

Abstract
Targeted therapy for lung squamous cell carcinoma (LUSC) remains a challenge due to the lack of robust targets. Here, we identified MECOM as a candidate of therapeutic target for LUSC by screening 38 genes that were commonly amplified in three pairs of primary tumors and patient-derived xenografts (PDXs) using a clustered regularly interspaced short palindromic repeats (CRISPR)-mediated approach. High MECOM expression levels were associated with poor prognosis. Forced expression of MECOM in LUSC cell lines promoted cancer stem cell (CSC) properties, and its knockout inhibited CSC phenotypes. Furthermore, systemic delivery of CRISPR-mediated MECOM depletion cassette using adenovirus with an adaptor, which is composed of a single-chain fragment variable (scFv) against epithelial cell adhesion molecules (EpCAM) fused to the ectodomain of coxsackievirus and adenovirus receptor, and a protector, which consists of the scFv connected to the hexon symmetry of the adenovirus, could specifically target subcutaneous and orthotopic LUSC and retard tumor growth. This study could provide a novel therapeutic strategy for LUSC with high efficacy and specificity.
Keywords: lung squamous cell carcinoma, CRISPR, cancer stem cells, MECOM
Graphical abstract
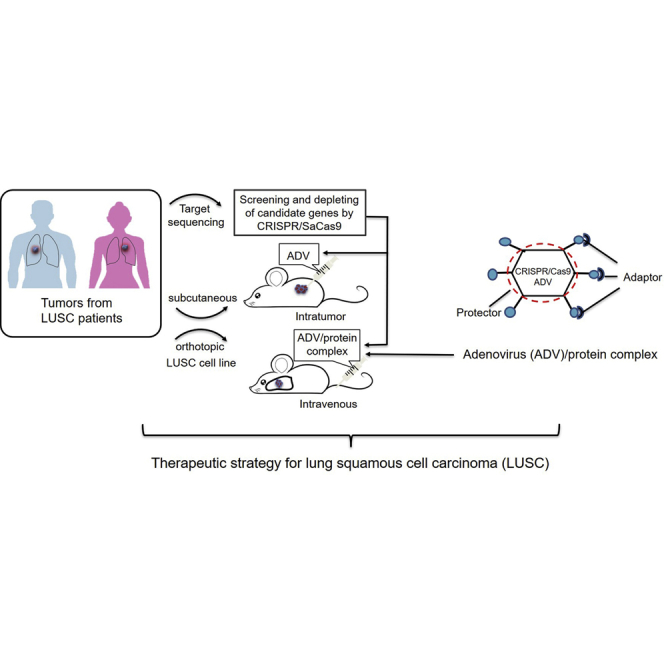
Ma et al. identified that MECOM frequently amplified in lung squamous cell carcinoma (LUSC) promoted cancer stem cell properties. Interestingly, CRISPR-medicated MECOM depletion had the antitumor effect using systemic delivery of adenovirus vector with the engineered adaptor and protector proteins, indicating a novel therapeutic strategy with high efficacy and specificity.
Introduction
Lung squamous cell carcinoma (LUSC) represents a major type of lung cancer, which is a highly prevalent malignancy and leading cause of cancer-related deaths worldwide.1 Targeted therapy against driver genes such as epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) dramatically improves the prognosis of lung adenocarcinoma.2 Nevertheless, existing targeted therapies have poor efficacy for LUSC, mainly due to its variety of genetic alterations and the lack of effective therapeutic targets and safe approaches.3,4,5 Thus, robust targets and optimal strategies for LUSC treatment are urgently required.
Currently, gene editing such as clustered regularly interspaced short palindromic repeats (CRISPR)/Staphylococcus aureus Cas 9 (SaCas9) are characterized to screen therapeutic targets and directly deplete the specific sites of target genes.6,7 Adenovirus-based delivery of CRISPR is an efficient method, of which species C adenovirus serotype 5 (ADV) is extensively used in gene therapy for malignancies in vivo.8,9 However, ADV-mediated treatment mostly results in off-target tissue tropism and immunogenicity through intravenous delivery, and this hampers clinical studies. The engineered protein with a single-chain fragment variable (scFv) against the specific antigens like EGFR and EGFR 2 (HER2) has the function to retarget ADV to cancer with high expression of the marker.10,11 In addition, the “shield” protein could block the linkage of coagulation factor X to the hexon protein on the surface of ADV, which leads to the reduction of liver tropism and immune neutralization.12
MDS1 and EVI1 complex locus protein (MECOM) has oncogenic properties that drive malignant transformation in leukemia and other solid cancers.13,14,15 This oncogene promotes cell proliferation and depresses apoptosis through the activation of the AKT/mammalian target of rapamycin (mTOR) pathway and interaction with polycomb proteins.16 According to previous studies,15,17,18 MECOM maintains the self-renewal property of hematopoietic stem cells and cancer stem cells (CSC) by transcriptional, post-translational, or epigenetic modulations. MECOM acts as a transcription factor binding to DNA sequences with its zinc finger, which is also called ecotropic viral integration site 1 (EVI1).14,19 EVI1 promotes Fos/Jun heterodime expression and activates its transcription by binding with its promoter.20 EVI1 reprograms hematopoietic stem cells toward acute myeloid leukemia by stimulating the transcriptional response of the transcription factor PU.1.14
In this investigation, we identify therapeutic targets in LUSC using CRISPR-SaCas9. Among them, MECOM depletion was found to suppress tumor growth and CSC characteristics by regulating SOX2′ transcription. Furthermore, we developed two proteins, one of which is an adaptor with scFv against epithelial cell adhesion molecules (EpCAM), which enhances the delivery of the ADV vector to the EpCAM-overexpressing LUSC cells under intravenous administration. The other is a protector carrying scFv, which covers the ADV to reduce off-target tissue tropism and immunogenicity. Importantly, CRISPR-SaCas9-mediated MECOM depletion in the ADV/adaptor/protector system shows significant antitumor effects in patient-derived xenograft (PDX) and orthotopic xenograft models for LUSC, indicating a highly efficient and specific approach for LUSC treatment.
Results
CRISPR-SaCas9 screens the therapeutic targets in LUSC
In light of the evidence that CRISPR-SaCas9 was used to identify candidate targets for treatment,3,21 we screened targets that can drive the cell growth of LUSC using the CRISPR-SaCas9 system both in vitro and in vivo (Figure 1A). We validated the antitumor effect of the target depletion using intratumoral delivery of the ADV-CRISPR-SaCas9 system. Furthermore, we developed the adaptor and protector proteins that were administered in PDX and orthotopic xenograft models by intravenous delivery of ADV-CRISPR-SaCas9.
Figure 1.

Identification of the therapeutic targets for LUSC
(A) Diagram of the present study for identification of the driver-gene targets and evaluation of therapeutic potential using CRISPR-SaCas9-mediated depletion in vitro and in vivo.
(B) Aberrant status of 38 driver genes was detected by target sequence in primary (T) and PDX tumors (P) of LUSC012, LUSC19, and LUSC21. AMP, amplification.
(C–E) The PDX tumor cells of LUSC012 (C), LUSC019 (D), and LUSC021 (E) were infected with the lentivirus expressing the corresponding sgRNA for 38 driver genes for 48 h. The inhibition rate of the sgRNA for the driver gene in PDX tumor cells was analyzed, compared to non-targeting sgRNA using CellTiter-Glo detection. The aberrant status was labeled as SNV, amplification (AMP), and SNV + AMP. The dotted lines indicate the inhibition rates of 50%.
(F) SNV and AMP frequencies of 10 candidate genes in 501 LUSC cases (The Cancer Genome Atlas [TCGA] database).
(G) The primary tumor cells, corresponding normal lung cells, and endothelial cells (CD31+) from 3 cases of LUSC patients were infected with lentivirus expressing non-targeting sgRNA and the sgRNA of MECOM depletion for 48 h by CellTiter-Glo detection. Cell inhibitory rates was calculated. n = 4. ∗p < 0.05, unpaired 2-tailed t test.
In our previous report, we identified candidate genes carrying frequent single-nucleotide variations (SNVs) and/or copy number variations (CNVs) in LUSC.22 Among them, we further analyzed 38 drive genes in the primary and PDX tumors, including LUSC012, LUSC019, and LUSC021, using target sequencing (Figure 1B). We observed a high consistency of SNV and CNV for these genes in both primary tumors and matched PDX tumors (Tables S1–S3).
Subsequently, we screened the effective targets for the inhibition of cell growth in PDX tumor cells by single-guide RNA (sgRNA) designed using the Benchling tool (https://benchling.com) in vitro. We confirmed the editing efficiency of the specific sgRNA for those 38 drive genes (Figure S1). Next, we depleted those genes in 3 PDX tumor cells, including 26 genes in LUSC012, 37 genes in LUSC019, and 31 genes in LUSC021 using lentivirus carrying the specific sgRNA. Compared with the non-targeting control, 10 gene depletions led to over 50% survival inhibition, including BRAF, CREBBP, GRM8, HDAC9, ITGB4, MECOM, MET, PIK3CG, PLXNA4, and SNAI1 in all 3 tumor cells (Figures 1C–1E). Among them, MECOM had the highest frequency of amplification, with 89.82% in 501 cases of LUSC (The Cancer Genome Atlas database, https://doi.org/10.7908/C14Q7TDH) (Figure 1F; Table S4). In addition, MECOM amplification was detected in five cases of LUSC tumors used in the present study (Table S5). Furthermore, MECOM depletion resulted in significant inhibition of cell survival in the primary tumor cells, when compared to that in the matched normal cells and CD31+ endothelial cells23,24 from three LUSC patients (Figure 1G).
These results suggest that MECOM may be a potential therapeutic target for the LUSC.
MECOM is correlated with a poor prognosis for LUSC
Since therapeutic targets are usually associated with the prognosis of cancer patients, we studied correlations between the expression levels of those 10 genes with tumor-inhibition ability and LUSC prognosis. The high mRNA levels of MECOM, BRAF, GRM8, MET, and SNAI1 were significantly correlated with poor overall survival (OS) in 524 cases of LUSC using the Kaplan-Meier plot (Figure S2). Moreover, we analyzed the correlation between MECOM protein levels in the tumor tissues of LUSC and disease-free survival (DFS) or OS. High MECOM expression levels were significantly correlated with shorter DFS and OS (Figures 2A–2C). Furthermore, the univariate and multivariate analyses indicated that MECOM was an independent factor affecting DFS and OS (Tables S6 and S7). The results were consistent with those of a previous study,25 in that MECOM was identified as an indicator of poor prognosis for LUSC.
Figure 2.

The role of MECOM in prognosis of LUSC patients and CSC properties
(A) Representative images of MECOM expression in the tumor tissues of LUSC patients by immunohistochemistry. Scale bars, 50 μm.
(B and C) DFS (B) and OS (C) curves of 150 LUSC cases with different expression levels of MECOM.
(D and E) The representative photographs (D) and statistical efficiencies (E) of primary (1°) and secondary (2°) spheroids in H520 and EBC-1 cells with forced expression of MECOM (MECOM-OE). n = 6. Scale bars, 100 μm.
(F and G) The representative images (F) and percentages (G) of CD44 and CD133 in H520 and EBC-1 cells with forced expression of MECOM (MECOM-OE). n = 6.
(H and I) The representative photographs (H) and statistical efficiencies (I) of spheroids in SKMES-1 cells with depleted MECOM (sg-MECOM). Scramble sgRNA (Sg-scramble) was as the control. n = 6. Scale bars, 100 μm.
(J) Spheroid efficiencies in PDX tumor cells of LUSC006, LUSC018, and LUSC021 with depleted MECOM. n = 3. ∗p < 0.05, unpaired 2-tailed t test in (E), (G), (I), and (J).
MECOM promotes tumor proliferation and the CSC phenotype in LUSC
CRISPR-SaCas9 could induce a gene-independent antiproliferative response when targeting amplified genomic regions.26 To avoid the selection of insignificant targets, we further clarified the biological characteristics of MECOM in LUSC cells. Based on the variable expression of MECOM in LUSC cell lines (Figure S3A), the forced expression of this gene was found to significantly promote cell proliferation and colony formation in the LUSC cell lines H520 and EBC-1 (Figures S3B and S3C). The forced expression of MECOM significantly increased primary and secondary spheroid formation (Figures 2D and 2E) and led to the increased expression of CSC markers, such as CD4427 and CD13328 (Figures 2F and 2G). In addition, sgRNA was used to deplete MECOM expression (Figure S3D), and dramatically inhibited cell proliferation, colony formation and spheroid formation in SKMES-1 cells (Figures 2H, 2I, S3E, and S3F). Compared to that in the control groups (sg-scramble), the sgRNA-MECOM also resulted in significant inhibition of cell viability in the PDX tumor cells, including LUSC006, LUSC018, and LUSC021 (Figure 2J). These results demonstrate that MECOM can enhance tumor proliferation and maintain the CSC phenotype in LUSC.
MECOM activates SOX2 by transcriptional regulation
To identify the altered genes that were regulated by MECOM, we performed RNA sequencing in the H520 cells with forced expression of this gene. As shown in Figure 3A, 1,217 and 428 genes were upregulated and downregulated in the H520 cells with forced expression of MECOM compared to those in the control cells. MECOM extensively modulated the genes involved in the signaling pathways of tumor proliferation and regulating pluripotency of stem cells (Figure 3B). Subsequently, we validated the role of MECOM in regulating these genes enriched in proliferation and stem cell pathways using reverse transcription and quantitative real-time PCR. The expression of FGFR3, FGFR4, FZD4, GLI1, SOX2, TCF7L1, and WNT3A were significantly upregulated by the forced expression of MECOM in both H520 and EBC-1 cells (Figures 3C and 3D). In addition, we found positive correlations between MECOM and SOX2, FGFR3, WNT4, WNT3A, FZD4, TCF7L1, JAK3, and MEIS1 in 501 of the LUSC cases in the database of LinkedOmics (Figure S4). Interestingly, we observed a strong relationship between MECOM and SOX2 (Spearman correlation: 0.5064).
Figure 3.

MECOM stimulates SOX2′ transcriptional response
(A and B) Volcano map (upregulation, 1,217; downregulation, 428; no difference, 20,317; padj < 0.05, log2 fold change >1) and functional enrichment (B) of differential genes regulated by MECOM, which is forcedly expressed in H520 cells using RNA sequencing analyses.
(C and D) The gene expression was examined in H520 (C) and EBC-1 (D) cells with forced expression of MECOM (MECOM-OE) by quantitative real-time-PCR analysis. n = 3.
(E) The wild-type (WT) and mutant sequences of predicted binding sites on the promoter of SOX2 with EVI1, using JASPAR online software.
(F) The ChIP assay was done in H520 cells with forced expression of MECOM using EVI1 antibody or immunoglobulin G (IgG). The specific primers were used to amplify the binding sequence on the promoter of SOX2 by quantitative real-time-PCR. n = 6.
(G) The control and forcedly expressed EVI1 (EVI1-OE) cells of H520 were transfected with the pGL3-basic vectors with WT or mutant (mut) EVI1′ binding sequences for 48 h. The relative binding activity of EVI1 with the SOX2′ promoter was analyzed using luciferase reporter assays. Renilla reporter was used as the internal control for normalizing luciferase values. n = 6.
(H) Western blots show expression of MECOM, SOX2, ABCG2, and CD44 in H520 and EBC-1 cells with forced expression of MECOM (MECOM-OE).
(I) Western blots demonstrate expression of the indicated protein in SKMES-1 cells with depleted MECOM (sg-MECOM). β-actin was used as loading control. The numerical value under the band shows densitometric analyses of the indicated protein expression, compared to the corresponding control, which was normalized as “1.0.” ∗p < 0.05, unpaired 2-tailed t test in (C), (D), (F), and (G).
Based on previous reports that MECOM (EVI1) functions as a transcriptional regulator,29,30 we predicted that EVI1 could bind to the sequences within the promoter of SOX2, which is a critical regulator that maintains the CSC phenotype31 (Figure 3E). We verified that EVI1 could bind to the sequences within the SOX2′ promoter region by chromatin immunoprecipitation (ChIP) following quantitative real-time-PCR (Figure 3F). Higher promoter activity of SOX2 was detected in the cells with forced expression of EVI1, and this increased effect was inhibited by the cells with mutant binding sequences (Figure 3G).
In addition, the forced expression of MECOM significantly upregulated the protein levels of the CSC-related markers, including SOX2, CD44, and ABCG231 in H520 and EBC-1 cells (Figure 3H). MECOM depletion downregulated the expression of these CSC factors (Figure 3I).
Therefore, MECOM promotes the expression of CSC-associated factors and stimulates SOX2′ transcription.
MECOM depletion restrains the tumor growth of LUSC using the ADV-mediated CRISPR-SaCas9 system in vivo
To explore the therapeutic potential of MECOM depletion in LUSC by the CRISPR-SaCas9 system,8,32 we selected a suitable delivery vector among the viruses, including lentivirus, ADV, and serotype 2 of adeno-associated virus (AAV2) in vivo. These three types of viruses expressing green fluorescent protein (GFP) were intratumorally injected into the LUSC021 PDX model. The infection efficiency of ADV was significantly higher than that of the other two vectors (Figures 4A and 4B). Hence, ADV was chosen as the delivery vector for the CRISPR-SaCas9 system in LUSC.
Figure 4.

ADV-CRISPR-SaCas9-mediated MECOM depletion inhibits tumor growth in LUSC PDX models
(A and B) Three types of viruses (ADV, 1 × 1010 VP; lentivirus, 1 × 109 VP; AAV2, 1 × 1012 VP) expressing GFP were intratumorally injected into the LUSC021 PDX model (NOD-SCID mice). The representative images (A) and infection efficiencies (B) of ADV, lentivirus, and AAV2 in the tumors isolated from those mice treated with different viruses by flow cytometry detection.
(C–F) A diagram (C) for intratumoral administration of ADV-CRISPR-SaCas9-mediated MECOM depletion in PDX models (NOD-SCID mice). The ADV system was intratumorally injected at days 0 and 12 with the amounts of 1.0 × 1010 VP and 0.5 × 1010 VP, respectively. Tumor growth curves and representative tumor photos of PDX models including LUSC006 (D), LUSC018 (E), and LUSC021 (F) treated with MECOM-depleted ADV (MECOM-KO) or control ADV. Scale bars, 1 cm. n = 5. ∗p < 0.05, unpaired 2-tailed t test.
Furthermore, we validated the antitumor effect of CRISPR-SaCas9-mediated MECOM depletion by ADV delivery. The MECOM depletion system was intratumorally delivered into LUSC006, LUSC018, and LUSC021 PDX mice using the ADV vector (Figure 4C). Compared with those in the non-targeting controls, the tumor growth and final tumor volumes were significantly inhibited by MECOM depletion (Figures 4D–4F).
The target site of MECOM depletion was significantly edited in PDX tumor samples (Figures 5A and 5B). The expression of MECOM, SOX2, CD44, and ABCG2 was significantly downregulated in MECOM-depleted tumors (Figures 5C–5E).
Figure 5.

MECOM depletion suppressed CSC-associated factors
(A and B) ADV-CRISPR-SaCas9 with MECOM depletion (MECOM-KO) was intratumorally injected into the PDX models. After 2 days, the tumors were isolated from the LUSC006, LUSC018, and LUSC021 cases for gene editing analyses using T7EI assay (A: LUSC006, LUSC018, and LUSC021) and Sanger sequence (B: LUSC021).
(C–E) After 24–28 days, the treated tumors were isolated from the LUSC006, LUSC018, and LUSC021 cases for western blot and immunohistochemistry assays. Western blots (C) show MECOM, SOX2, ABCG2, CD44, and β-actin expression in the tumor samples of LUSC006, LUSC018, and LUSC021 models with depleted MECOM (MECOM-KO). β-Actin was used as the loading control. The numerical value under the band shows densitometric analyses of the indicated protein expression, compared to the corresponding control, which was normalized as “1.0.” Photographs (D) and IRS scores (E) of MECOM, CD44, and ABCG2 in the tumor samples of LUSC006, LUSC018, and LUSC021 models with MECOM-depleted ADV (MECOM-KO) by immunohistochemistry analyses. Scale bars, 50 μm. ∗p < 0.05, unpaired 2-tailed t test.
These data indicate that MECOM depletion has antitumor effects for LUSC when using the ADV-CRISPR-SaCas9 system.
Engineered proteins retarget ADV to EpCAM+ LUSC cells with high specificity and low immunogenicity
To avoid off-target tissue tropism and host immune response caused by the intravenous administration of ADV, we constructed adaptor and protector proteins (Figure 6A). According to previous studies,11,33 the overexpression of EpCAM in approximately 86% of LUSC cases34 and human LUSC cell line H520 (Figures S5A and S5B), a humanized scFv against EpCAM, and coxsackievirus and adenovirus receptor (ECXADR) were connected by a phage T4 fibritin, which formed a trivalent adaptor protein (CFS). This CFS interacted with EpCAM+ cells and the knob protein of ADV through the specific scFv and ECXADR domain, respectively, that could increase targeting efficiency. Another trivalent protector protein (HF) was generated with a humanized scFv against hexon12 and the phage T4 fibritin. This HF covered the ADV surface to reduce the immune response.
Figure 6.

The function of adaptor and protector
(A) Construction of adaptor (CFS) and protector (HF) proteins. The CFS was composed of a humanized anti-EpCAM scFv, the ectodomain of CXADR, and a phage T4 fibritin polypeptide. The HF was composed of a humanized scFv against the hexon protein of ADV and a phage T4 fibritin polypeptide.
(B–E) A diagram (B) for intravenous administration of ADV with or without CFS and HF proteins on NOD-SCID mice with H520 xenograft. ADV was incubated with CFS, HF, and CFS + HF for 2 h, and this ADV/protein complex was injected into the mice via the tail vein. The amount of ADV was 1.0 × 1010 VP and concentration of the protein was 1.0 × 10−7 pmol/VP. After administration of ADV, ADV + CFS, ADV + HF, and ADV + CFS + HF for 48 h, the tumor, liver, lung, and kidney isolated from the mice were analyzed for distribution of ADV expressing GFP and SaCas9. The representative images (C) and relative abundance (D) of GFP bioluminescence and quantitative real-time-PCR analyses of SaCas9 (E) were demonstrated. n = 3.
(F) ADV expressing GFP was incubated with CFS and HF proteins for 2 h, followed by adding serum of C57BL/6N mice for 1 h. The amounts of proteins and serum were 1.0 × 10−7 pmol/VP and 5 μL, respectively. The mixture was added in the LEWIS cells (mouse lung cancer cell line) for 48 h. The multiplicity of infection (MOI) of ADV was 1:500. The relative infection efficiencies were detected by comparing the ratios of GFP by flow cytometry.
(G–I) A diagram (G) for intratumoral and intravenous administration of ADV with or without CFS and HF on LEWIS xenograft (C57BL/6N mice). ADV was incubated with CFS, HF, and CFS + HF for 2 h, and this ADV/protein complex was injected into the mice via intratumor and tail vein. The amount of ADV was 1.0 × 1010 VP and concentration of the protein was 1.0 × 10−7 pmol/VP. The ADV distribution in the isolated tumor, liver, lung, and kidney of C57BL/6N mice was detected after 2 days by quantitative real-time-PCR analyses of SaCas9 (H). The abundance of anti-ADV antibody in serum of C57BL/6N mice was administrated with ADV/protein complex as indicated by ELISA analyses after 7 days (I). The control mice were untreated. i.t., intratumor; i.v., intravenous, via tail vein. n = 3. ∗p < 0.05, unpaired 2-tailed t test in (D–(F), (H), and (I).
The CFS and HF incubating with ADV expressing CRISPR-SaCas9 and GFP formed a complex. This complex was delivered into the H520 xenograft in non-obese diabetic (NOD)-server combined immunodeficiency (SCID) mice via tail vein (Figure 6B). Based on a previous study,12 the intravenous administration of ADV was expected to show organ damage, especially for the liver. The ADV-GFP signal was increased in the tumor and was decreased in the livers of NOD-SCID mice treated with the CFS or HF. The ADV was even induced in the tumor and was reduced in the livers of mice with the CFS and HF complex (Figures 6C and 6D). The same effect as with CFS and HF on ADV distribution was found when analyzing SaCas9 expression (Figure 6E).
To prevent the immune neutralization induced by the existing antibody in the host12 and block the anti-ADV antibody in the sera of human lung cancer patients and C57BL6 mice (Figure S5C), we elucidated the effects of CFS and HF on the complement neutralization. CFS and HF with ADV-GFP were incubated with the serum of C57BL6 mice, and this mixture was infected into the mouse lung cancer cell line (LEWIS). CFS enhanced the infection efficiency of ADV, and this increase was inhibited by mouse serum containing anti-ADV antibody (Figure 6F). This effect of immune neutralization could be attenuated by HF, showing a high efficiency of infection (Figure 6F).
To further examine the delivery efficiency and immunogenicity of the ADV in the model with an intact immune system, the mixture including CFS, HF, and ADV expressing CRISPR-SaCas9 and GFP was injected into the LEWIS xenograft in C57BL6 mice via intratumor and tail-vein methods (Figure 6G). After administration for 2 and 7 days, the tumor tissue and serum samples were collected for further experiments (Figure 6G). The CFS and HF both elevated ADV delivery into the tumor, and a complex of CFS and HF significantly enhanced the distribution of ADV into the tumor (Figure 6H). In addition, the immunogenicity of the anti-ADV antibody was tested in the serum of C57BL/6N after complex administration for 7 days. ADV or the combination of ADV and CFS upregulated the anti-ADV antibody, and HF significantly downregulated this immunogenicity (Figure 6I).
Together, the adaptor and protector enhanced tumor tropism and attenuated immunogenicity for ADV delivery, indicating that this ADV/protein complex may be an optimal system for gene depletion through CRISPR-SaCas9 under intravenous administration.
MECOM depletion had antitumor effects with intravenous administration via the ADV/adaptor/protector system
To evaluate the antitumor effects of MECOM depletion mediated by the ADV/adaptor/protector system, we administrated the complex of CFS, HF, and ADV expressing CRISPR-SaCas9 into the PDX model via tail-vein injection (Figure 7A). MECOM depletion with the ADV/CFS/HF complex significantly suppressed tumor growth in the LUSC021 model (Figure 7B). This ADV/protein complex mediated efficient MECOM depletion (Figure 7C) and reduced the expression of CSC factors, including SOX2, CD44, and ABCG2 in the LUSC tumors (Figures 7D, 7E, and S6A). In addition, SOX2 increased tumor growth and tumor volume, which were inhibited by the depletion of MECOM (Figures 7F–7H). Therefore, MECOM depletion inhibits tumor growth in the PDX model of LUSC via the intravenous administration of the ADV, adaptor, and protector complex.
Figure 7.

Intravenous administration of MECOM depletion by ADV, adaptor, and protector complex
(A and B) A diagram (A) for therapeutic treatment with the complex CFS, HF, and ADV with depleted MECOM in NOD-SCID mice (LUSC021 PDX model) via tail-vein injection. MECOM-depleted ADV was incubated with CFS and HF to form ADV/protein complex for 2 h. The complex was injected into the NOD-SCID mice via tail vein for 4 rounds with a 12-day interval. The ADV amount was 0.7 × 1010 VP, and the protein concentration was 1.0 × 10−7 pmol/VP. The growth curve (B) of LUSC021 PDX mice with the ADV/protein complex (MECOM-KO) via tail-vein injection. The ADV/protein complex expressing non-targeting sgRNA was used as the control. n = 4, ∗p < 0.05, unpaired 2-tailed t test.
(C) The image of T7EI detection in the ADV/protein-treated tumors after administration for 48 h.
(D) Immunohistochemistry staining shows MECOM, ABCG2, and CD44 expression in the tumors with treatment of ADV/protein. Scale bar, 50 μm.
(E) Western blots demonstrate the indicated protein expression in the tumors with treatment of ADV/protein. β-Actin was used as loading control. The numerical value under the band shows densitometric analyses of the indicated protein expression, compared to the corresponding control, which was normalized as “1.0.”
(F–H) The ADV viruses including control, MECOM depletion (MECOM-KO), and MECOM-KO plus SOX2 overexpression (MECOM-KO + SOX2) were intratumorally injected into the NOS-SCID mice (LUSC021). The ADV was intratumorally injected at days 0 and 12 with the amounts of 1.0 × 1010 VP and 0.5 × 1010 VP, respectively. Tumor image (F), growth curve (G), and tumor weight graph (H) of LUSC021 PDX model with indicated treatment. Scale bar, 1 cm. n = 5, ∗p < 0.05, unpaired 2-tailed t test.
MECOM depletion via the ADV/adaptor/protector system in an orthotopic xenograft model of LUSC
To further validate the therapeutic potential of MECOM depletion via the CRISPR-SaCas9 and ADV/protein system, an orthotopic xenograft model of H520 cells was established. The ADV vector for MECOM depletion with or without CFS and HF was then intravenously administered via the tail vein in the orthotopic model (Figure 8A). Compared with the gradual proliferation of LUSC in the control mice, the tumor growth with MECOM depletion was significantly inhibited (Figures 8B and 8C). The ADV/protein complex could mediate efficient MECOM depletion and downregulation of the expression of CSC factors, including SOX2, CD44, and ABCG2 (Figures 8D, 8E, and S6B).
Figure 8.

Intravenous administration of MECOM-depleted ADV/protein complex in orthotopic xenograft model
(A–C) A diagram (A) for intravenous administration of ADV/protein complex with MECOM depletion in H520 orthotopic xenograft (NOD-SCID mice). MECOM-depleted ADV was incubated with CFS and HF proteins to form ADV/protein complex for 2 h. The complex was injected into the mice via tail vein for 2 rounds with a 14-day interval. The ADV amount was 0.7 × 1010 VP, and the protein concentration was 1.0 × 10−7 pmol/VP. The bioluminescent imaging (BI) was taken on days 0, 14, and 28 after ADV delivery to demonstrate the tumor growth. Bioluminescent images (B) and growth curves (C) of orthotopic tumors treated with MECOM-depleted ADV/protein complex (MECOM-KO) via tail-vein injection. n = 4. ∗p < 0.05, unpaired 2-tailed t test.
(D) Immunohistochemistry staining demonstrates MECOM, ABCG2, and CD44 expression in the H520 orthotopic xenografts treated with ADV/protein complex including control and MECOM depletion groups. Scale bar, 100 μm.
(E) Western blots show the indicated proteins in the H520 orthotopic xenografts treated with ADV/protein complex, including control and MECOM depletion groups. β-Actin was used as loading control. The numerical value under the band shows densitometric analyses of the indicated protein expression, compared to the corresponding control, which was normalized as “1.0.”
(F) On-target and off-target mutations detected by WES sequencing in tumor tissues of orthotopic models with MECOM depletion. The frequencies and locations of various on-target and off-target mutations were respectively presented in bar plots with different colors.
(G) The charts show the numbers of SNVs and indels detected in the tumors derived from H520 orthotopic xenograft with ADV/protein treatment.
To investigate whether the CRISPR-SaCas9 system resulted in on-target and off-target mutations, whole-exome sequencing (WES) was done in the orthotopic H520 tumor treated with the MECOM-depleted ADV/protein complex. In total, three types of on-target indels with mutation alleles over 2% were detected in the guide genomic regions of MECOM. The frequency of on-target mutations was 29% (Figure 8F). There were no off-target mutations detected among the 49 potential off-target loci of MECOM that had been predicted by the benchling tool. Furthermore, the total SNV and indel detected in the control and MECOM-depleted samples were almost equal (SNV: 62,004 versus 61,685; indel: 8,068 versus 7,937, respectively) (Figure 8G). These results suggest that minor off-target mutagenesis is induced by MECOM depletion via the ADV/protein complex.
In total, the MECOM depleting system composed of the CRISPR-SaCas9 and ADV/protein complex could be a therapeutic strategy for LUSC with both high efficiency and specificity.
Discussion
Here, we identify the candidate targets for LUSC treatment using CRISPR-SaCas9, in which MECOM depletion has antitumor effects both in vitro and in vivo. Furthermore, we have developed an adaptor against EpCAM that could retarget ADV to the LUSC with high specificity, and a protector against hexon on the ADV surface that reduces liver tropism and immunogenicity, conferring a significantly effective and safe strategy for targeted therapy.
MECOM has been extensively studied in acute myeloid leukemia, as it was found to stimulate cell proliferation, drug resistance, and delay the cell cycle and apoptosis.35,36,37 Its function and molecular mechanism in solid cancers, especially LUSC, have not yet been fully elucidated. Recently, MECOM was shown to play an essential role in the occurrence and progression of ovarian, colorectal, breast, and lung cancers.38,39,40,41,42 MECOM reportedly maintained the self-renewal of hematopoietic stem cells17,18 and promoted CSC properties in acute myeloid leukemia17,43 and nasopharyngeal carcinoma.15,44 Consistent with the results of these previous studies, we detected enriched proliferation and stem cell-associated signaling in LUSC cells with the forced expression of MECOM. Moreover, MECOM depletion significantly restrained the tumor growth of LUSC both in vitro and in vivo.
MECOM encodes several protein isoforms containing myelodysplastic syndrome 1 (MDS1)/EVI1, MDS1, and EVI1 and truncated EVI1, of which EVI1 is a transcriptional factor with specific DNA-binding sites.45,46 It upregulated Pbx1 transcription, which was involved in maintaining hematopoietic stem/progenitors and leukemogenesis.47 EVI1 stimulated Spi1 transcription with a DNA-binding function that mediates the hematopoiesis program.14 In addition, EVI1 was reported to increase the CSC factors SOX2, NANOG, c-MYC, and EMT-associated signal pathways in nasopharyngeal carcinoma.15 In the present study, we found that MECOM promoted CSC properties, including the increased spheroid formation of self-renewal and expression of CSC-associated factors, including CD133, CD44, SOX2, and ABCG2. Furthermore, we found that EVI1 could bind to sequences within the SOX2 promoter region and stimulate its transcription.
Based on our analysis, the ratio of LUSC cases with MECOM amplification was found to be approximately 90%, and high MECOM protein expression levels were associated with poor outcomes in LUSC patients. Hence, MECOM, as a potential target, could be administered in our oncogene-depleting system for most LUSC cases. Since there is no available inhibitor for MECOM as a transcription factor located in the nucleus, the CRISPR system could be a promising tool by which to block this driver gene.48 To induce specificity and reduce immunogenicity for the ADV vector under intravenous administration, we constructed an adaptor and a protector, forming an ADV/protein complex. As we expected, this complex system effectively retargeted ADV to tumor cells with high EpCAM expression, significantly decreased the off-target response, and obviously overcame the liver toxicity of ADV. The adaptor against EpCAM could be replaced with other scFv fragments against membrane markers in malignant cancers. Moreover, the protector protein dramatically antagonized the antibody-dependent neutralization of ADV to maintain infection efficiency, similar to or better than the performance of polymers such as polyethylene glycol.12
Overall, MECOM depletion hampers the tumor growth of LUSC, and this gene stimulates CSC characteristics by activating SOX2′ transcription and upregulating the CSC-associated factors. CRISPR-SaCas9-mediated MECOM depletion via the ADV/adaptor/protector system may be a potentially therapeutic strategy for LUSC with both high efficiency and specificity.
Materials and methods
LUSC clinical samples and cell lines
A total of 150 case samples using paraffin sections for the immunohistochemistry assay and 3 case samples from fresh tumor tissues and corresponding normal tissues for cell viability assay were collected from LUSC patients who had undergone resection at the Department of Thoracic Surgery II of the Peking University Cancer Hospital & Institute. Informed consent was obtained from all of the participants with the approval of the ethics committee in Peking University Cancer Hospital & Institute.
The LUSC cell lines H520 and SKMES-1 were obtained from the American Type Culture Collection, and EBC-1 was retrieved from Deutsche Sammlung von Mikroorganismen und Zellkulturen. These cells were maintained in RPMI-1640 or DMEM medium with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) in 5% CO2 at 37°C. After cleaning any mycoplasma contamination, the cell lines were authenticated using polymorphic short tandem repeat loci analyses.
Vector construction and virus packaging
The sequences of vector containing SaCas9-2A-GFP and sgRNA were synthesized from Sangon Biotech, and this vector was designated CMV (cytomegalovirus):SaCas9-2A-GFP; U6:BsaI-sgRNA. The Gateway recombination reaction was conducted using the donor vector (CMV:SaCas9-2A-GFP; U6:BsaI-sgRNA) flanked by attL sequences and the destination vector pLEX_305 (41390, Addgene) with attR sequences. For lentivirus packaging, this plasmid expressing the specific sgRNA and packaging plasmids VSVG and psPAX2 (8454 and 12260, Addgene) were transfected into HEK293T cells using calcium phosphate precipitation, followed by centrifuging the culture medium at 25,000 rpm and 4°C for 2 h. The lentivirus was collected and stored at −80°C for the in vitro assay.
The donor vector CMV:SaCas9-2A-GFP; U6:BsaI-sgRNA was inserted into the destination vector pAdeno-MCMV (Obio Technology) using the Gateway recombination reaction. The ADV and AAV2 packages were supplied using an Obio Technology assay for the in vivo assay.
The full length for MECOM was synthetized and cloned into lentiCRISPR v2 (52961, Addgene) using the restriction sites of AgeⅠ and BamHⅠ for the construction of the lentiviral vector. The MECOM-expressing vector as well as the VSVG and psPAX2 plasmids were transfected into HEK293T cells for lentivirus packaging.
CRISPR-SaCas9 screening
The PDX tumors, including LUSC012, LUSC019, and LUSC021, were subcutaneously injected into NOD-SCID mice. The formed tumors were cut into pieces and digested using a tumor dissociation kit (130-095-929, Miltenyi Biotec). Cell suspensions of the PDX tumor were then prepared using MACS SmartStrainers (130-098-462, Miltenyi Biotec). The most efficient sgRNA for the 38 driver genes was chosen using the Benchling tool (https://benchling.com). The LUSC PDX tumor cells was transfected with the lentivirus containing a specific sgRNA in the pLEX_305-CMV:SaCas9-2A-GFP; U6:BsaI-sgRNA vector for 48 h. Cell viability was then evaluated using a CellTiter-Glo 2.0 kit (Promega). The cell inhibition rate was calculated in the PDX tumor cells depleted with a specific driver gene and normalized to a non-targeting control.
Cell sorting
The fresh matched normal lung tissues from the three LUSC cases were dissociated into single-cell suspensions, according to the manufacturer’s guidelines for the tumor dissociation kit (130-095-929, Miltenyi Biotec). CD31 labeled fluorescein isothiocyanate (FITC) (E-AB-F1050C, Elabscience) was added into the cell suspensions for 30 min on ice. The CD31+ endothelial cells49 were sorted on a FACSAria II flow cytometer (BD Biosciences) and collected for further study.
Cell viability
Cell viability was assessed using a CellTiter-Glo 2.0 kit (Promega). The cell suspensions derived from the PDX tumors, primary tumors, paired normal lung tissues, and endothelial cells (CD31+) were planted into 96-well ultralow attachment plates and were infected with lentivirus for 48 h. The CellTiter-Glo Reagent was added with equal volume to the cell culture medium present in each well. The luminescent signal was evaluated in the Spark multimode microplate reader (Infinite 200 Pro, TECAN).
Target sequence and WES
DNA was extracted using the EasyPure Genomic DNA kit (TransGen Biotech). According to our previous study,22 target sequencing of a custom panel for 56 genes was performed on the BGI-Seq500 platform by the Beijing Genomics Institute (BGI).
The on-target and off-target mutations in the tumor tissues with MECOM depletion were analyzed by WES on the BGI-Seq500 platform using two 100-bp paired-end reads. Only reads that completely spanned the 35-bp window, 17 bp upstream and 17 bp downstream of the predicted target site, were retained by Samtools. SNVs and indels were identified using the Bamcount tool. Allele frequencies for the SNVs and indels were computed by counting every corresponding mutation type. Mutations with a frequency <2% were pooled (=“sum of alleles <2%”).
Reverse transcription and quantitative real-time-PCR
Total RNA was extracted from LUSC cells with the Trizol kit (Thermo Fisher Scientific). cDNA was obtained by reverse transcription and was subjected to quantitative real-time-PCR, using GoTaq qPCR Master Mix (A6001, Promega). The PCR primers used for this experiment are listed in Table S8. The expression level of the gene was normalized to that of the internal control (β-actin).
RNA sequencing
Altered genes in the H520 cells with forced expression of MECOM were screened using RNA sequencing. The total mRNA (≥200 ng) was extracted and applied to Novogene Corporation. A paired-end 150-bp sequencing strategy was performed using the state-of-the-art Illumina NovaSeq platforms. The gene expression level was normalized as RPKM (reads per kilobase of transcript per million mapped reads). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed by the Novogene Corporation.
Immunohistochemistry staining
A total of 150 paraffin section samples of LUSC tumor tissues were fixed with formalin. Briefly, the slides were blocked with normal goat serum and stained with the primary antibodies of MECOM (28100002, Novus Biologicals), CD44 (15675-1-AP, Proteintech), and ABCG2 (27286-1-AP, Proteintech). After washing with phosphate-buffered saline (PBS), the secondary antibody labeled with horseradish peroxidase (HRP) from Sigma-Aldrich was incubated. All of the immunohistochemical images were captured with a Zyla 5.5 complementary metal oxide semiconductor (CMOS) camera attached to a Leica microscope (DM6000B). Expression levels were evaluated using the immunoreactive score (IRS) method.50,51 Scoring was reviewed in parallel by two experienced pathologists in a double-blind manner.
Flow cytometry analysis
Single cells were suspended in cold PBS buffer. The cell suspension was stained with the anti-human CD133 labeled with phycoerythrin (130-098-046, Miltenyi Biotec) and anti-human CD44 labeled with allophycocyanin (559942, BD Pharmingen). The cells stained with CD133 and CD44 or those cells with a GFP-expressing vector were analyzed using CytoFlex flow cytometry (Beckman Coulter). The results were analyzed using FlowJo VX software.
Spheroid formation
Single cells of LUSC were seeded in ultralow attachment plates. DMEM/F-12 serum-free medium with B27, EGF (20 ng/mL, Invitrogen), bFGF (20 ng/mL, Invitrogen), and 1% methylcellulose (Sigma-Aldrich) was used to culture spheroid for 2–3 weeks.
ChIP
A ChIP assay was carried out in H520 cells with forced expression of MECOM, according to the manufacturer’s instructions (Millipore). The cells were cross-linked by 1% formaldehyde for 10 min and then sonicated in the lysis buffer. Next, the lysed cells were incubated with a MECOM antibody or control immunoglobulin G (IgG) at 4°C overnight. The complex was rinsed, and DNA was purified using a PCR Purification Kit (Qiagen). The primers were used to amplify the specific sequence (Table S8).
Western blotting
A radioimmunoprecipitation assay (RIPA) buffer containing complete protease inhibitor cocktail (Roche) was used to extract proteins, followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After transferring the protein to polyvinylidene fluoride membranes, the proteins were blocked with 5% non-fat milk. The primary antibodies used in this experiment were as follows: MECOM (2593, Cell Signaling), CD44 (15675-1-AP, Proteintech), ABCG2 (27286-1-AP, Proteintech), SOX2 (3579S, Cell Signaling), and β-actin (66009-1, Proteintech). Signals were visualized using a chemiluminescence instrument (Millipore). The ImageJ 2× software was used for densitometric analyses.
Luciferase reporter assay
The wild-type or mutant-binding sequences within the SOX2′ promoters by EVI1 were cloned into the pGL3-Basic vector (Promega). The LUSC cell lines were transfected with wild-type or mutant-binding sequences using the Lipofectamine 3000 transfection kit (L3000015, Thermo Fisher Scientific). The activity of the pGL3-Basic firefly was assessed using the dual-luciferase reporter assay system (Promega), and Renilla was used as the normalized control.
T7 endonuclease I enzyme (T7EI) assay
Cells were transfected with specific sgRNA to deplete the genomic sequences of the target gene. After DNA was isolated from these cells, specific primers (Table S8) were used to amplify the target sequence by PCR. The product was digested by T7 endonuclease I enzyme (NEB). The efficiency of the targeted editing disrupted by the specific sgRNA was evaluated by quantifying the electrophoresis results using ImageJ 2× software.
Protein purification
Nucleotide sequences of the adaptor (CFS: a humanized scFv against EpCAM, the ectodomain of CXADR, and a phage T4 fibritin polypeptide) and protector (HF: a humanized scFv against hexon and a phage T4 fibritin polypeptide) were synthesized and cloned into the pcDNA3.1 with a His tag. The CFS and HF proteins were purified by Applied Biological Materials. In brief, vectors with CFS or HF were transfected into HEK293FT and the proteins in the culture medium were purified using nickel-nitrilotriacetic acid (NTA) affinity chromatography. The sequences for the CFS and HF proteins were applied in Table S9.
Enzyme-linked immunosorbent assay (ELISA)
A total of 7.5 × 107 ADV vectors were immobilized and blocked. The immobilized vectors were incubated with sequentially diluted human and mouse sera for 2 h, followed by incubation with HRP-conjugated goat anti-human and anti-mouse antibodies (CWBIO, 1:2,000). Then the assay was developed with a tetramethylbenzidine substrate (CWBIO), and the optical density was measured at 450 nm.
Establishment of LUSC xenografts and treatment
The animal studies were approved by the ethics committee of the Peking University Cancer Hospital & Institute. NOD-SCID and C57BL/6N mice were obtained from Vitalriver. Human LUSC cell line H520 or PDX tumors were subcutaneously injected into NOD-SCID mice, and the mouse lung cancer cell line LEWIS was subcutaneously inoculated into C57BL/6N mice for xenograft establishment. In addition, the orthotopic model was established in the lung of NOD-SCID mice using H520 cells with the forced expression of MECOM and firefly luciferase. When the tumor volume reached 50–200 mm, the mice were used for further experiments.
NOD-SCID mice with H520 tumors and C57BL/6N mice with LEWIS tumors were treated with ADV (1.0 × 1010 viral particle [VP]) and CFS and HF protein complex (1.0 × 10−7 pmol/VP) via the tail vein to analyze the distribution, specificity, and immunogenicity of ADV. To determine the therapeutic potential of ADV (0.7 × 1010 VP) and protein complex (1.0 × 10−7 pmol/VP), 2–4 rounds (12–14 days intervals) of the complex were administered by intratumoral or intravenous injection.
Bioluminescent imaging (BI)
The tumor, liver, lung, and kidney were isolated from NOD-SCID mice treated with ADV expressing GFP and protein complex for 48 h, and the GFP signals were obtained for bioluminescent analysis. In addition, orthotopic tumors in the lung of NOD-SCID mice were recorded by BI, using firefly d-luciferin (200 mg/kg, PerkinElmer), which was intraperitoneally injected into the mice. An IVIS Lumina system (Xenogen Corporation) was used to capture the bioluminescent images. Live Image data acquisition software from Xenogen Corporation was used to analyze and quantify the bioluminescence signal.
Statistical analysis
IBM SPSS Statistics version 23 or GraphPad Prism 8 software were used to perform statistical analysis, and data are shown as the means ± standard deviations. Student’s t test and one-way ANOVA were used to assess two groups and more than two groups, respectively. The Kaplan-Meier method with a log rank test was used to compare results from survival analyses. Spearman rank correlation was done to analyze the relationship between MECOM and other gene expression. p < 0.05 was considered statistically significant.
Data availability
The data that support the findings of this study have been deposited into the CNGB Sequence Archive (CNSA)52 of the China National GeneBank DataBase (CNGBdb)53 with the accession number CNP0000324 (https://db.cngb.org/).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81972842 and 81903159), China; the Beijing Natural Science Foundation (7212009), China; the Beijing Nova Program (Z201100006820092) from Beijing Municipal Science & Technology Commission, China; the Capital’s Funds for Health Improvement and Research (2020-2-2154), China; the Hospital’s Ascent Plan (DFL20191101), China; the Beijing Municipal Administration of Hospitals’ Youth Program (QMS20191107), China; the Beijing Excellent Individual Program from Beijing Municipal Organization Department (2018000021469G270), China; the Guangdong Provincial Academician Workstation of BGI Synthetic Genomics (no. 2017B090904014), China; the Guangdong Provincial Key Laboratory of Genome Read and Write (no. 2017B030301011), China; and the Guangdong Enterprise Key Laboratory of Human Disease Genomics (2020B1212070028), China. We would like to thank the China National GeneBank for providing sequencing services for this project. We would like to thank Dr. Liang Chen from Wuhan University for his valuable suggestions on this manuscript.
Author contributions
Y.M., B.K., Y.G., Y.Y., and N.W. conceptualized the study; Y.M., B.K., S.L., J.B., G.A., B.L., J.L., X.X., and H.Y., performed the experiments; G.X., F.L., and Y.S. performed the bioinformatics analysis; N.W., Y.Y., Y.G., Y.M., B.K., S.L., and G.X. wrote the original draft; all of the authors approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.06.011.
Contributor Information
Yue Yang, Email: zlyangyue@bjmu.edu.cn.
Ying Gu, Email: guying@genomics.cn.
Nan Wu, Email: nanwu@bjmu.edu.cn.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch F.R., Scagliotti G.V., Mulshine J.L., Kwon R., Curran W.J., Jr., Wu Y.L., Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311. doi: 10.1016/S0140-6736(16)30958-8. [DOI] [PubMed] [Google Scholar]
- 3.Behan F.M., Iorio F., Picco G., Gonçalves E., Beaver C.M., Migliardi G., Santos R., Rao Y., Sassi F., Pinnelli M., et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568:511–516. doi: 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 4.Gandara D.R., Hammerman P.S., Sos M.L., Lara P.N., Jr., Hirsch F.R. Squamous cell lung cancer: from tumor genomics to cancer therapeutics. Clin. Cancer Res. 2015;21:2236–2243. doi: 10.1158/1078-0432.CCR-14-3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murty T., Mackall C.L. Gene editing to enhance the efficacy of cancer cell therapies. Mol. Ther. 2021;29:3153–3162. doi: 10.1016/j.ymthe.2021.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon A.R., Hong J., Kim S.W., Yun C.O. Redirecting adenovirus tropism by genetic, chemical, and mechanical modification of the adenovirus surface for cancer gene therapy. Expert Opin. Drug Deliv. 2016;13:843–858. doi: 10.1517/17425247.2016.1158707. [DOI] [PubMed] [Google Scholar]
- 9.Kalyuzhniy O., Di Paolo N.C., Silvestry M., Hofherr S.E., Barry M.A., Stewart P.L., Shayakhmetov D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:5483–5488. doi: 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dreier B., Honegger A., Hess C., Nagy-Davidescu G., Mittl P.R.E., Grütter M.G., Belousova N., Mikheeva G., Krasnykh V., Plückthun A. Development of a generic adenovirus delivery system based on structure-guided design of bispecific trimeric DARPin adapters. Proc. Natl. Acad. Sci. USA. 2013;110:E869–E877. doi: 10.1073/pnas.1213653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kashentseva E.A., Seki T., Curiel D.T., Dmitriev I.P. Adenovirus targeting to c-erbB-2 oncoprotein by single-chain antibody fused to trimeric form of adenovirus receptor ectodomain. Cancer Res. 2002;62:609–616. [PubMed] [Google Scholar]
- 12.Schmid M., Ernst P., Honegger A., Suomalainen M., Zimmermann M., Braun L., Stauffer S., Thom C., Dreier B., Eibauer M., et al. Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat. Commun. 2018;9:450. doi: 10.1038/s41467-017-02707-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang B., Wang J. EVI1 in leukemia and solid tumors. Cancers (Basel) 2020;12:2667. doi: 10.3390/cancers12092667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayoub E., Wilson M.P., McGrath K.E., Li A.J., Frisch B.J., Palis J., Calvi L.M., Zhang Y., Perkins A.S. EVI1 overexpression reprograms hematopoiesis via upregulation of Spi1 transcription. Nat. Commun. 2018;9:4239. doi: 10.1038/s41467-018-06208-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Y., Liang Y., Zheng X., Deng X., Huang W., Zhang G. EVI1 promotes epithelial-to-mesenchymal transition, cancer stem cell features and chemo-/radioresistance in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2019;38:82. doi: 10.1186/s13046-019-1077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshimi A., Goyama S., Watanabe-Okochi N., Yoshiki Y., Nannya Y., Nitta E., Arai S., Sato T., Shimabe M., Nakagawa M., et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood. 2011;117:3617–3628. doi: 10.1182/blood-2009-12-261602. [DOI] [PubMed] [Google Scholar]
- 17.Paredes R., Kelly J.R., Geary B., Almarzouq B., Schneider M., Pearson S., Narayanan P., Williamson A., Lovell S.C., Wiseman D.H., et al. EVI1 phosphorylation at S436 regulates interactions with CtBP1 and DNMT3A and promotes self-renewal. Cell Death Dis. 2020;11:878. doi: 10.1038/s41419-020-03099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paredes R., Schneider M., Stevens A., White D.J., Williamson A.J.K., Muter J., Pearson S., Kelly J.R., Connors K., Wiseman D.H., et al. EVI1 carboxy-terminal phosphorylation is ATM-mediated and sustains transcriptional modulation and self-renewal via enhanced CtBP1 association. Nucleic Acids Res. 2018;46:7662–7674. doi: 10.1093/nar/gky536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurokawa M., Mitani K., Irie K., Matsuyama T., Takahashi T., Chiba S., Yazaki Y., Matsumoto K., Hirai H. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature. 1998;394:92–96. doi: 10.1038/27945. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka T., Nishida J., Mitani K., Ogawa S., Yazaki Y., Hirai H. Evi-1 raises AP-1 activity and stimulates c-fos promoter transactivation with dependence on the second zinc finger domain. J. Biol. Chem. 1994;269:24020–24026. doi: 10.1016/s0021-9258(19)51041-9. [DOI] [PubMed] [Google Scholar]
- 21.Sachdeva M., Sachdeva N., Pal M., Gupta N., Khan I.A., Majumdar M., Tiwari A. CRISPR/Cas9: molecular tool for gene therapy to target genome and epigenome in the treatment of lung cancer. Cancer Gene Ther. 2015;22:509–517. doi: 10.1038/cgt.2015.54. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P., Kang B., Xie G., Li S., Gu Y., Shen Y., Zhao X., Ma Y., Li F., Si J., et al. Genomic sequencing and editing revealed the GRM8 signaling pathway as potential therapeutic targets of squamous cell lung cancer. Cancer Lett. 2019;442:53–67. doi: 10.1016/j.canlet.2018.10.035. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Y., Ruan Z., Lin Z., Long H., Zhao R., Sun B., Cheng L., Tang L., Xia Z., Li C., Zhao S. The association between CD31(hi)Emcn(hi) endothelial cells and bone mineral density in Chinese women. J. Bone Miner. Metab. 2019;37:987–995. doi: 10.1007/s00774-019-01000-4. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y., Dong X., Shirazi J., Gleghorn J.P., Lingappan K. Pulmonary endothelial cells exhibit sexual dimorphism in their response to hyperoxia. Am. J. Physiol. Heart Circ. Physiol. 2018;315:H1287–H1292. doi: 10.1152/ajpheart.00416.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu X., Liu S., Ji X. Overexpression of ecotropic viral integration site-1 is a prognostic factor of lung squamous cell cancer. Onco Targets Ther. 2017;10:2739–2744. doi: 10.2147/OTT.S132410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguirre A.J., Meyers R.M., Weir B.A., Vazquez F., Zhang C.Z., Ben-David U., Cook A., Ha G., Harrington W.F., Doshi M.B., et al. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016;6:914–929. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang C., Wang H., Wang X., Zhao C., Wang H. CD44, a marker of cancer stem cells, is positively correlated with PD-L1 expression and immune cells infiltration in lung adenocarcinoma. Cancer Cell Int. 2020;20:583. doi: 10.1186/s12935-020-01671-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eramo A., Lotti F., Sette G., Pilozzi E., Biffoni M., Di Virgilio A., Conticello C., Ruco L., Peschle C., De Maria R., et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 29.Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene. 2007;396:346–357. doi: 10.1016/j.gene.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 30.Tsukahara T., Nabeta Y., Kawaguchi S., Ikeda H., Sato Y., Shimozawa K., Ida K., Asanuma H., Hirohashi Y., Torigoe T., et al. Identification of human autologous cytotoxic T-lymphocyte-defined osteosarcoma gene that encodes a transcriptional regulator, papillomavirus binding factor. Cancer Res. 2004;64:5442–5448. doi: 10.1158/0008-5472.CAN-04-0522. [DOI] [PubMed] [Google Scholar]
- 31.Queisser A., Hagedorn S., Wang H., Schaefer T., Konantz M., Alavi S., Deng M., Vogel W., von Massenhausen A., Kristiansen G., et al. Ecotropic viral integration site 1, a novel oncogene in prostate cancer. Oncogene. 2017;36:1573–1584. doi: 10.1038/onc.2016.325. [DOI] [PubMed] [Google Scholar]
- 32.Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–360. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- 33.Raum T., Gruber R., Riethmüller G., Kufer P. Anti-self antibodies selected from a human IgD heavy chain repertoire: a novel approach to generate therapeutic human antibodies against tumor-associated differentiation antigens. Cancer Immunol. Immunother. 2001;50:141–150. doi: 10.1007/pl00006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pak M.G., Shin D.H., Lee C.H., Lee M.K. Significance of EpCAM and TROP2 expression in non-small cell lung cancer. World J. Surg. Oncol. 2012;10:53. doi: 10.1186/1477-7819-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niu Y., Yang X., Chen Y., Jin X., Li L., Guo Y., Li X., Xie Y., Zhang Y., Wang H. EVI1 induces autophagy to promote drug resistance via regulation of ATG7 expression in leukemia cells. Carcinogenesis. 2020;41:961–971. doi: 10.1093/carcin/bgz167. [DOI] [PubMed] [Google Scholar]
- 36.Hinai A.A., Valk P.J.M. Review: aberrant EVI1 expression in acute myeloid leukaemia. Br. J. Haematol. 2016;172:870–878. doi: 10.1111/bjh.13898. [DOI] [PubMed] [Google Scholar]
- 37.Glass C., Wilson M., Gonzalez R., Zhang Y., Perkins A.S. The role of EVI1 in myeloid malignancies. Blood Cells Mol. Dis. 2014;53:67–76. doi: 10.1016/j.bcmd.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Wu L., Wang T., He D., Li X., Jiang Y. EVI1 acts as an oncogene and positively regulates calreticulin in breast cancer. Mol. Med. Rep. 2019;19:1645–1653. doi: 10.3892/mmr.2018.9796. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Wang H., Schaefer T., Konantz M., Braun M., Varga Z., Paczulla A.M., Reich S., Jacob F., Perner S., Moch H., et al. Prominent oncogenic roles of EVI1 in breast carcinoma. Cancer Res. 2017;77:2148–2160. doi: 10.1158/0008-5472.CAN-16-0593. [DOI] [PubMed] [Google Scholar]
- 40.Li H., Xiao N., Wang Y., Wang R., Chen Y., Pan W., Liu D., Li S., Sun J., Zhang K., et al. Smurf1 regulates lung cancer cell growth and migration through interaction with and ubiquitination of PIPKIγ. Oncogene. 2017;36:5668–5680. doi: 10.1038/onc.2017.166. [DOI] [PubMed] [Google Scholar]
- 41.Dutta P., Bui T., Bauckman K.A., Keyomarsi K., Mills G.B., Nanjundan M. EVI1 splice variants modulate functional responses in ovarian cancer cells. Mol. Oncol. 2013;7:647–668. doi: 10.1016/j.molonc.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng X., Cao Y., Liu Y., Li F., Sambandam K., Rajaraman S., Perkins A.S., Fields A.P., Hellmich M.R., Townsend C.M., Jr., et al. Overexpression of Evi-1 oncoprotein represses TGF-beta signaling in colorectal cancer. Mol. Carcinog. 2013;52:255–264. doi: 10.1002/mc.21852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen C.H., Grandits A.M., Vassiliou G.S., Staber P.B., Heller G., Wieser R. Evi1 counteracts anti-leukemic and stem cell inhibitory effects of all-Trans retinoic acid on Flt3-ITD/Npm1c-driven acute myeloid leukemia cells. Biomedicines. 2020;8:385. doi: 10.3390/biomedicines8100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan L.S., Lung H.L., Ngan R.K.C., Lee A.W.M., Tsao S.W., Lo K.W., Kahn M., Lung M.L., Wieser R., Mak N.K. Role of miR-96/EVI1/miR-449a Axis in the nasopharyngeal carcinoma cell migration and tumor sphere formation. Int. J. Mol. Sci. 2020;21:5495. doi: 10.3390/ijms21155495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lopingco M.C., Perkins A.S. Molecular analysis of Evi1, a zinc finger oncogene involved in myeloid leukemia. Curr. Top. Microbiol. Immunol. 1996;211:211–222. doi: 10.1007/978-3-642-85232-9_21. [DOI] [PubMed] [Google Scholar]
- 46.Funabiki T., Kreider B.L., Ihle J.N. The carboxyl domain of zinc fingers of the Evi-1 myeloid transforming gene binds a consensus sequence of GAAGATGAG. Oncogene. 1994;9:1575–1581. [PubMed] [Google Scholar]
- 47.Shimabe M., Goyama S., Watanabe-Okochi N., Yoshimi A., Ichikawa M., Imai Y., Kurokawa M., Yoshimi A. Pbx1 is a downstream target of Evi-1 in hematopoietic stem/progenitors and leukemic cells. Oncogene. 2009;28:4364–4374. doi: 10.1038/onc.2009.288. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H., Qin C., An C., Zheng X., Wen S., Chen W., Liu X., Lv Z., Yang P., Xu W., et al. Application of the CRISPR/Cas9-based gene editing technique in basic research, diagnosis, and therapy of cancer. Mol. Cancer. 2021;20:126. doi: 10.1186/s12943-021-01431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crouch E.E., Doetsch F. FACS isolation of endothelial cells and pericytes from mouse brain microregions. Nat. Protoc. 2018;13:738–751. doi: 10.1038/nprot.2017.158. [DOI] [PubMed] [Google Scholar]
- 50.Fedchenko N., Reifenrath J. Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue - a review. Diagn. Pathol. 2014;9:221. doi: 10.1186/s13000-014-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koerdt S., Siebers J., Bloch W., Ristow O., Kuebler A.C., Reuther T. Immunohistochemial study on the expression of von Willebrand factor (vWF) after onlay autogenous iliac grafts for lateral alveolar ridge augmentation. Head Face Med. 2013;9:40. doi: 10.1186/1746-160X-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo X., Chen F., Gao F., Li L., Liu K., You L., Hua C., Yang F., Liu W., Peng C., et al. CNSA: a data repository for archiving omics data. Database (Oxford) 2020;2020:baaa055. doi: 10.1093/database/baaa055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen F.Z., You L.J., Yang F., Wang L.N., Guo X.Q., Gao F., Hua C., Tan C., Fang L., Shan R.Q., et al. CNGBdb: China National GeneBank DataBase. Yi Chuan. 2020;42:799–809. doi: 10.16288/j.yczz.20-080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study have been deposited into the CNGB Sequence Archive (CNSA)52 of the China National GeneBank DataBase (CNGBdb)53 with the accession number CNP0000324 (https://db.cngb.org/).