

Abstract
目的
探讨利用导管损伤制备钙化性主动脉瓣疾病小鼠模型的最优时间。
方法
通过向8周龄小鼠右颈总动脉插入导管, 在超声引导下将导管送至主动脉瓣口处损伤主动脉瓣膜, 对导管损伤主动脉瓣膜后不同时间段的C57BL6小鼠主动脉瓣膜进行比较, 将小鼠分为对照组、造模后4周、造模后8周与造模后16周组, 从适应性喂养至造模结束取材均给予普通饲料饮食。使用超声检测小鼠心脏短轴缩短率、主动脉瓣膜峰值流速及主动脉瓣口面积, 使用苏木素-伊红染色观察小鼠主动脉瓣膜厚度变化及瓣膜组织结构变化, 利用茜素红染色检测小鼠主动脉瓣膜钙盐沉积情况, 使用免疫荧光方法检测瓣膜成骨相关蛋白碱性磷酸酶表达。
结果
小鼠从造模8周后开始出现心脏短轴缩短率明显下降。导管损伤4周、8周、16周后小鼠主动脉瓣膜明显增厚(P=0.002), 4周、8周和16周比较无统计学差异; 导管损伤4周、8周、16周后钙盐沉积明显增多(P < 0.0001), 4周、8周和16周比较无统计学差异; 4周、8周、16周后成骨相关蛋白ALP指标表达量明显升高(P=0.0016)。导管损伤4周、8周和16周ALP表达量相比无统计学差异。
结论
在保证小鼠生存率的情况下, 导管损伤主动脉瓣膜4周后的小鼠模型即可模拟钙化性主动脉瓣膜疾病病理生理改变。
Keywords: 钙化性主动脉瓣膜疾病, 动物模型, 导管损伤
Abstract
Objective
To investigate the optimal time window for observation of catheter-induced valve injury that mimics calcified aortic valve disease in mice.
Methods
A catheter was inserted into the right common carotid artery of 8-week-old C57BL6 mice under ultrasound guidance, and aortic valve injury was induced using the guide wire.At 4, 8 and 16 weeks after modeling, the mice were subjected to ultrasound measurement of the heart short axial shortening rate, aortic valve peak velocity and aortic valve orifice area.Grain-Eosin staining was used to observe the changes in the thickness of the aortic valve, and calcium deposition in the aortic valve was assessed using Alizarin red staining.Immunofluorescence assay was performed to detect the expression of alkaline phosphatase (ALP) in the aortic valve.
Results
At 4, 8 and 16 weeks after modeling, valve thickness (P=0.002), calcium deposition (P < 0.0001) and the expression of osteogenic protein ALP (P=0.0016) were significantly increased, but their increments were comparable at the 3 time points of observation.
Conclusion
In mouse models of calcific aortic valve disease induced by catheter valve injury, 4 weeks after the injury appears to be the optimal time window for observation of pathophysiological changes in the aortic valves to avoid further increase of the death rate of the mice over time.
Keywords: calcific aortic valve disease, mouse model, catheter injury
钙化性主动脉瓣疾病(CAVD)是如今发病率最高的心脏瓣膜疾病,并随人口老龄化进展持续升高[1, 2]。钙化性主动脉瓣膜病是一种与年龄有关的非感染性退行性病变,病程初期主要表现为主动脉瓣硬化,即主动脉瓣膜的纤维化增厚伴轻度钙化,无明显的血流动力学改变及临床表现。病程后期瓣膜钙化加重,瓣口面积减小,导致明显的主动脉狭窄,主动脉与左心室之间收缩期的压力阶差明显,主动脉峰值流速升高,且大多数退行性病变造成主动脉瓣狭窄的患者同时具有主动脉关闭不全的临床表现[3],主要可能因为瓣膜钙化造成瓣膜功能不全,无法正常关闭,表现为主动脉瓣处出现返流,左室射血时间延长、左室舒张末压进行性升高,临床症状表现为劳力性呼吸困难、胸痛甚至心力衰竭[4]。由于钙化性主动脉瓣疾病目前尚无有效药物治疗,出现症状后主要靠手术或介入治疗更换主动脉瓣膜作为治疗手段[5],因此寻找可以模拟钙化性主动脉瓣疾病发病过程的动物模型并从中探讨其发病机制、寻找可能具有缓解甚至治疗的手段是研究这一疾病的重要手段[6]。
目前模拟钙化性主动脉瓣模病发病机制的小鼠模型主要包括高脂喂养ApoE-/-小鼠、高脂喂养LDLR-/-小鼠、老年鼠、基因编辑鼠、机械损伤模型等[7-9]。但高脂喂养ApoE-/-小鼠、高脂喂养LDLR-/-小鼠存在血脂谱与钙化性主动脉瓣膜病患者差别大、价格昂贵等问题[10-12],且临床试验表明他汀类药物虽然可以改善动脉粥样硬化预后,但不能逆转或治疗钙化性主动脉瓣膜病;老年鼠存在喂养需1~1.5年,购买费用昂贵等问题[13];基因编辑鼠存在价格昂贵、造模时间长、单独的基因编辑并不能完全代表钙化性主动脉瓣膜疾病病理生理过程等问题[14, 15]。因此,寻找一种相对短时间内造成主动脉瓣钙化的动物模型尤为重要。
研究证明,在主动脉瓣不断开合的过程中,血流动力学应力引起的机械损伤是主动脉瓣钙化的重要危险因素之一[16, 17]。Honda等[18]构建了一种通过向小鼠右颈总动脉插入导管,在超声引导下用其中的导丝损伤主动脉瓣膜造成小鼠主动脉瓣增厚与钙化,用于模拟钙化性主动脉瓣膜病患者疾病发生发展的动物模型。他们发现小鼠在造模16后出现主动脉峰值流速明显升高,瓣膜明显增厚,相关炎症因子、成骨因子、细胞衰老蛋白表达明显升高,钙盐沉积及钙结节明显增多的现象,与钙化性主动脉瓣膜病患者的心功能改变以及主动脉瓣膜钙化过程较为重合。但该模型同样存在造模时间长的问题(16周),且Honda等的结果显示,小鼠主动脉瓣膜在损伤1周后即出现炎症因子表达量升高,4周后即出现主动脉瓣口狭窄等变化。因此我们推测小鼠在损伤早期即出现主动脉瓣处血流动力学改变、主动脉瓣膜增厚等病理生理改变。
与Honda等[18]的结论不同,我们发现在导管损伤小鼠瓣膜后,主动脉处立即出现血流动力学改变,且钙化性主动脉瓣膜病初期出现的瓣膜增厚、代表骨形成的钙盐沉积、成骨蛋白ALP表达升高均在4周出现并与对照组有统计学差异,并维持至16周。另外,我们发现造模成功小鼠在造模后8周~16周时容易出现心力衰竭而死亡。鉴于我们发现导管损伤模型小鼠心功能在短轴缩短率逐渐下降的情况下不伴有心悸肥厚,因此该模型可以进一步用于模拟主动脉瓣狭窄伴关闭不全造成的心力衰竭。
1. 材料和方法
1.1. 动物模型的制备
1.1.1. 设计
随机对照动物实验。
1.1.2. 时间及地点
实验于2021年8月~2022年3月在南方医科大学南方医院器官衰竭防治重点实验室完成。
1.1.3. 材料
1.1.3.1. 实验动物
SPF级雄性C57/BL6小鼠24只,体质量20 g左右,8周龄[广东志远生物医药科技有限公司,SYXK(粤)2021-0057]。本研究动物实验在南方医科大学南方医院动物实验中心进行,经南方医科大学实验动物伦理委员会批准(批准号NFYY-2020-06045)。室内保持良好通风,室内温度为25 ± 2 ℃,湿度(50 ± 10)%,12 h循环光照下普通饲料分笼(4笼)饲养,48 h/ 次更换垫料和饲料,自由饮水。
1.1.3.2. 实验用主要试剂、材料
FST血管穿刺引导系统;苏木素伊红(HE)染色试剂盒(Solarbio);茜素红S染色液(0.2%,pH4.2,北京雷根DS0058);兔抗人ALP抗体(Abclonal);Cy3标记的羊抗兔IgG H & L(Bioss)。
1.1.4. 实验方法
1.1.4.1. 模型构建及分组
将24只C57/BL6小鼠适应性饲养1周后随机分为4组,6只/组:假手术组、造模4周组、造模8周组、造模16周组。小鼠造模前禁食禁水8 h,以0.1 mL/g的浓度向小鼠腹腔注射0.2%戊巴比妥钠,脱掉颈部、胸前区毛发,并用碘伏擦拭常规消毒,于颈正中稍偏右纵向剪约1 cm切口,用镊子钝性分离颈前区筋膜并分离出右侧颈总动脉,远心端结扎,近心端打活结,在中间剪口并插入导管,超声引导导丝损伤主动脉瓣,在超声下确认导丝通过每只鼠的瓣膜并来回损伤100次,超声多普勒血流显示返流达到1000 mm/s左右,如未达到1000 mm/s,则继续损伤20次,查看超声多普勒血流直至返流达到1000 mm/s左右。假手术组仅插入导管,不进行瓣膜损伤(图 1)。
图 1.

超声引导下将导管送至主动脉瓣膜处损伤瓣膜
Ultrasound-guided catheter injury of aortic valve in mice. A: The catheter was inserted in the ascending aorta via the common carotid artery. B: The guide wire was pulled out of the catheter repeatedly to induced aortic valve injury.
1.1.4.2. 主动脉瓣膜组织的制备
造模达相应时间后麻醉下颈椎脱臼法处死各小鼠,注射器吸取10 mL生理盐水缓慢冲洗左心室及右心室,将心脏同升主动脉一起取下,固定于4%多聚甲醛,经脱水、常规石蜡包埋,进行5 μm连续切片,65 ℃烤箱烘烤2 h后存放。
1.2. 主要观察指标
1.2.1. 超声心动图
小鼠在造模4周、8周、16周时进行超声心动图检测。以1%~2%浓度异氟烷进行气体麻醉,分别在左室长轴、短轴、流出道平面检测小鼠各项心脏指标。
1.2.2. 苏木精-伊红染色
取小鼠瓣膜组织切片进行脱蜡、水化,按照苏木精-伊红试剂盒进行染色,透明后干燥,中性树胶封片后在倒置荧光显微镜下观察组织形态并拍摄。
1.2.3. 茜素红染色
取小鼠瓣膜组织切片进行脱蜡、水化,完全干燥后用茜素红染液(0.2%,pH 4.2)染色两分钟,透明后干燥,中性树胶封片后在倒置荧光显微镜下观察组织形态并拍摄。
1.2.4. 免疫荧光
取小鼠瓣膜组织切片进行脱蜡、水化,柠檬酸钠抗原修复后,4% 多聚甲醛固定后0.4% TritonX破膜,5% BSA封闭后一抗低温敷育过夜,PBS清洗后敷育荧光二抗、DAPI并用抗荧光淬灭剂封片,避光低温保存。共聚焦显微镜拍照。
1.3. 统计学处理
使用GraphPad Prism 8.0软件对所有数据进行统计分析。数据表达为均数±标准差,使用Shapiro-Wilk法检验数据正态性,多组数据符合正态分布且方差齐,使用单因素方差分析(one way ANOVA),并选择Tukey HSD法进行两两比较;多组数据符合正态分布但方差不齐,使用Brown-Forsythe检验,并选择Games-Howell法进行两两比较。当P < 0.05时认为差异有统计学意义。
2. 结果
2.1. 心脏功能变化
超声结果显示,小鼠在造模时即出现主动脉峰值流速升高以及瓣口返流(图 2、3),1周后出现主动脉瓣峰值流速以及主动脉瓣峰值压力升高,直至2周上升至最高流速与最高压力,与对照组比较差异有显著性差异,之后主动脉瓣峰值流速略微降低但差异无统计学意义;左心室射血分数缓慢降低,直至造模12周出现心力衰竭;主动脉瓣口面积在造模1周后减小,之后主动脉瓣口面积略微上升但无统计学差异(表 1)。小鼠生存率造模后不同时间点小鼠生存曲线显示不同组之间生存率无明显区别(图 4)。
图 2.
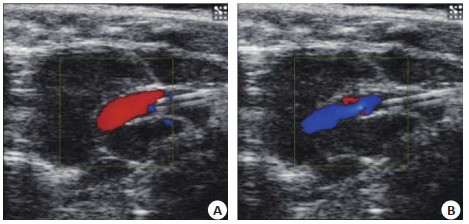
主动脉瓣膜损伤前后多普勒超声显示主动脉瓣口处血流
Blood flow at the aortic valve before (A) and after (B) valve injury.
图 3.

主动脉瓣膜损伤前后多普勒超声显示主动脉瓣峰值流速
AV peak velocity before (A) and after (B) valve injury.
表 1.
小鼠不同周数各种指标数值
Heart rate, body weight, FS, AV peak velocity, aortic valve orifice area (AVA) and aortic valve thickness in C57BL6 mice at different weeks
| Parameter | Sham | 4 week | 8 week | 16 week |
| *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs Sham group. | ||||
| Heart rate | 413±40 | 425±32 | 417±25 | 423±9 |
| Weight | 20.55±0.5 | 22.11±0.8**** | 22.5±0.87**** | 24.82±0.5**** |
| FS | 51.12±5.4 | 47.32±11.1 | 40.99±7.8*** | 27.95±4.8**** |
| AV peak velocity | 1112±115 | 1963±446**** | 1719±553** | 1849±274* |
| AVA | 1.28±0.2 | 0.87±0.2**** | 0.9±0.3**** | 0.82±0.03**** |
| Valve thickness | 37.13±2.24 | 101.12±31.31* | 103.33±35.48* | 122.69±51.19*** |
图 4.

不同组小鼠生存率曲线
Survival curves of the mice in different groups. A: Comparison of survival rates among Sham, 4-week, 8-week, and 16-week groups. B: Comparison of survival rates between Sham group and the injured group.
2.2. 主动脉瓣膜组织形态学变化
小鼠在造模4周、8周、16周后都出现瓣膜增厚(图 5,表 1)。造模4周后瓣膜增厚,与对照组相比有统计学差异且维持至16周,其中4周与8周(P>0.9)、8周与16周的瓣膜厚度相比无统计学差异(P=0.86)。损伤后4周的瓣膜中出现粉色的胶原沉积,并在之后的周数中增多,与对照组相比有统计学差异。茜素红染色示在造模4、8、16周后出现瓣膜钙盐沉积明显增多(图 6),与对照组比较差异有统计学差异(P < 0.0001)。
图 5.

造模后不同周数小鼠瓣膜厚度
Aortic valve thickness in C57BL6 mice at different weeks after injury (Original magnification: ×100, Grain-Eosin staining).
图 6.

造模后不同周数小鼠瓣膜钙盐沉积
Aortic valve calcium deposition at different weeks after injury (×400, Alizarin red staining).
2.3. 主动脉瓣膜成骨指标变化
免疫荧光显示,对照组成骨蛋白ALP表达量低,造模4周、8周、16周后ALP表达量升高(图 7),与对照组相比有统计学差异(P=0.0016)。4周、8周、16周比较无统计学差异(P=0.3138)。
图 7.

造模后不同周数小鼠瓣膜ALP表达量
Expression of ALP in the aortic valves at different weeks after injury (×200, Immunofluorescence).
3. 讨论
研究表明,由于主动脉瓣膜的结构与位置的特殊性,其机械损伤是钙化性主动脉瓣疾病的主要原因之一[16, 19]。而局部血流动力学改变是造成主动脉瓣膜机械损伤的重要因素[20]:局部湍流造成瓣膜内皮细胞损伤,出现内皮-间质转化,瓣膜成纤维细胞开始增生,并转化为肌成纤维细胞[20-22]。Panizzi等[23]报道了一种小鼠血浆凝固酶阳性的金黄色葡萄球菌心内膜炎模型,该模型不依赖超声引导将特殊材料插入心脏引起心内膜炎。虽然他们的方法与Honda等[18]的导管损伤模型相似,但他们的模型不会发展成AVS。Honda等[18]构建的种通过直接向野生型C57/BL6小鼠右颈总动脉插入导管至升主动脉损伤主动脉瓣,喂养小鼠普通饮食,在16周内即可造成主动脉瓣膜增厚、钙化的小鼠主动脉瓣膜损伤模型。但我们发现,造模成功小鼠在造模后8周~ 16周时容易出现心力衰竭而死亡,造成实验过程中动物脱失,需要持续补加模型数量,而且在实验过程中施加刺激如病毒、药物等干预的持续时间较短,因此在保证动物存活率的基础上探讨主动脉瓣膜损伤模型成功率的时间尤为重要。
钙化性主动脉疾病患者往往因为气短、心悸等原因就诊,行心脏彩超检查发现主动脉瓣处存在返流与流速增快而确诊[24]。我们发现在导管损伤小鼠瓣膜后,主动脉处立即出现血流动力学改变,包括主动脉瓣处返流以及主动脉峰值流速与峰值压力升高,小鼠在导管损伤1周后即出现主动脉峰值流速与峰值压力升高等血流动力学升高改变,至2周达到最高,并维持至16周。主动脉瓣口面积在损伤2周后缩小,4周即开始出现主动脉瓣膜增厚及钙盐沉积。高脂饮食喂养的小鼠及其他模型在以往的研究中被用于模拟主动脉瓣膜钙化性疾病,但这些小鼠往往不出现明显的血流动力学改变,瓣膜增厚的同时伴随心室肥大、冠脉粥样硬化等改变[25, 26]。与其他模型相比,代表骨形成的钙盐沉积也在4周即出现[27, 28]。钙化性主动脉瓣膜疾病中,主动脉瓣膜形态改变主要表现为瓣膜的增厚与钙结节形成[29]。我们发现造模4周后瓣膜增厚,与对照组相比有统计学差异且维持至16周,其中4周以后的周数中,瓣膜厚度无统计学差异。需要特别注意的是,我们在造模后4周小鼠的瓣膜HE染色切片中发现粉红色的胶原沉积,推测可能是损伤后坏死并由瘢痕组织修复的产物,在之后的周数中胶原沉积不断增多,这与钙化性主动脉瓣膜病发病过程中的营养不良性钙化有共同之处:主动脉瓣钙化时瓣膜发生内皮损伤、基底膜破坏、瓣膜内异常胶原沉积等多种改变,细胞发生凋亡,瓣膜间质细胞被降解,作为钙和磷酸盐沉积的底物聚集成为钙结节[28, 30-32]。茜素红染色示在造模4、8、16周后出现瓣膜钙盐沉积明显增多,与对照组相比有统计学差异。在人钙化的主动脉瓣膜中能观察到表达升高的成骨因子,与瓣膜软骨细胞增多、骨形成及钙盐沉积有关[32]。我们的结果表明,对照组成骨蛋白ALP表达量低,造模4周、8周、16周后ALP表达量升高(图 6),与对照组相比有统计学差异。4周、8周、16周比较无统计学差异。造模过程中我们观察到小鼠体质量上升有统计学差异,但通过与高脂饮食诱导的APOE-/-小鼠对比[33],导管损伤的C57BL6小鼠体质量只轻微增长,原因可能是小鼠的正常生长造成的体质量增加,因此我们认为该模型对小鼠体质量无影响。以上结果均显示,小鼠瓣膜在导管损伤4周后即出现增厚、钙盐沉积与成骨蛋白表达升高,并维持至16周。
与Honda等[18]的研究结论不同的是,我们发现在导管损伤小鼠瓣膜后,主动脉处立即出现血流动力学改变,且钙化性主动脉瓣膜病初期出现的瓣膜增厚、代表骨形成的钙盐沉积、成骨蛋白ALP表达升高均在4周出现并与对照组有统计学差异,并维持至16周。另外,我们发现造模成功小鼠在造模后8周~16周时容易出现心力衰竭而死亡,可以为后续因主动脉瓣返流出现心力衰竭的动物模型提供选择。
综上所述,我们认为在保证小鼠生存率的情况下,导管损伤主动脉瓣膜4周后的小鼠模型即可模拟钙化性主动脉瓣膜疾病病理生理改变,该动物模型对分析钙化性主动脉瓣膜疾病发病机制和制定治疗策略有一定参考价值。另外,鉴于我们发现在导管损伤过程中小鼠的短轴缩短率是逐渐下降的趋势,且过程中左心室无明显肥厚,因此该模型可以进一步用于模拟主动脉瓣狭窄伴关闭不全造成的心力衰竭。
Biography
曾婧欣,在读硕士研究生,E-mail: 136972065@qq.com
Funding Statement
国家自然科学基金(82070403)
Supported by National Natural Science Foundation of China(82070403)
Contributor Information
曾 婧欣 (Jingxin ZENG), Email: 136972065@qq.com.
曾 庆春 (Qingchun ZENG), Email: qingchunzeng@smu.edu.cn.
References
- 1.Pawade TA, Newby DE, Dweck MR. Calcification in aortic Stenosis: the skeleton key. J Am Coll Cardiol. 2015;66(5):561–77. doi: 10.1016/j.jacc.2015.05.066. [DOI] [PubMed] [Google Scholar]
- 2.Yutzey KE, Demer LL, Body SC, et al. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol. 2014;34(11):2387–93. doi: 10.1161/ATVBAHA.114.302523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung JJ, Jadbabaie F, Sadeghi MM. Molecular imaging of calcific aortic valve disease. J Nucl Cardiol. 2018;25(4):1148–55. doi: 10.1007/s12350-017-1158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostyunin AE, Yuzhalin AE, Ovcharenko EA, et al. Development of calcific aortic valve disease: do we know enough for new clinical trials? J Mol Cell Cardiol. 2019;132:189–209. doi: 10.1016/j.yjmcc.2019.05.016. [DOI] [PubMed] [Google Scholar]
- 5.Mathieu P, Boulanger MC. Basic mechanisms of calcific aortic valve disease. Can J Cardiol. 2014;30(9):982–93. doi: 10.1016/j.cjca.2014.03.029. [DOI] [PubMed] [Google Scholar]
- 6.Zeng QC, Song R, Fullerton DA, et al. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc Natl Acad Sci USA. 2017;114(7):1631–6. doi: 10.1073/pnas.1619667114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plump AS, Smith JD, Hayek T, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71(2):343–53. doi: 10.1016/0092-8674(92)90362-G. [DOI] [PubMed] [Google Scholar]
- 8.宗 永辉, 刘 新灿. 钙化性主动脉瓣膜病小鼠模型比较及应用研究进展. 中国比较医学杂志. 2020;30(11):133-9, 145. doi: 10.3969/j.issn.1671-7856.2020.11.022. [DOI] [Google Scholar]
- 9.Guerraty M, Mohler Iii ER. Models of aortic valve calcification. J Investig Med. 2007;55(6):278–83. doi: 10.2310/6650.2007.00012. [DOI] [PubMed] [Google Scholar]
- 10.Jung JJ, Razavian M, Kim HY, et al. Matrix metalloproteinase inhibitor, doxycycline and progression of calcific aortic valve disease in hyperlipidemic mice. Sci Rep. 2016;6:32659. doi: 10.1038/srep32659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Annabi MS, Clisson M, Fleury MA, et al. Sex-differences in echocardiographic assessment of aortic valve in young adult LDLr-/-/ ApoB 100/100/IGF-Ⅱ +/- mice. Exp Gerontol. 2020;140:111075. doi: 10.1016/j.exger.2020.111075. [DOI] [PubMed] [Google Scholar]
- 12.Qian JY, Chen ZW, Ge JB, et al. Relationship between aortic valve calcification and the severity of coronary atherosclerotic disease. J Heart Valve Dis. 2010;19(4):466–70. [PubMed] [Google Scholar]
- 13.Vander Roest M, Krapp C, Thorvaldsen JL, et al. H19 is not hypomethylated or upregulated with age or sex in the aortic valves of mice. Physiol Rep. 2019;7(19):e14244. doi: 10.14814/phy2.14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez-Stallons MV, Wirrig-Schwendeman EE, Hassel KR, et al. Bone morphogenetic protein signaling is required for aortic valve calcification. Arterioscler Thromb Vasc Biol. 2016;36(7):1398–405. doi: 10.1161/ATVBAHA.116.307526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Husseini D, Boulanger MC, Mahmut A, et al. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: implication for calcific aortic valve disease. J Mol Cell Cardiol. 2014;72:146–56. doi: 10.1016/j.yjmcc.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171(5):1407–18. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C, Xu SY, Gotlieb AI. The response to valve injury. A paradigm to understand the pathogenesis of heart valve disease. Cardiovasc Pathol. 2011;20(3):183–90. doi: 10.1016/j.carpath.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Honda S, Miyamoto T, Watanabe T, et al. A novel mouse model of aortic valve Stenosis induced by direct wire injury. Arterioscler Thromb Vasc Biol. 2014;34(2):270–8. doi: 10.1161/ATVBAHA.113.302610. [DOI] [PubMed] [Google Scholar]
- 19.Grande KJ, Cochran RP, Reinhall PG, et al. Stress variations in the human aortic root and valve: the role of anatomic asymmetry. Ann Biomed Eng. 1998;26(4):534–45. doi: 10.1114/1.122. [DOI] [PubMed] [Google Scholar]
- 20.Gould ST, Srigunapalan S, Simmons CA, et al. Hemodynamic and cellular response feedback in calcific aortic valve disease. Circ Res. 2013;113(2):186–97. doi: 10.1161/CIRCRESAHA.112.300154. [DOI] [PubMed] [Google Scholar]
- 21.Hjortnaes J, Shapero K, Goettsch C, et al. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis. 2015;242(1):251–60. doi: 10.1016/j.atherosclerosis.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leopold JA. Cellular mechanisms of aortic valve calcification. Circ Cardiovasc Interv. 2012;5(4):605–14. doi: 10.1161/CIRCINTERVENTIONS.112.971028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panizzi P, Nahrendorf M, Figueiredo JL, et al. In vivo detection of Staphylococcus aureus endocarditis by targeting pathogen-specific prothrombin activation. Nat Med. 2011;17(9):1142–6. doi: 10.1038/nm.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beckmann E, Grau JB, Sainger R, et al. Insights into the use of biomarkers in calcific aortic valve disease. J Heart Valve Dis. 2010;19(4):441–52. [PMC free article] [PubMed] [Google Scholar]
- 25.Niizeki T, Takeishi Y, Kitahara T, et al. Diacylglycerol kinase-Epsilon restores cardiac dysfunction under chronic pressure overload: a new specific regulator of Galpha(q) signaling cascade. Am J Physiol Heart Circ Physiol. 2008;295(1):H245–55. doi: 10.1152/ajpheart.00066.2008. [DOI] [PubMed] [Google Scholar]
- 26.Kitahara T, Takeishi Y, Harada M, et al. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc Res. 2008;80(1):40–6. doi: 10.1093/cvr/cvn163. [DOI] [PubMed] [Google Scholar]
- 27.Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. Int J Inflam. 2011;2011:364310. doi: 10.4061/2011/364310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller JD, Weiss RM, Heistad DD. Calcific aortic valve Stenosis: methods, models, and mechanisms. Circ Res. 2011;108(11):1392–412. doi: 10.1161/CIRCRESAHA.110.234138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaser MC, Kraler S, Lüscher TF, et al. Multi-omics approaches to define calcific aortic valve disease pathogenesis. Circ Res. 2021;128(9):1371–97. doi: 10.1161/CIRCRESAHA.120.317979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez-Stallons MV, Tretter JT, Hassel K, et al. Calcification and extracellular matrix dysregulation in human postmortem and surgical aortic valves. Heart. 2019;105(21):1616–21. doi: 10.1136/heartjnl-2019-314879. [DOI] [PubMed] [Google Scholar]
- 31.Yip CYY, Chen JH, Zhao RG, et al. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29(6):936–42. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 32.Small A, Kiss D, Giri J, et al. Biomarkers of calcific aortic valve disease. Arterioscler Thromb Vasc Biol. 2017;37(4):623–32. doi: 10.1161/ATVBAHA.116.308615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YJ, Han D, Zhou TW, et al. Melatonin ameliorates aortic valve calcification via the regulation of circular RNA CircRIC3/miR-204-5p/DPP4 signaling in valvular interstitial cells. J Pineal Res. 2020;69(2):e12666. doi: 10.1111/jpi.12666. [DOI] [PubMed] [Google Scholar]