya et al., 2015; Wang et al., 2020b). However, there was no significant upregulation observed in AQP4-A25Q mutant mice in water intoxication and MCAO, which may as well be attributed to alleviated astrocyte swelling.
In addition, astrocytes activated by edema can induce neuroinflammation and subsequently activate microglia (Goshi et al., 2020). The microglial morphology was observed, and the results indicated that AQP4-A25Q mutation reduced the level of microglial activation in the mice with water intoxication (Fig. 5F). Together, these results suggested that neuroinflammation was inhibited in AQP4-A25Q mutant mice after water intoxication.
Next, we used another model of cellular edema, MCAO/R, because of its relevance to ischemic hemispheric stroke in humans (Hossmann, 1998). The MCAO/R model leads to cellular and vasogenic edema, as well as serious brain injury, and is likely to result in neurologic impairments or even death (Q. Li et al., 2020). After 2 h of ischemia followed by 24 h of reperfusion, no mice showed any surgical complications, such as subarachnoid hemorrhage. There were no significant differences in rCBF between the AQP4+/+ and AQP4A25Q/A25Q mice, suggesting that both groups were exposed to the same degree of ischemia (Extended Data Fig. 6-1). The AQP4A25Q/A25Q mice showed a lower mortality rate (Fig. 6A) and mean deficit score (20.00 ± 4.10 vs 13.83 ± 4.31, n = 6, p < 0.05) than the AQP4+/+ mice at 24 h after MCAO/R (Fig. 6B). The AQP4A25Q/A25Q mice showed higher survival rates and relieved neurologic impairment compared with the AQP4+/+ mice.
Figure 6.
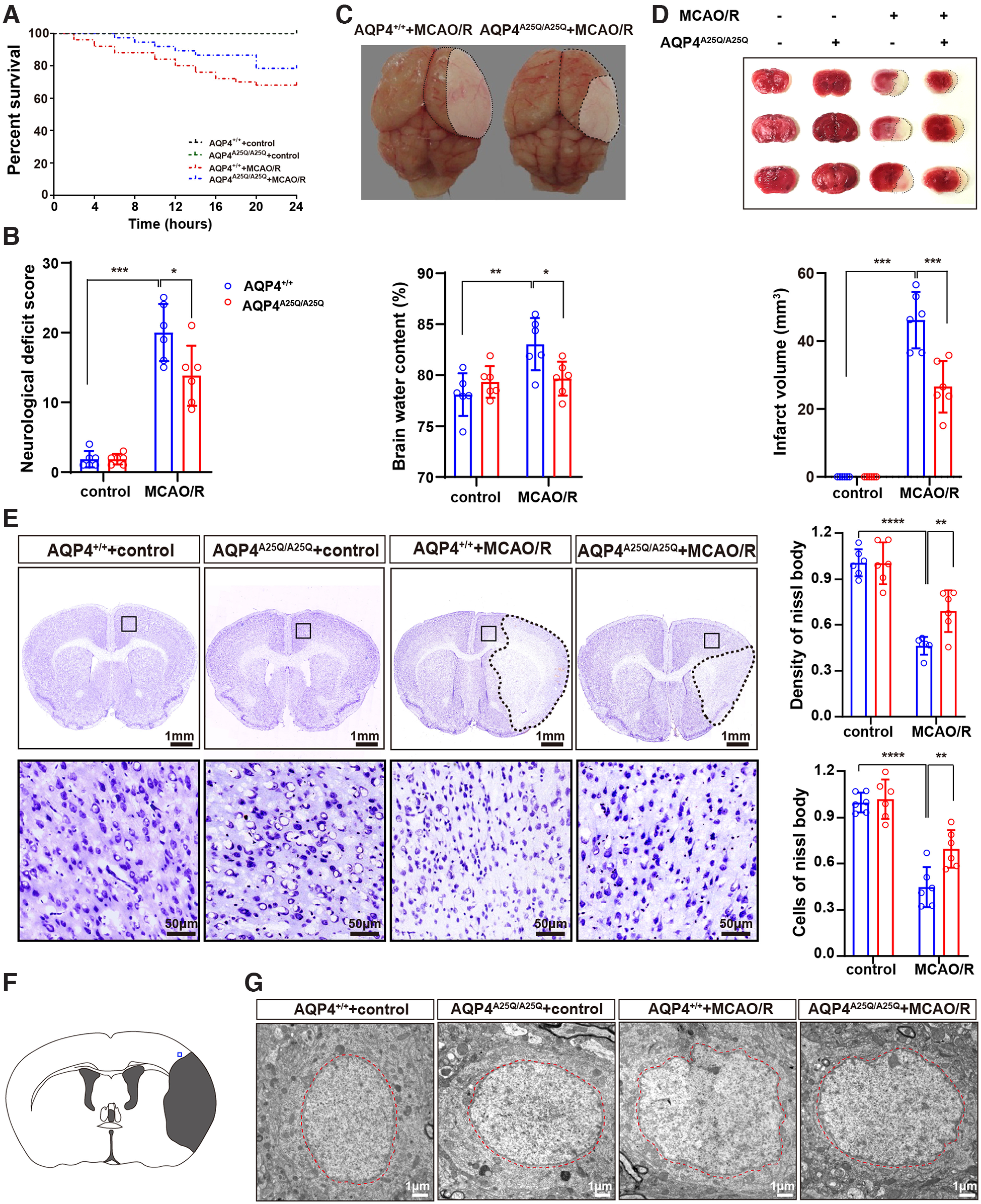
AQP4A25Q/A25Q mice exhibited improved brain edema, neurologic outcomes, and survival after MCAO/R. A, Cumulative survival in AQP4+/+ and AQP4A25Q/A25Q mice with and without MCAO/R; n = 24 mice for AQP4+/+ without MCAO/R, n = 50 mice for AQP4+/+ with MCAO/R, n = 24 mice for AQP4A25Q/A25Q without MCAO/R, and n = 37 for AQP4A25Q/A25Q mice with MCAO/R. Changes of CBF in mice with MCAO/R are provided in Extended Data Figure 6-1. B, Neurologic scores of AQP4+/+ and AQP4A25Q/A25Q mice with and without MCAO/R; n = 6 mice for each group. C, Representative photographs of brain edema in mice with MCAO/R. Quantitative analysis of brain water contents were determined in mice with and without MCAO/R; n = 6 mice for each group. D, Representative photographs of brain slices stained with TTC. Red tissue is healthy, and white tissue is infarcted. Quantitative analysis of infarct volume was performed in mice with and without MCAO/R; n = 6 mice for each group. E, Representative photographs of Nissl body staining of AQP4+/+ and AQP4A25Q/A25Q mice with and without MCAO/R. Scale bars: 1 mm, 50 μm. Quantitative analysis of the density and cells in Nissl body staining was performed; n = 6 mice for each group. F, Blue square represents the anatomic location of images in G in the ipsilateral peri-infarct regions. G, Representative electron microscope photographs of neuron nuclei in AQP4+/+ mice and AQP4A25Q/A25Q mice with and without MCAO/R. Scale bar, 1 μm. A, Log-rank (Mantel–Cox) test was used. B-D, Data are mean ± SD. *p < 0.05; **p < 0.01; ***p<0.001; ****p<0.0001; two-way ANOVA with Tukey's post hoc test.
Previous studies have demonstrated correlations between neurologic deficits and the degree of cerebral edema measured according to hemispheric enlargement after MCAO/R (Kondo et al., 1997). Figure 6C shows the typical swelling of brain tissues in the AQP4+/+ and AQP4A25Q/A25Q mice 24 h after MCAO/R. The image shows more severe tissue infarct and hemispheric enlargement in AQP4+/+ MCAO/R mice. Quantitative analysis indicated decreased brain water content in the AQP4A25Q/A25Q mice compared with that in the AQP4+/+ mice after MCAO/R (p < 0.05) (Fig. 6C). Extensive infarction was detected by TTC staining in the cerebral cortical and subcortical areas from sections of the ipsilateral hemisphere in mice subjected to MCAO/R, with red color representing healthy tissue and white representing infarcted tissue (Fig. 6D). Compared with the AQP4+/+ mice, the AQP4A25Q/A25Q mice showed a reduced infarct volume in the MCAO/R model (p < 0.01) (Fig. 6D).
Cresyl violet staining was performed to evaluate neuronal death in the MCAO/R model. We found that the neuron bodies were much larger, the color was shallower, and, specifically, the number of neurons was significantly greater in the AQP4A25Q/A25Q versus AQP4+/+ mice (p < 0.01) (Fig. 6E). Figure 6F shows the anatomic location of images in Figure 6G in the ipsilateral peri-infarct regions. Notably, cell nuclei pyknosis in the AQP4A25Q/A25Q mice was less obvious than that in the AQP4+/+ mice with MCAO/R, as detected by electron microscopy (Fig. 6G).
Similarly, the area of swollen astrocytic foot process in the AQP4A25Q/A25Q mice was also remarkably smaller (p = 0.0086, two-way ANOVA followed by Tukey's post hoc test) than that in the AQP4+/+ mice following MCAO/R (Fig. 7A). We also observed the time course to demonstrate the causative role of astrocytes and microglia play in the brain injury in MCAO/R model. The results showed an increased number of astrocytes and microglia reaching a peak by 24 and 48 h, respectively (Extended Data Fig. 7-1). Figure 7B shows the anatomic location of images in Figure 7C, D in the ipsilateral peri-infarct regions. The fluorescence intensity and area of GFAP and S100β were significantly lower in AQP4A25Q/A25Q versus AQP4+/+ mice with MCAO/R (Fig. 7C). The total number of GFAP-positive cells was also significantly lower in AQP4A25Q/A25Q (Extended Data Fig. 7-2). The results indicated less swelling and activation of astrocytes in AQP4A25Q/A25Q mice in the MCAO/R model. In addition, morphologic analysis indicated that AQP4-A25Q mutation also reduced microglial activation in MCAO/R mice (Fig. 7D), suggesting that neuroinflammation was inhibited in AQP4-A25Q mutant mice after MCAO/R.
Figure 7.
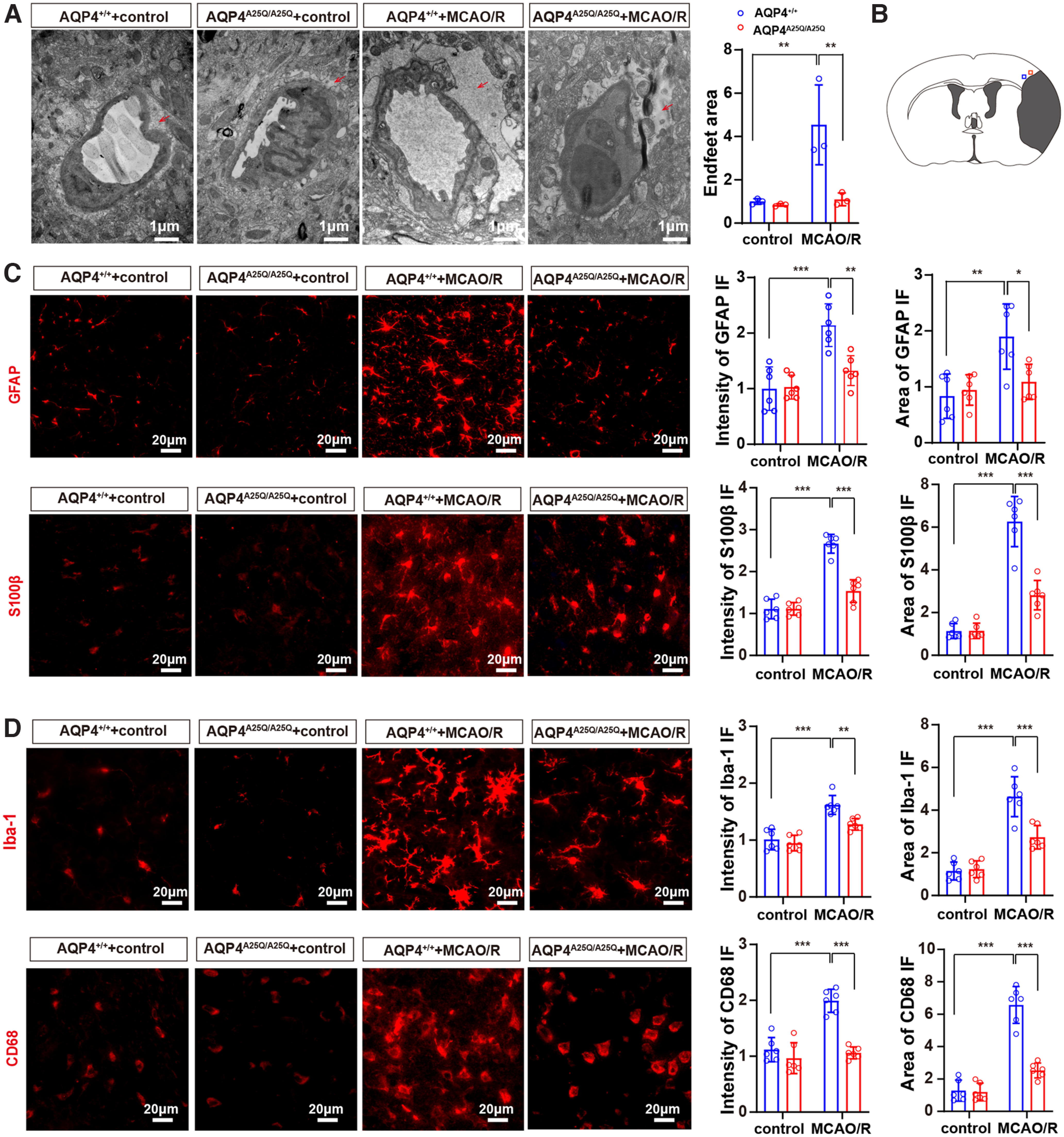
AQP4A25Q/A25Q mice exhibited decreased swelling and activation of astrocytes after MCAO/R model. A, Transmission electron micrograph showing the edematous cerebral cortex at 24 h after MCAO/R. Scale bar, 1 μm. Quantification of the pericapillary astrocyte foot process area was performed with 3 fields/mouse; n = 3 mice. B, Blue square represents the anatomic location of images in C in the ipsilateral peri-infarct regions. Red square represents the anatomic location of images in D in the ipsilateral peri-infarct regions. A time course of astrocyte and microglia playing in the brain injury in MCAO/R model is provided in Extended Data Figure 7-1. C, Representative photographs of GFAP and S100β in astrocytes of AQP4+/+ and AQP4A25Q/A25Q mice with and without MCAO/R. The quantified intensity and area of GFAP and S100β in the brain; n = 6 mice for each group. Scale bar, 20 μm. Total number of GFAP-positive cells in control and mutant mice with MCAO/R are provided in Extended Data Figure 7-2. D, Representative photographs of Iba-1 and CD68 in microglia of AQP4+/+ and AQP4A25Q/A25Q mice with and without MCAO/R. The quantified intensity and area of Iba-1 and CD68 in the brain, n = 6 mice for each group. Scale bar, 20 μm. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p<0.001; two-way ANOVA with Tukey's post hoc test. Antibodies used are provided in Extended Data Table 2-1.
These results further demonstrated that decreased AQP4 distribution in astrocytic end processes because of diminished OAP structures can reduce cerebral edema and alleviate glial activation and brain damage caused by ischemia-reperfusion injury (Fig. 8).
Figure 8.
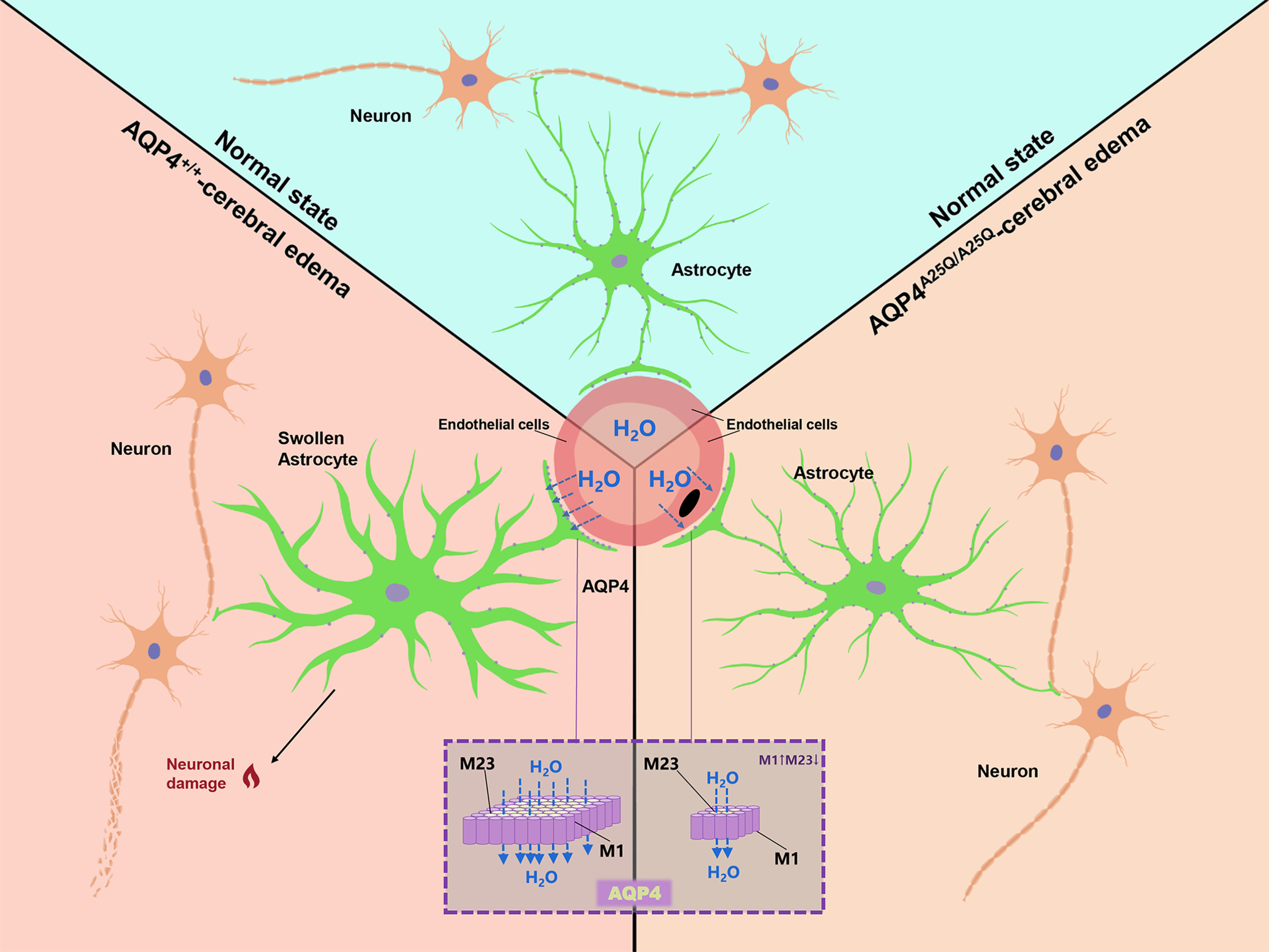
Mechanisms of AQP4-A25Q mutation-dependent neuroprotection in cerebral edema. In the normal brain, astrocytes occupy a strategic position between capillaries and neurons. Perivascular astrocytic endfeet at BBB maintain brain ion and water homeostasis. In the swollen brain, AQP4 facilitates water movement across the BBB into astrocytes, resulting in astrocyte swelling and astrocyte activation. The aquaporin 4-A25Q mutation causes OAP depolymerization and inhibits supramolecular clustering, resulting in less AQP4 anchored at glial endfeet, consequently decreasing water movement from the vasculature into astrocytes and alleviating neuronal injury.
Discussion
AQP4 is a water-selective channel strongly expressed in astrocytes and plays a crucial role in the regulation of water homeostasis in the brain (Trachtman, 1992; Kondo et al., 1997; Hossmann, 1998; Crane and Verkman, 2009; Lisjak et al., 2017). OAPs on the cell membrane are a characteristic structure of AQP4 formed mainly by M23-AQP4, but their size can be affected by the integration of M1-AQP4 because the aggregation of tetramers can be prevented by M1-AQP4 through coating of the outer surface of the OAPs (Papadopoulos and Verkman, 2013). The biological importance of AQP4 OAP formation has attracted great interest in the aquaporin field. It was postulated that OAP formation is associated with the polarized distribution of the AQP4 protein in the endfeet processes at the BBB. A recent study reported an “OAP-null” mouse model that selectively lacks the OAP-forming M23-AQP4 isoform because of targeted removal of its start codon in the AQP4 genomic sequence (de Bellis et al., 2021). The consequence of this mutation is a massive reduction in the overall membrane and intracellular expression of the AQP4 protein in the mouse brain. In this null model, M1-AQP4 tetramers cannot be used by astrocytes as an alternative to OAPs. However, the study did not address the relationship of AQP4 OAP structure with its polarized expression at astrocytic endfeet.
In the present study, we generated a transgenic knock-in mouse model by introducing an AQP4-A25Q mutation that was reported to depolymerize OAPs efficiently (Crane and Verkman, 2009). They found that hydrophobic and aromatic residues downstream of Met23 play a key role in AQP4 OAP formation. Hydrophobic interactions involving residues Ala25 and Phe26 have greatest influence on OAP formation and glutamine substitution of Ala25 (M23-A25Q) or Phe26 (M23-F26Q) reduced the OAP content by ∼80% on average, which was similar to the result obtained for the M1 isoform alone in transfected cell models. Other amino acids substitutions, such as A25L and A25S, have much less impact on the OAP formation (reduced ∼30%). Leucine substitution of Phe26 (M23-F26L) had no effect on OAP formation, and tyrosine substitution at this position (M23-F26Y) even facilitated OAP formation. Therefore, we chose the A25Q point mutation to generate the OAP-less mouse model. Analyses of brain tissue and cultured astrocytes indicated that the AQP4-A25Q mutation did not change the mRNA, total protein, and membrane protein levels of AQP4 in the brain but remarkably decreased the distribution of AQP4 protein in the endfeet processes of astrocytes, providing the first evidence that the OAP structure determines the polarized expression of AQP4 in astrocytic endfeet at the BBB.
Several experiments were performed to determine the effects of the AQP4-A25Q mutation on OAP formation both in vivo and in vitro. First, although the AQP4-A25Q mutation had no obvious effect on the overall and membrane AQP4 protein expression, the amount of AQP4 tetramers in the AQP4A25Q/A25Q mouse brain increased ∼5-fold alongside diminished higher-molecular weight bands corresponding to different sizes of OAPs compared with AQP4+/+ mice, as determined by BN–PAGE followed by immunoblotting. Second, the average size of OAPs was 3.5 times smaller in the AQP4A25Q/A25Q mice than in the AQP4+/+ mice. Third, immunofluorescence labeling using the NMO-IgG antibody (only recognizes OAPs) indicated dramatically decreased AQP4 OAP structure in AQP4A25Q/A25Q mouse brains, as indicated by a decreased overall fluorescence signal. Based on these results, we successfully established a transgenic mouse model of diminished OAP structure of AQP4 protein at astrocytic membrane without affecting the overall AQP4 mRNA and protein expression.
It has been speculated that OAPs might be involved in AQP4 polarization in astrocyte endfeet because a single link is required to tether an entire AQP4 array to the plasma membrane, whereas scattered AQP4 tetramers also require a tether (Stokum et al., 2015). We compared the distributions of AQP4 protein in AQP4A25Q/A25Q and AQP4+/+ mouse brains using confocal microscopy and IEM. The results confirmed remarkably decreased localization of AQP4 in the glial endfeet area at the BBB and increased AQP4 labeling in the non-BBB region of the AQP4A25Q/A25Q mouse brain, demonstrating diminished polarized AQP4 distribution at the endfeet surrounding microvessels at the BBB. Immunofluorescence labeling of the AQP4 protein in the BBB region using an NMO-IgG antibody was also dramatically decreased in AQP4A25Q/A25Q mice. Based on the results in Figures 2D, 3C, and 4 and the quantification of immune-gold particles at the endfeet and other parts of astrocytes plasma membrane, AQP4-A25Q mutant protein probably dispersed to other parts of astrocytes plasma membrane. This distribution change of the AQP4-A25Q mutant protein may be related to the faster diffusion of its tetrameric forms and altered AQP4-cytoskeleton interaction because of the diminished OAPs.
Previous studies have demonstrated protective effects of AQP4 KO in brain edema formation in ischemic and ischemia-reperfusion brain injuries in mice (Manley et al., 2000; Hirt et al., 2017; Clement et al., 2020). However, because no small-molecule AQP4 inhibitors are available to date, the therapeutic effect on brain edema by targeting AQP4 has not been evaluated (Verkman et al., 2017; Fang et al., 2020). Our findings in this study provide a novel therapeutic strategy involving interfering with AQP4 OAP formation and polarized expression in astrocytic endfeet at the BBB. The AQP4A25Q/A25Q mice did not show changes in overall protein expression in the brain, therefore providing a model to investigate whether AQP4 protein redistribution affects brain edema formation. We found that the AQP4-A25Q mice exhibited remarkably reduced cerebral edema, as indicated by alleviated astrocytic endfeet swelling, reduced brain water content, and inhibited neuroinflammation in response to water intoxication and MCAO/R, with improved clinically relevant indices, including survival, neurologic status, along with cerebral edema.
In conclusion, the AQP4-A25Q point mutation results in depolymerization of the AQP4 OAP structure without affecting overall AQP4 protein expression, which inhibits supramolecular clustering, decreases glial endfeet localization, and changes the water transport properties of astrocytes in the brain. The polarized expression of AQP4 in the OAP form at BBB is responsible for cellular edema formation in astrocytes during ischemic brain injury. Therefore, our study may provide a new insight in the intervention of cerebral cellular edema by reducing AQP4 OAP formation.
Extended table 1-1. Summary of PCR primers used. Summary of PCR primers used illustrate the application, target mRNA and primer sequence of primers. Download Table 1-1, docx file (20.2KB, docx) .
Extended table 2. Summary of commercial antibodies used. Summary of commercial antibodies used illustrate the application, host, dilution and source of antibodies. Download Table 2-1, docx file (21.4KB, docx) .
Total number of GFAP positive cells in control and mutant mice with MCAO/R. The total number of GFAP positive astrocytes in AQP4+/+ and AQP4A25Q/A25Q mice with or without MCAO/R. n = 6 mice for each group. The data are represented as the mean ± SD, **p < 0.01, ****p<0.0001 (two-way ANOVA with Tukey's post hoc test). Download Figure 7-2, TIF file (2.6MB, tif) .
A time course of astrocyte and microglia playing in the brain injury in MCAO/R model. A Representative immunofluorescence images of GFAP labeled astrocytes (left panel) and the relative number of GFAP positive astrocytes (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. B Representative immunofluorescence images of s100β labeled astrocytes (left panel) and the relative number of s100β positive astrocytes (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. C Representative immunofluorescence images of Iba-1 labeled microglia (left panel) and the relative number of Iba-1 positive microglia (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. D Representative immunofluorescence images of CD68 labeled microglia (left panel) and the relative number of CD68 positive microglia (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. In A, B, C and D the data are represented as the mean ± SD, *p < 0.05, **p < 0.01, ***p<0.001 versus 0 h. (one-way ANOVA with Tukey's post hoc test). Download Figure 7-1, TIF file (12.6MB, tif) .
Images monitoring the changes of CBF in mice with MCAO/R. A. Doppler flow imaging showed reduced perfusion of the ipsilateral hemisphere in the circle of Willis between sham and MCAO group. B Representative perfusion images of the AQP4+/+ and AQP4A25Q/A25Q mice acquired by laser speckle flowmetry at baseline, 2 hours after ischemia, and 24 hours after reperfusion. C Relative rCBF in AQP4+/+ and AQP4A25Q/A25 mice with MCAO/R were presented as the percentage by normalizing baseline. No difference was observed between the two groups. n = 10 mice per group. D Representative graphs displayed mean perfusion at baseline, ischemia and reperfusion. Download Figure 6-1, TIF file (38.8MB, tif) .
Expression of AQP4 in different group of mice. A. Representative bands and densitometry analysis of the AQP4 protein expression level in AQP4+/+ and AQP4A25Q/A25Q mouse brains with water intoxication (WI). n = 6 mice for each group. B. Representative bands and densitometry analysis of the AQP4 protein expression level in AQP4+/+ and AQP4A25Q/A25Q mouse brains with MCAO/R. n = 6 mice for each group. In A and B, the data are represented as the mean ± SD, ***p<0.001 (two-way ANOVA with Tukey's post hoc test). Download Figure 5-2, TIF file (10.6MB, tif) .
Total number of GFAP positive cells in control and mutant mice with water intoxication. The total number of GFAP positive astrocytes in AQP4+/+ and AQP4A25Q/A25Q mice with water intoxication (WI). n = 9 mice for each group. The data are represented as the mean ± SD, ***p<0.001 (two-way ANOVA with Tukey's post hoc test). Download Figure 5-1, TIF file (2.8MB, tif) .
Footnotes
This work was supported by Liaoning Revitalization Talents Program XLYC1902044 and XLYC1808031; Liaoning Provincial Key R&D Program 2019020048-JH2/103; National Natural Sciences Foundation of China 81571061, U1908208, and 81671111; and Natural Science Foundation of Liaoning Province 2021-MS-280.
The authors declare no competing financial interests.
References
- Alikina TY, Illarionova NB, Zelenin SM, Bondar AA (2012) Identification of new M23A mRNA of mouse aquaporin-4 expressed in brain, liver, and kidney. Biochemistry (Mosc) 77:425–434. [DOI] [PubMed] [Google Scholar]
- Clark WM, Lessov NS, Dixon MP, Eckenstein F (1997) Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurol Res 19:641–648. 10.1080/01616412.1997.11740874 [DOI] [PubMed] [Google Scholar]
- Clement T, Rodriguez-Grande B, Badaut J (2020) Aquaporins in brain edema. J Neurosci Res 98:9–18. [DOI] [PubMed] [Google Scholar]
- Crane JM, Verkman AS (2009) Determinants of aquaporin-4 assembly in orthogonal arrays revealed by live-cell single-molecule fluorescence imaging. J Cell Sci 122:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bellis M, Cibelli A, Mola MG, Pisani F, Barile B, Mastrodonato M, Banitalebi S, Amiry-Moghaddam M, Abbrescia P, Frigeri A, Svelto M, Nicchia GP (2021) Orthogonal arrays of particle assembly are essential for normal aquaporin-4 expression level in the brain. Glia 69:473–488. 10.1002/glia.23909 [DOI] [PubMed] [Google Scholar]
- Dong X, Gao J, Zhang CY, Hayworth C, Frank M, Wang Z (2019) Neutrophil membrane-derived nanovesicles alleviate inflammation to protect mouse brain injury from ischemic stroke. ACS Nano 13:1272–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Shi H, Ren R, Huang L, Okada T, Lenahan C, Gamdzyk M, Travis ZD, Lu Q, Tang L, Huang Y, Zhou K, Tang J, Zhang J, Zhang JH (2020) Pituitary adenylate cyclase-activating polypeptide attenuates brain edema by protecting blood–brain barrier and glymphatic system after subarachnoid hemorrhage in rats. Neurotherapeutics 17:1954–1972. 10.1007/s13311-020-00925-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshi N, Morgan RK, Lein PJ, Seker E (2020) A primary neural cell culture model to study neuron, astrocyte, and microglia interactions in neuroinflammation. J Neuroinflammation 17:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Weigand SD, Popescu BF, Lennon VA, Parisi JE, Pittock SJ, Parks NE, Clardy SL, Howe CL, Lucchinetti CF (2017) Pathogenic implications of cerebrospinal fluid barrier pathology in neuromyelitis optica. Acta Neuropathol 133:597–612. 10.1007/s00401-017-1682-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan-Olive MM, Enger R, Hansson HA, Nagelhus EA, Eide PK (2019) Loss of perivascular aquaporin-4 in idiopathic normal pressure hydrocephalus. Glia 67:91–100. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, Fallier-Becker P, Noell S, Lennon VA (2012) Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci USA 109:1245–1250. 10.1073/pnas.1109980108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, Mizoguchi A, Fujiyoshi Y (2006) Implications of the aquaporin-4 structure on array formation and cell adhesion. J Mol Biol 355:628–639. 10.1016/j.jmb.2005.10.081 [DOI] [PubMed] [Google Scholar]
- Hirt L, Fukuda AM, Ambadipudi K, Rashid F, Binder D, Verkman A, Ashwal S, Obenaus A, Badaut J (2017) Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J Cereb Blood Flow Metab 37:277–290. 10.1177/0271678X15623290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann KA (1998) Experimental models for the investigation of brain ischemia. Cardiovasc Res 39:106–120. 10.1016/S0008-6363(98)00075-3 [DOI] [PubMed] [Google Scholar]
- Jiang R, Wu XF, Wang B, Guan RX, Lv LM, Li AP, Lei L, Ma Y, Li N, Li QF, Ma QH, Zhao J, Li S (2020) Reduction of NgR in perforant path decreases amyloid-beta peptide production and ameliorates synaptic and cognitive deficits in APP/PS1 mice. Alzheimers Res Ther 12:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Park J, Kwon BS, Park JW, Lee HJ, Choi JH, Nam K (2017) Radiculopathy in neuromyelitis optica: How does anti-AQP4 Ab involve PNS? Mult Scler Relat Disord 18:77–81. [DOI] [PubMed] [Google Scholar]
- Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH (1997) Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci 17:4180–4189. 10.1523/JNEUROSCI.17-11-04180.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, Liang W, Thomson AW, Chen J, Hu X (2013) Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol 74:458–471. 10.1002/ana.23815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Cao Y, Dang C, Han B, Han R, Ma H, Hao J, Wang L (2020) Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol Med 12:e11002. 10.15252/emmm.201911002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisjak M, Potokar M, Rituper B, Jorgačevski J, Zorec R (2017) AQP4e-based orthogonal arrays regulate rapid cell volume changes in astrocytes. J Neurosci 37:10748–10756. 10.1523/JNEUROSCI.0776-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6:159–163. 10.1038/72256 [DOI] [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS (1997) Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest 100:957–962. 10.1172/JCI231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Yang B, Verkman AS (1996) Gene structure, cDNA cloning, and expression of a mouse mercurial-insensitive water channel. Genomics 33:382–388. [DOI] [PubMed] [Google Scholar]
- Karmacharya MB, Kim KH, Kim SY, Chung J, Min BH, Park SR, Choi BH (2015) Low intensity ultrasound inhibits brain oedema formation in rats: potential action on AQP4 membrane localization. Neuropathol Appl Neurobiol 41:e80–e94. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Ottersen OP (2013) Physiological roles of aquaporin-4 in brain. Physiol Rev 93:1543–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP (1997) Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci 17:171–180. 10.1523/JNEUROSCI.17-01-00171.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntim M, Li QF, Zhang Y, Liu XD, Li N, Sun HL, Zhang X, Khan B, Wang B, Wu Q, Wu XF, Walana W, Khan K, Ma QH, Zhao J, Li S (2020) TRIM32 deficiency impairs synaptic plasticity by excitatory-inhibitory imbalance via notch pathway. Cereb Cortex 30:4617–4632. 10.1093/cercor/bhaa064 [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nat Rev Neurosci 14:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Bennett JL, Verkman AS (2014) Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol 10:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosito S, Nicchia GP, Palazzo C, Lia A, Buccoliero C, Pisani F, Svelto M, Trojano M, Frigeri A (2018) Supramolecular aggregation of aquaporin-4 is different in muscle and brain: correlation with tissue susceptibility in neuromyelitis optica. J Cell Mol Med 22:1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman A (2012) Neuromyelitis optica IgG does not alter aquaporin-4 water permeability, plasma membrane M1/M23 isoform content, or supramolecular assembly. Glia 60:2027–2039. 10.1002/glia.22417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Verkman AS (2015) Superresolution imaging of aquaporin-4 cluster size in antibody-stained paraffin brain sections. Biophys J 109:2511–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Jin BJ, Ratelade J, Verkman AS (2014) Aggregation state determines the localization and function of M1–and M23–aquaporin-4 in astrocytes. J Cell Biol 204:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner E, Enzmann GU, Lin S, Ghavampour S, Hannocks MJ, Zuber B, Rüegg MA, Sorokin L, Engelhardt B (2012) Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia 60:1646–1659. 10.1002/glia.22383 [DOI] [PubMed] [Google Scholar]
- Stokum JA, Mehta RI, Ivanova S, Yu E, Gerzanich V, Simard JM (2015) Heterogeneity of aquaporin-4 localization and expression after focal cerebral ischemia underlies differences in white versus grey matter swelling. Acta Neuropathol Commun 3:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Wang J, Zhou Y, Wang Z, Jiang Y, Li M (2016) Isotetrandrine reduces astrocyte cytotoxicity in neuromyelitis optica by blocking the binding of NMO-IgG to aquaporin 4. Neuroimmunomodulation 23:98–108. [DOI] [PubMed] [Google Scholar]
- Trachtman H (1992) Cell volume regulation: a review of cerebral adaptive mechanisms and implications for clinical treatment of osmolal disturbances: II. Pediatr Nephrol 6:104–112. 10.1007/BF00856852 [DOI] [PubMed] [Google Scholar]
- Verkman AS, Smith AJ, Phuan PW, Tradtrantip L, Anderson MO (2017) The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin Ther Targets 21:1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XG, Zhu DD, Li N, Huang YL, Wang YZ, Zhang T, Wang CM, Wang B, Peng Y, Ge BY, Li S, Zhao J (2020a) Scorpion venom heat-resistant peptide is neuroprotective against cerebral ischemia-reperfusion injury in association with the NMDA-MAPK pathway. Neurosci Bull 36:243–253. 10.1007/s12264-019-00425-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zheng X, Jin J, Zheng L, Guan T, Huo Y, Xie S, Wu Y, Chen W (2020b) LncRNA MALAT1 silencing protects against cerebral ischemia-reperfusion injury through miR-145 to regulate AQP4. J Biomed Sci 27:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZT, Lu MH, Zhang Y, Ji WL, Lei L, Wang W, Fang LP, Wang LW, Yu F, Wang J, Li ZY, Wang JR, Wang TH, Dou F, Wang QW, Wang XL, Li S, Ma QH, Xu RX (2019) Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer's disease as a mitophagy receptor. Aging Cell 18:e12860. 10.1111/acel.12860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon V, Pittock S, Lucchinetti C, Weinshenker B (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66:1485–1489. [DOI] [PubMed] [Google Scholar]
- Yang Y, Liu H, Zhang H, Ye Q, Wang J, Yang B, Mao L, Zhu W, Leak RK, Xiao B, Lu B, Chen J, Hu X (2017) ST2/IL-33-dependent microglial response limits acute ischemic brain injury. J Neurosci 37:4692–4704. 10.1523/JNEUROSCI.3233-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended table 1-1. Summary of PCR primers used. Summary of PCR primers used illustrate the application, target mRNA and primer sequence of primers. Download Table 1-1, docx file (20.2KB, docx) .
Extended table 2. Summary of commercial antibodies used. Summary of commercial antibodies used illustrate the application, host, dilution and source of antibodies. Download Table 2-1, docx file (21.4KB, docx) .
Total number of GFAP positive cells in control and mutant mice with MCAO/R. The total number of GFAP positive astrocytes in AQP4+/+ and AQP4A25Q/A25Q mice with or without MCAO/R. n = 6 mice for each group. The data are represented as the mean ± SD, **p < 0.01, ****p<0.0001 (two-way ANOVA with Tukey's post hoc test). Download Figure 7-2, TIF file (2.6MB, tif) .
A time course of astrocyte and microglia playing in the brain injury in MCAO/R model. A Representative immunofluorescence images of GFAP labeled astrocytes (left panel) and the relative number of GFAP positive astrocytes (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. B Representative immunofluorescence images of s100β labeled astrocytes (left panel) and the relative number of s100β positive astrocytes (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. C Representative immunofluorescence images of Iba-1 labeled microglia (left panel) and the relative number of Iba-1 positive microglia (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. D Representative immunofluorescence images of CD68 labeled microglia (left panel) and the relative number of CD68 positive microglia (right panel) in AQP4+/+ mice ischemic penumbra with MCAO/R at 0, 6, 12, 24, 48 and 72 h after reperfusion of AQP4+/+ mice. Scale bar = 50 μm. n = 6 mice for each group. In A, B, C and D the data are represented as the mean ± SD, *p < 0.05, **p < 0.01, ***p<0.001 versus 0 h. (one-way ANOVA with Tukey's post hoc test). Download Figure 7-1, TIF file (12.6MB, tif) .
Images monitoring the changes of CBF in mice with MCAO/R. A. Doppler flow imaging showed reduced perfusion of the ipsilateral hemisphere in the circle of Willis between sham and MCAO group. B Representative perfusion images of the AQP4+/+ and AQP4A25Q/A25Q mice acquired by laser speckle flowmetry at baseline, 2 hours after ischemia, and 24 hours after reperfusion. C Relative rCBF in AQP4+/+ and AQP4A25Q/A25 mice with MCAO/R were presented as the percentage by normalizing baseline. No difference was observed between the two groups. n = 10 mice per group. D Representative graphs displayed mean perfusion at baseline, ischemia and reperfusion. Download Figure 6-1, TIF file (38.8MB, tif) .
Expression of AQP4 in different group of mice. A. Representative bands and densitometry analysis of the AQP4 protein expression level in AQP4+/+ and AQP4A25Q/A25Q mouse brains with water intoxication (WI). n = 6 mice for each group. B. Representative bands and densitometry analysis of the AQP4 protein expression level in AQP4+/+ and AQP4A25Q/A25Q mouse brains with MCAO/R. n = 6 mice for each group. In A and B, the data are represented as the mean ± SD, ***p<0.001 (two-way ANOVA with Tukey's post hoc test). Download Figure 5-2, TIF file (10.6MB, tif) .
Total number of GFAP positive cells in control and mutant mice with water intoxication. The total number of GFAP positive astrocytes in AQP4+/+ and AQP4A25Q/A25Q mice with water intoxication (WI). n = 9 mice for each group. The data are represented as the mean ± SD, ***p<0.001 (two-way ANOVA with Tukey's post hoc test). Download Figure 5-1, TIF file (2.8MB, tif) .
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.