

Abstract
Caged morpholino oligonucleotides (cMOs) are synthetic tools that allow light-inducible gene silencing in live organisms. Previously reported cMOs have utilized hairpin, duplex, and cyclic structures, as well as caged nucleobases. While these antisense technologies enable efficient optical control of RNA splicing and translation, they can have limited dynamic range. A new caging strategy was developed where the two MO termini are conjugated to an internal position through a self-immolative trifunctional linker, thereby generating a bicyclic cMO that is conformationally resistant to RNA binding. The efficacy of this alternative cMO design has been demonstrated in zebrafish embryos and compared to linear MOs and monocyclic constructs.
Keywords: Antisense agents, nucleic acids, photoactivatable, morpholino oligonucleotides, zebrafish
Graphical Abstract
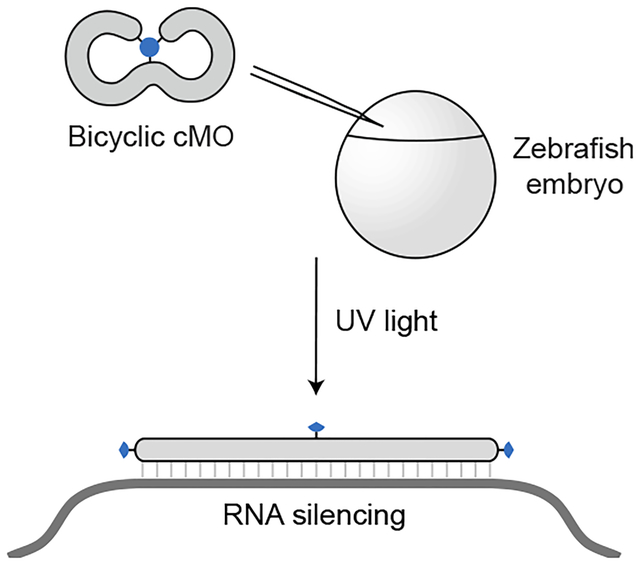
Caged morpholino oligonucleotides allow light-dependent gene silencing in whole organisms. In this study, a new type of caged morpholino with a bicyclic geometry was developed. Ultraviolet light linearizes the bicyclic structure through self-immolative linker cleavage, leading to targeted RNA silencing in zebrafish embryos.
INTRODUCTION
Light-gated technologies have emerged as powerful research tools to decipher the molecular mechanisms that underlie complex biological systems.[1, 2] Because specific tissues can be illuminated at precise timepoints, photoactivatable reagents are well-suited for inducing molecular perturbations in whole organisms with spatial and temporal control.[3, 4] These technologies are particularly useful when applied in optically transparent model organisms that develop ex utero such as zebrafish,[5] sea urchins,[6] and ascidians.[7] Oligonucleotide-based reagents are especially versatile probes as they can be readily designed to specifically target any gene,[8, 9] and phosphorodiamidate morpholino oligonucleotides (MOs) have been most commonly used to inhibit gene expression in vivo.[10–12] These synthetic DNA/RNA mimetics are composed of nucleobase-bearing morpholine rings connected with neutral phosphorodiamidate linkages (Figure 1A)[13] and constitute three out of the eleven FDA-approved oligonucleotide-based drugs in the last six years.[14] MOs are generally designed as 25-mer sequences that hybridize with their cognate RNAs to inhibit protein synthesis or alter splicing. These reagents typically are injected into early-stage embryos (e.g., one- to four-cell zebrafish zygotes) and become uniformly distributed throughout the animal.[15] As a result, standard MOs constitutively and ubiquitously silence their targeted genes, and in zebrafish embryos these reagents can be effective for up to five days.[16] To achieve spatiotemporal gene regulation, we and others have developed caged morpholino oligonucleotides (cMOs) that remain inactive until released by irradiation with light of specific wavelengths.
Figure 1. Morpholino oligonucleotides and their caged derivatives.
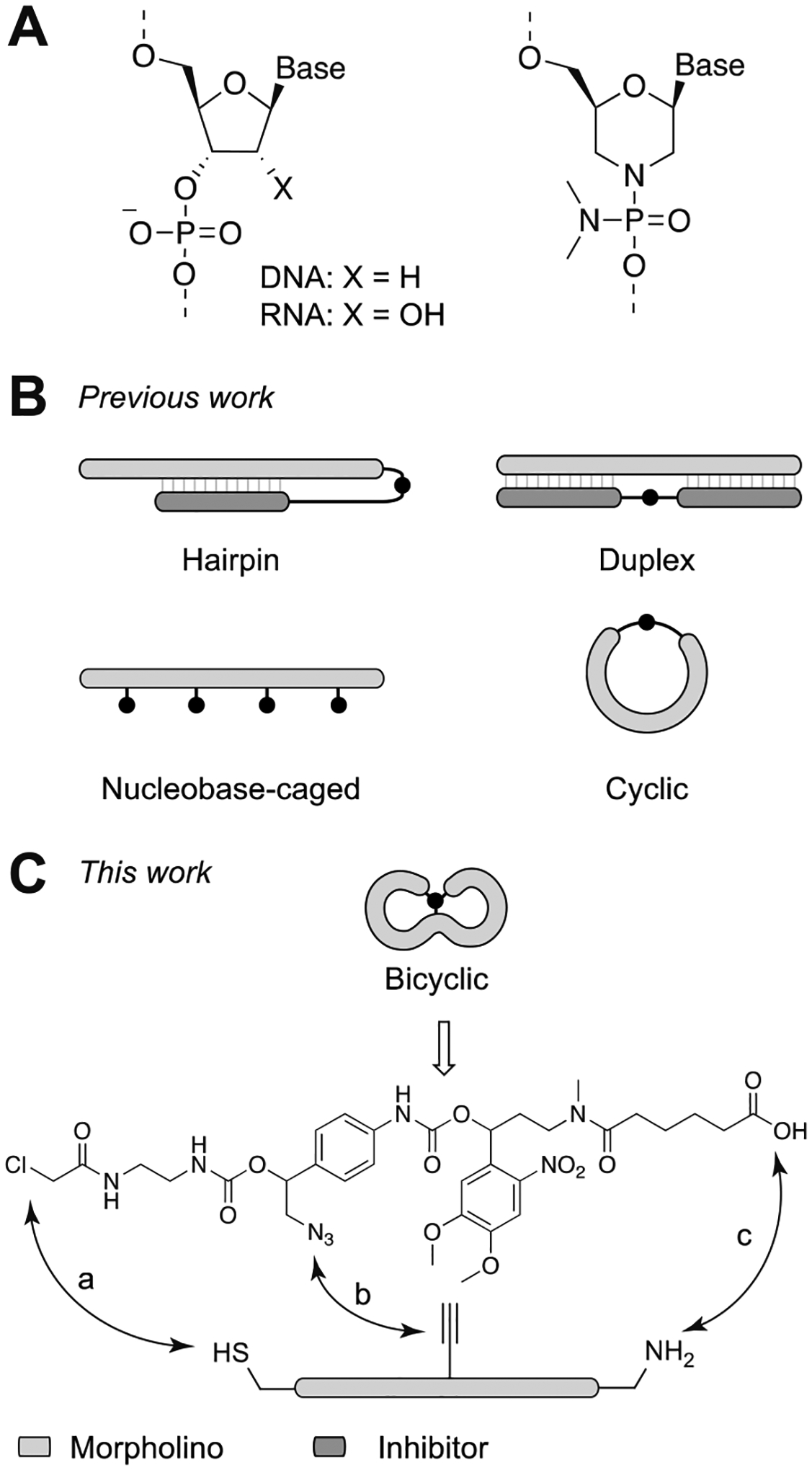
A) Comparison of DNA/RNA and MO chemical structures. B) Current caged morpholino structural types. C) Bicyclic cMO structure and overview of the synthesis strategy. The bicyclic geometry can be achieved by three orthogonal bioconjugation reactions: a) thiol-halogen exchange, b) CuAAC click reaction, c) amide coupling.
The first generation of photocaged morpholinos had a hairpin structure, in which a shorter inhibitory MO was used to restrict the 25-mer MO from binding to its target mRNA (Figure 1B).[17, 18] Multiple versions of the MO caging strategy were subsequently developed. For example, duplexes between the full-length MO and cleavable complementary oligonucleotides have been employed as cMOs (Figure 1B).[19, 20] In another design, nucleobases were protected with photoremovable groups to prevent them from Watson-Crick base-pairing (Figure 1B).[21, 22] However, each of these designs has certain drawbacks that can limit their dynamic range. For instance, hairpin cMOs require careful tuning of the inhibitor binding energetics to minimize basal activity,[18] and the inhibitory MO can impart additional toxicity. The complementary oligonucleotides released by photoactivated duplex cMOs are also potentially toxic, and the nucleobase-caged reagents require photolysis of multiple caging groups, which can be challenging to achieve quantitatively. To address these issues, we and the Tang laboratory independently developed cyclic cMOs by tethering the oligonucleotide termini with a photocleavable linker (Figure 1B).[23, 24] The curvature introduced by cyclization hinders cMO-RNA hybridization because oligonucleotide duplexes have limited conformational flexibility.[25]
Cyclic cMOs addressed several shortcomings of the previous generations.[23] This design does not require optimization of thermodynamic parameters, employs a single light-cleavable group, does not liberate any oligonucleotide byproducts, and is readily synthesized from commercially available reagents. In addition, the modular linker chemistry of cyclic cMOs facilitates the application of other uncaging methods. For instance, while the first generation of cyclic cMOs was designed to be activated by ultraviolet (UV) light, we have subsequently reported cyclic cMOs that can be uncaged with blue light,[26, 27] enzymes,[28] and small molecules[29]. Other versions of cyclic cMOs that contain ruthenium- or BHQ-based linkers have also been reported.[30, 31] Cyclic cMOs have enabled optochemical control of multiple genes in zebrafish, including no tail-a (tbxta), t-box gene 16 (tbx16), pancreas transcription factor 1 alpha (ptf1a), ets variant gene 2 (etv2), and focal adhesion kinase (fak).[23, 32] Taking advantage of the spatiotemporal control afforded by cMOs, we have further demonstrated how these optochemical tools can be combined with photoactivatable lineage tracers, fluorescence-activated cell sorting, and transcriptome profiling to gain new insights into tissue-patterning mechanisms.[33, 34]
Despite these successes, all current cMO structural types suffer from limited dynamic range. cMOs can inhibit their RNA targets to varying degrees prior to photoactivation, and finding a working dose that minimizes ‘leak’ and maximizes post-irradiation phenotypic penetrance can be challenging. The simultaneous application of two or more cMOs is even more likely to have significant basal activity. For example, while individual cyclic tbxta and tbx16 cMOs work effectively, we were unable to combine these two reagents to study how these transcription factors act in concert to regulate mesoderm development in zebrafish embryos.[35] The combined ‘leakiness’ of these cyclic cMOs resulted in light-independent mesodermal defects in all tested concentrations, including those lower than the effective dose of an individual tbxta or tbx16 MO. Overcoming these limitations will be critical for achieving the full potential of cMOs as tools for elucidating the mechanisms that regulate development, physiology, and disease.
To address these issues and build upon the cyclic cMO design, we report herein a new class of cMOs that tethers the MO termini to an internal position through a single photocleavable moiety, thereby generating a bicyclic structure. The photocleavable linker was designed to undergo light-dependent self-immolative cleavage, allowing release of the termini and MO linearization. We hypothesized that the smaller macrocycles and increased local curvature of these reagents would further limit RNA hybridization compared to standard cyclic cMOs.
RESULTS AND DISCUSSION
The cyclic cMO strategy validated that MO activity can be conformationally gated by macrocyclization.[23] In principle, a smaller macrocycle size would increase caging efficacy due to greater oligonucleotide curvature. To test this hypothesis, we previously synthesized non-photocleavable cyclic tbxta MOs of varying size (21-, 23-, and 25-mers) and measured their melting temperatures (Tm’s) with a complementary 25-base RNA.[23] While the smaller cyclic MOs exhibited reduced affinities towards the target RNA than the conventional 25-mer reagent, their uncaged products also were less potent in zebrafish embryos, as gauged by their ability to reduce Tbxta protein levels. Thus, it is essential to have 25-mer MOs to achieve efficient RNA targeting. We envisioned that a bicyclic caging strategy would preserve the required 25-mer length while forming smaller macrocycles, which could be achieved by tethering both MO termini to an internal nucleotide (Figure 1C).
While cyclic cMOs can be generated with bifunctional linkers, a trifunctional crosslinker is required for the bicyclic structure. Moreover, all three attachment points need to be released by a single photolytic reaction. To achieve both goals, we devised a novel self-immolative linker (Figure 1C) bearing a single photoremovable group upon photolysis which would trigger a spontaneous 1,6-elimination reaction to linearize the caged oligonucleotide (Figure 2). To generate the tandem-release core, we chose a photolabile 4,5-dimethoxy-2-nitrobenzyl (DMNB) group, which we used in our previous hairpin and cyclic cMO design.[18, 23] In this case, the DMNB moiety was strategically linked with a masked hydroxymethylaniline scaffold through a carbamate linkage, which was also employed in our nitroreductase- and small molecule-activatable cMOs.[28, 29] The spontaneous 1,6-elimination reaction has been reported to be rapid (t1/2 ~ 90 s),[36] enabling cMO activation to occur within minutes after linker photocleavage. The methylenetriazole moiety in the benzylic position is further expected to accelerate the reaction.[37] We also selected three orthogonal reactive moieties for the self-immolative linker: a chloroacetamide, an azide-bearing hydroxymethylaniline, and a carboxylic acid-functionalized DMNB group. This trifunctional linker can undergo three sequential reactions with a linear MO bearing a thiol, internal alkyne, and amine, respectively (Figure 1C). After light-dependent linearization, the final MO would bear an aminophenyltriazole scar in the phosphorodiamidate linkage (Figure 2). MOs with substituted interlinkages have been shown to be tolerated for antisense applications.[38, 39]
Figure 2. Bicyclic cMO photoactivation.
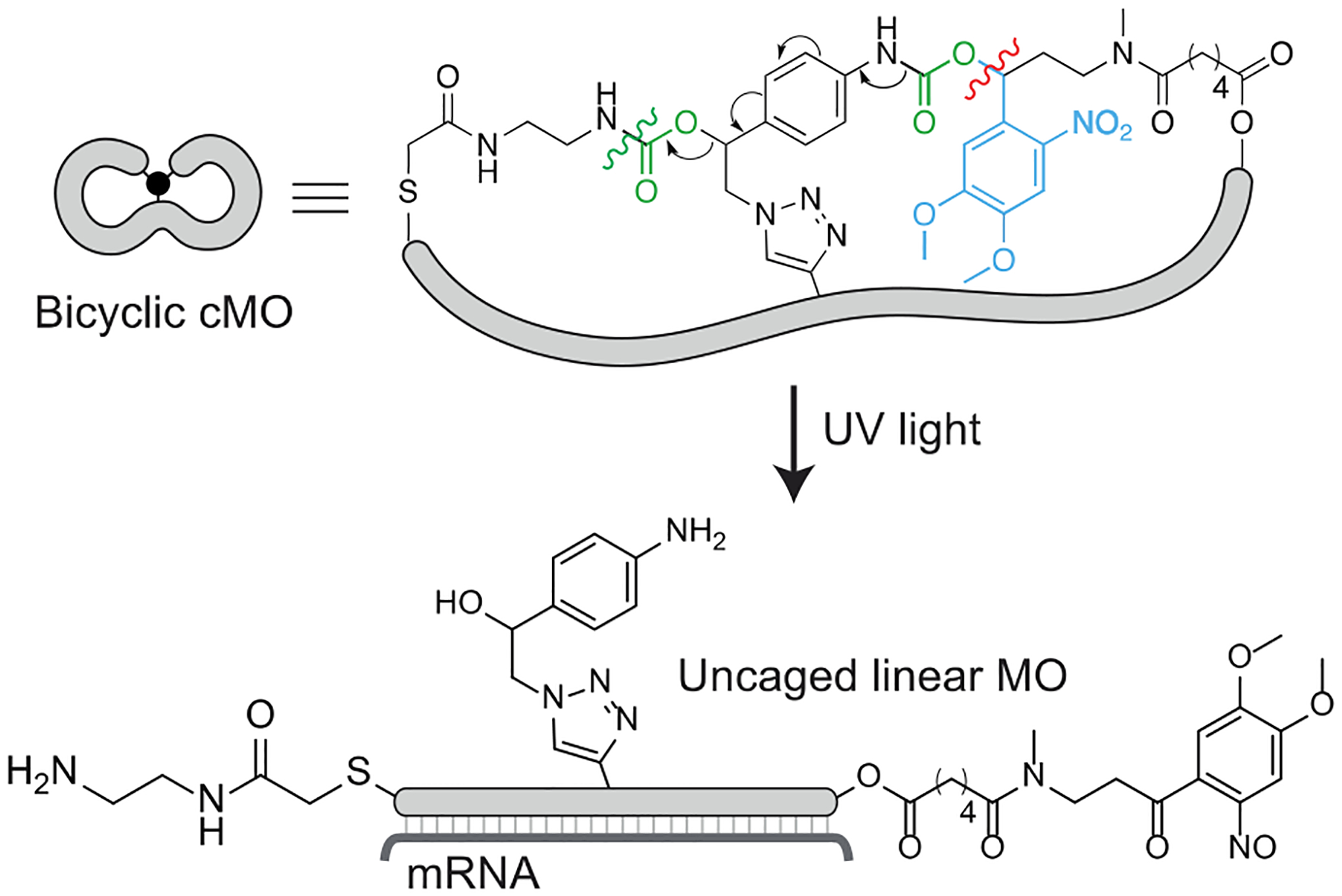
Proposed uncaging mechanism and subsequent hybridization to the targeted mRNA. The photocleavage site (red), 1,6-elimination cascade (arrows), and CO2 eliminations (green) have been shown.
According to our design, the self-immolative core in the linker (Figure 1C, and 10 in Scheme 1) includes a hydroxymethylaniline moiety with the aryl amine masked by a carbamate-linked DMNB photolabile group. Since most substituents at the benzylic position of the aniline are unstable due to the 1,6-elimination reaction, we planned to install the chloroacetamide moiety on the hydroxymethylaniline after building the self-immolative core. Accordingly, we first synthesized the TBS-protected azido hydroxymethylaniline 4 from commercially available 2-bromo-4’-nitroacetophenone 1 in six steps (Scheme 1). Sodium borohydride-mediated reduction of the α-bromoketone 1 and subsequent TBS protection provided protected α-bromo alcohol 2. For the alcohol protection, the TBS group was chosen because it has been shown to be stable in other self-immolative systems.[40] Palladium-mediated hydrogenation of the nitro group of 2 yielded the aryl amine, which was subsequently protected with trifluoroacetic anhydride to obtain 3. Substitution of this bromo compound with sodium azide afforded the corresponding azido derivative, which was then treated with methanolic ammonia to remove the trifluoroacetyl protection to afford the azide-bearing hydroxymethylaniline 4.
Scheme 1. Synthesis of the self-immolative linker for the bicyclic cMO.
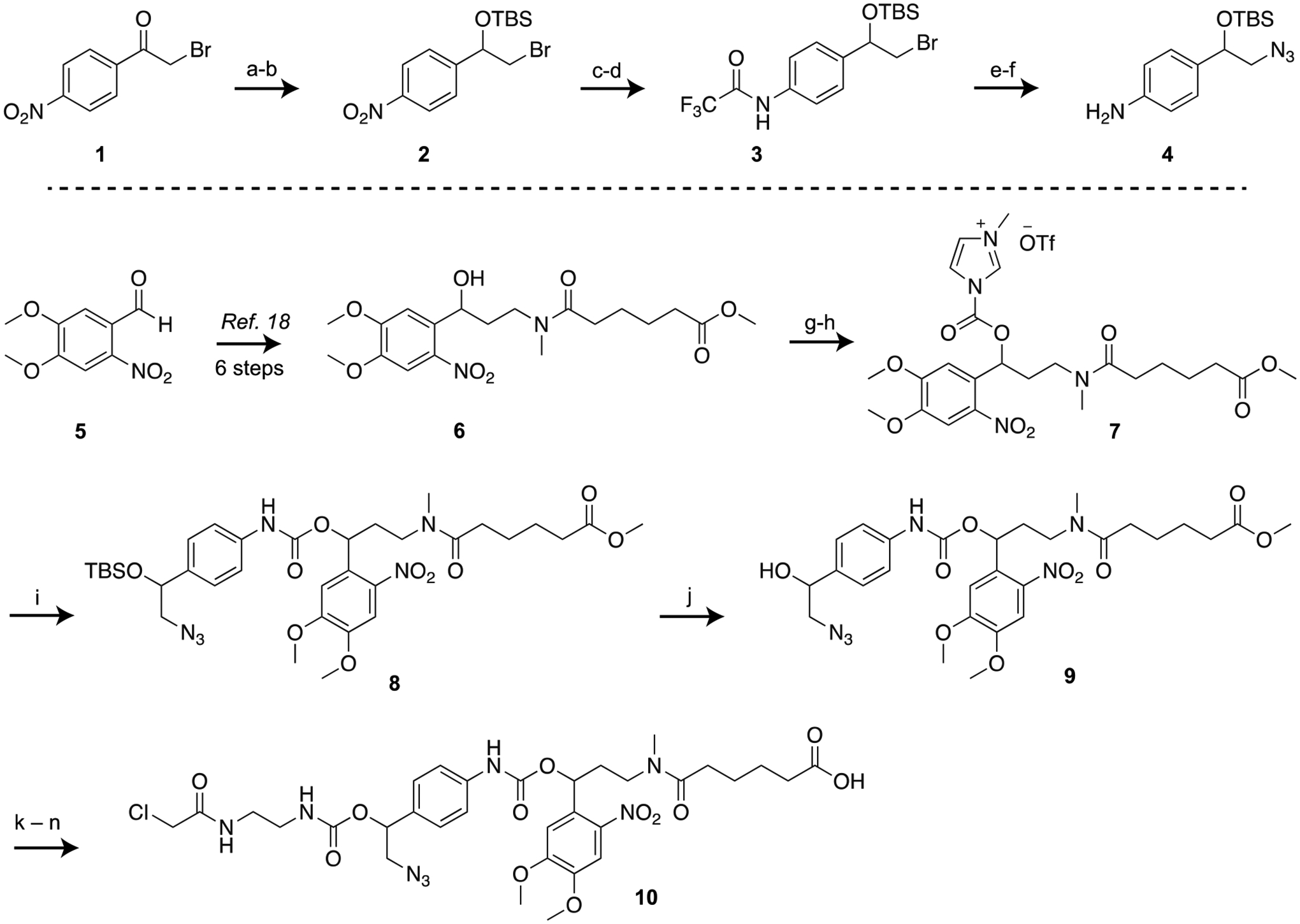
(a) NaBH4, MeOH, 69%; (b) TBSCl, imidazole, DMF, 62%; (c) 5% Pd-C, H2, THF, 87%; (d) (CF3CO)2O, DCM, 71%; (e) NaN3, TBAI, DMF, 60 °C, 66%; (f) 7 M NH3 in MeOH, 84%; (g) CDI, DCM; (h) MeOTf, DCM; (i) azido aniline 4, DCM, 24% over three steps; (j) TBAF, THF, 80%; (k) CDI, DCM; (l) ethylenediamine, 91% over two steps; (m) chloroacetyl chloride, 48%; (n) LiOH, THF-H2O, 75%.
To synthesize the rest of the trifunctional linker (Scheme 1), we prepared the DMNB-functionalized ester 6 from commercially available 4,5-dimethoxy-2-nitrobenzaldehyde 5, following our earlier reports.[18] To form the self-immolative core, we then attempted to couple the secondary alcohol functionality of 6 with the aryl amine of azido hydroxymethylaniline 4 through a carbamate linkage. This coupling, however, posed a significant hurdle,[41] likely due to weak nucleophilicity of the aniline and steric hindrance of the secondary alcohol. Our attempts to form the carbamate linkage by activating the alcohol as an N-hydroxysuccinimide carbonate, chloroformate, or imidazolium carbamate were unreliable and only afforded a complex mixture of products. Activation of the aniline functionality of 4 by isocyanate formation with phosgene proceeded smoothly, but subsequent coupling with 6 was unsuccessful under several conditions. We finally successfully coupled these two components by activating the alcohol 6 as an N-methylimidazolium salt 7 (Rapoport’s reagent type activation),[42] which to the best of our knowledge is the first example of hindered carbamate synthesis through N-methylimidazolium salt formation. The activated reagent 7 is not stable and had to be prepared in situ by a two-step sequence using carbonyl diimidazole (CDI) and methyl triflate immediately prior to use. Subsequent reaction of 7 with hydroxymethylaniline 4 afforded the product 8 containing the self-immolative core. To install the chloroacetamide side chain, the TBS protection from 8 was first removed with fluoride treatment to obtain the alcohol 9. The secondary alcohol 9 was then activated with CDI and conjugated with ethylenediamine followed by capping with chloroacetyl chloride to yield the methyl ester intermediate. Subsequent hydrolysis of the methyl ester with lithium hydroxide provided the fully functionalized bicyclic linker 10. We then confirmed that 10 can be cleaved by UV light by irradiating a dilute solution of the linker and monitoring the resulting reaction by HPLC. We observed that the linker formed photodegraded products within 30 seconds of UV light exposure (Figure S1).
Unlike MOs with terminal disulfides and amines, internally modified MOs are not commercially available. To develop trifunctionalized MOs with an additional internal alkyne handle, we decided to incorporate the alkyne group into monomeric MO building blocks through an N-methylpropargylamine modification in place of a standard dimethylamine moiety (Scheme 2). Accordingly, we synthesized 5’-hydroxyl adenine and thymine morpholino monomers (13 and 14, respectively)[43, 44] and developed the new propargylphosphoramidic dichloride reagent 16. Activation of the hydroxyl monomers with DBU and lithium bromide afforded the corresponding chlorophosphoramidites 17 and 18.[45] These monomers were then incorporated into the MOs during standard solid-phase synthesis. Since MOs are typically 25 nucleotides in length, we decided to include the alkyne moiety at the 12th or the 13th phosphoroamidate linkage, placing the alkyne handle in a central position. We chose to prepare two alkyne-functionalized MOs targeting zebrafish tbxta and tbx16, composed of the nucleotide sequences 5′-GACTTGAGGCAGACATATTTCCGAT-3′ and 5′-CTCTGATAGCCTGCATTATTTAGCC-3′, respectively (alkyne-functionalized monomers denoted in bold font). Having made the alkyne-functionalized MOs, we sought to confirm if the aminophenyltriazole scar in the final photocleavage product would interfere with gene inhibition in zebrafish embryos. We therefore prepared a model photocleavage product by copper-mediated azide-alkyne cycloaddition (CuAAC) or ‘click’ reaction of tbxta MO 19 and the azide-bearing hydroxymethylaniline 4 (Figure S2A). We then assessed its efficacy to inhibit the tbxta gene compared to a non-functionalized linear MO, as determined by the resulting notochord phenotypes. There was no appreciable difference between the model photocleavage product MO and the linear MO in their efficacy and toxicity (Figure S2B).
Scheme 2. Synthesis of the trifunctionalized linear morpholinos.
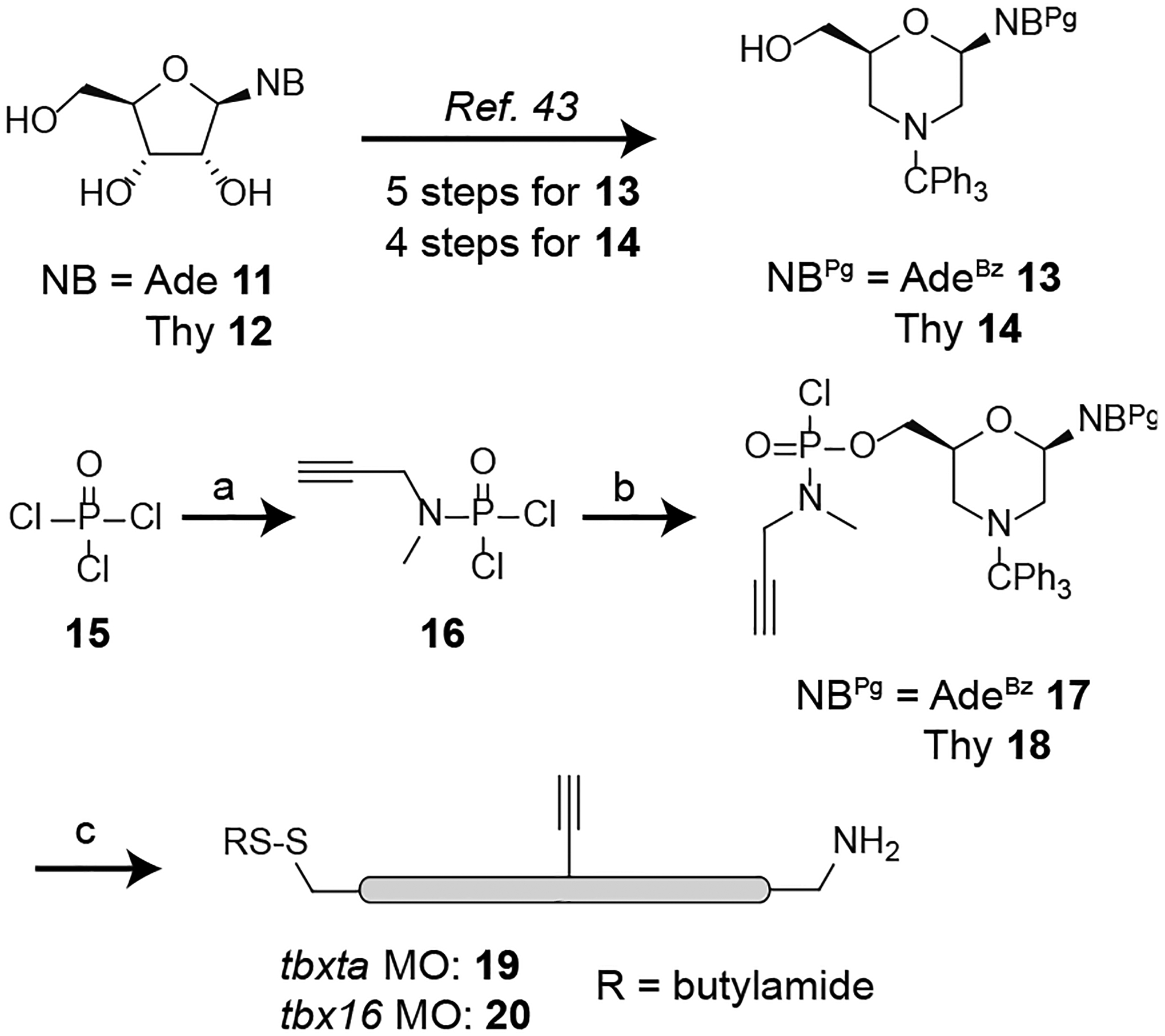
(a) N-methylpropargylamine, triethylamine, DCM, 78%; (b) 19, LiBr, DBU, DCM-acetonitrile, 46% for adenine (Ade); 51% for thymine (Thy); (c) Incorporation into MO sequences by solid-phase synthesis.
Having synthesized both the trifunctionalized linker and morpholinos, we next focused on developing an efficient synthetic protocol for MO bicyclization. While our cyclic cMO version used amide coupling followed by a thiol-halogen exchange reaction to close the ring, we first decided to perform the CuAAC click reaction for the bicyclic cMO synthesis (Scheme 3). This would allow us to monitor the product formation with mass spectrometry, whereas conducting this cycloaddition reaction at a later stage would not lead to a change in molecular weight. In our optimized protocol (see Supporting Information), the alkyne-functionalized linear MOs (tbxta MO 19 or tbx16 MO 20) and the linker 10 were click-conjugated using copper iodide catalyst. Reducing agents such as sodium ascorbate were avoided because the disulfide end group was unstable in these conditions.[46] Formation of the cycloaddition product (tbxta MO 21 and tbx16 MO 22) was monitored by HPLC and mass spectrometry. Any unreacted linear morpholino was removed by further click reaction with azide-functionalized agarose beads.
Scheme 3. Synthesis of the bicyclic cMO.
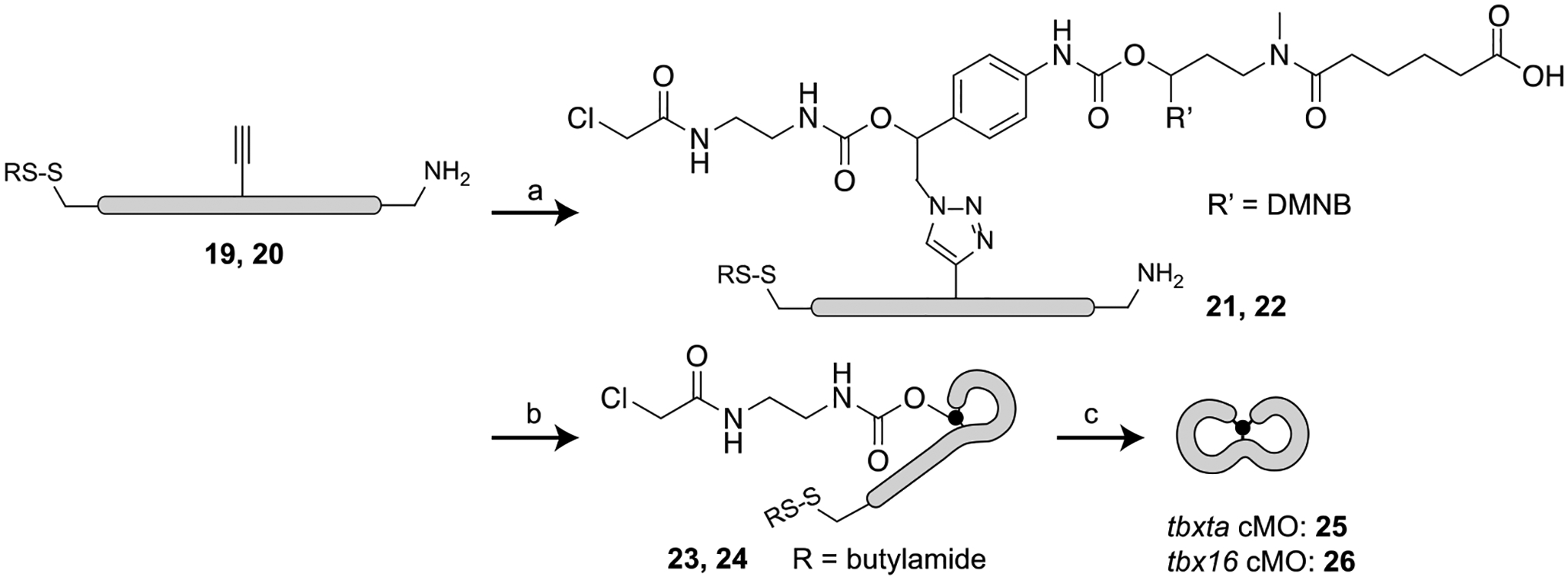
(a) Linker 10, CuI, tris(benzyltriazolylmethyl)amine (TBTA), DMSO, 0.5 M triethylammonium acetate (TEAA) buffer, 37 °C, 50–72%; (b) DMTMM, 100 mM MOPS - 1 M NaCl buffer, pH 7.5, 37 °C, 40–55%; (c) tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl), 0.1 M Tris-HCl buffer, pH 7.5 then pH 8.5, 37 °C, 45–60%.
To generate the first macrocycle (tbxta MO 23 or tbx16 MO 24), we next performed the amide-coupling reaction by activating the carboxylic acid with a large excess of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM)[47] (Scheme 3). Assuming the carboxylic acid was quantitatively converted to activated ester, the reaction mixture was treated with ω-aminohexyl agarose resin to remove any linear morpholino. This method of removing the unreacted amine morpholino was more effective than N-hydroxysuccinimide-functionalized resin treatment. The monocyclic oligonucleotides (tbxta MO 23 or tbx16 MO 24) were then isolated with a Nap-25 (Sephadex G-25) column. Although we were unable to find a suitable HPLC condition to separate the single lariat product from the linear conjugate (tbxta MO 21 or tbx16 MO 22) (see Supporting Information), we could confirm the purity of the products 23 and 24 by mass spectrometry. Finally, to prepare the second macrocycle, the terminal disulfide was reduced with brief treatment with tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl). The reducing agent was removed by dialysis to avoid side reactions with the chloroacetamide group, and the resulting thiol reacted spontaneously with the chloroacetamide group by thiol-halogen exchange reaction to form the desired bicyclic cMO (tbxta cMO 25 or tbx16 cMO 26). Unlike our previously reported cyclic cMO synthesis,[23] treatment with TCEP-HCl solution provided a cleaner reaction profile than resin-immobilized TCEP. The reaction mixture was then treated with maleimide-functionalized agarose beads to remove any unreacted single-lariat MO, and the final bicyclic cMO was purified with Nap-5 columns and confirmed by mass spectroscopy.
We next assessed the ability of the bicyclic tbxta cMO to hybridize to a complementary oligonucleotide, using a cyanine 5 (Cy5) dye-tagged DNA sequence and gel-shift assays. The bicyclic construct had weaker affinity towards the 25-base DNA target in comparison to its linear counterpart, with a half maximal effective concentrations (EC50’s) of 310 nM and 27 nM, respectively (Figure S3A–B). To determine if UV irradiation would linearize the bicyclic structure, we exposed a solution of the bicyclic cMO to 365-nm light and analyzed its interactions with the Cy5-tagged DNA sequence by gel electrophoresis (Figure S3C). As expected, the bicyclic cMO exhibited light-dependent duplex formation with its DNA target within 20 seconds of UV irradiation. To further confirm the linearization of the bicyclic cMO, we irradiated an aqeuous solution of the tbxta cMO with UV light and analyzed the resulting product by mass spectrometry (Figure S4). The observed molecular weight of the photocleaved product (9510 Da) was consistent with that predicted for the linear MO bearing an aminophenyltriazole scar (9516 Da).
To test our new caging strategy in vivo, we evaluated the bicyclic tbxta and tbx16 cMOs in zebrafish embryos. Tbxta, the zebrafish orthologue of mammalian Brachyury, has an essential role in the differentiation of axial mesoderm progenitors into notochord cells.[48] Tbxta deficiency results in the loss of notochord and posterior mesoderm, as well as abnormal U-shaped somites due to the loss of notochord-derived signals.[49, 50] We have previously found that a 1 ng/embryo (~115 fmol) dose of the linear tbxta MO fully recapitulates the tbxta mutant phenotype in zebrafish embryos.[17, 18] Accordingly, we injected an equivalent amount of the bicyclic cMO into one-cell zebrafish zygotes and either cultured the embryos in the dark or UV-irradiated them for 15 seconds at 3.5 hours post-fertilization (hpf) for 15 seconds. We have previously demonstrated that this irradiation regimen does not cause any toxicity to the embryos.[17, 51] The embryos were then scored at 24 hpf using a morphology-based system that includes four mesodermal phenotypes.[18] We also compared basal activity of the bicyclic tbxta cMOs to that of a cyclic tbxta cMO (Figure 3A), which was prepared as previously described.[23] We found that the bicyclic cMO was at least comparable to the cyclic reagent in terms of basal activity. Approximately 85% of the embryos developed normally in both cases without any developmental defects when raised in the dark. However, the bicyclic cMO exhibited lower activity recovery upon illumination; about 80% of embryos injected with this reagent exhibited a fully penetrant tbxta morphant phenotype, as opposed to the 90% observed with the cyclic cMO.
Figure 3. In vivo efficacy of bicyclic cMOs.
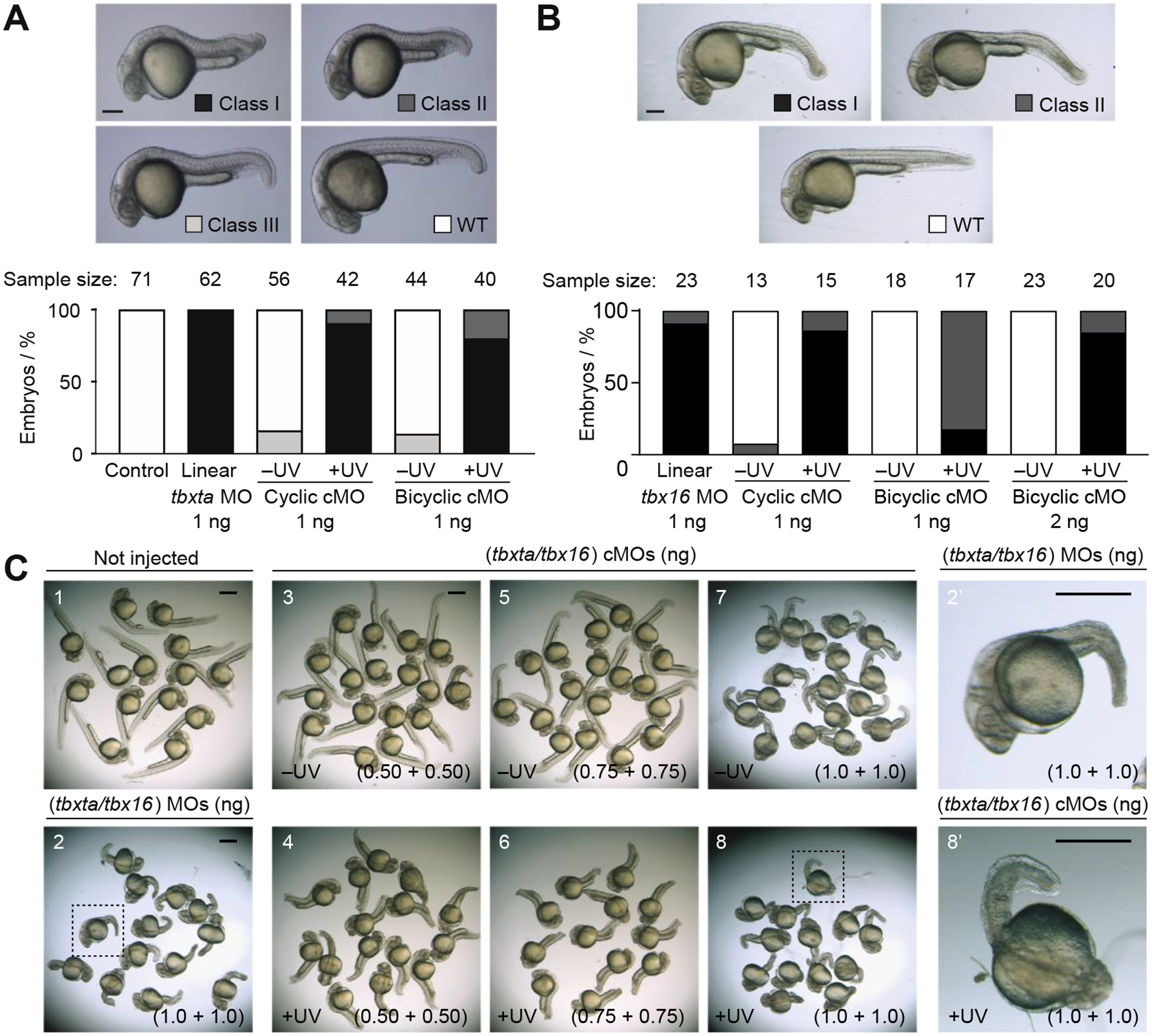
Phenotypic distributions for zebrafish embryos injected with the indicated oligonucleotides and either cultured in the dark or globally irradiated with UV light at 3 – 3.5 hpf. A) Comparison of cyclic and bicyclic tbxta cMOs. Classification of tbxta loss-of-function phenotypes (Class I = most severe, WT = wildtype). 24-hpf embryos are shown. Scale bar = 200 μm. B) Comparison of cyclic and bicyclic tbx16 cMOs. Classification of tbx16 loss-of-function phenotypes (Class I = most severe, WT = wildtype). 30-hpf embryos are shown. Scale bar = 200 μm. C) Comparison of wild-type zebrafish embryos (1) and those co-injected with linear MOs (2) or bicyclic cMOs (3–8) targeting tbxta and tbx16. Embryos injected with the bicyclic cMOs and cultured in the dark developed normally (3 and 5) with mild defects at the highest dose (7). UV irradiation of the bicyclic cMO-injected embryos replicated the tbxta/tbx16 double mutant phenotype (loss of notochord, trunk, and tail mesoderm) in a dose-dependent manner (4, 6, and 8). Insets provide close-up views of representative embryos co-injected with linear tbxta/tbx16 MOs or bicyclic tbxta/tbx16 cMOs and then irradiated (dashed boxes in 2 and 8; 2’ and 8’). Scale bars: 500 μm.
The T-box gene, tbx16 regulates paraxial mesoderm patterning. Zebrafish with loss-of-function tbx16 alleles (also known as spadetail mutants) fail to form trunk somites, and the undifferentiated mesodermal progenitor cells accumulate in the tailbud form a spade-like structure.[52, 53] In analogy to the experiments described above, we injected one-cell zebrafish zygotes with bicyclic or cyclic tbx16 cMOs and scored their phenotypes at 30 hpf (Figure 3B). As with the tbxta cMOs, the bicyclic tbx16 cMO exhibited less efficient functional recovery upon UV illumination than the cyclic version. A 1 ng/embryo dose of the linear tbx16 MO phenocopied the tbx16 mutant, and zebrafish embryos injected with an equivalent dose of the bicyclic tbx16 cMO developed normally when cultured in the dark. However, recovery of MO activity upon UV irradiation was incomplete. About 18% of the irradiated embryos lacked trunk somites and exhibited a spade-like tailbud (Class I), and the remainder had intermediate tbx16 loss-of-function phenotypes (Class II). To fully recapitulate the tbx16 mutant phenotype upon cMO uncaging, we needed to inject 2 ng/embryo of the bicyclic reagent. Nevertheless, even this higher dose of the bicyclic cMO had no detectable light-independent activity. In comparison, a 1 ng/embryo dose of the cyclic tbxta cMO led to developmental defects in 8% of the embryos under the same conditions. These findings demonstrate the general efficacy of bicyclic cMOs and confirm that these more conformationally constrained reagents can have lower basal activity.
To conclude our studies, we explored the feasibility of using bicyclic cMOs to simultaneously inactivate two genes. Our tbxta- and tbx16-targeting reagents are ideal for assessing combinatorial applications since the two T-box transcription factors have distinct roles during mesoderm differentiation (notochord vs. somite development). Zebrafish embryos with tbxta and tbx16 mutations are phenotypically distinct from the single mutants, exhibiting loss of notochord, trunk, and tail mesoderm.[54] We injected one-cell zebrafish zygotes with varying amounts of the bicyclic tbxta and tbx16 cMOs and then either maintained the embryos in the dark or globally UV-irradiated them for 15 seconds at 3.5 hpf. Most of the resulting embryos developed normally in the dark when each bicyclic cMO was injected at up to a 0.75 ng/embryo dose. This markedly contrasts the light-independent mesoderm defects we previously observed when cyclic tbxta and tbx16 cMOs were co-injected into zebrafish embryos.[35] Moreover, the bicyclic cMO-injected zebrafish (0.75 ng/embryo dose) exhibited substantial mesodermal deficits upon exposure to UV light, albeit with less severity that that obtained with linear tbxta and tbx16 MOs. Embryos injected with 1 ng/embryo of each bicyclic cMO and then UV-irradiated recapitulated the double mutant phenotype; however, this dose also caused a basal level of intermediate notochord defects, likely due to the residual ‘leakiness’ of the tbxta-targeting reagent. Taken together, our studies establish our bicyclic cMOs as valuable reagents for optical gene silencing, with reduced basal activity but also lower functional recovery in comparison to cyclic cMOs.
CONCLUSION
In summary, we have designed and synthesized cMOs with a bicyclic structure, utilizing a self-immolative linker that releases three components with a single photolysis event. With its smaller macrocycles, the bicyclic cMOs can exhibit lower dark-state activity than their cyclic counterparts, thereby validating the concept that increased MO curvature is associated with reduced RNA binding. Finally, the best of our knowledge, this is the first example of a short oligonucleotide (~25 bases) with a bicyclic oligonucleotide geometry. While short bicyclic peptides have been shown to have better drug-like properties and improved cell-penetration capabilities,[40, 55] with the exception of a few larger bi/multicyclic structures (36 bases[56] and 90 bases[57]), analogous bicyclic oligonucleotides have not been reported in the literature.
Taking advantage of this diminished ‘leakiness,’ we could use bicyclic cMOs targeting tbxta or tbx16 in combination to achieve light-dependent defects in both axial and paraxial mesoderm derivatives. The extent to which this basal activity is reduced varied between the tbxta- and tbx16-targeting reagents, and we surmise that this reflects differences in the secondary and tertiary structures or binding proteins of the targeted mRNAs. We also noted that the bicyclic cMOs do not fully reach the uncaging level of cyclic cMOs. This is possibly due to non-quantitative linearization of the caged structure after linker photolysis, which could result from incomplete 1,6-elimination. In addition, although our studies indicate that the internal aminophenyltriazole scar does not significantly affect tbxta MO function, this modification might reduce hybridization efficiencies with other endogenous RNA targets. Consistent with this idea, the functional recovery of the photolyzed bicyclic tbx16 cMO was lower than that of the tbxta-targeting reagent. Our findings therefore suggest ways in which cMOs could be further improved. For example, basal activities can be further diminished by tethering the MO termini to an internal nucleobase rather than phosphorodiamidate linkage, impeding RNA hybridization by both conformational constraints and loss of Watson-Crick base-pairing. Alternative self-immolative linkers with more rapid elimination kinetics or scarless photorelease chemistry could also improve the functional recovery of bicyclic cMOs after photolysis.
We anticipate that further development of bicyclic cMOs will lead to versatile reagents for controlling multiple genes in whole organisms. These conformationally constrained oligonucleotides could not only enable the study of complex genetic networks that regulate tissue formation but might also find applications in oligonucleotide-based drug design.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yongfu Li, Ph.D. at Gene Tools, LLC for incorporating the alkyne-functionalized monomers into MO oligomers. This work was supported by the National Institutes of Health (R35 GM127030 and R01 GM108952 to J.K.C.) and a Stanford Maternal and Child Health Research Institute Postdoctoral Support Award (B.R.S.).
Footnotes
Supporting Information containing supplementary figures, experimental procedures, NMR data, and mass spectra.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- [1].Forlani G, Di Ventura B, Curr. Opin. Syst. Biol 2021, 28, 100356. [Google Scholar]
- [2].Ankenbruck N, Courtney T, Naro Y, Deiters A, Angew. Chem. Int. Ed. Engl 2018, 57, 2768–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hartmann D, Smith JM, Mazzotti G, Chowdhry R, Booth MJ, Biochem. Soc. Trans 2020, 48, 1645–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hughes RM, Crit. Rev. Biochem. Mol. Biol 2018, 53, 453–474. [DOI] [PubMed] [Google Scholar]
- [5].Shestopalov IA, Chen JK, Zebrafish 2010, 7, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bardhan A, Deiters A, Ettensohn CA, Dev. Biol 2021, 475, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hozumi A, Horie T, Sasakura Y, Dev. Dyn 2015, 244, 1375–1393. [DOI] [PubMed] [Google Scholar]
- [8].Darrah KE, Deiters A, Chem. Soc. Rev 2021, 50, 13253–13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Housden BE, Muhar M, Gemberling M, Gersbach CA, Stainier DY, Seydoux G, Mohr SE, Zuber J, Perrimon N, Nat. Rev. Genet 2017, 18, 24–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Corey DR, Abrams JM, Genome Biol 2001, 2, REVIEWS1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Blum M, De Robertis EM, Wallingford JB, Niehrs C, Dev. Cell 2015, 35, 145–149. [DOI] [PubMed] [Google Scholar]
- [12].Morpholino Oligomers: Methods and Protocols, Vol. 1565, (Eds.: Moulton HM, Moulton JD), Humana Press, New York, 2017, pp. 1–277. [Google Scholar]
- [13].Summerton J, Weller D, Antisense Nucleic Acid Drug Dev 1997, 7, 187–195. [DOI] [PubMed] [Google Scholar]
- [14].Shadid M, Badawi M, Abulrob A, Expert Opin. Drug Metab. Toxicol 2021, 17, 1281–1292. [DOI] [PubMed] [Google Scholar]
- [15].Ekker SC, Larson JD, Genesis 2001, 30, 89–93. [DOI] [PubMed] [Google Scholar]
- [16].Shestopalov IA, Chen JK, Chem. Soc. Rev 2008, 37, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shestopalov IA, Sinha S, Chen JK, Nat. Chem. Biol 2007, 3, 650–651. [DOI] [PubMed] [Google Scholar]
- [18].Ouyang X, Shestopalov IA, Sinha S, Zheng G, Pitt CL, Li WH, Olson AJ, Chen JK, J. Am. Chem. Soc 2009, 131, 13255–13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tallafuss A, Gibson D, Morcos P, Li Y, Seredick S, Eisen J, Washbourne P, Development 2012, 139, 1691–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tomasini AJ, Schuler AD, Zebala JA, Mayer AN, Genesis 2009, 47, 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Deiters A, Garner RA, Lusic H, Govan JM, Dush M, Nascone-Yoder NM, Yoder JA, J. Am. Chem. Soc 2010, 132, 15644–15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu Q, Deiters A, Acc. Chem. Res 2014, 47, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yamazoe S, Shestopalov IA, Provost E, Leach SD, Chen JK, Angew. Chem. Int. Ed. Engl 2012, 51, 6908–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Y, Wu L, Wang P, Lv C, Yang Z, Tang X, Nucleic Acids Res 2012, 40, 11155–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ramstein J, Lavery R, Proc. Natl. Acad. Sci. U. S. A 1988, 85, 7231–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yamazoe S, Liu Q, McQuade LE, Deiters A, Chen JK, Angew. Chem. Int. Ed. Engl 2014, 53, 10114–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pattanayak S, Vázquez-Maldonado LA, Deiters A, Chen JK, Methods Enzymol 2019, 624, 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yamazoe S, McQuade LE, Chen JK, ACS Chem. Biol 2014, 9, 1985–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Darrah K, Wesalo J, Lukasak B, Tsang M, Chen JK, Deiters A, J. Am. Chem. Soc 2021, 143, 18665–18671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Griepenburg JC, Rapp TL, Carroll PJ, Eberwine J, Dmochowski IJ, Chem. Sci 2015, 6, 2342–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Deodato D, Dore TM, Molecules 2020, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gutzman JH, Graeden E, Brachmann I, Yamazoe S, Chen JK, Sive H, Biol. Open 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shestopalov IA, Pitt CL, Chen JK, Nat. Chem. Biol 2012, 8, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Payumo AY, McQuade LE, Walker WJ, Yamazoe S, Chen JK, Nat. Chem. Biol 2016, 12, 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Payumo AY, Walker WJ, McQuade LE, Yamazoe S, Chen JK, ACS Chem. Biol 2015, 10, 1466–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Carl PL, Chakravarty PK, Katzenellenbogen JA, J. Med. Chem 1981, 24, 479–480. [DOI] [PubMed] [Google Scholar]
- [37].Alouane A, Labruère R, Le Saux T, Schmidt F, Jullien L, Angew. Chem. Int. Ed. Engl 2015, 54, 7492–7509. [DOI] [PubMed] [Google Scholar]
- [38].Weller DD, Hassinger JN, Cai BZ (AVI Biopharma), U.S. Patent 7,943.762, 2011.
- [39].Mellbye BL, Weller DD, Hassinger JN, Reeves MD, Lovejoy CE, Iversen PL, Geller BL, J. Antimicrob. Chemother 2010, 65, 98–106. [DOI] [PubMed] [Google Scholar]
- [40].Davies S, Oliveira BL, Bernardes GJL, Org. Biomol. Chem 2019, 17, 5725–5730. [DOI] [PubMed] [Google Scholar]
- [41].Shahsavari S, Gooding J, Wigstrom T, Fang S, ChemistrySelect 2017, 2, 3959–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Saha AK, Rapoport H, Schultz P, J. Am. Chem. Soc 1989, 111, 4856–4859. [Google Scholar]
- [43].Pattanayak S, Paul S, Nandi B, Sinha S, Nucleosides Nucleotides Nucleic Acids 2012, 31, 763–782. [DOI] [PubMed] [Google Scholar]
- [44].Bhadra J, Pattanayak S, Sinha S, Curr. Protoc. Nucleic Acid Chem 2015, 62, 4.65.61–26. [DOI] [PubMed] [Google Scholar]
- [45].Pattanayak S, Sinha S, Tetrahedron Lett 2012, 53, 6714–6717. [Google Scholar]
- [46].Giustarini D, Dalle-Donne I, Colombo R, Milzani A, Rossi R, Nitric Oxide 2008, 19, 252–258. [DOI] [PubMed] [Google Scholar]
- [47].Li X, Gartner ZJ, Tse BN, Liu DR, J. Am. Chem. Soc 2004, 126, 5090–5092. [DOI] [PubMed] [Google Scholar]
- [48].Schulte-Merker S, van Eeden FJ, Halpern ME, Kimmel CB, Nüsslein-Volhard C, Development 1994, 120, 1009–1015. [DOI] [PubMed] [Google Scholar]
- [49].Halpern ME, Ho RK, Walker C, Kimmel CB, Cell 1993, 75, 99–111. [PubMed] [Google Scholar]
- [50].Showell C, Binder O, Conlon FL, Dev. Dyn 2004, 229, 201–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shestopalov IA, Chen JK, Methods Cell Biol 2011, 104, 151–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kimmel CB, Kane DA, Walker C, Warga RM, Rothman MB, Nature 1989, 337, 358–362. [DOI] [PubMed] [Google Scholar]
- [53].Griffin KJ, Amacher SL, Kimmel CB, Kimelman D, Development 1998, 125, 3379–3388. [DOI] [PubMed] [Google Scholar]
- [54].Amacher SL, Draper BW, Summers BR, Kimmel CB, Development 2002, 129, 3311–3323. [DOI] [PubMed] [Google Scholar]
- [55].Lian W, Jiang B, Qian Z, Pei D, J. Am. Chem. Soc 2014, 136, 9830–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chaudhuri NC, Kool ET, J. Am. Chem. Soc 1995, 117, 10434–10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Seyfried P, Eiden L, Grebenovsky N, Mayer G, Heckel A, Angew. Chem. Int. Ed. Engl 2017, 56, 359–363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.