

Abstract
Reactive oxygen species (ROS) derived from oxygen-dependent mitochondrial metabolism are the essential drivers of cardiomyocyte (CM) cell-cycle arrest in adulthood. Mitochondria-localized circular RNAs (circRNAs) play important roles in regulating mitochondria-derived ROS production, but their functions in cardiac regeneration are still unknown. Herein, we investigated the functions and underlying mechanism of mitochondria-localized circSamd4 in cardiac regeneration. We found that circSamd4 was selectively expressed in fetal and neonatal CMs. The transcription factor Nrf2 controlled circSamd4 expression by binding to the promoter of circSamd4 host gene. CircSamd4 overexpression reduced while circSamd4 silenced increased mitochondrial oxidative stress and subsequent oxidative DNA damage. Moreover, circSamd4 overexpression induced CM proliferation and prevented CM apoptosis, which reduced the size of the fibrotic area and improved cardiac function after myocardial infarction (MI). Mechanistically, circSamd4 reduced oxidative stress generation and maintained mitochondrial dynamics by inducing the mitochondrial translocation of the Vcp protein, which downregulated Vdac1 expression and prevented the mitochondrial permeability transition pore (mPTP) from opening. Our findings suggest that circSamd4 is a novel therapeutic target for heart failure after MI.
Keywords: circRNA, cardiac regeneration, mitochondria, ROS, myocardial infarction
Graphical abstract
A mitochondria-located circRNA Samd4 promotes CM cell-cycle progression and inhibits CM apoptosis by preventing the mitochondrial permeability transition pore from opening and repressing mitochondrial ROS production in CMs, providing a valuable therapeutic target for improving prognosis after myocardial infarction.
Introduction
Reactive oxygen species (ROS) signaling is a critical regulator of cardiac regeneration that potentially offers innovative strategies for preventing or treating heart failure after myocardial infarction (MI). One of many factors shared by organisms capable of cardiac regeneration is a hypoxic environment accompanied by relatively low ROS levels in cardiomyocytes (CMs).1 In contrast, adult mammalian CMs exist in an oxygen-rich environment, which triggers an increase in intracellular ROS production.2 Excessive ROS are thought to cause cellular toxicity by inducing oxidative damage to DNA, such as oxidized bases and DNA strand breaks, leading to adult CM cell-cycle arrest and even CM death or senescence. The systemic scavenging of ROS or DNA damage response (DDR) inhibition has been confirmed to induce postnatal CM cell-cycle re-entry, indicating the therapeutic potential of targeting ROS metabolism to induce cardiac repair.2,3 However, clinical studies on general antioxidant therapies have failed to reveal any beneficial effects in that regard. Thus, it is necessary to explore novel targets for antioxidant therapy to effectively restore cardiac function and prevent heart failure post MI.
It is known that intracellular ROS are generated in multiple compartments via the contributions of various macromolecules. Mitochondria, the core organelles of cell metabolism and redox balance, are a major source of ROS production in adult CMs.4 Nearly 90% of ROS, including mitochondrial superoxide, which is believed to be largely responsible for DNA damage, are estimated to be generated by the leakage of electrons from mitochondria. Excessive mitochondrial ROS generation in CMs has been causally linked to cell-cycle arrest and disease pathophysiology. Interestingly, accumulating evidence shows that other sources of ROS could also play protective and signaling roles in the heart. A recent study reported that nicotinamide adenine dinucleotide phosphate (NADPH) oxidases from the epicardium produce H2O2 to recruit inflammatory leukocytes during heart regeneration.5 Additionally, NADPH-induced oxidative stress in endothelial cells mediates vascular endothelial growth factor (VEGF)-induced endothelial cell activation and angiogenesis.6 The different functions of mitochondrial ROS and other sources of ROS indicate that precisely scavenging mitochondria-derived ROS might improve the efficacy of antioxidant therapy and promote cardiac regeneration. Despite the clinical significance, scavengers targeting mitochondria-derived ROS for cardiac regeneration are still lacking.
Circular RNAs (circRNAs) are a newly identified class of noncoding RNAs that circularize by joining at their 3′ and 5′ ends.7 It is known that circRNAs are more likely to be stable than their parent linear transcripts due to their structure.8 Emerging studies have proposed that circRNAs have fundamental roles in physiological and pathophysiological processes in the heart. Several circRNAs have also been reported to regulate CM cell-cycle progression, apoptosis, and survival by affecting oxidative stress injury after MI.9,10 Recent studies have demonstrated that mitochondria-localized circRNAs act as key regulators of mitochondrial function and dynamics. They also regulate mitochondrial ROS production by controlling mitochondrial membrane potential, indicating their potential role in alleviating the mitochondrial ROS burden.11,12 Moreover, these regulators tend to have few effects on nonmitochondrial ROS due to their special localization in cells.11 Therefore, targeting mitochondria-localized circRNAs might be an attractive strategy for promoting cardiac regenerative repair due to their unique effect on mitochondrial ROS. Herein, we hypothesized that mitochondria-localized circRNAs highly expressed in neonatal CMs promote cardiac regeneration by precisely scavenging mitochondria-derived ROS in CMs.
In line with this hypothesis, we first identified the mitochondria-localized circRNA Samd4 (circSamd4) as an important regulator of mitochondrial oxidative stress in CMs. Crucially, circSamd4 overexpression increased CM proliferation and induced structural and functional recovery after MI. Our results indicate that circSamd4 may serve as a valuable therapeutic target for promoting heart regenerative repair and improving the outcome after MI.
Results
CircSamd4 is located in CM mitochondria and involved in the antioxidant response
It is known that the heart of neonatal mouse possesses the ability to regenerate lost myocardium, partially mediated by the activation of antioxidant response.3 The comparison of transcriptome between neonatal CMs and adult CMs is likely to uncover the antioxidant factors involved in cardiac regeneration. To identify the potential circRNAs associated with antioxidant response during cardiac regeneration, we used previously published transcriptome datasets from mammalian hearts13 and selected 87 circRNAs that are homologously conserved and have more than 30 reads supporting a circular junction (Figure S1A). Among them, 13 circRNAs were differentially expressed in neonatal and adult mouse hearts (Figure 1A). The distinct tendencies of circular and linear transcripts suggest the important biological roles of circRNAs in cells.14 We further filtered four circRNAs with expression profiles that were opposite those of the linear transcripts: circSamd4, circHipk3, circRyr2, and circStrn3 (Figure 1A). With the combination of differential expression between adult and neonatal hearts, we speculated these four circRNAs were involved into antioxidant response during cardiac regeneration. Accordingly, we detected their expression in CMs treated with H2O2. We found that circSamd4 and circRyr2 were upregulated in CMs after treatment with 20 μM H2O2 (Figure 1B). We also investigated the expression profiles of these circRNAs. Notably, qRT-PCR analyses showed that, compared with other circRNAs, circSamd4 and circRyr2 were highly expressed in mouse hearts (Figures 1C and S1B–S1D). Moreover, compared with the other circRNAs, circSamd4 was the most abundant circRNA in the mouse heart (Figure S1E). We also investigated the expression of these circRNAs in different types of heart cells, revealing that circSamd4 and circRyr2 were highly expressed in CMs (Figure S1F). In addition, we isolated cytosol, nucleus, and mitochondria fractions, and then investigated the subcellular distribution of circSamd4 in CMs by qRT-PCR assays. CircHipk3, circRyr2, and circStrn3 was also detected in cytoplasm and mitochondria extracts of CM. Interestingly, we revealed the predominant expression of circSamd4 in the mitochondria of CMs (Figures 1D and S1G). We also performed in situ hybridization staining of circSamd4, circHipk3, circRyr2, and circStrn3 in CMs ex vivo. The colocalization of circSamd4 and mitochondria in CMs was observed (Figures 1E, S1H, and S1I). The in situ hybridization staining of circSamd4 in heart tissue also revealed the colocalization of circSamd4 and mitochondria (Figure S1J). These results suggest that circSamd4 is a mitochondria-localized circRNA and that its expression is associated with the antioxidant response in CMs. We subsequently focused on circSamd4 for detailed expression profiling and functional investigation.
Figure 1.

A mitochondria-localized circRNA, circSamd4, is highly expressed in the neonatal heart
(A) Heatmap of conserved circRNAs that were differentially expressed in adult and neonatal rat hearts and their linear transcripts. Changes in the expression profile of circRNAs were opposite to those in the linear transcripts. (B) The relative expression levels of the indicated circRNAs in P1 CMs treated with 20 μM H2O2 or PBS for 8 h; ∗p < 0.05, n = 6. (C) qPCR analysis showing the circSamd4 expression levels in multiple tissues of neonatal mice; ∗p < 0.05 versus heart, n = 6. (D) The indicated circRNAs were detected in cytoplasm and mitochondria extract of CM. Gapdh and mt-Co2 was used as the cytoplasm and mitochondria marker, respectively. ∗p < 0.05, n = 6. (E) RNA-fluorescence ISH (FISH) assay of circSamd4 and coimmunostaining of Cox4 in P1 CMs. (F) Amplification of circSamd4 in cDNA but not genomic DNA using divergent primers. gDNA, genomic DNA. (G) Comparison of circSamd4 and Samd4 mRNA expression levels by RNase R digestion analysis; ∗p < 0.05 versus mock treatment, n = 6. (H) Detection of the expression levels of circSamd4 in neonatal and adult mouse hearts using qRT-PCR assays; ∗p < 0.05, n = 6. (I) Detection of the expression levels of circSamd4 in adult, P7, and P1 mouse CMs using qRT-PCR assays; ∗p < 0.05, n = 6. (J) ISH assays showing circSamd4 expression in the rat and mouse hearts; ∗p < 0.05, n = 6. (K) ChIP-seq analysis of H3K27ac at the promoter of the circSamd4 host gene. TSS, transcription start site. (L) ChIP-PCR assays detecting the H3K27ac levels in the promoter of the circSamd4 host gene; ∗p < 0.05 versus anti-IgG, n = 6. (M) Detection of the expression levels of the circSamd4 pre-mRNA and Samd4 mRNA in P1 CMs after Nrf2 knockdown or control treatment using qRT-PCR assays; ∗p < 0.05 versus negative control siRNA (si-NC), n = 6. (N) The predicted transcription factor (TF) motif of Nrf2 and binding sites on the circSamd4 host gene promoter, which was provided by the JASPAR database. (O) ChIP-PCR assays detecting the binding of Nrf2 to the promoter of the circSamd4 host gene; ∗p < 0.05 versus anti-IgG, n = 6. (P and Q) Detection of cytosolic (P) and mitochondrial (Q) ROS in P1 CMs by confocal microscopy; ∗p < 0.05 versus si-NC, n = 6.
CircSamd4 is derived from the third exon of the Samd4 gene (Figure S2A). The sequence of circSamd4 is highly conserved among the human, mouse, and rat genomes (Figures S2A–S2C). The circRNA ID of circSamd4 in the circBase database (circBase: http://www.circbase.org/) is mmu_circ_0000529. The predicted head-to-tail junction of circSamd4 was validated in CMs by a PCR assay and subsequent Sanger sequencing (Figures 1F and S2A). RNase R digestion analysis also supported the circularized structure of circSamd4 (Figure 1G). In accordance with the RNA-seq results, qRT-PCR assays confirmed that circSamd4 expression is significantly decreased during heart development (Figure 1H). We then isolated CMs from P1, P7, and adult hearts and found that the circSamd4 expression in P1 CMs was higher than that in P7 or adult CMs (Figure 1I). Furthermore, cardiac circSamd4 expression was validated in rats and mice of different ages via in situ hybridization (ISH) assays. We found that circSamd4 expression was approximately 3-fold higher in the neonatal stage than in the adult stage (Figure 1J). These results also indicate that the expression level of circSamd4 might be dynamically downregulated in CMs over time.
It is known that posttranslational modifications of histones mediate several important biological processes, including chromatin modification, which affects the expression or repression of target genes.15 The dynamics of H3K27ac in the DNA regulatory element are a fundamental issue controlling the precise timing and level of gene transcription. Moreover, a previous study revealed that H3K27ac could be a marker for active promoters, which supports the transcriptional process and the high expression of circRNAs.16 Consistent with the reduced circSamd4 expression in the myocardium with age, both chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) and ChIP-PCR assays showed that the levels of the active histone marker H3K27ac at the Samd4 promoter were obviously decreased (Figures 1K and 1L). A previous study confirmed that the transcription factor Nrf2 is involved in the antioxidant response during cardiac repair.3 qRT-PCR analysis revealed that circSamd4 was downregulated in CMs after Nrf2 knockdown (Figure 1M). We also detected the changes of circSamd4 pre-mRNA and Samd4 mRNA expression after Nrf2 knockdown (Figure 1M). We found that Nrf2 knockdown significantly reduced the expression level of circSamd4 pre-mRNA and Samd4 mRNA, which indicated that Nrf2 might bind to the promoter of circSamd4 host gene and controls the Samd4 transcription, thereby regulating the expression the circSamd4 indirectly. Bioinformatics analysis using the JASPAR program (JASPAR: http://jaspar.genereg.net) revealed two binding sites of Nrf2 on the circSamd4 promoter (Figure 1N). ChIP-PCR assays further confirmed the interaction between Nrf2 and the circSamd4 host gene promoter (Figure 1O). Then, we detected the expression level of Nrf2 in CMs of different developmental stages. The western blot results revealed that the Nrf2 expression level was significantly higher in P1 CMs than in P7 or adult CMs (Figure S3A). To further investigate the effect of Nrf2 overexpression, adeno-associated virus 9 (AAV9)-Nrf2 was injected into the myocardial tissue of adult mice (Figure S3B). Nrf2 overexpression increased the circSamd4 expression and the extent of CM proliferation (Figures S3C–S3E). These results suggest that circSamd4 expression is controlled by Nrf2.
CircSamd4 reduces oxidative stress and promotes CM proliferation in vitro
To explore the effect of circSamd4 on the antioxidant response, we designed two siRNAs specifically targeting the circSamd4 back-splicing site. qRT-PCR assays confirmed that the transfection of the siRNAs significantly reduced circSamd4 expression (Figure S4A), but had no obvious effect on Samd4 mRNA expression. The intracellular ROS production in CMs transfected with circSamd4 siRNAs was significantly upregulated compared with that in CMs transfected with the control siRNA (Figure 1P). We also quantified cellular oxidative stress by 2',7'-Dichlorodihydrofluorescein diacetate (DCFH-DA) assay using flow cytometry, and the results also revealed that circSamd4 knockdown increased oxidative stress in CMs (Figure S4B). ROS levels have been shown to influence the expression of key genes involved in regulating cellular and systemic oxidative stress.17 We performed qRT-PCR assays to investigate the expression of these genes after circSamd4 knockdown, including Sod1, Sod2, Ucp3, Catalase, Ant1, and Gpx1. We found that circSamd4 knockdown significantly increased the expression of these genes (Figure S4C). Moreover, western blotting assay and immunofluorescence assay were used to detect phosphorylated ataxia telangiectasia mutated (pATM), an index of DNA damage caused by oxidative stress. It was shown that circSamd4 knockdown also significantly increased pATM level (Figures S4D–S4F). Immunofluorescence assay showed that another index of DNA damage, 8-hydroxyguanosine (8-OHG), was also significantly elevated in CMs after circSamd4 downregulation (Figure S4G). Previous studies demonstrated that mitochondria-localized circRNAs play important roles in mitochondrial functions and dynamics.11 We also found an increased mitochondria-derived ROS level after circSamd4 downregulation in CMs (Figure 1Q). We then investigated the effect of circSamd4 downregulation on mitochondrial permeability transition pore (mPTP) opening, which is crucial for mitochondrial membrane potential (MMP) maintenance and ROS generation in CMs. We recorded the mPTP opening after circSamd4 downregulation in vitro by detecting alterations in the JC-1 fluorescence signal over time (Figure S5A). The opening status of the mPTP was also assessed by the calcein-AM/CoCl2 method using confocal microscopy (Figure S5B). Downregulation of circSamd4 promoted mPTP opening in CMs (Figures S5A and S4B). We also transduced CMs with an adenovirus (Adv) vector to overexpress circSamd4 (Figures S6A and S6B). Northern blot analysis was performed to prove the successful formation of the back-splicing junction after transfection of circSamd4 overexpression vector. It was observed that Adv-circSamd4 transfection increased circSamd4 that was resistant to RNase R in CMs (Figure S6C). We also performed PCR amplification using the divergent primers, and the products were detected by agarose gel electrophoresis. The results also indicated the successful formation of back-splicing junction after circSamd4 overexpression plasmid transfection (Figure S6D). Functionally, circSamd4 overexpression significantly decreased the level of total cellular ROS as well as mitochondria-derived ROS production (Figures 2A–2C). We also examined the mitochondrial DNA content in CMs after circSamd4 overexpression. qRT-PCR assays showed that the value obtained by diving the mitochondrial DNA copy number by the nuclear DNA copy number was increased after H2O2 exposure, which was reversed by circSamd4 overexpression (Figure 2D). Next, we detected the mitochondrial crista density, a predictor of the oxygen uptake rate and metabolic power in mitochondria.18 Transmission electron microscopy revealed that the mitochondrial crista density was increased after H2O2 exposure but decreased after circSamd4 overexpression (Figure 2E). It is widely known that ROS are likely to cause widespread damage to nucleic acids. We therefore assessed and quantified oxidative DNA base modifications by detecting 8-OHG. We found that the nuclear 8-OHG level was significantly increased when CMs were treated with H2O2 and decreased by ectopic circSamd4 expression (Figure 2F). In concert, pATM, an index of DDR activation, was lower in the CMs overexpressing circSamd4 in combination with H2O2 than in those exposed to H2O2 only (Figure 2G). H2O2 induced mPTP opening and decreased the MMP, which was reversed by circSamd4 overexpression (Figures 2H and S6E). Moreover, TdT-mediated 2'-deoxyuridine 5'-triphosphate nick-end labeling (TUNEL) staining, trypan blue exclusion, and flow cytometry analysis of Annexin V-FITC/PI staining further indicated that circSamd4 overexpression could mitigate H2O2-induced apoptosis (Figures S6F–S6H). These findings indicate that circSamd4 reduces mitochondrial ROS generation and protects CMs from oxidative injury.
Figure 2.

CircSamd4 reduces mitochondrial ROS and oxidative injury in CMs
(A–C) CircSamd4 reduced the total cellular ROS (A) and mitochondrial ROS (B and C) levels in CMs. P1 CMs were transduced with Adv for 24 h and then incubated with H2O2 (20 μM) for 8 h. Total cellular ROS were detected by DCFH-DA staining. Mitochondrial ROS were detected by mitoSOX staining. ∗p < 0.05, n = 6. (D) CircSamd4 decreased the mitochondrial DNA content in P1 CMs treated with H2O2. ∗p < 0.05, n = 6. (E) CircSamd4 repressed mitochondrial crista maturation in P1 CMs treated with H2O2. The mitochondrial ultrastructure was detected by transmission electron microscopy. ∗p < 0.05, n = 6. (F and G) CircSamd4 alleviated DNA damage (F) and the DDR (G) induced by H2O2. ∗p < 0.05, n = 6. (H) CircSamd4 overexpression repressed the H2O2-induced decrease in the MMP. ∗p < 0.05, n = 6. JC-1 monomer, green; J-aggregate, red.
Next, we assessed the effect of circSamd4 on CM proliferation by detecting several cell-cycle markers, including 5-Ethynyl-2’-deoxyuridine (EdU), Ki67, pH3, and Aurora B. First, we explored whether circSamd4 is required for P1 CM proliferation in vitro. We found that circSamd4 knockdown decreased the proportion of P1 CMs expressing EdU, Ki67, and pH3 (Figures S7A–S7F). Then, we investigated whether circSamd4 overexpression could protect CMs from cell-cycle arrest under oxidative stress. H2O2 significantly decreased the proportion of P1 CMs expressing EdU, Ki67, pH3, and Aurora B, while circSamd4 overexpression reversed this repression (Figures 3A–3D). Importantly, the mitochondrial dye tetramethylrhodamine ethyl ester (TMRE) was used to capture time-lapse images of CMs after treatment. In the H2O2-only group, CMs hardly underwent cell division. In contrast, CMs overexpressing circSamd4 in combination with H2O2 underwent karyokinesis and cytokinesis (Figures 3E; Videos S1, S2, S3, and S4). Additionally, flow cytometry assays revealed that H2O2 prevented P1 CMs from entering the S and G2/M phases, while circSamd4 increased the percentage of CMs in the S and G2/M phases (Figure 3F). CircSamd4 also reversed the negative effect of Nrf2 downregulation on CM proliferation (Figures S3F and S3G). Moreover, circSamd4 overexpression significantly increased the proportion of P7 CMs expressing EdU, Ki67, pH3, and Aurora B (Figures S8A–S8D).
Figure 3.

CircSamd4 induces CM cell-cycle progression and proliferation in vitro
(A) Detection of P1 CM proliferation by an EdU incorporation assay. P1 CMs were transduced with Adv for 24 h and then incubated with H2O2 (20 μM) for 8 h. EdU+ CMs are indicated by arrows, ∗p < 0.05, n = 6. (B) Evaluation of P1 CM proliferative activity by Ki-67 immunostaining. Ki67+ CMs are indicated by arrows, ∗p < 0.05, n = 6. (C) Evaluation of P1 CM proliferative activity by pH3 immunostaining. ∗p < 0.05, n = 6. (D) Evaluation of P1 CM proliferative activity by Aurora B immunostaining. ∗p < 0.05, n = 6. (E) Representative images obtained from a time-lapse video of CM cell division. The arrows indicate CMs undergoing division events. Immunostaining of cTnT was then performed to determine whether the proliferating cells were CMs. ∗p < 0.05, n = 6. (F) Flow cytometry analysis of CMs undergoing cell-cycle events. ∗p < 0.05, n = 6.
The Adv vectors encoding for Samd4 mRNA and Samd4 pre-mRNA fragments were also used as the control. After transfection of these vectors, we detected circSamd4 and Samd4 mRNA level (Figure S9A). It was observed that vector encoding for circSamd4 specially overexpressed circSamd4 but not Samd4 mRNA, and vector encoding for Samd4 mRNA specially overexpressed Samd4 mRNA (Figure S9B). The vector with Samd4 pre-mRNA fragment containing exon3 and its flanking sequences also successfully overexpressed circSamd4 but not Samd4 mRNA (Figure S9B). Interestingly, we found that transfection of vector for Samd4 pre-mRNA fragment could reduce intracellular oxidative stress and increase cell-cycle markers, while transfection of vector for Samd4 mRNA had no significant effect (Figures S9C–S9G). These results further indicated that circSamd4, but not Samd4 mRNA, play an important role on antioxidative stress role in cardiac regeneration.
CircSamd4 overexpression induces adult CM proliferation in vivo
To investigate the pro-proliferative effect of circSamd4 in vivo, AAV9-circSamd4 was delivered to the myocardial tissues of adult mice to overexpress circSamd4 (Figures S10A and S10B). We also performed northern blot analysis and found that AAV9-circSamd4 infection increased circSamd4 that was resistant to RNase R in heart tissue (Figure S10C). By detecting GFP- and cTnT-colocalized cells, we found that AAV9 targeted CM with high efficiency and specificity, as determined by an exclusive GFP signal in approximately 80% of cells that were assigned as a CM by morphology and cTnT signals (Figure S10D). To further determine the rate of AAV9 infection in non-CMs, we isolated CMs and non-CMs from adult mouse hearts 4 weeks after AAV9-GFP injection, as described in a previous study.19 Immunostaining of cTnT, CD31, and vimentin was used to determine the individual cell types. The proportion of GFP+ CMs was approximately 40%, while that of GFP+ endothelial cells and fibroblasts was less than 10% (Figures S10E–S10G). Our results are consistent with previous studies reporting the CM-specific transduction of AAV9.20 To determine whether AAV9-circSamd4 induced CM-specific circSamd4 overexpression, we further detected circSamd4 expression in CMs and non-CMs isolated from the AAV9-circSamd4 and AAV9-negative control (NC) groups. The qRT-PCR assay results confirmed that AAV9-circSamd4 specifically increased circSamd4 expression in CMs but not in non-CMs (Figure S10H). Immunofluorescence assays revealed that circSamd4 overexpression significantly increased the proportion of adult CMs expressing proliferative markers, including Ki67, pH3, and Anillin, both in vivo and ex vivo (Figures 4A–4C and S11A–S11C). Moreover, circSamd4 overexpression decreased the intracellular ROS level and the number of 8-OHG sites (Figures 4D and 4E). Quantification analysis revealed an increase in the total number of CMs after circSamd4 overexpression (Figure S11D). We detected heart weight/body weight ratios at 4 weeks after the injection of AAV9-circSamd4 or AAV9-NC. Consistently, circSamd4 overexpression significantly increased heart weight/body weight ratios (Figure S11E). In addition, wheat germ agglutinin (WGA) staining revealed that circSamd4 overexpression had no effect on cardiac hypertrophy (Figure 4F).
Figure 4.

CircSamd4 overexpression reduces oxidative injury and promotes CM proliferation in vivo
(A) Evaluation of adult CM proliferation by Ki67 immunostaining. CMs were isolated from adult mouse hearts 14 days after AAV9-circSamd4 or AAV9-NC injection. ∗p < 0.05, n = 6. (B) Evaluation of adult CM proliferation by pH3 immunostaining. ∗p < 0.05, n = 6. (C) Evaluation of adult CM proliferation by Anillin immunostaining. ∗p < 0.05, n = 6. (D) Detection of intracellular ROS levels in the myocardium 14 days after AAV9-circSamd4 or AAV9-NC injection; ∗p < 0.05, n = 6. (E) Detection of oxidative DNA damage in the adult myocardium 14 days after AAV9-circSamd4 or AAV9-NC injection; ∗p < 0.05, n = 6. (F) WGA staining of adult mouse hearts 14 days after AAV9-circSamd4 or AAV9-NC injection. (G) Detection of Ki67+ adult CMs in the peri-infarcted area 14 days post MI. Ki67+ CMs are indicated by arrows, ∗p < 0.05, n = 6. (H) Detection of pH3+ adult CMs in the peri-infarcted area 14 days post MI; ∗p < 0.05, n = 6. (I) Detection of intracellular ROS levels in the peri-infarcted area 14 days post MI; ∗p < 0.05, n = 6. (J) Detection of oxidative DNA damage in adult CMs 14 days post MI; ∗p < 0.05, n = 6.
On the basis of these in vivo results, we explored whether circSamd4 overexpression induces adult cardiac repair after MI. We injected AAV9-circSamd4 and AAV9-NC into the peri-infarcted areas of MI model animals to overexpress circSamd4 (Figure S12A). CircSamd4 overexpression significantly increased the proportion of CMs expressing Ki67 and pH3 (Figures 4G and 4H). Moreover, upregulation of circSamd4 reduced the intracellular ROS production, the number of 8-OHG sites, and the DDR in the CMs of the infarct border zone (Figures 4I, 4J, and S11B). TUNEL staining revealed that circSamd4 also protected CMs from apoptosis (Figure S11C). We also found that circSamd4 overexpression significantly increased the heart weight/body weight ratios after MI (Figure S11D).
We also used AAV9 to overexpress Samd4 mRNA and Samd4 pre-mRNA fragment in myocardium tissue. Similar to the results from Adv transduction, it was observed that vector encoding for circSamd4 specially overexpressed circSamd4 but not Samd4 mRNA, and vector encoding for Samd4 mRNA specially overexpressed Samd4 mRNA (Figure S13A). The vector with Samd4 pre-mRNA fragment containing exon3 and its flanking sequences also successfully overexpressed circSamd4 but not Samd4 mRNA (Figure S13A). We subsequently studied the effect of circSamd4 overexpression on cardiac function improvement post MI. It was found that AAV9 overexpressing circSamd4 and AAV9 overexpressing Samd4 pre-mRNA fragments significantly restored cardiac functional post MI (Figures 5A and S13B). Furthermore, quantitative assessment of cardiac perfusion by positron emission tomography (PET)/computed tomography (CT) revealed that the infarct size in the AAV9-circSamd4 group was significantly reduced compared with that in the control group at 4 weeks after MI (Figure 5B). Histological examination by Masson trichrome staining revealed that, compared with the control treatment, circSamd4 overexpression significantly reduced the size of the cardiac fibrotic area after MI (Figure 5C).21 Triphenyltetrazolium chloride (TTC) staining further confirmed that the proportion of viable myocardium was higher in the AAV9-circSamd4 group than in the AAV9-NC group (Figure 5D).22
Figure 5.

CircSamd4 overexpression restores cardiac function after MI
(A) Evaluation of the left ventricular ejection fraction (LVEF), left ventricular end-systolic dimension (LVESD), left ventricular end-diastolic dimension (LVEDD), and fractional shortening (FS) post MI after circSamd4 overexpression. The echocardiography analysis was performed on days 1, 7, 14, and 28 after surgery. ∗p < 0.05 versus the sham group, #p < 0.05 versus the AAV9-NC group, and ampersand (&) indicates p < 0.05 versus the AAV9-Samd4 group, n = 6. (B) PET-CT of the adult mouse heart 28 days post MI; ∗p < 0.05 versus the AAV9-NC group, n = 6. (C) Masson trichrome staining of cross sections from adult mouse hearts at 28 days post MI. ∗p < 0.05 versus the AAV9-NC group, n = 6. (D) TTC staining of cross sections from adult mouse hearts at 28 days post MI. ∗p < 0.05 versus the AAV9-NC group, n = 6.
Collectively, these results indicate that circSamd4 overexpression could inhibit cardiac remodeling and improve the recovery of cardiac performance after MI.
CircSamd4 is required for cardiac regeneration in neonatal mice
To determine the important role of circSamd4 in cardiac regeneration in vivo, we injected Adv encoding a short hairpin RNA (shRNA) into the myocardia of P1 neonatal mice (Figure S14A). By detecting GFP fluorescence in isolated CMs, we confirmed that Adv infected the myocardial tissues of P1 neonatal mice with high efficiency (Figure S14B). Both qRT-PCR assays and ISH assays confirmed that, compared with mice treated with Adv-shRNA-control, neonatal mice treated with Adv-shRNA-CircSamd4 had successful downregulation of circSamd4 in the myocardial tissues (Figures S14C and S14D).
To determine whether the increase in ROS resulting from circSamd4 knockdown contributed to damaged cardiogenesis, we administered an ROS inhibitor, N-acetyl-L-cysteine (NAC), to circSamd4-knockdown neonates after MI. CircSamd4 knockdown led to the upregulation of oxidative stress and DNA damage levels in the peri-infarcted zone of neonates (Figures 6A and 6B). Accordingly, the number of proliferative CMs in the peri-infarcted area was reduced in the Adv-shRNA-circSamd4 group compared with the Adv-shRNA-control treated group (Figures 6C and 6D). Moreover, echocardiographic analysis showed that circSamd4 knockdown impaired cardiac functional recovery after MI (Figure 6E). Masson trichrome staining also revealed an increase in the size of the fibrotic area after circSamd4 knockdown (Figure 6F). Collectively, these findings demonstrate that circSamd4 functions in endogenous cardiac regeneration by regulating the antioxidant pathway.
Figure 6.

CircSamd4 downregulation increases oxidative injury and impairs regenerative repair in the neonatal mouse heart post MI
(A) Detection of intracellular ROS levels in the myocardium with dihydroethidium (DHE) 7 days post MI; ∗p < 0.05, n = 6. (B) Detection of oxidative DNA damage in neonatal CMs with 8-OHG staining 7 days post MI; ∗p < 0.05, n = 6. (C) In vivo evaluation of CM proliferation by Ki67 immunostaining 7 days post MI. Ki67+ CMs are indicated by arrows; ∗p < 0.05, n = 6. (D) In vivo evaluation of CM proliferation by pH3 immunostaining 7 days post MI. pH3+ CMs are indicated by arrows; ∗p < 0.05, n = 6. (E) Evaluation of left ventricular function 7 days post MI; ∗p < 0.05, n = 6. (F) Evaluation of the fibrotic area 7 days post MI; ∗p < 0.05, n = 6.
CircSamd4 reduces mitochondria-derived ROS by recruiting Vcp to the mitochondria
Mitochondria-derived circRNAs have been reported to regulate mitochondrial function by interacting with proteins.11 Thus, we conducted RNA pull-down assays to detect potential circSamd4-binding proteins in CMs using a biotin-labeled circSamd4-specific probe. The results of mass spectrometry analysis showed that Vcp is the most abundant protein that has been reported to affect mitochondrial functions in heart. Previous studies confirmed that it could be recruited to mitochondria and participate in several mitochondrial functions, such as promoting mPTP opening, increasing mitochondrial respiration capacity, leading to ATP synthesis and so on.23,24 Moreover, functional inhibition of Vcp induced cardiac dilation and dysfunction, indication the important role of Vcp in maintaining cardiac function and homeostasis.25 All things considered, we speculated that Vcp might be the downstream protein of circSamd4. The following western blotting assays confirmed that Vcp interacts with circSamd4 (Figure 7A; Table S3). Moreover, RNA immunoprecipitation (RIP) assays showed that anti-Vcp antibodies enriched circSamd4 in CMs, further demonstrating the direct interaction between circSamd4 and the Vcp protein (Figure 7B). To identify the specific region of circSamd4 that directly bound to Vcp, various regions of circSamd4 were subcloned into pcDNA3.1 plasmid to generate missing constructs, which were separately transfected into CMs (Figure S15A). RNA pull-down assays revealed that deletion of circSamd4 184-320 bp, but not circSamd4 47-133 bp, 376–519 bp, disrupted the interaction between the circSamd4 and Vcp, indicating that circSamd4 184–320 bp may play a critical role in the circSamd4-Vcp bond (Figure S15A).
Figure 7.

CircSamd4 decreases mitochondria-derived ROS by recruiting Vcp to the mitochondria
(A) The proteins precipitated by a specific biotin-labeled circSamd4 probe were resolved by SDS-PAGE; bottom, the Vcp protein was detected by western blotting. CircSamd4 probe, biotin-labeled probe targeting circSamd4 back-splicing sequence; control probe, non-labeled probe targeting circSamd4 back-splicing sequence. (B) RIP assays revealed that the Vcp protein enriched circSamd4 in P1 CMs; ∗p < 0.05, n = 6. (C) qPCR assays were performed to detect the Vcp mRNA expression levels after circSamd4 knockdown; n = 6. (D) Western blot analysis of the Vcp protein levels in CMs. P1 CMs were transduced with Adv for 24 h and then incubated with H2O2 (20 μM) for 8 h, ∗p < 0.05, n = 6. (E) Western blot analysis of the Vcp protein levels in the cytoplasmic and mitochondrial fractions of CMs. ∗p < 0.05, n = 6. (F) Western blot analysis of the Vcp protein levels in the cytoplasmic and mitochondrial fractions of CMs. ∗p < 0.05, n = 6. (G) Western blot analysis of the Vcp protein levels in the cytoplasmic and mitochondrial fractions of CMs. ∗p < 0.05, n = 6. (H) Evaluation of P1 CM proliferative activity by Ki67 immunostaining after Adv-Vcp or Adv-NC transduction. Ki67+ CMs are indicated by arrows, ∗p < 0.05, n = 6. (I) Evaluation of P1 CM proliferative activity by pH3 immunostaining after Adv-Vcp or Adv-NC transduction. pH3+ CMs are indicated by arrows, ∗p < 0.05, n = 6. (J) Evaluation of the MMP in P1 CMs after Adv-Vcp or Adv-NC transduction. ∗p < 0.05, n = 6. JC-1 monomer, green; J-aggregate, red. (K) Evaluation of mitochondria-derived ROS levels in P1 CMs after Adv-Vcp or Adv-NC transduction. ∗p < 0.05, n = 6. (L) Evaluation of oxidative DNA damage in P1 CMs after Adv-Vcp or Adv-NC transduction. ∗p < 0.05, n = 6. (M) Detection of intracellular ROS levels in the myocardium 14 days after AAV9-circSamd4 transduction or ML240 treatment; ∗p < 0.05, n = 6. (N) Detection of oxidative DNA damage in adult CMs 14 days after AAV9-circSamd4 transduction or ML240 treatment; ∗p < 0.05, n = 6. (O) Evaluation of CM proliferation by pH3 immunostaining 14 days after AAV9-circSamd4 transduction or ML240 treatment; ∗p < 0.05, n = 6. (P) Masson trichrome staining of cross sections from adult mouse hearts 28 days after AAV9-circSamd4 transduction or ML240 treatment; ∗p < 0.05, n = 6.
Next, we explored the biological effects of the interaction between circSamd4 and the Vcp protein. First, we detected the expression level of Vcp after circSamd4 interference. qRT-PCR assays and western blotting assays revealed that circSamd4 knockdown had no significant effect on the protein or mRNA expression of Vcp (Figures 7C and 7D). Subsequently, we investigated whether circSamd4 affects the subcellular location of the Vcp protein. H2O2 promoted the mitochondrial translocation of the Vcp protein in CMs (Figure 7E), which was consistent with the results of previous studies.26,27 We found that circSamd4 overexpression promoted the mitochondrial translocation of the Vcp protein, while circSamd4 silencing repressed this phenomenon (Figures 7F, 7G, and S16), indicating that circSamd4 acted as a mitochondrial recruitment factor for Vcp in CMs. Previous studies demonstrated an important role of the Vcp protein in mitochondrial function and dynamics.26,28 Indeed, once cells are subjected to DNA damage, cell-cycle checkpoint pathways are activated to arrest cells in the G1 or G2 phase.29 Recent studies have revealed that Wee1 is the key negative regulator of the G2-to-M transition and prevents cycle entry into mitosis in response to ROS-induced DNA damage in CMs.2 Inactivation of Wee1-dependent signaling is sufficient to promote CM proliferation and cardiac regeneration.2 Thus, we hypothesized that Wee1 is the essential effector that functions downstream of the circSamd4-induced antioxidant response and cardiac regeneration. The Wee1 expression level might be negatively correlated with the oxidation status and regenerative capacity of the heart. Importantly, we observed that the expression level of Wee1 was also upregulated by circSamd4, which was reversed by a selective ATP-competitive inhibitor of Vcp, ML240 (Figure S15B). To explore the role of Vcp, we overexpressed Vcp in CMs using an Adv vector and detected the Vcp protein level (Figure S15C). The increase in the proportion of proliferative CMs after Vcp overexpression further suggested a link between the Vcp protein and CM proliferation (Figures 7H and 7I). We also observed that Vcp overexpression inhibited mPTP opening and MMP decline in CMs upon H2O2 treatment (Figure 7J). Moreover, we found that Vcp overexpression reduced the intracellular ROS levels and subsequent DNA damage in H2O2-treated CMs (Figures 7K, 7L, S15D, and S15E).
In vivo studies were then performed to determine the role of Vcp in circSamd4-mediated cardiac repair. The administration of the Vcp inhibitor ML240 significantly reversed the circSamd4-induced reduction in oxidative stress and DNA damage in the adult myocardium post MI (Figures 7M and 7N). Consistently, ML240 administration alleviated circSamd4-induced CM proliferation and fibrosis reduction post MI (Figures 7O, 7P, and S17). In addition, we investigated the effect of ML240 on neonatal CM and observed that the administration of ML240 promoted mPTP opening in CMs in vitro (Figures S15F and S15G). Furthermore, ML240 treatment in vivo increased ROS production, induced oxidative DNA damage, and even repressed CM proliferation in the neonatal myocardium (Figures S15H–S15K). Collectively, the effects of circSamd4 on CM proliferation and cardiac regeneration are likely mediated by the Vcp protein.
Vcp protein attenuates mPTP opening by decreasing Vdac1 expression
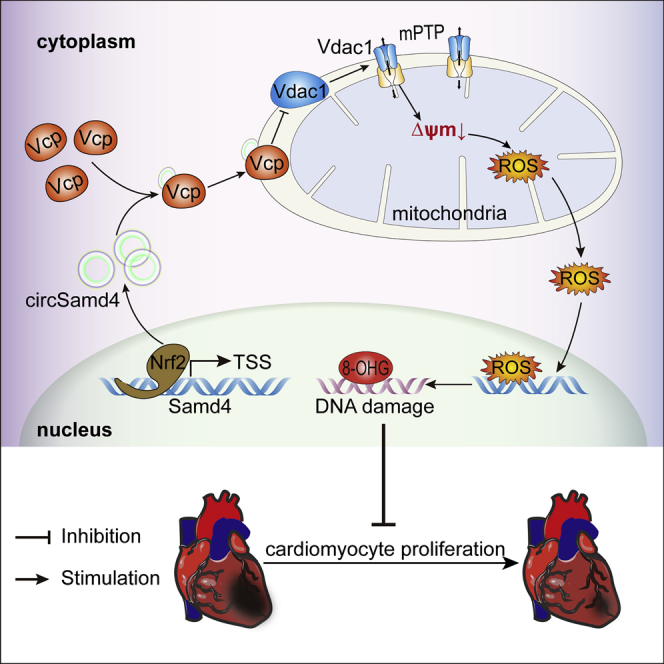
Next, we further investigated the underlying mechanism by which Vcp regulates mPTP opening in CMs. Previous studies demonstrated that mPTP opening and MMP maintenance in CMs are regulated by Vdac1, a channel protein located in the outer mitochondrial membrane.30 We performed co-immunoprecipitation (coIP) and RIP assays using myocardial tissues isolated from the border zones of infarcted hearts. CoIP assays using an antibody against Vcp revealed that Vcp bound to Vdac1 (Figure S18A), and the subsequent coIP assay using an antibody targeting Vdac1 also showed an interaction between Vdac1 and Vcp (Figure S18B). The following RIP assays using an antibody targeting Vcp revealed that Vcp effectively immunoprecipitated with circSamd4 (Figure S18C). These results confirmed the ternary interaction of circSamd4/Vcp/Vdac1 in the peri-infarcted zone. As Vdac1 was reported to be degraded in a ubiquitin-dependent manner, we assumed that Vcp might promote the degradation of the Vdac1 protein. We assessed the expression level of Vdac1 after Vcp or circSamd4 interference. As expected, western blotting assays showed that Vcp or circSamd4 overexpression decreased Vdac1 expression (Figures 8A and S18D), while inhibition of Vcp or circSamd4 downregulation increased Vdac1 expression (Figures 8B and S18E). Moreover, the application of the Vcp inhibitor ML240 reversed the effect of circSamd4 overexpression on Vdac1 (Figure 8C). We also performed qRT-PCR assays to assess the mRNA expression of Vdac1 in CMs transduced with Adv-Vcp or Adv-NC. The Vdac1 mRNA levels did not significantly differ between the Adv-Vcp group and Adv-NC group, indicating that Vcp overexpression had no effect on the transcription of Vdac1 (Figure S18F). Next, we investigated the effect of Vdac1 on MMP and mPTP opening, which was determined by the JC-1 fluorescence signal. We found that treatment with VBIT-12, a Vdac1 oligomerization inhibitor,31 suppressed mPTP opening and the decrease in the MMP (Figures 8D and 8E). Additionally, VBIT-12 treatment reduced the DDR (Figures 8F and 8G) and promoted CM proliferation (Figures 8H and 8I). Moreover, VBIT-12 treatment reversed the inhibitory effect of ML240 treatment on mPTP opening, ROS generation, oxidative DNA injury, and CM proliferation (Figures 8J–8N). Taken together, these results demonstrate that the circSamd4/Vcp/Vdac1s pathway plays an essential role in reducing oxidative stress and inducing CM proliferation (Figure 8O).
Figure 8.


Vcp attenuates mPTP opening by reducing Vdac1 expression
(A) Changes in the expression level of the Vdac1 protein after Vcp overexpression; ∗p < 0.05, n = 6. Mitochondria were extracted from P1 CMs, and western blotting was then performed to detect the Vdac1 protein. (B) Changes in the expression level of the Vdac1 protein after 5 μΜ ML240 treatment; ∗p < 0.05, n = 6. (C) Changes in the expression level of the Vdac1 protein after circSamd4 overexpression and/or 5 μΜ ML-240 treatment; ∗p < 0.05, n = 6. (D) Detection of the MMP in P1 CMs treated with VBIT-12 using the flow cytometry assay. P1 CMs were incubated with VBIT-12 (20 μM) for 24 h and then incubated with H2O2 (20 μM) for 8 h. JC-1 monomer, green; J-aggregate, red. ∗p < 0.05, n = 6. (E) Detection of the MMP in P1 CMs treated with VBIT-12 using an immunofluorescence assay. (F) Detection of mitochondrial ROS in CMs treated with VBIT-12. ∗p < 0.05, n = 6. (G) Detection of DDR activation in CMs treated with VBIT-12. ∗p < 0.05, n = 6. (H) Detection of Ki67+ CMs after VBIT-12 treatment. Ki67+ CMs are indicated by arrows, ∗p < 0.05, n = 6. (I) Detection of pH3+ CMs after VBIT-12 treatment. ∗p < 0.05, n = 6. (J) Detection of the MMP in P1 CMs after circSamd4 overexpression, 5 μΜ ML-240 and/or 20 μΜ VBIT-12 treatment using the flow cytometry assay. The CMs were treated with 20 μΜ H2O2 prior to detection. ∗p < 0.05, n = 6. (K) Detection of the MMP in P1 CMs after circSamd4 overexpression, 5 μΜ ML-240 and/or 20 μΜ VBIT-12 treatment using the immunofluorescence assay. JC-1 monomer, green; J-aggregate, red. (L) Detection of mitochondrial ROS after circSamd4 overexpression, 5 μΜ ML-240 and/or 20 μΜ VBIT-12 treatment. ∗p < 0.05, n = 6. (M) Detection of DDR activation after circSamd4 overexpression, 5 μΜ ML-240 and/or 20 μΜ VBIT-12 treatment. ∗p < 0.05, n = 6. (N) Detection of pH3+ P1 CMs after circSamd4 overexpression, 5 μΜ ML-240 and/or 20 μΜ VBIT-12 treatment. ∗p < 0.05, n = 6. (O) Schematic illustrating the main finding of this study: Nrf2-upregulated circSamd4 binds to the Vcp protein and promotes Vcp mitochondrial translocation, thereby reducing Vdac1 expression and attenuating mPTP opening. CircSamd4 overexpression decreases oxidative stress production and triggers CM proliferation, leading to cardiac repair after MI.
We also performed RNA-seq to understand the global effect of circSamd4 overexpression. The expression levels of 189 genes were significantly increased and those of 616 genes were significantly reduced after circSamd4 overexpression (Figures S19A and S19B; Table S4). Enriched Gene Oncology (GO) analysis of these differentially upregulated genes revealed processes related to cell proliferation, such as the cell cycle, cell division, and mitosis (Figure S19C). Further qRT-PCR assays validated the expression levels of several genes involved in cell-cycle progression (Figure S19D). The results of RNA-seq analysis also demonstrated the important role of circSamd4 in inducing CM proliferation.
Discussion
In this study, we originally proposed a novel antioxidant therapeutic strategy to achieve cardiac regeneration by targeting mitochondria-localized circSamd4 in CMs. In vivo and in vitro studies demonstrated that circSamd4 synergistically promoted CM proliferation and CM apoptosis inhibition, thereby effectively restoring cardiac function and reducing the size of the fibrotic area after MI. These findings suggested that circSamd4 is a valuable therapeutic target to improve the outcome of MI.
The current study revealed the potential of circSamd4 as a crucial regulator of cardiac regeneration by alleviating mitochondrial oxidative injury. Mitochondria-derived ROS are thought to be important signals that block CM cell-cycle re-entry in adulthood.2 We confirmed the mitochondrial localization of circSamd4 and found that circSamd4 exerted a marked effect on maintaining the MMP and preventing mPTP opening. By scavenging CM mitochondrial ROS, circSamd4 overexpression reduced oxidative DNA damage and subsequent DDR pathway activation while increasing DNA synthesis and mitosis markers in CMs both in vitro and in vivo. The proliferative rate of CMs was similar to that seen in previous studies using other strategies, indicating the efficacy of mitochondria-based strategies.2 Moreover, we found that circSamd4 overexpression prevented CMs from undergoing apoptosis, which is also essential for regenerative repair after MI. The beneficial effects of circSamd4 overexpression work together to decrease the size of the infarct area and restore cardiac function after MI, indicating that circSamd4 might be an effective target for cardiac regeneration. Furthermore, conservation of the circSamd4 sequence among different species indicates the clinical translational value of circSamd4.
In this study, we demonstrated that circSamd4 reduces mitochondria-derived ROS by recruiting Vcp to mitochondria. A previous study reported that cytoplasmic circRNAs function by redistributing proteins from the nucleus to the cytoplasm.32 Our studies revealed a novel mechanism of action via which mitochondria-localized circRNAs promote protein mitochondrial translocation. As a member of the AAA+ (ATPase associated with diverse cellular activities) family of proteins, Vcp is ubiquitously expressed in mammals and can be detected in multiple tissues and cells.33 Prior research has demonstrated that the Vcp protein is expressed in various subcellular organelles and cell types.34,35,36 The Vcp protein is reported to have many biological functions, including playing roles in protein degradation and homeostasis, the assembly of organelle membranes, cell proliferation, and apoptosis.34,35,36 In our study, Vcp was recruited by circSamd4 and translocated to mitochondria, thereby regulating mPTP opening and mitochondrial ROS generation. Our results were consistent with those of previous studies reporting that Vcp participates in mitochondrial functions and combats oxidative stress in CMs.26,28 Thus, Vcp might serve as the main downstream effector of circSamd4 during cardiac regeneration.
We also revealed the mechanism by which Vcp exerted its functions in CMs. The downregulation of Vdac1 was further found to be responsible for the antioxidant activity of Vcp. The Vdac1 protein is also ubiquitously expressed in the heart, duodenum, and other tissues.37 This protein functions as the gatekeeper on the outer mitochondrial membrane and regulates the flow of ions, nucleotides, and metabolites.38 Previous studies have confirmed that Vdac1 plays essential roles in controlling cell fate transition by mediating metabolism and energy crosstalk in cells.39 It was reported that Vdac1 participates in the process of cell apoptosis by permitting the release of apoptotic proteins and mitochondrial DNA from mitochondria.39 Moreover, it also maintains the MMP, cristae organization, and mitochondrial morphology, thereby controlling mitochondrial ROS generation.40 We assumed that the effect of Vcp on Vdac1 expression was mediated by ubiquitin-dependent degradation. Previous studies also reported that targeting Vdac1 could restore the dissipated MMP and decrease ROS production.40 Taken together, the results of these mechanistic studies confirm that the circSamd4/Vcp/Vdac1 pathway plays an antioxidative stress role in cardiac regeneration.
There are several limitations to the current study. First, this study focused on the association between circSamd4 and CM proliferation, but circSamd4 might also function in non-CMs. Considering the indispensable role of non-CMs in cardiac regeneration, the effect of circSamd4 on non-CM ROS generation should be explored in the future. Second, in our study, overexpression and knockdown of circSamd4 expression in the heart was conducted via AAV9 injection, which has been widely applied in gene therapy. Although circSamd4 expression was significantly changed after AAV9 administration, there are still several limitations affecting the results, such as the instability of AAV transduction, the potential effect on inflammation, and safety concerns.41 The application of CM-specific knockout and overexpression transgenic mice might be helpful for determining the role of circSamd4 in cardiac regeneration.
In conclusion, mitochondria-localized circRNA Samd4, regulated by the transcription factor Nrf2, reduced CM mitochondrial oxidative stress and induced cell-cycle progression by promoting Vcp mitochondrial translocation and reducing Vdac1 expression. CircSamd4 overexpression triggered cardiac repair and decreased the infarct size after MI, which significantly improved cardiac function. Our findings suggest that circSamd4 is a promising therapeutic target for heart failure after MI.
Materials and methods
MI model establishment
C57BL/6 mice were obtained from Guangdong Medical Laboratory Animal Center. Experiments were approved by the Animal Research Committee of Southern Medical University and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. MI of neonatal and adult mouse hearts was performed according to previous studies.16,42 Briefly, adult mice (8–10 weeks old, male) were anesthetized by inhalation of isoflurane (5% for induction and 2% for maintenance). The mice were also endotracheally intubated and ventilated using a volume-controlled ventilator to maintain the airway. Neonatal mice were anesthetized by hypothermia with an ice box. MI was conducted by ligation of the left coronary artery (left anterior descending [LAD]) at a level of 1 mm below the left atrium. An analogous surgical operation was performed without occlusion of the LAD for sham-operated animals.
Transthoracic echocardiography
Transthoracic echocardiography was conducted under light anesthesia with inhaled isoflurane (0.4%–1.5%). A Vevo 2100 high-resolution imaging system equipped with a 30-MHz transducer (RMV-707B, VisualSonics, Toronto, ON, Canada) was utilized as previously reported.16
Histology
Hearts were isolated and fixed in 4% paraformaldehyde for 48 h at room temperature and then processed for paraffin sectioning. Masson’s trichrome staining was performed according to the manufacturer’s instructions (catalog no. MST-8004, MXB Biotechnologies). TUNEL staining was performed using an In-Situ Cell Death Detection Kit (catalog no. 11684795910, Roche). ImageJ Software (NIH, Bethesda, MD) was used to measure the percentage of fibrotic area in the left ventricle.21
Cell isolation and culture
Neonatal CMs from 1-day-old (P1) and 7-day-old (P7) C57BL/6 mice were isolated as previously described.32,42 Briefly, the ventricles of the mice were isolated, cut into pieces, digested with 0.25% trypsin (Sigma) at 4°C for approximately 12 h, and digested again with type II collagenase (Roche) at 37°C for 15 min two or three times. The supernatant was collected and centrifuged, and the collected cells were resuspended in DMEM/F12 medium (Life Technologies) supplemented with 10% FBS. The cell suspension was subsequently plated onto uncoated 100-mm plastic dishes for 90 min at 37°C and 5% CO2 in a humidified atmosphere. Due to the differential adhesion between CMs and fibroblasts, the supernatant, composed mostly of CMs, was then collected, resuspended in the aforementioned culture medium, and seeded at the appropriate density.
Adult CMs were isolated from 56- to 70-day-old mice as previously described.32,42 Briefly, adult hearts were isolated, placed on a Langendorff apparatus, and perfused with calcium-free perfusion buffer for 5 min and digestion buffer for 10 min. The perfusion buffer contained NaCl (113 mM), KCl (4.7 mM), KH2PO4 (0.6 mM), Na2HPO4 (0.6 mM), MgSO4 (1.2 mM), Na-HEPES (10 mM), NaHCO3 (12 mM), KHCO3 (10 mM), phenol red (0.032 mM), taurine (30 mM), BDM (10 mM), and glucose (5.5 mM, pH 7.0). The digestion buffer contained 15,000 U of type II collagenase (Roche) and 50 μM CaCl2. Then, the hearts were cut into small pieces and triturated with a Pasteur pipette to separate individual cells. The adult CMs were then centrifuged at a low speed and seeded onto culture slides coated with laminin ( 1 mg/mL Life Technologies, 23017015) in DMEM/F12 medium (Life Technologies) supplemented with 10% FBS. Primary mouse cardiac endothelial cells and fibroblasts were obtained from Cell Biologics (C57-6024 and C57-6049, respectively) and handled according to the company’s instructions. Descriptions of the isolation and quality control analyses of these cells are presented at the company’s website (https://cellbiologics.com/).
qRT-PCR assays
RNA isolation, reverse transcription, and qRT-PCR assays were performed as previously described.16,32,42 The primers used for qRT-PCR assays are listed in Table S1.
For mitochondrial DNA (mtDNA) quantification, mtDNA was extracted from samples with a Cell Mitochondria Isolation Kit (#C3601, Beyotime). mtDNA was quantified with qRT-PCR with primers using SYBR Green PCR Master Mix. The relative mtDNA copies were calculated from the ratio of mtDNA copies to nuclear DNA copies.
Small interfering RNA, adenovirus (Adv) and AAV9 transduction
Small interfering RNAs (siRNAs) were purchased from RiboBio (Guangzhou, China). The target sequences of the siRNAs are presented in Table S2. A scrambled siRNA was used as an NC. Transfections were conducted with a LipoFilter Kit (MicroAire). SiRNAs were transfected at a final concentration of 50 nM, and subsequent experiments were performed 48 h after transfection.
The Adv and AAV9 vectors for gene knockdown or overexpression were purchased from Vigene (Shandong, China). Neonatal CMs were transduced with Adv for 48 h. The multiplicity of infection (MOI) ranged from 10 to 20. The delivery of Adv and AAV9 vectors to mice was conducted as previously reported.16,32,42 Briefly, adult mice were intramyocardially injected at three to five sites with AAV9 vectors expressing circSamd4 or an empty vector (AAV9-circSamd4 or AAV9-vector) at a dose of 10 × 1011 viral genome particles per mouse (approximately 30 μL) using an insulin syringe with a 30-gauge needle. For the infarcted mice, AAV9 was injected into myocardial tissue close to the infarct zone immediately after LAD coronary artery ligation. Myocardial tissues were collected 28 days after injection. In P1 neonatal mice, an Adv vector expressing an shRNA targeting circSamd4 or an empty vector (Adv-shcircSamd4 or Adv-NC) was injected into three sites in the left ventricle at a dose of 5 × 109 viral genome particles per animal (approximately 5 μL) by using an insulin syringe with a 30-gauge needle. Adv was also injected into the peri-infarcted myocardium immediately after LAD ligation. The hearts of the mice were isolated 1 week after the Adv injection. Real-time PCR (qRT-PCR) assays were used to determine successful transduction.
Subcellular fractionation
To determine subcellular distribution of circRNAs, three distinct fractions were isolated from CMs (mitochondria, cytoplasmic, and nuclear) using a Cell Fractionation Kit (#9038, Cell Signaling Technology) according to the manufacturer’s instructions.
Immunostaining
Immunostaining assays were performed as previously described.43 The primary antibodies used were as follows: anti-Ki67 antibody (ab15580, Abcam), anti-p-histone H3 (pH3) antibody (ab170904, Abcam), anti-c-TnT (ab33589, Abcam), anti-aurora B (ab2254, Abcam), anti-Anillin (ab211872, Abcam), anti-CD31 (AF3628, R&D), anti-α-actinin (ab68167, Abcam), anti-vimentin (60330-1-Ig, Proteintech), anti-p-Atm (sc-47739, Santa Cruz), and anti-8-OHG (ab62623, Abcam). To detect EdU incorporation, cells were stained with a Click-it EdU Imaging Kit (Life Technologies, #C10638) according to the manufacturer’s instructions. Image acquisition was performed using confocal laser scanning microscopy (Leica).
ISH
ISH assays were performed as previously reported.16,32,42 Briefly, samples were incubated in 3% pepsin diluted in fresh citrate buffer at 37°C for 30 min and then prehybridized in prehybridization solution for 2 h at 37°C. Digoxigenin (DIG)-labeled RNA probes (Biosense Bioscience, Guangzhou, China) were used for hybridization at 37°C overnight. Samples were then washed with a graded series of saline sodium citrate (SSC) buffer, blocked with 3% BSA, and incubated with alkaline phosphatase-conjugated sheep anti-DIG Fab fragments.
ROS detection
Total cellular ROS and mitochondria-derived ROS were detected by DCFH-DA (catalog no. D6883, Sigma-Aldrich) and MitoSOX (catalog no. M36008, Invitrogen), respectively. CMs at a density of 10 × 105 cells/well in a confocal dish were cultured in 10 mM DCFH-DA or 5 mM mitoSOX working solution at 37°C for 30 min in the dark. Images were captured by Zeiss LSM 800 confocal microscopy. The fluorescent density was also detected via a flow cytometry as previously described.44
RNase R digestion
RNase R digestion was performed as previously reported.45 Briefly, 2 μg of RNA was incubated with or without 3 U of RNase R (Sigma) at 37°C for 30 min. Then, the treated RNA was purified using the RNeasy MinElute Cleanup Kit (Qiagen).
Drug treatments
A Vcp inhibitor (ML240, MedChemExpress) was dissolved in diluted DMSO. In vitro, CMs were incubated with 5 μM ML240 for 24 h; in vivo, mice were injected with ML240 (1.2 mg/kg) intraperitoneally every 2 days from day 1 to day 28. A Vdac1 inhibitor (VBIT-12, 20 μM, Selleck Chemicals) was added to CMs for 2 h. To explore the effect of H2O2 on CMs, CMs were transduced with Adv for 24 h and then incubated with H2O2 (20 μM, Sigma) for 8 h as indicated.
Western blot analysis
Total cells or tissue extracts were lysed by RIPA lysis buffer (BestBio) containing Protease Inhibitor Cocktail Set I (BestBio). Proteins were resolved in 10% SDS-PAGE buffer, fractioned by polyacrylamide gels, and transferred onto nitrocellulose membranes. The following primary antibodies were used: anti-Vcp (ab11433, Abcam), anti-Vdac1 (55259-1-AP, Proteintech), anti-Wee1 (ab137377, Abcam), anti-Nrf2 (ab19867, Abcam), anti-Sod2 (ab137037, Abcam), and anti-Gpx1 (29329-1-AP, Proteintech). A rabbit anti-Gapdh antibody (1:1000 dilution; bs-0755R, Bioss) and anti-Cox4 antibody (11242-1-AP, Proteintech) were used as controls. Alexa Fluor 680-conjugated anti-mouse IgG (1:10,000 dilution; Abcam, USA) was used as the secondary antibody. The signal was detected by an Odyssey detection system (LI-COR Biosciences, Lincoln, NE, USA).
RNA fluorescence ISH
Cy3-labeled RNA probes against the circSamd4 back-splice sequence were purchased from RiboBio (Guangzhou, China). Cultured cells and isolated tissues were permeabilized with 0.5% Triton X-100 and then incubated with RNA probes in hybridization buffer (RiboBio, Guangzhou, China). Nuclei were stained with DAPI.
RNA pull-down assays
RNA pull-down assays were performed as previously reported.46 Briefly, cell lysates were incubated with biotinylated DNA oligo probes at room temperature for 4 h. Then, cell lysates were incubated with washed streptavidin-coated magnetic beads (SA1004; Invitrogen) at room temperature for 1 h. The precipitated components were extracted by SDS-PAGE and then subjected to silver staining and mass spectrometry analysis. The biotin-labeled probe targeting circSamd4 back-splicing sequence was used for RNA pull-down assay, and the probe without biotin labeling was used as the control group.
TTC staining
TTC assays were performed as previously reported.16,32,42 Briefly, the mouse hearts were isolated and sectioned at a thickness of 3 mm. The slices were incubated in 1% TTC buffer (Sigma-Aldrich) diluted in PBS for 30 min at room temperature. Then, the slices were washed with PBS and photographed. Image-Pro Plus 6.0 (Media Cybernetics, Bethesda, MD, USA) was utilized to calculate the infarcted area.
ChIP assay
ChIP assays were performed as reported previously.16,32,42 An EpiQuik Chromatin Immunoprecipitation Assay Kit (EpiGentek, Brooklyn, NY) was used according to the manufacturer’s recommendations. An anti-Nrf2 (#12721, CST) antibody or IgG antibody was used for immunoprecipitation. Then, qRT-PCR and PCR gel electrophoresis were performed to detect the enrichment of DNA fragments at potential Nrf2 binding sites.
CoIP assay
CoIP assays were performed according to a previous study.16 Briefly, cell lysates were collected and incubated with antibodies at 4°C for 12 h. Then, they were incubated with protein A/G magnetic beads (Thermo Scientific, USA). Finally, the beads were collected and subjected to western blotting.
RIP assay
RIP assays were performed according to previous studies.16,32,42 Briefly, a Magna RIP RNA-binding Protein Immunoprecipitation Kit (Millipore, Stafford, VA) was used according to the manufacturer’s instructions. We used an anti-Vcp antibody (ab11433, Abcam) to precipitate RNA, and PCR assays were then performed to assess the binding of RNA to Vcp.
MMP measurement
MMP changes were measured by a mitochondrial membrane potential assay kit with JC-1 (Beyotime) according to the manufacturer’s instructions. Briefly, CMs were harvested and washed with PBS. Then, these CMs were resuspended in a mixture of culture medium and JC-1 staining fluid for 20 min in the dark at 37°C. Next, CMs were washed with cold staining buffer prior to flow cytometry (BD, USA). The MMP was indicated by the ratio of red to green fluorescence intensity. When the MMP is low in cells, JC-1 remains in the monomeric form and emits a green fluorescence. In cells with high MMP, JC-1 forms complexes known as J-aggregates. Aggregates of JC-1 emit an orange-red fluorescence. The higher ratios of red to green fluorescence were correlated with higher mitochondrial membrane polarization.
Annexin V-FITC/PI staining
The apoptosis ratio of CMs was assessed by the Annexin V-FITC/PI apoptosis detection kit according to the manufacturer’s instructions. Briefly, CMs were resuspended in 0.1 mL of binding buffer and then incubated with Annexin V-FITC/PI buffer in the dark for 10 min. The fluorescence intensity was determined by flow cytometry (FACScan, BD Biosciences), and the data were analyzed by FlowJo 7.6.1.
Trypan blue exclusion
Trypan blue exclusion was used to determine cell viability. Cells were trypsinized, pelleted, resuspended in a 1:1 mixture of PBS and 0.5% trypan blue, and manually counted using a hemocytometer. The number of dead (blue-stained) cells was then expressed as a ratio of the total (stained and unstained) cells counted.
Calcein-AM/CoCl2 assays
mPTP opening was assessed by calcein-AM/CoCl2 assays as described previously.47 Briefly, the isolated CMs were incubated with 5 μM calcein-AM for 30 min at 37°C and then incubated with 40 μM CoCl2 for 20 min at 37°C. When the mPTP was unopened, calcein fluorescence was maintained in mitochondria in the presence of CoCl2. When the mPTP opened, calcein was lost from the mitochondria. Fluorescence intensities were determined by confocal microscopy.
PET/CT imaging
MicroPET-CT was used to investigate glucose uptake in myocardial tissue, according to previous studies.48,49 Briefly, 18F-fluorodeoxyglucose (18F-FDG) was intraperitoneally injected into animals. Then, the mice were scanned by using a Focus 220 microPET scanner (Siemens Medical Solutions USA, Knoxville, TN, USA). Dynamic scans were conducted for 1 h. PET images were reconstructed using the microPET-CT manager (Siemens Medical Solutions USA).
Northern blot
Northern blots were performed as previously described.11 Briefly, RNA with or without RNase R treatments was added into agarose gels, and electrophoresis for 3 h. Then, RNA was transferred to a nylon membrane and UV-crosslinked for 2 min. After hybridization with digoxigenin-labeled oligonucleotide probes overnight, membrane was incubated in anti-digoxigenin-AP and exposed onto chemiluminescence solution. The images were captured by a Typhoon 9500 scanner (GE Healthcare). The sequence of the probe is ATTCCTGTTGCCATTGGTTAATGATTCTCGTGCTTGCGCCGGTTCCAATCGTGT.
RNA isolation and library preparation
Total RNA was extracted using TRIzol reagent according to the manufacturer’s protocol. RNA purity and quantification were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). RNA integrity was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Then, libraries were constructed using the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Transcriptome sequencing and analysis were conducted by OE Biotech (Shanghai, China).
RNA sequencing and differentially expressed gene analysis
The libraries were sequenced on an Illumina HiSeq X Ten platform, and 150-bp paired-end reads were generated. Raw data (raw reads) in the fastq format were first processed using Trimmomatic,50 and the low-quality reads were removed to obtain clean reads. The clean reads were mapped to the mouse genome (mm10) using HISAT2.51 The fragments per kilobase of exon model per million mapped fragments (FPKM)52 values of each gene were calculated using Cufflinks,53 and the read counts of each gene were obtained by HTSeq count.54 Differential expression analysis was performed using the DESeq R package.55 The adjusted p value <0.05 and a fold change >2 or <0.5 were set as the thresholds for significantly different expression. Enriched GO analysis of differentially expressed genes was performed using R based on the hypergeometric distribution.56
Data availability
CircRNA and gene expression profiles in adult and neonatal hearts were obtained from previous studies.13 The thresholds for up- and downregulated circRNAs and genes were a fold change ≥2 and a false discovery rate (FDR) <0.05.
The ChIP-seq data are available through the Gene Expression Omnibus (GEO) under accession numbers GSM1264370 (embryonic day 11.5, E11.5 mouse heart H3K27ac), GSM1264378, (postnatal day 0, P0 mouse heart H3K27ac), and GSM1264384 (postnatal day 56, P56 mouse heart H3K27ac).
Statistical analysis
The results were statistically analyzed using SPSS 22.0 software. All data are shown in graphs as the mean ± SD. For the statistical comparison of two groups, an unpaired, two-tailed Student’s t test was utilized. One-way ANOVA, followed by Bonferroni’s multiple comparisons test, was used for the comparison of more than two groups. The p value was calculated, and p < 0.05 indicated statistical significance.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (nos. 81771857, 82070315, 82000282, and 82000248), Guangzhou Regenerative Medicine and Health Laboratory of Guangdong (no. 2018GZR110105009), Guangdong Basic and Applied Basic Research Foundation (nos. 2019A1515110687 and 2021A1515220026), Natural Science Foundation of Guangdong Province (no. 2020A151501302), Guangzhou Basic and Applied Basic Research Foundation (no. 202002030198), and Outstanding Youths Development Scheme of Nanfang Hospital, Southern Medical University (nos. 2019J003 and 2019J012).
Author contributions
J.B., S.H., and H.Z. conceived the study. S.H. wrote the original manuscript. S.H., H.Z., and J.B. reviewed and edited the manuscript. H.Z., G.W., and Y.S. performed in vitro experiments. H.Z., C.L., and Y.C. performed in vivo experiments. X.S., Z.T., X.L., and Y.C. analyzed the data. Y.L., W.L., and J.B. supervised the experiments. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.06.016.
Supplemental information
References
- 1.Nakada Y., Canseco D.C., Thet S., Abdisalaam S., Asaithamby A., Santos C.X., Shah A.M., Zhang H., Faber J.E., Kinter M.T., et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–227. doi: 10.1038/nature20173. [DOI] [PubMed] [Google Scholar]
- 2.Puente B.N., Kimura W., Muralidhar S.A., Moon J., Amatruda J.F., Phelps K.L., Grinsfelder D., Rothermel B.A., Chen R., Garcia J.A., et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tao G., Kahr P.C., Morikawa Y., Zhang M., Rahmani M., Heallen T.R., Li L., Sun Z., Olson E.N., Amendt B.A., et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–123. doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deshwal S., Antonucci S., Kaludercic N., Di Lisa F. Measurement of mitochondrial ROS formation. Methods Mol. Biol. 2018;1782:403–418. doi: 10.1007/978-1-4939-7831-1_24. [DOI] [PubMed] [Google Scholar]
- 5.Han P., Zhou X.H., Chang N., Xiao C.L., Yan S., Ren H., Yang X.Z., Zhang M.L., Wu Q., Tang B., et al. Hydrogen peroxide primes heart regeneration with a derepression mechanism. Cell Res. 2014;24:1091–1107. doi: 10.1038/cr.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li K., Liu Y.Y., Lv X.F., Lin Z.M., Zhang T.T., Zhang F.R., Guo J.W., Hong Y., Liu X., Lin X.C., et al. Reduced intracellular chloride concentration impairs angiogenesis by inhibiting oxidative stress-mediated VEGFR2 activation. Acta Pharmacol. Sin. 2021;42:560–572. doi: 10.1038/s41401-020-0458-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilusz J.E. A 360° view of circular RNAs: from biogenesis to functions. Wiley Interdiscip. Rev. RNA. 2018;9:e1478. doi: 10.1002/wrna.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia S., Feng J., Lei L., Hu J., Xia L., Wang J., Xiang Y., Liu L., Zhong S., Han L., et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief. Bioinform. 2017;18:984–992. doi: 10.1093/bib/bbw081. [DOI] [PubMed] [Google Scholar]
- 9.Wang K., Gan T.Y., Li N., Liu C.Y., Zhou L.Y., Gao J.N., Chen C., Yan K.W., Ponnusamy M., Zhang Y.H., et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017;24:1111–1120. doi: 10.1038/cdd.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S., Chen J., Yu W., Deng F. Circular RNA DLGAP4 ameliorates cardiomyocyte apoptosis through regulating BCL2 via targeting miR-143 in myocardial ischemia-reperfusion injury. Int. J. Cardiol. 2019;279:147. doi: 10.1016/j.ijcard.2018.09.023. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Q., Liu J., Deng H., Ma R., Liao J.Y., Liang H., Hu J., Li J., Guo Z., Cai J., et al. Targeting mitochondria-located circRNA SCAR alleviates NASH via reducing mROS output. Cell. 2020;183:76–93.e22. doi: 10.1016/j.cell.2020.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Wu Z., Sun H., Wang C., Liu W., Liu M., Zhu Y., Xu W., Jin H., Li J. Mitochondrial genome-derived circRNA mc-COX2 functions as an oncogene in chronic lymphocytic leukemia. Mol. Ther. Nucleic Acids. 2020;20:801–811. doi: 10.1016/j.omtn.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werfel S., Nothjunge S., Schwarzmayr T., Strom T.M., Meitinger T., Engelhardt S. Characterization of circular RNAs in human, mouse and rat hearts. J. Mol. Cell. Cardiol. 2016;98:103–107. doi: 10.1016/j.yjmcc.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Guarnerio J., Zhang Y., Cheloni G., Panella R., Mae Katon J., Simpson M., Matsumoto A., Papa A., Loretelli C., Petri A., et al. Intragenic antagonistic roles of protein and circRNA in tumorigenesis. Cell Res. 2019;29:628–640. doi: 10.1038/s41422-019-0192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stillman B. Histone modifications: insights into their influence on gene expression. Cell. 2018;175:6–9. doi: 10.1016/j.cell.2018.08.032. [DOI] [PubMed] [Google Scholar]
- 16.Si X., Zheng H., Wei G., Li M., Li W., Wang H., Guo H., Sun J., Li C., Zhong S., et al. circRNA Hipk3 induces cardiac regeneration after myocardial infarction in mice by binding to Notch1 and miR-133a. Mol. Ther. Nucleic Acids. 2020;21:636–655. doi: 10.1016/j.omtn.2020.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locato V., Cimini S., De Gara L. ROS and redox balance as multifaceted players of cross-tolerance: epigenetic and retrograde control of gene expression. J. Exp. Bot. 2018;69:3373–3391. doi: 10.1093/jxb/ery168. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen J., Gejl K.D., Hey-Mogensen M., Holmberg H.C., Suetta C., Krustrup P., Elemans C.P.H., Ørtenblad N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 2017;595:2839–2847. doi: 10.1113/JP273040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlüter K.D., Schreiber D. Adult ventricular cardiomyocytes: isolation and culture. Methods Mol. Biol. 2005;290:305–314. doi: 10.1385/1-59259-838-2:305. [DOI] [PubMed] [Google Scholar]
- 20.Ramanujam D., Sassi Y., Laggerbauer B., Engelhardt S. Viral vector-based targeting of miR-21 in cardiac nonmyocyte cells reduces pathologic remodeling of the heart. Mol. Ther. 2016;24:1939–1948. doi: 10.1038/mt.2016.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iles L.M., Ellims A.H., Llewellyn H., Hare J.L., Kaye D.M., Mclean C.A., Taylor A.J. Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. Eur. Heart J. Cardiovasc. Imaging. 2015;16:14–22. doi: 10.1093/ehjci/jeu182. [DOI] [PubMed] [Google Scholar]
- 22.Freeman I., Grunwald A.M., Robin B., Rao P.S., Bodenheimer M.M. Effect of early reperfusion on use of triphenyltetrazolium chloride to differentiate viable from non-viable myocardium in area of risk. Cardiovasc. Res. 1990;24:109–114. doi: 10.1093/cvr/24.2.109. [DOI] [PubMed] [Google Scholar]
- 23.Guo X., Sun X., Hu D., Wang Y.J., Fujioka H., Vyas R., Chakrapani S., Joshi A.U., Luo Y., Mochly-Rosen D., et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease. Nat. Commun. 2016;7:12646. doi: 10.1038/ncomms12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo X., Qi X. VCP cooperates with UBXD1 to degrade mitochondrial outer membrane protein MCL1 in model of Huntington's disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:552–559. doi: 10.1016/j.bbadis.2016.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun X., Zhou N., Ma B., Wu W., Stoll S., Lai L., Qin G., Qiu H. Functional inhibition of valosin-containing protein induces cardiac dilation and dysfunction in a new dominant-negative transgenic mouse model. Cells. 2021;10:2891. doi: 10.3390/cells10112891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang T., Mishra P., Hay B.A., Chan D., Guo M. Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. Elife. 2017;6:e17834. doi: 10.7554/eLife.17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim N.C., Tresse E., Kolaitis R.M., Molliex A., Thomas R.E., Alami N.H., Wang B., Joshi A., Smith R.B., Ritson G.P., et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. 2013;78:65–80. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lizano P., Rashed E., Stoll S., Zhou N., Wen H., Hays T.T., Qin G., Xie L.H., Depre C., Qiu H. The valosin-containing protein is a novel mediator of mitochondrial respiration and cell survival in the heart in vivo. Sci. Rep. 2017;7:46324. doi: 10.1038/srep46324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heijink A.M., Krajewska M., van Vugt M.A.T.M. The DNA damage response during mitosis. Mutat. Res. 2013;750:45–55. doi: 10.1016/j.mrfmmm.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Yang X., Zhou Y., Liang H., Meng Y., Liu H., Zhou Y., Huang C., An B., Mao H., Liao Z. VDAC1 promotes cardiomyocyte autophagy in anoxia/reoxygenation injury via the PINK1/Parkin pathway. Cell Biol. Int. 2021;45:1448–1458. doi: 10.1002/cbin.11583. [DOI] [PubMed] [Google Scholar]
- 31.Niu B., Lei X., Xu Q., Ju Y., Xu D., Mao L., Li J., Zheng Y., Sun N., Zhang X., et al. Protecting mitochondria via inhibiting VDAC1 oligomerization alleviates ferroptosis in acetaminophen-induced acute liver injury. Cell Biol. Toxicol. 2022;38:505–530. doi: 10.1007/s10565-021-09624-x. [DOI] [PubMed] [Google Scholar]
- 32.Huang S., Li X., Zheng H., Si X., Li B., Wei G., Li C., Chen Y., Chen Y., Liao W., et al. Loss of super-enhancer-regulated circRNA nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation. 2019;139:2857–2876. doi: 10.1161/CIRCULATIONAHA.118.038361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halawani D., Latterich M. p97: the cell's molecular purgatory? Mol. Cell. 2006;22:713–717. doi: 10.1016/j.molcel.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Jentsch S., Rumpf S. Cdc48 (p97): a "molecular gearbox" in the ubiquitin pathway? Trends Biochem. Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Chia W.S., Chia D.X., Rao F., Bar Nun S., Geifman Shochat S. ATP binding to p97/VCP D1 domain regulates selective recruitment of adaptors to its proximal N-domain. PLoS One. 2012;7:e50490. doi: 10.1371/journal.pone.0050490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bar-Nun S. The role of p97/Cdc48p in endoplasmic reticulum-associated degradation: from the immune system to yeast. Curr. Top. Microbiol. Immunol. 2005;300:95–125. doi: 10.1007/3-540-28007-3_5. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto T., Yamada A., Watanabe M., Yoshimura Y., Yamazaki N., Yoshimura Y., Yamauchi T., Kataoka M., Nagata T., Terada H., et al. VDAC1, having a shorter N-terminus than VDAC2 but showing the same migration in an SDS-polyacrylamide gel, is the predominant form expressed in mitochondria of various tissues. J. Proteome Res. 2006;5:3336–3344. doi: 10.1021/pr060291w. [DOI] [PubMed] [Google Scholar]
- 38.Shoshan-Barmatz V., Nahon-Crystal E., Shteinfer-Kuzmine A., Gupta R. VDAC1, mitochondrial dysfunction, and Alzheimer's disease. Pharmacol. Res. 2018;131:87–101. doi: 10.1016/j.phrs.2018.03.010. [DOI] [PubMed] [Google Scholar]
- 39.Shoshan-Barmatz V., Shteinfer-Kuzmine A., Verma A. VDAC1 at the intersection of cell metabolism, apoptosis, and diseases. Biomolecules. 2020;10:E1485. doi: 10.3390/biom10111485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gatliff J., East D., Crosby J., Abeti R., Harvey R., Craigen W., Parker P., Campanella M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy. 2014;10:2279–2296. doi: 10.4161/15548627.2014.991665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hudry E., Vandenberghe L.H. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron. 2019;101:839–862. doi: 10.1016/j.neuron.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y., Li X., Li B., Wang H., Li M., Huang S., Sun Y., Chen G., Si X., Huang C., et al. Long non-coding RNA ECRAR triggers post-natal myocardial regeneration by activating ERK1/2 signaling. Mol. Ther. 2019;27:29–45. doi: 10.1016/j.ymthe.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D'Uva G., Aharonov A., Lauriola M., Kain D., Yahalom-Ronen Y., Carvalho S., Weisinger K., Bassat E., Rajchman D., Yifa O., et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- 44.du Plessis S.S., Hagenaar K., Lampiao F. The in vitro effects of melatonin on human sperm function and its scavenging activities on NO and ROS. Andrologia. 2010;42:112–116. doi: 10.1111/j.1439-0272.2009.00964.x. [DOI] [PubMed] [Google Scholar]
- 45.Zheng Q., Bao C., Guo W., Li S., Chen J., Chen B., Luo Y., Lyu D., Li Y., Shi G., et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L., Long H., Zheng Q., Bo X., Xiao X., Li B. Circular RNA circRHOT1 promotes hepatocellular carcinoma progression by initiation of NR2F6 expression. Mol. Cancer. 2019;18:119. doi: 10.1186/s12943-019-1046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petronilli V., Miotto G., Canton M., Colonna R., Bernardi P., Di Lisa F. Imaging the mitochondrial permeability transition pore in intact cells. Biofactors. 1998;8:263–272. doi: 10.1002/biof.5520080314. [DOI] [PubMed] [Google Scholar]
- 48.Fragkouli A., Tsilibary E.C., Tzinia A.K. Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer's 5xFAD mice. Neurobiol. Dis. 2014;70:179–189. doi: 10.1016/j.nbd.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 49.Brewer S., Mcpherson M., Fujiwara D., Turovskaya O., Ziring D., Chen L., Takedatsu H., Targan S.R., Wei B., Braun J. Molecular imaging of murine intestinal inflammation with 2-deoxy-2-[18F]fluoro-D-glucose and positron emission tomography. Gastroenterology. 2008;135:744–755. doi: 10.1053/j.gastro.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts A., Trapnell C., Donaghey J., Rinn J.L., Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong S., Xie D. Gene Ontology analysis in multiple gene clusters under multiple hypothesis testing framework. Artif. Intell. Med. 2007;41:105–115. doi: 10.1016/j.artmed.2007.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CircRNA and gene expression profiles in adult and neonatal hearts were obtained from previous studies.13 The thresholds for up- and downregulated circRNAs and genes were a fold change ≥2 and a false discovery rate (FDR) <0.05.
The ChIP-seq data are available through the Gene Expression Omnibus (GEO) under accession numbers GSM1264370 (embryonic day 11.5, E11.5 mouse heart H3K27ac), GSM1264378, (postnatal day 0, P0 mouse heart H3K27ac), and GSM1264384 (postnatal day 56, P56 mouse heart H3K27ac).