

Abstract
Background
Von Willebrand disease (VWD) is a common inherited bleeding disorder, however the diagnosis can be complicated by a subjective bleeding history and issues with some current von Willebrand factor (VWF) laboratory assays.
Objectives
In the Zimmerman Program, we sought to determine how often a type 1 diagnosis was based on a single low VWF ristocetin cofactor (VWF:RCo) level resulting from the common genetic variant p.D1472H or an isolated assay issue, if that low value was corroborated by the VWF glycoprotein‐IbM (VWF:GPIbM) assay, and if retesting confirmed original levels.
Methods
New patients being evaluated for bleeding were consented. Analysis included VWF sequencing, bleeding scores, and comparisons of local VWF antigen (VWF:Ag) and VWF:RCo to central VWF:Ag and VWF:GPIbM.
Results
A total of 18% of VWD subjects had a low local VWF:RCo, but normal VWF:Ag and normal central testing including VWF:GPIbM. Seventy percent of the low VWF:RCo cohort had no pathogenic VWF variants; however, 33% carried p.D1472H. Low VWF:RCo subjects with follow‐up local testing within 2 years showed those with p.D1472H continued to have low VWF:RCo and VWF:RCo/VWF:Ag ratio with normal VWF:GPIbM. Subjects without p.D1472H had an increase mean VWF:RCo, resulting in 59% with normal levels on repeat testing.
Conclusions
The diagnosis of VWD based on a single low VWF:RCo but normal VWF:Ag, was often attributed to p.D1472H or variability in VWF:RCo that was eliminated with VWF:GPIbM. Our study suggests that using VWF:RCo alone for diagnostic purposes may be insufficient while repeat VWF:RCo or VWF:GPIbM testing can be valuable in establishing a VWD diagnosis.
Keywords: clinical laboratory tests, genetic polymorphism, Ristocetin cofactor, von Willebrand disease, von Willebrand factor
Essentials.
Von Willebrand Disease (VWD) is a bleeding disorder due to abnormal von Willebrand factor (VWF).
Von Willebrand factor (VWF) activity is often measured by a ristocetin cofactor assay (VWF:RCo).
Relying on one low VWF:RCo value is problematic in those with the common p.D1472H VWF variant.
Using a ristocetin‐less assay or repeat VWF:RCo testing is valuable for a VWD diagnosis.
1. INTRODUCTION
Von Willebrand disease (VWD) is a common inherited bleeding disorder, though diagnosis can be complicated because of a subjective bleeding history and issues with current von Willebrand factor (VWF) function assays. VWF activity is commonly measured in the United States using a ristocetin‐dependent cofactor (VWF:RCo) assay, although this assay is highly variable and has poor sensitivity. 1 The gain‐of‐function VWF glycoprotein‐IbM (VWF:GPIbM) is a ristocetin‐independent activity assay that provides both better precision and sensitivity and avoids the false low values seen with the common p.D1472H variant. 2 p.D1472H causes decreased binding of ristocetin to the VWF A1 domain and has been previously identified in 63% African American and 17% Caucasian healthy controls; however, it is not associated with an increased bleeding risk. 3 , 4
In the Zimmerman Program on the Biology of VWD (Zimmerman Program) prospective study, we sought to determine how often a diagnosis of type 1 or low VWF was based on a single low VWF:RCo because of the p.D1472H variant or an isolated assay issue, if that low value was corroborated by the VWF:GPIbM assay, and if retesting confirmed original levels.
2. STUDY DESIGN
New patients being evaluated for bleeding were consented and enrolled in the Zimmerman Program prospective cohort study at 12 hematology centers across the United States and Canada as previously described. 5 The study was open to any patients who were new to the hematology clinic and being evaluated for a bleeding disorder. Informed consent was obtained from all subjects and the study protocol was approved by each local clinical acquisition center's institutional review board. Hematology centers diagnosed subjects based on local laboratory values, and those with VWD continued in the study and were given the opportunity to participate in follow‐up research blood draws. Central testing was performed at Versiti Blood Research Institute (BRI) including VWF antigen (VWF:Ag), VWF activity by VWF:GPIbM, and exonic VWF Sanger sequencing. 6 Rare VWF genetic variants (present <1% of healthy controls 7 ) were considered potentially causative. The bleeding score was calculated according to the ISTH bleeding assessment tool (ISTH‐BAT). 8 Although the local centers had different normal ranges for VWF:Ag and VWF:RCo, a level of 50 was used as a cutoff in this analysis for type 1 VWD. GraphPad Prism was used to perform all statistical analyses. Comparisons of mean laboratory values and median bleeding scores used the nonparametric Mann–Whitney test.
3. RESULTS AND DISCUSSION
A total of 1860 subjects were enrolled, including 677 with a new diagnosis of VWD and 1183 without VWD. A total of 454 VWD subjects with suspected type 1 or low VWF and complete laboratory and genetic results were analyzed (Figure 1). Most of this group was female (67%), self‐identified as White (86%), non‐Latino (86%), and was pediatric (69%) (Table 1). Eighty‐two (18%) of these VWD subjects had a low local VWF:RCo (mean 43 IU/dl), but normal VWF:Ag (mean 62 IU/dl) and normal central testing with a mean VWF:Ag of 63 IU/dl (p = 0.9732) and mean VWF:GPIbM of 69 IU/dl (p < 0.0001) (Figure 2). This low VWF:RCo cohort had a median ISTH‐BAT bleeding score of 3, which was not different from subjects without VWD (n = 1005; p = 0.2784), although there was a slight difference in age between these groups (median 12 vs. 14 years; p = 0.0195) (Table 2).
FIGURE 1.
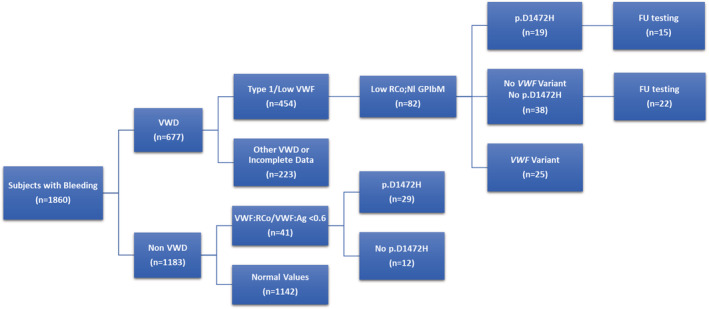
Flow chart of study subjects analyzed. Eighty‐two of 454 newly diagnosed von Willebrand disease (VWD) subjects with type 1 or low von Willebrand factor (VWF) had low local ristocetin cofactor (RCo), but normal (Nl) local and central VWF antigen (VWF:Ag), normal VWF glycoprotein‐IbM activity (GPIbM). Nineteen of the 82 subjects with low VWF:RCo had the common p.D1472H variant, which causes decreased binding of ristocetin to VWF A1 domain. 3 Forty‐one of non VWD subjects had low VWF:Rco/VWF:Ag ratio <0.6 and 29 (71%) also had the p.D1472H variant. Follow‐up testing (FU) was available on 37 subjects with and without p.D1472H.
TABLE 1.
Demographics of study population
| Demographics | Non‐VWD (n = 1005) | VWD (n = 454) | Low VWF:RCo cohort (n = 82) | Low VWF:RCo FU cohort (n = 37) |
|---|---|---|---|---|
| Race, n | ||||
| White | 833 (83%) | 390 (86%) | 66 (80%) | 31 (84%) |
| African American | 114 (11%) | 35 (8%) | 9 (11%) | 5 (14%) |
| Asian | 19 (2%) | 12 (3%) | 3 (4%) | 1 (3%) |
| Other or unknown | 39 (4%) | 17 (4%) | 4 (4%) | – |
| Ethnicity, n | ||||
| Latino | 116 (12%) | 57 (13%) | 11 (13%) | 7 (19%) |
| Non‐Latino | 882 (88%) | 389 (86%) | 71 (87%) | 30 (81%) |
| Unknown or not reported | 7 (1%) | 8 (2%) | – | |
| Sex (two unreported), n | ||||
| Female | 711 (71%) | 304 (67%) | 49 (60%) | 25 (68%) |
| Male | 294 (29%) | 148 (33%) | 33 (40%) | 12 (32%) |
| Adult vs pediatric, n | ||||
| Adult (>18 y) | 317 (32%) | 141 (31%) | 23 (28%) | 11 (30%) |
| Pediatric (<18 y) | 688 (68%) | 313 (69%) | 59 (72%) | 26 (70%) |
Note: Self‐reported race, ethnicity, sex, and distribution of adult (over 18 years of age; >18 year) and pediatric (under 18 years of age; <18 year) populations at time of enrollment in the non‐von Willebrand disease (non‐VWD) and VWD cohorts as well as subjects with a single low VWF ristocetin cofactor (VWF:Co) and those with available follow‐up (FU) testing.
FIGURE 2.
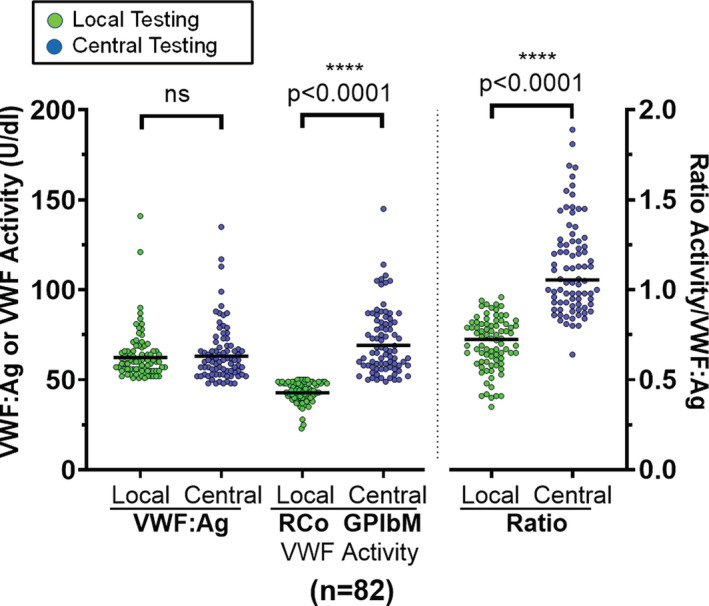
Eighty‐two subjects of 454 had a low local von Willebrand factor (VWF) ristocetin cofactor activity (VWF:RCo), but normal local and central VWF antigen (VWF:Ag), normal VWF glycoprotein‐IbM activity (VWF GPIbM), which resulted in abnormal VWF:RCo/VWF:Ag vs. VWF:GPIbM ratio (p < 0.0001)
TABLE 2.
Laboratory values and bleeding scores in study subjects
| VWF mean values (IQR) | Non‐VWD (n = 1005) | VWD (n = 454) | Low VWF:RCo cohort (n = 82) | Low VWF:RCo + p.D1472H (n = 19) | Low VWF:RCo + p.D1472H FU (n = 15) | Low VWF:RCo ‐ p.D1472H FU (n = 22) |
|---|---|---|---|---|---|---|
| Local VWF:Ag IU/dl | 98 (78–124) | 50 (41–63) | 62 (55–66) | 63 (58–69) | 68 (59–74) | 67 (55–74) |
| Local VWF:RCo IU/dl | 88 (66–114) | 42 (36–52) | 43 (40–48) | 44 (41–49) | 46 (39–58) | 57 (43–68) |
| Local VWF:RCo/VWF:Ag | 0.9 (0.8–1.0) | 0.8 (0.7–1.0) | 0.7 (0.6–1.0) | 0.7 (0.6–0.8) | 0.7 (0.5–0.8) | 0.9 (0.8–1.0) |
| BRI VWF:Ag IU/dl | 94 (74–117) | 50 (42–62) | 63 (53–69) | 63 (59–69) | 62 (54–70) | 62 (46–72) |
| BRI VWF:GPIbM IU/dl | 110 (84–142) | 56 (46–75) | 69 (58–82) | 61 (52–72) | 65 (57–74) | 80 (55–112) |
| BRI VWF:GPIbM/VWF:Ag | 1.2 (1.0–1.4) | 1.1 (1.0–1.4) | 1.1 (0.9–1.3) | 1.0 (0.9–1.0) | 1.1 (0.9–1.3) | 1.3 (1.1–1.6) |
| Median ISTH‐BAT BS (range) | 3 (0–24) | 4 (0–34) | 3 (0–21) | 2 (0–21) | − | − |
Note: Mean and interquartile range (IQR) of local von Willebrand factor (VWF) antigen (VWF:Ag), VWF ristocetin cofactor activity (VWF:RCo), central (BRI) VWF antigen (VWF:Ag) and VWF glycoprotein‐IbM activity (VWF:GPIbM) and median ISTH‐BAT bleeding score (BS) in subjects without VWD (non‐VWD), with suspected VWD and those with a single low VWF:RCo with (+) and without (−) common p.D1472H variant at time of study entry and follow‐up (FU).
When we analyzed VWF sequencing results, we found that 70% of the single low VWF:RCo cohort had no potentially causative rare variant identified; however, 19 (33%) of these subjects did carry the common p.D1472H variant (17 heterozygous; 2 homozygous). Eight of these subjects had a reduced VWF:RCo/VWF:Ag ratio (<0.7) that could be considered type 2M VWD; however, all had normal VWF:GPIbM/VWF:Ag and no type 2M genetic variant identified. In the subjects with p.D1472H, 58% self‐identified as White (including three Latino), 29% African American, and 10% Asian. Subjects with p.D1472H had a lower median bleeding score of 2 compared with 4 in those without p.D1472H (p = 0.2136) and those with a VWF variant (p = 0.2552); however, that difference was not significant.
In the Zimmerman Program prospective study, we hypothesized that repeat testing is critical for diagnosing VWD. To determine if subsequent retesting was valuable to confirm original findings, we examined a convenience sample including 37 of the 82 subjects (45%) with available follow‐up testing performed locally and centrally within 2 years of the initial diagnostic draw (Figure 1). There were 15 subjects with p.D1472H who had follow‐up testing where we observed a normal follow‐up VWF:Ag in both the local and central laboratory testing. As expected, we found a low follow‐up VWF:RCo (mean 46 IU/dl) similar to baseline, and a normal VWF:GPIbM (mean 65 IU/dl). Follow‐up laboratory values were consistent with the presence of p.D1472H in these subjects resulting in low VWF:RCo (Figure S1). The other 22 subjects without p.D1472H had a follow‐up VWF:Ag (mean 67 IU/dl) that was similar to baseline and central testing. However, we observed an increase in mean VWF:RCo (57 IU/dl) compared with 44 IU/dl at baseline and a normal VWF:GPIbM consistent with baseline (Table 2). Retesting in this small sample group revealed an increase in mean VWF:RCo and a normal mean VWF:RCo/VWF:Ag ratio (0.9), with approximately one‐half of these subjects presenting with a normal VWF:RCo at follow‐up.
Next, we identified 41 subjects who were enrolled in the study and categorized as non‐VWD by their local hematology center, yet had a low VWF:RCo/VWF:Ag level <0.6 (Figure 1). All these subjects had normal VWF:Ag (mean 127 IU/dl), slightly reduced VWF:RCo (mean 63 IU/dl), and a normal VWF:GPIbM (90 IU/dl) and VWF:GPIbM/VWF:Ag ratio (0.9). Sequencing was performed where p.D1472H was detected in 71% (29) of these non‐VWD subjects, which would explain their low VWF:RCo/VWF:Ag ratio. Twenty‐one (72%) were heterozygous and eight (28%) were homozygous. Those with p.D1472H comprised 48% White, 38% African American, and 3% Asian participants. There was no difference in median bleeding score (4) between those with or without p.D1472H (p = 0.6149).
In addition to the small sample size, there are several limitations to this study. Although all new patients being evaluated for bleeding were approached for the study, we estimate that 75% agreed to participate, which could bias the composition of the cohort. Another limitation is the bleeding score may not accurately reflect all bleeding symptoms, especially in the pediatric population. It is also possible that genetic variants outside of VWF could be contributing to the phenotype in cases where the low VWF:RCo value could not be explained by p.D1472H or variability in the assay. Finally, in the non‐VWD subjects with both a low VWF:RCo/VWF:Ag ratio and p.D1472H, clinical VWF exon 28 sequencing may have been performed and an erroneous VWD diagnosis avoided.
This report demonstrates that the reliance on VWF:RCo alone for diagnostic purposes can be insufficient, especially in those with the p.D1472H variant. Francis et al. 9 showed similar results from a single institution study in which their retrospective analysis uncovered six patients diagnosed with type 1 VWD who had normal VWF:Ag levels and borderline low VWF:RCo, which could be explained by the presence of p.D1472H. Our data expand on this study and highlight the value of repeat testing along with using the VWF:GPIbM assay to avoid the potential pitfalls of the VWF:RCo assay. This study supports the recent VWD diagnostic guidelines which recommends using abnormal bleeding, VWF:Ag <50, and newer assays that measure VWF activity for diagnosing type 1 VWD. 10 We also demonstrate that for patients who present with a disproportionate decrease in VWF:RCo compared with VWF:Ag, the presence of p.D1472H should be investigated to avoid misclassification as type 2M VWD.
In conclusion, 18% of our cohort had a VWD diagnosis based on a single low VWF:RCo but had normal VWF:GPIbM activity. The discrepancy between VWF:RCo and VWF:Ag was often because of the p.D1472H variant or variability in VWF:RCo that was improved with the VWF:GPIbM assay. Reliance on one VWF:RCo value alone for diagnostic purposes may be problematic, especially in those who have p.D1472H. Our findings suggest that performing VWF:GPIbM or repeat VWF:RCo testing can be valuable to ensure an accurate VWD diagnosis.
AUTHOR CONTRIBUTIONS
P. A. C. designed the research, analyzed data, and wrote the manuscript; U. O. S. performed research and analyzed data, S. L. H., V. H. F., and T. C. A. designed the research and edited the manuscript; R. R. M. conceived the original study, designed the research, and edited the manuscript. All authors have read and approved the final version of the manuscript.
RELATIONSHIP DISCLOSURE
The authors do not have any relevant conflicts of interest to declare.
Supporting information
Appendix S1
Figure S1
ACKNOWLEDGMENTS
The authors thank all the subjects, hematology centers, and laboratory personnel who were all critical to this study. This research was supported by a grant from the National Institutes of Health NHLBI for the Zimmerman Program and multiple investigators under award numbers P01HL081588, P01HL144457, R01HL112614, R01HL136430, and R01HL126810. A complete list of Zimmerman Program investigators and contributing centers are included in the Appendix S1.
Christopherson PA, Haberichter SL, Flood VH, et al. Ristocetin dependent cofactor activity in von Willebrand disease diagnosis: Limitations of relying on a single measure. Res Pract Thromb Haemost. 2022;6:e12807. doi: 10.1002/rth2.12807
Handling Editor: Pantep Angchaisuksiri
A complete list of members of the Zimmerman Program Investigators appears in Appendix S1.
Contributor Information
Pamela A. Christopherson, Email: pchristopherson@versiti.org, @pamchristophers.
Veronica H. Flood, @VeronicaFlood18.
the Zimmerman Program Investigators:
H. Weiler, D. Lillicrap, P. James, J. O’Donnell, C. Ng, J. Di Paola, B. Sadler, C. Bennett, R. Sidonio, M. Manco‐Johnson, C. Ng, J. Journeycake, A. Zia, J. Lusher, M. Rajpurkar, A. Shapiro, S. Lentz, J. Gill, C. Leissinger, M. Ragni, M. Tarantino, and J. Roberts
REFERENCES
- 1. Kitchen S, Jennings I, Woods T, Kitchen D, Walker I, Preston F. Laboratory tests for measurement of von Willebrand factor show poor agreement among different centers: results from the United Kingdom National External Quality Assessment Scheme for Blood Coagulation. Semin Thromb Hemost. 2006;32(5):492‐498. [DOI] [PubMed] [Google Scholar]
- 2. Flood VH, Gill JC, Morateck PA, et al. Gain‐of‐function GPIb ELISA assay for VWF activity in the Zimmerman Program for the Molecular and Clinical Biology of VWD. Blood. 2011;117(6):e67‐e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flood VH, Gill JC, Morateck PA, et al. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood. 2010;116(2):280‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Flood VH, Friedman KD, Gill JC, et al. No increase in bleeding identified in type 1 VWD subjects with D1472H sequence variation. Blood. 2013;121(18):3742‐3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DiGiandomenico S, Christopherson PA, Haberichter SL, et al. Laboratory variability in the diagnosis of type 2 VWD variants. J Thromb Haemost. 2021;19(1):131‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flood VH, Christopherson PA, Gill JC, et al. Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood. 2016;127(20):2481‐2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bellissimo DB, Christopherson PA, Flood VH, et al. VWF mutations and new sequence variations identified in healthy controls are more frequent in the African‐American population. Blood. 2012;119(9):2135‐2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063‐2065. [DOI] [PubMed] [Google Scholar]
- 9. Francis JC, Hui SK, Mahoney D Jr, et al. Diagnostic challenges in patients with bleeding phenotype and von Willebrand exon 28 polymorphism p.D1472H. Haemophilia. 2014;20(3):e211‐e214. [DOI] [PubMed] [Google Scholar]
- 10. James PD, Connell NT, Ameer B, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021;5(1):280‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Figure S1