

Abstract
BET inhibition has been shown to have a promising antitumor effect in multiple tumors. However, the impact of BET inhibition on antitumor immunity was still not well documented in HNSCC. In this study, we aim to assess the functional role of BET inhibition in antitumor immunity and clarify its mechanism. We show that BRD4 is highly expressed in HNSCC and inversely correlated with the infiltration of CD8+ T cells. BET inhibition potentiates CD8+ T cell-based antitumor immunity in vitro and in vivo. Mechanistically, BRD4 acts as a transcriptional suppressor and represses the expression of MHC class I molecules by recruiting G9a. Pharmacological inhibition or genetic depletion of BRD4 potently increases the expression of MHC class I molecules in the absence and presence of IFN-γ. Moreover, compared to PD-1 blocking antibody treatment or JQ1 treatment individually, the combination of BET inhibition with anti-PD-1 antibody treatment significantly enhances the antitumor response in HNSCC. Taken together, our data unveil a novel mechanism by which BET inhibition potentiates antitumor immunity via promoting the expression of MHC class I molecules and provides a rationale for the combination of ICBs with BET inhibitors for HNSCC treatment.
Keywords: antitumor immunity, BET inhibition, head and neck squamous cell carcinoma, MHC class I molecules, PD-1
Graphical abstract
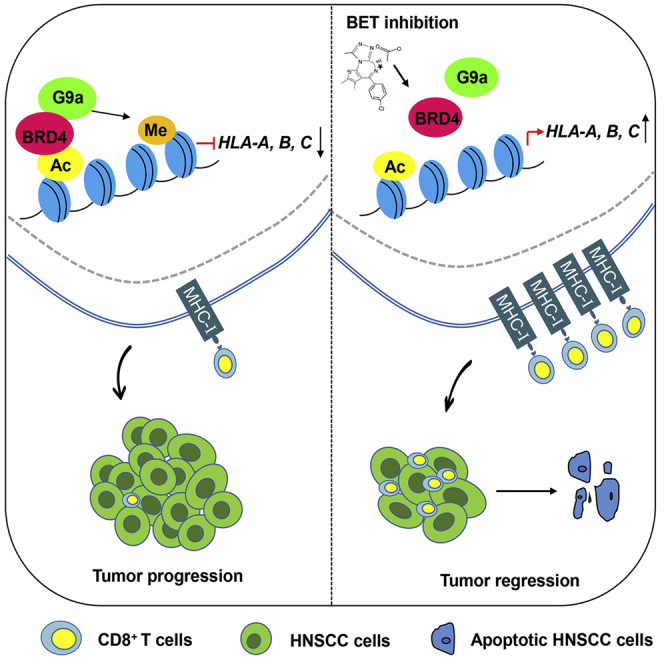
Zhang and colleagues show that BET inhibition triggers CD8+ T cell-mediated antitumor immunity by upregulating the expression of MHC class I molecules. Combination of BET inhibition with anti-PD-1 treatment significantly enhances the antitumor response in HNSCC.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, accounting for 90% of all head and neck tumors, which is characterized by high invasion and metastasis potential. Although HNSCC patients initially respond well to surgery, radiotherapy, and chemotherapy, tumors frequently recur and spread, with a low 5-year survival rate.1,2 Recently, increasing evidence has shown that immunotherapy is a promising therapeutic option for cancer patients.3,4 Immune checkpoint blockers (ICBs), such as anti-PD-1/PD-L1 and anti-CTLA-4 reagents, have been developed and are achieving clinical success in the management of various cancers, including HNSCC.5,6,7,8 However, only approximately 15%–40% of patients respond to ICB treatment in HNSCC.9 Therefore, it is crucial to better understand the determinants of a favorable response to ICBs and identify rational combinations to improve the efficacy and response rates.
BRD4 is a member of the bromodomain and extraterminal (BET) protein family and is associated with acetylated chromatin, facilitating transcriptional activation through increasing the recruitment of transcriptional activators.10,11 Specific BET inhibitors (iBETs), such as JQ1 and iBET-151, have been reported to not only induce direct antitumor effects, such as apoptosis and cell-cycle arrest but also alter the tumor microenvironment (TME) through modulating immune effector cell activities.12,13,14,15 Of note, a study on the therapeutic effect of JQ1 on lymphoma has shown that the antitumor responses were more effective in immunocompetent mice as compared to immunodeficient ones.14 These findings suggested that BET inhibition may provoke antitumor immunity in immunocompetent mice. However, the functional role of immune cells and their underlying mechanisms remain poorly understood in BET inhibition-mediated antitumor effects in HNSCC.
In this study, we show that CD8+ T cells are involved in the BET inhibition-mediated antitumor reaction. In a 4-nitroquinoline 1-oxide (4NQO)-induced mouse model, targeting BRD4 with JQ1 effectively impedes tumor growth with an increase in CD8+ T cell infiltration in HNSCC. Importantly, combining BET inhibition with anti-PD-1 antibody results in a synergistic antitumor effect and enhances the antitumor response of anti-PD-1 antibody in HNSCC. Mechanistically, we reveal that BRD4 can function as a transcriptional repressor of major histocompatibility complex (MHC) class I molecule to promote the immune escape of HNSCC by inhibiting MHC class I molecule expression via recruiting G9a.
Results
BRD4 expression is increased in HNSCC and BET inhibition reinvigorates antitumor immunity
The expression of BRD4 was evaluated in HNSCC by using immunohistochemistry (IHC) and The Cancer Genome Atlas (TCGA) database. As shown in Figure S1A, BRD4 mRNA expression was upregulated in HNSCC in comparison with the normal tissue. Immunostaining of the tissue microarray was performed and showed that BRD4 protein expression levels were significantly increased in HNSCC as compared to the normal tissue (Figures S1B and S1C). Similar results were also observed in 8 HNSCC tissues and their corresponding normal tissues (Figure S1D). These findings implicated that BRD4 may serve as a therapeutic target for HNSCC, which was further verified by using a 4NQO-induced HNSCC mouse model treated with the BRD4 inhibitor, JQ1 (Figures S1E and S1F).
To address the potential impact of JQ1 on immune response to tumors, murine primary HNSCC samples were harvested for immune profiling 10 days after JQ1 treatment, as shown in Figures 1A and 1B. A notable increase in CD8+ tumor-infiltrated lymphocytes (TILs), CD4+ TILs and macrophages was observed in the mice upon JQ1 treatment. Importantly, the percentage of granzyme B (GZMB)+CD8+ TILs and interferon-γ (IFN-γ)+CD8+ TILs was also increased in JQ1-treated mice. In addition, significantly more CD8+ T cells and CD4+ T cells expressed PD-1 in TILs from mice upon JQ1 treatment (Figure S1G). However, there was no significant change in the frequencies of natural killer (NK) cells and myeloid-derived suppressor cells (MDSCs). These results suggest that JQ1 treatment modifies the tumor-immune infiltrate in HNSCC, and may promote increased T cell activation and potentially enhance responsiveness to anti-PD-1 blockade. Next, we further profiled the immune cells in the spleen and periphery blood to examine whether JQ1 systemically enhanced the immune cell-mediated antitumor reaction. As shown in Figures 1C and 1D, the CD8+ T cell-mediated antitumor immunity was predominately enhanced. Taken together, these findings show that JQ1 treatment induces the coordinate alterations in CD8+ T cells, CD4+ T cells, and macrophages to achieve optimal antitumor immune response, but centered on CD8+ T cells. Then, we further investigated the association of BRD4 expression and the infiltration of CD8+ T cells. We analyzed the correlation of BRD4 mRNA expression with CD8A, CD8B, IFNG, GZMA, and GZMB gene expressions in HNSCC based on TCGA HNSCC database. As evident in Figures S1H–S1L, BRD4 mRNA expression was significantly inversely correlated with CD8A, CD8B, IFNG, GZMA, and GZMB expression in HNSCC. To reinforce these findings, immunostaining for BRD4 and CD8+ T cells were performed in HNSCC tissue samples. As shown in Figures 1E and 1F, high expression of BRD4 was significantly correlated with less infiltration of CD8+ T cells in human HNSCC tissue samples. In addition, the expression of BRD4 was also inversely correlated with the infiltration of CD8+ T cells and CD8+ effector memory T cells in HNSCC (Figures 1G and 1H) based on TCGA dataset. Taken together, these findings indicate that BRD4 plays a critical role in immune surveillance during tumor progression and may function by excluding CD8+ T cells in HNSCC.
Figure 1.

BET inhibition reinvigorates antitumor immunity in HNSCC
(A) Experimental design for JQ1 treatment and immune profiling. (B) Flow cytometry analysis of the immune infiltrates from the murine primary HNSCC upon JQ1 treatment for 10 days. Values are means ± SDs from the pool of 3 independent experiments. n = 8. ∗p < 0.05 and ∗∗p < 0.01 by Student’s t test. ns, non-significant. (C) Flow cytometry analysis of the immune cells in the spleen from the 4NQO-induced HNSCC mice upon JQ1 treatment for 10 days. Values are means ± SDs from the pool of 3 independent experiments. n = 8. ∗p < 0.05 by Student’s t test. ns, non-significant. (D) Flow cytometry analysis of the immune cells in the blood from the 4NQO-induced HNSCC mice upon JQ1 treatment for 10 days. Values are means ± SDs from the pool of 3 independent experiments. n = 8. ∗p < 0.05 and ∗∗p < 0.01 by Student’s t test. ns, non-significant. (E) Representative immunostaining of BRD4 and CD8+ T cells in human HNSCC samples. Scale bar, 50 μm. (F) BRD4 expression was inversely correlated with the infiltration of CD8+ T in human HNSCC samples. (G) BRD4 expression was negatively correlated with the infiltration of CD8+ T cells in the TCGA HNSCC database. (H) BRD4 expression was negatively correlated with the infiltration of CD8+ effector memory T cells in the TCGA HNSCC database.
Single-cell RNA sequencing (scRNA-seq) reveals the alterations of tumor-infiltrating immune cells upon BET inhibition
To define the impact of BET inhibition on the tumor immune microenvironment, we performed scRNA-seq analysis on murine HNSCC from 4NQO-induced mice. After quality filtering and doublet removal, we obtained 7,029 cells in the control group and 12,286 cells in JQ1 treatment. Seven clusters were identified according to graph-based clustering and dimensionally reduction with uniform manifold approximation and projection (UMAP) and then further annotated based on the expression of characteristic marker genes and assessment using the SingleR package (Figure 2A). We identified cancer cells expressing the epithelial cell adhesion molecule (Epcam), lymphoid cells expressing Cd3d, endothelial cells expressing Pecam1, neurons cells expressing Plp1, myeloid cells expressing Lyz2, fibroblasts expressing Col1a1, and myofibroblasts cells expressing Acta2 (Figure 2B).
Figure 2.

Single-cell transcriptomic profiling of the tumor immune microenvironment upon BET inhibition
(A) UMAP plot showing identified cell populations from 2 merged groups that were treated with control and JQ1. (B) UMAP plot showing the expression of selected marker genes in the 7 clusters. (C) UMAP plot of the identified intratumoral infiltrating immune cells. (D) UMAP plot showing the expression of selected marker genes in immune cells. (E) Proportion of different subsets of immune cells in response to JQ1 treatment. (F) UMAP plot of T cells from merged conditions. (G) UMAP plot of selected markers in T cells from merged conditions. (H) Proportion of different T cell subpopulations in response to JQ1 treatment.
To clarify an overall immune landscape remodeling associated with JQ1 treatment, the alterations of the subpopulations of the tumor-infiltrating immune cells were characterized. After re-clustering immune cells, including myeloid and lymphoid cells, 6 immune subclusters were observed and annotated as neutrophils expressing S100a8, γδ T cells expressing Trdc, αβ T cells expressing Trac, macrophage expressing Apoe, dendritic cells (DCs) expressing Cst3, and mast cells expressing Tpsb2 (Figures 2C and 2D). In comparison to control, JQ1 treatment led to an increase in the proportion of αβ T cells, γδ T cells, and macrophages (Figure 2E).
Our in vivo immune profiling suggests that JQ1 therapy provokes a robust antitumor immune program centered on CD8+ T cells. To obtain a more refined view of T cell states upon JQ1 treatment, we separated T cells and analyzed the data at a higher resolution. Overall, 9 distinct T cell subpopulations (c0–c8) were defined by the distribution of classical marker genes, representing high plasticity and complexity (Figures 2F and 2G). C0 expressed high levels of Foxp3 and Ctla4, which are markers for T regulatory cells (Tregs). C1 displayed a high expression of Tcf7, Klf2, and Klf3, representing a naive-like T cell. Cells in c2 and c6 both expressed Cd3d, Trdc, and Tcrg-C1, but lack the expression of Cd4 and Cd8, which were defined as γδ T cells. In addition, c2 expressed a high level of Il17f and Il17a, while AP-1-associated genes were enriched in c6, including Fos and Junb. Then, we defined c2 as γδ T cells_1 subset and c6 as γδ T cells_2 subset.16 Cells in c3 identified a unique cluster of T cells exhibiting heat-shock stress-activated pathway genes, including Hspa1a, Hspa1b, Dnajb1, Ifng, and Fos,17 suggesting that they may be involved in the pathway of stress response and implying the activation of inflammation in the TME. Cells in c4 expressed genes associated with T cell activation and cytotoxicity (Ccl4, Ccl3, Nkg7, and Klrd1) and Cd8b1 and Cd8a, which are annotated as cytotoxic CD8+ T cells. C5 expressed effector gene markers (Tnfsf8, Tnfsf11, and Bhlhe40) and Cd4, representing an effector CD4+ T cell signature.18,19 Cells in c7 expressed high levels of Xcl1, Ccl5, Arl4d, Klrb1b, and Ifng, appearing to be called Xcl1+ T cells.20 Notably, Xcl1+ T cells were reported to be associated with high tumor mutational burden status and contribute to activating antitumor T cell responses by enhancing antigen presentation.21 Cells in c8 expressed high levels of Gata3 and Kit, identified as ILC2. Of note, in comparison with mice treated with control, JQ1 treatment resulted in an increase in the percentage of c4 (CD8+ cytotoxic T cells) and c7 (Xcl1+ T cells), indicating an activated tumor immune microenvironment in primary HNSCC upon JQ1 treatment (Figure 2H). These findings were consistent with our immune cell profiling data and further confirmed that JQ1 promoted T cell activation centered on CD8+ T cells.
CD8+ T cells are required for BET inhibition-mediated antitumor immunity
To assess the functional role of CD8+ T cells in BET inhibition-mediated antitumor effect, we established a 4NQO-induced HNSCC mouse model, which was then treated with JQ1 and/or depletion of CD8+ T cells. As illustrated in Figure 3A, C57BL/6 mice were administered 4NQO in their drinking water for 16 weeks and then provided with normal drinking water. At 22 weeks from the initial 4NQO treatment, the tumor-bearing mice were randomly divided into 4 groups and treated with vehicle control, JQ1, anti-CD8, and JQ1 plus anti-CD8 for 4 weeks. The animals were then sacrificed, and tongues were collected for further analysis. As expected, JQ1 treatment significantly inhibited tumor growth in the tongues of the mice when compared to those treated with vehicle control (Figures 3B and 3C). Interestingly, the depletion of CD8+ T cells significantly impaired the JQ1-mediated inhibition of tumor growth by using anti-CD8 (Figures 3B and 3C). In addition, similar results were also observed for SCC number counting and invasive depth (Figures 3D–3F). In agreement with these findings, immunostaining showed that the tumor-infiltrating CD8+ T cells were significantly increased in mice treated with JQ1 but abrogated by the administration of anti-CD8 (Figures 3G and 3H). These findings support a notion that CD8+ T cells were required for the BET inhibition-mediated antitumor effect.
Figure 3.

CD8+ T cells are involved in pharmacological inhibition of BRD4-mediated tumor regression
(A) Experimental design for JQ1 treatment and depletion of CD8+ T cells in 4NQO-induced HNSCC mice. (B) Representative image of tongue lesions in 4NNQ-induced HNSCC mice. Scale bar, 3 mm. (C) Quantification of lesion areas from mice treated with different conditions as indicated. Values are means ± SDs from the pool of 3 independent experiments. n = 8. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 1-way ANOVA. (D) Representative H&E staining of HNSCC from 4NQO-induced HNSCC mice treated as indicated. Scale bar, 200 μm. Enlarged images are shown in the lower panels. Scale bar, 50 μm. (E) Quantification of microscopic SCC number from 4NQO-induced HNSCC mice treated as indicated. Values are means ± SDs from the pool of 3 independent experiments. n = 8. ∗p < 0.05 and ∗∗p < 0.01 by 1-way ANOVA. (F) Quantification of invasive depths in murine HNSCC samples. n = 8. ∗p < 0.05 and ∗∗∗p < 0.001 by 1-way ANOVA. (G) Immunohistochemistry of CD8+ T infiltration in murine HNSCC samples. Scale bar, 50 μm. (H) Quantification of infiltrated CD8+ T cells in murine HNSCC samples treated as indicated. The values are means ± SDs from the pool of 3 independent experiments. n = 8. ∗p < 0.05 and ∗∗∗p < 0.001 by 1-way ANOVA. (I) Representative image and quantification of cleaved caspase-3+ cells in SCC1 cells with genetic depletion BRD4 cocultured with or without T cells. ∗∗∗p < 0.001 by 2-way ANOVA. ns, non-significant. (J) Representative image and quantification of cleaved caspase-3+ cells in CAL27 cells with genetic depletion BRD4 co-cultured with or without T cells. ∗∗∗p < 0.001 by 2-way ANOVA. ns, non-significant.
To further explore whether targeting BRD4 in tumor cells can regulate T cell function, we performed co-culture experiments to evaluate the ability of T cell-mediated killing of tumor cells by using activated human peripheral blood T cells and HNSCC cells. As shown in Figures 3I, 3J, and S2A, significantly more cleaved caspase 3+ cells were observed in genetically depleted BRD4 HNSCC cells co-cultured with activated T cells, indicating that the depletion of BRD4 in HNSCC cells can enhance the antitumor function of T cells. As expected, the overexpression of BRD4 in HNSCC cells could significantly inhibit the antitumor function of T cells (Figures S2B and S2C). Collectively, these results suggest that CD8+ T cells are involved in BRD4 inhibition-mediated growth suppression of HNSCC.
BRD4 inhibition enhances the expression of MHC class I antigen presentation molecules
To explore the intrinsic regulatory mechanism of BRD4 on immunity, RNA-seq was performed in SCC1 cells treated with JQ1 and iBET-151 with or without IFN-γ exposure. We identified a subset of genes whose expression was commonly upregulated by BET inhibition in both the absence and presence of IFN-γ exposure (Figure S3A). Ingenuity Pathway Analysis (IPA) of 398 commonly upregulated genes revealed that the upregulated genes were enriched for the MHC class I antigen presentation pathway (Figure S3B). Notably, MHC class I antigen presentation molecules, including HLA-A, HLA-B and HLA-C, were significantly upregulated in cells treated with both JQ1 and iBET-151 in the presence or absence of IFN-γ exposure (Figure 4A). As expected, we examined our scRNA-seq data and found that JQ1 treatment significantly enhanced antigen processing and presentation of peptide antigen via MHC class I in vivo (Figures S3C and S3D). The expression of H2-k1, murine MHC class I molecule, was also increased in cancer cells from murine HNSCC upon JQ1 treatment (Figure S3E).
Figure 4.

BRD4 inhibition enhances MHC class I antigen presentation molecules in HNSCC
(A) Heatmap showing a subset of BET inhibition upregulated genes involved in antigen presentation pathways.
(B and C) RT-qPCR showing the expression of HLA-A, HLA-B, and HLA-C in SCC1 and CAL27 cells treated with JQ1 and iBET-151 in the presence or absence of IFN-γ. Means ± SDs were shown. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 1-way ANOVA. (D) Immunoblot showing the expression of MHC class I in SCC1 and CAL27 cells treated with JQ1 and iBET-151 in the presence or absence of IFN-γ. (E and F) Flow cytometry displayed cell surface MHC class I expression in SCC1 and CAL27 cells treated with JQ1 and iBET-151 in the presence or absence of IFN-γ. Means ± SDs are shown. ∗p < 0.05, ∗∗∗p < 0.001 by 1-way ANOVA. (G and H) RT-qPCR showed that the expression of HLA-A, HLA-B and HLA-C in SCC1 and CAL27 cells transfected with sgBRD4. Means ± SDs were shown. ∗∗∗p < 0.001 by 1-way ANOVA. (I) Immunoblot showed that the expression of MHC class I in SCC1 and CAL27 cells transfected with sgBRD4. (J and K) Flow cytometry displayed cell surface MHC class I expression in SCC1 and CAL27 cells transfected with sgBRD4. Means ± SDs were shown. ∗∗∗p < 0.001 by 1-way ANOVA.
To confirm the above results, 2 HNSCC cell lines, SCC1 and CAL27, were treated with JQ1 and iBET-151, followed by RT-qPCR. The RT-qPCR results showed a significant increase in MHC class I antigen presentation molecules in cells treated with BET inhibitors in the presence or absence of IFN-γ (Figures 4B and 4C). Similar results were also observed in western blot and flow cytometry analyses (Figures 4D–4F). In addition, the endogenous expression of BRD4 was depleted with 2 validated single-guide RNA (sgRNA) in 2 cell lines, and we showed that the genetic depletion of BRD4 promoted the mRNA and protein expression of MHC class I antigen presentation molecules (Figures 4G–4K). Similar results were also observed in additional HN6 cells (Figures S4A–S4F). As expected, the ectopic overexpression of BRD4 suppresses the expression of MHC class I in HNSCC cells (Figure S4G). Importantly, the expression of MHC class I was also increased in murine primary HNSCC from 4NQO-induced mice treated with JQ1 as compared to control mice (Figure S4H). In agreement with these findings, the expression of BRD4 was inversely correlated with HLA-A, HLA-B, and HLA-C expression in TCGA HNSCC database (Figure S4I). Collectively, these data confirmed that the expression of a subset of MHC class I antigen presentation molecules was regulated by BRD4.
BET inhibition enhances MHC class I antigen presentation molecules through suppressing G9a recruitment in HNSCC
BRD4 has been shown to be a transcriptional activator that establishes super enhancers associated with multiple biological processes.11,22 However, we showed that BRD4 inhibited the expression of MHC class I antigen presentation molecules, which inspired us to explore the molecular mechanism by which BRD4 repressed MHC class I expression. G9a, also called euchromatic histone methyltransferase 2 (EHMT2) or lysine methyltransferase 1C (KMT1C), is a common histone methyltransferase that catalyzes the methylation of H3K9 with S-adenosylmethionine (SAM) as a methyl donor,23 which plays a significant role in transcriptional silencing.24 Interestingly, it has been reported that BRD4 suppressed a subset of autophagy gene expression via recruiting G9a.25
To determine whether BRD4 inhibits MHC class I expression through G9a-mediated H3K9 methylation, chromatin immunoprecipitation (ChIP)-qPCR was performed to detect the recruitment of BRD4, G9a, H3K9me1, H3K9me2, and H3K9me3 to HLA-A, HLA-B, and HLA-C promoter regions. As shown in Figures 5A–5E, BRD4, G9a, H3K9me1, H3K9me2, and H3K9me3 were enriched at HLA-A, HLA-B, and HLA-C promoter regions as compared to immunoglobulin G (IgG) controls in SCC1 cells. As expected, the enrichment of BRD4 to HLA-A, HLA-B, and HLA-C promoter regions was significantly suppressed in HNSCC cells upon JQ1 treatment in SCC1 cells. Similar repression of BRD4 enrichment was also observed in SCC1 cells treated with IFN-γ. In addition, a dramatic reduction in BRD4 at HLA-A, HLA-B, and HLA-C promoter regions was found in SCC1 cells given JQ1 treatment in the presence of IFN-γ. Moreover, G9a recruitment to promoters of antigen presentation genes was abolished by JQ1 treatment. Notably, the recruitment of H3K9me1, H3K9me2, and H3K9me3 to these regions was also decreased upon JQ1 administration in both the absence and presence of IFN-γ in SCC1 cells. Similar ChIP-qPCR results were also observed in CAL27 cells (Figures S5A–S5E).
Figure 5.

BRD4 inhibition enhances MHC class I antigen presentation molecules through suppressing G9a in HNSCC
(A–E) SCC1 cells treated with JQ1 and/or IFN-γ were subjected to ChIP assay with BRD4, G9a, H3K9me1, H3K9me2, and H3K9me3 antibodies. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 1-way ANOVA. ns, non-significant. (F) Nuclear proteins extracted from SCC1 cells were subjected to reciprocal immunoprecipitation with G9a and BRD4 antibodies. (G) Nuclear proteins extracted from SCC1 cells treated with or without JQ1 were subjected to immunoprecipitation with G9a and BRD4 were detected. (H and I) RT-qPCR showed that the expression of HLA-A, HLA-B, and HLA-C in SCC1 and CAL27 cells transfected with siRNA targeting G9a. Means ± SDs were shown. ∗∗∗p < 0.001 by 1-way ANOVA. (J) Western blot showed the expression of MHC class I in SCC1 and CAL27 cells transfected with siRNA targeting G9a. (K and L) RT-qPCR showed the expression of HLA-A, HLA-B, and HLA-C in SCC1 and CAL27 cells treated with JQ1 and siRNA targeting G9a. Means ± SDs are shown. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 1-way ANOVA.
Besides ChIP results, the physical interaction between BRD4 and G9a was confirmed by the reciprocal immunoprecipitation assay (Figure 5F). Importantly, we observed that the interaction between BRD4 and G9a was inhibited by JQ1 administration (Figure 5G). Consistent with the functional role of G9a in the regulation of gene expression, G9a silencing enhanced MHC class I molecule expression (Figures 5H–5J). Furthermore, knockdown of G9a with JQ1 treatment did not cause further enhancement of MHC class I molecule expression at mRNA levels as compared to a single JQ1 treatment, but could slightly enhance the expression of MHC class I molecules when compared with single G9a depletion (Figures 5K and 5L). This indicates that G9a acts on the same pathway of BRD4 and may serve as a downstream regulator of BRD4. In addition, the overexpression of BRD4 significantly repressed MHC class I molecule expression and G9a silencing completely rescued BRD4-mediated inhibition of MHC class I molecules (Figures S5F and S5G). These data suggest that BET bromodomain inhibition activates the expression of MHC class I antigen presentation molecules by suppressing the recruitment of G9a in HNSCC.
Pharmacological inhibition and genetic ablation of BRD4 in tumor cells enhance T cell-mediated killing by modulating MHC class I in vitro
To explore the effects of MHC class I during BET inhibition-mediated T cell killing of HNSCC, we depleted the MHC class I expression by using small interfering RNAs (siRNAs) and assessed BET inhibition-mediated T cell killing efficacy. The knockdown efficiency of MHC class I was detected by flow cytometry (Figure 6A). As shown in Figure 6B, the enhanced T cell-killing efficacy resulting from JQ1 and iBET-151 treatment was significantly inhibited by MHC class I knockdown. In addition, similar results were observed by genetic depletion of both BRD4 and MHC class I molecules (Figure 6C). To further clarify the effect of BET inhibition on MHC class I-restricted, CD8+ T cell-mediated cytotoxicity, a classical in vitro model was used by co-culturing OVA expressing murine tumor cells and CD8+ T cells from OT-1 transgenic mice. Due to the lack of murine HNSCC cell lines in our lab, B16 expressing OVA and OT-1 CD8+ T cells were used to perform the co-culture assay. As shown in Figure S6A, we showed that the expression of MHC class I molecule (H2-K) was also increased in B16 cells treated with JQ1, iBET-151 and Brd4 siRNAs. Importantly, our co-culture assay confirmed that JQ1 and iBET-151treatment or silencing of Brd4 enhanced the killing ability of CD8+ T cells. Notably, silencing of MHC class I (H2-K) could significantly hindered the killing activity of CD8+ T cells enhanced by JQ1, iBET-151 treatment and Brd4 knockdown (Figures S6B and S6C). These results support the notion that targeting BRD4 in tumor cells enhance T cell-mediated killing by modulating MHC class I expression. In agreement with this notion, we found that BRD4 expression was inversely correlated with MHC class I molecule expression and infiltration of CD8+ T cells in HNSCC tissue samples, but MHC class I expression was positively correlated with the infiltration of CD8+ T cells (Figures 6D–6F). These findings imply that BET inhibition indeed potentiates CD8+ T cell-mediated antitumor immunity by modulating MHC class I expression.
Figure 6.

Pharmacological inhibition and genetic ablation of BRD4 in tumor cells enhance T cell-mediated killing by modulating MHC class I in vitro
(A) Flow cytometry displayed cell surface MHC class I in HNSCC cells transfected with siRNA targeting MHC class I. ∗∗∗p < 0.001 by 1-way ANOVA. (B) Quantification of cleaved caspase-3+ cells in HNSCC cells treated with JQ1, iBET-151 and siMHC class I co-cultured with T cells. ∗∗∗p < 0.001 by 2-way ANOVA; ns, non-significant. (C) Quantification of cleaved caspase-3+ cells in HNSCC cells treated with sgBRD4 and siMHC class I co-cultured with T cells. ∗p < 0.05, ∗∗p < 0.01 by 2-way ANOVA; ns, non-significant. (D) Representative images of immunostaining for BRD4, MHC class I, and CD8 in human HNSCC samples. Scale bar, 50 μm. (E) BRD4 expression was inversely correlated with the expression of MHC class I in human HNSCC samples. (F) MHC class I expression was positively correlated with infiltration of CD8+ T in human HNSCC samples.
Targeting BRD4 enhances the sensitivity of anti-PD-1 therapy in HNSCC
Since BET inhibition can promote CD8+ T cell-mediated antitumor immunity, we next investigated whether BET inhibition can sensitize anti-PD-1 therapy in HNSCC. We performed the co-culture experiment by using T cells and HNSCC cells with BRD4 knockout (KO) in the presence or absence of anti-PD-1 neutralizing antibodies in vitro. As shown in Figures 7A and 7B, the depletion of BRD4 or anti-PD-1 treatment could enhance the killing efficacy of activated T cells on tumor cells as compared to the control group. More important, the BRD4 depletion combined with anti-PD-1 treatment could further enhance the killing efficacy of activated T cells on tumor cells when compared with single BRD4 depletion or anti-PD-1 treatment alone. These findings suggest that the KO of BRD4 does enhance the sensitivity of tumor cells to anti-PD-1 treatment in HNSCC cells in vitro.
Figure 7.

Targeting BRD4 enhances the sensitivity of anti-PD-1 therapy in HNSCC
(A) Image and quantification of cleaved caspase-3+ cells in SCC1 cells treated with sgBRD4 co-cultured with T cells in the presence or absence of anti-PD-1. ∗∗∗p < 0.001 by 2-way ANOVA; ns, non-significant. (B) Image and quantification of cleaved caspase-3+ cells in CAL27 cells treated with sgBRD4 co-cultured with T cells in the presence or absence of anti-PD-1. ∗∗∗p < 0.001by 2-way ANOVA; ns, non-significant. (C) Experimental design for BET inhibition with anti-PD-1 treatment in 4NQO-induced HNSCC mice. (D) Representative image of tongue lesions in 4NQO-induced HNSCC mice upon different treatment as indicated. Scale bar, 3 mm. (E) Quantification of lesion areas from mice treated with different conditions as indicated. Values are means ± SDs from the pool of 4 independent experiments. n = 10. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 1-way ANOVA. (F) Representative H&E staining of HNSCC from mice with treatment as indicated. Scale bar, 200 μm. Enlarged images are shown in the lower panels. Scale bar, 50 μm. (G) Quantification of microscopic SCC number from the tongues of 4NQO-induced mice with different treatment as indicated. Values are means ± SDs from the pool of 4 independent experiments. n = 10. ∗p < 0.05 and ∗∗∗p < 0.001 by 1-way ANOVA. (H) Quantification of invasive depths in murine HNSCC samples. n = 10. ∗p < 0.05 and ∗∗∗p < 0.001 by 1-way ANOVA. (I) Immunohistochemistry of CD8+ T infiltration in murine HNSCC samples. Scale bar, 50 μm. (J) Quantification of infiltrated CD8+ T cells in murine HNSCC samples from mice upon different treatment as indicated. The values are means ± SDs from the pool of 4 independent experiments. n = 10. ∗p < 0.05 and ∗∗∗p < 0.001 by 1-way ANOVA.
Next, the 4NQO-induced tumor-bearing mice were randomly divided into 4 groups and treated with intraperitoneal injection of either an isotype control, JQ1, anti-PD-1, or JQ1 plus anti-PD-1, respectively (Figure 7C). As demonstrated in Figures 7D and 7E, JQ1 treatment led to a moderate reduction in tumor size, and anti-PD-1 caused a slight tumor growth suppression. Notably, JQ1 and anti-PD-1 combination therapy dramatically inhibited tumor growth when compared with those treated with JQ1 or anti-PD-1 alone. Histological analysis revealed that JQ1 combined with anti-PD-1 treatment significantly reduced the SCC number and invasive depth in mice as compared to those administered with JQ1 or anti-PD-1 alone (Figures 7F–7H). Immunostaining showed that JQ1 or anti-PD-1 treatment could induce CD8+ T cell infiltration in HNSCC, which was significantly increased in HNSCC-bearing mice treated with JQ1 coupled with anti-PD-1 (Figures 7I and 7J). In addition, analysis of an immune subset within the spleen of the mice revealed that the combination of JQ1 and anti-PD-1 therapy dramatically increased the percentage of CD8+ T cells as compared to the other groups (Figure S7A). Importantly, the percentages of IFN-γ+ and GZMB+ CD8+T lymphocytes were also significantly increased in mice upon dual JQ1 and anti-PD-1 treatment as compared to those treated with JQ1 or anti-PD-1 alone (Figures S7B and S7C). Similar results were also observed for the analysis of peripheral blood in 4NQO-induced HNSCC mice (Figures S7D–S7F). These results suggest that BET inhibition triggers an effective CD8+ T cell-mediated antitumor immunity, which provides a rationale for combining BET inhibitors with ICBs to treat HNSCC.
To verify the effect of cancer-specific Brd4 depletion on anti-PD-1 therapy and immune infiltrates, a syngeneic B16 cell-implantable mice model was used (Figure 8A). As presented in Figures 8B and 8C, Brd4 depletion displayed mild antitumor activity, but anti-PD-1 did not inhibit the growth of B16 xenografts, while the Brd4 depletion combined with anti-PD-1 treatment significantly suppressed tumor growth. These findings suggest that the KO of Brd4 does enhance the sensitivity of tumor cells to anti-PD-1 treatment. Consistent with our results from the 4NQO-induced HNSCC mice model, cancer-specific Brd4 depletion can also remodel the tumor-infiltrated immune cells in vivo. The percentages of CD8+ T cells and GZMB+ CD8+ T cells were significantly increased in mice bearing Brd4-KO tumor xenografts as compared to control mice, which can be further augmented by anti-PD-1 treatment (Figures 8D and 8E). A significant increase in CD4+ T cells and IFN-γ+CD4+ T cells and a decrease in MDSCs were also observed in Brd4 depletion combined with anti-PD-1 treatment as compared to the control mice (Figures 8F–8H). There was no change in the proportion of macrophages and NK cells (Figures 8I and 8J). These data showed that cancer-specific Brd4 depletion did trigger CD8+ T cell-mediated antitumor immunity in the B16 syngeneic model, which is consistent with the 4NQO-induced HNSCC mouse model upon JQ1 treatment. Taken together, these results showed that BET inhibition did enhance the sensitivity of anti-PD-1 therapy and provoke CD8+ T cell-mediated antitumor immunity.
Figure 8.

Cancer-specific depletion of Brd4 enhances the sensitivity of anti-PD-1 therapy in vivo
(A) Experimental design for Brd4-KO B16/F10 cells implantable mice treated with anti-PD-1 antibody. (B) Tumor growth curve of B16/F10 xenografts upon Brd4-KO following treatment with anti-PD-1 antibody. n = 7. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 2-way ANOVA. (C) Tumor weight of B16/F10 xenografts upon Brd4-KO following treatment with anti-PD-1 antibody. n = 7. ∗p < 0.05 and ∗∗∗p < 0.001 by 1-way ANOVA. (D) Percentage of CD8+ T cells in tumor-infiltrating lymphocytes (TILs) for each group. n = 7, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by one1way ANOVA. ns, non-significant. (E) Percentage of GZMB+CD8+ T cells in total CD8+ T cells. n = 7, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 1-way ANOVA. ns, non-significant. (F) Percentage of CD4+ T cells in TILs for each group. n = 7, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by 1-way ANOVA. ns, non-significant. (G) Percentage of IFN-γ+CD4+ T cells in total CD4+ T cells. n = 7, ∗∗p < 0.01 by 1-way ANOVA. ns, non-significant. (H–J) Percentage of macrophage, NK, and MDSC in TILs for each group. n = 7, ∗p < 0.05 and ∗∗∗p < 0.001 by 1-way ANOVA. ns, non-significant.
Discussion
BET family members are epigenetic readers that bind acetylated histones to regulate gene transcription, including BRD2, BRD3, BRD4, and BRDT.26 Due to the ability of BET family members, particularly BRD4, to regulate transcription and chromatin states, the inhibition of BET family members has been considered to be a promising therapeutic strategy, and has already shown a profound antitumor efficacy in preclinical models.27,28,29 Interestingly, several studies suggested that immune cells are involved in BET inhibition-mediated antitumor efficacy.12,13,14,30 However, the effect of BRD4 on the immune escape of HNSCC and its underling mechanism is still not well documented.
In this study, we revealed that BET inhibition remodeled tumor-infiltrated immune cells to achieve optimal antitumor immune response in HNSCC, specifically involving CD8+ T cells. We also showed that BRD4 was highly expressed and inversely correlated with the infiltration of CD8+ T cells in HNSCC based on TCGA dataset and immunostaining. Strikingly, the expression of BRD4 was positively associated with the immune infiltrating levels of CD8+ T cells in hepatocellular carcinoma.31 These findings imply that the functions of BRD4 on immune cells are cell and tissue dependent and may promote immune escape by modulating the function of CD8+ T cells in HNSCC.
To validate the functional role of CD8+ T cells in BET inhibition-mediated antitumor efficacy, an immunocompetent 4NQO-induced HNSCC mouse model was established and treated with JQ1 and anti-CD8. The results showed that targeting BRD4 with JQ1 significantly suppresses the growth of HNSCC via provoking CD8+ T cell-mediated antitumor immunity. Similar results were also observed in T cell-killing assays in vitro. In agreement with our findings, several studies suggest that BET inhibition promotes antitumor immunity involved in DCs, macrophages, cytotoxic T cells, and Treg cells, but mostly in CD8+ T cells.12,14,30,32,33 scRNA-seq analysis of tumor-infiltrating immune landscapes in murine HNSCC also revealed the connection of BET inhibition with cytotoxic CD8+ T cells. Then, we further validated that CD8+ T cells are required in BET inhibition-mediated antitumor effects by direct blockade of CD8+ T cells in an immunocompetent HNSCC mouse model. These results support the notion that BET inhibition enhances CD8+ T cell-mediated antitumor immunity in HNSCC.
It has been confirmed that BET inhibition modulates the tumor immune microenvironment by suppressing PD-L1 and IDO-1.13,14,34 To investigate how BET inhibition promotes CD8+ T cell-mediated antitumor immunity, an unbiased RNA-seq analysis was performed in HNSCC cells, and we identified that the MHC class I molecules HLA-A, HLA-B, and HLA-C are selectively upregulated upon treatment of BRD4 inhibitors JQ1 and iBET-151 in the absence or presence of IFN-γ, which was further verified by qPCR, western blot, and flow cytometry. A similar result was observed in our scRNA-seq data, which showed that the murine MHC class I molecule (H2-K1) was also upregulated in cancer cells from 4NQO-induced murine HNSCC upon JQ1 treatment.
MHC class I molecules are key components for immune surveillance, and the defect or loss of MHC class I on the surface of tumor cells has been regarded as one of the major mechanisms for tumor immune evasion and resistance against immunotherapy in many cancers, including HNSCC.35,36,37,38 Of note, a recent report showed that targeting BRD4 promoted the anticancer efficacy of anti-CTLA-4 in prostate cancer; Mao et al. found that the inhibition of the BET bromodomain BRD4 can reduce PD-L1 expression and increase MHC class I expression on the surface of prostate tumor cells.30 However, the exact regulatory mechanism of how BRD4 regulates MHC class I expression was not clarified.
In our study, we identified that BRD4 can function as a transcriptional repressor of MHC class I antigen presentation molecules, which binds to the promoter regions of MHC class I genes and then recruits G9a to modulate H3K9 methylation and inhibit the expression of MHC class I genes. Consistent with our findings, the repressive role of BRD4 on gene expression was also found in autophagy-related genes.25 More important, functional studies showed that the cytotoxic T cell-mediated cell lysis enhanced by BET inhibition was abolished when MHC class I molecules were silenced. Taken together, these results strongly suggest that BET inhibition promotes the expression of MHC class I antigen presentation molecules by disassociating G9a occupancy at the promoters of HLA-A, HLA-B, and HLA-C, which in turn promotes T-cell based antitumor immunity.
Accumulating evidence has shown that the PD-1 monoclonal antibody is a useful therapeutic strategy for the treatment of refractory or metastatic HNSCC. However, objective response rates of anti-PD-1 treatment are extremely low in HNSCC due to immunoresistance.39,40,41 To improve the efficacy of tumor immunotherapy, ICB-based combination therapies were developed and have shown promising results, including the combinations of anti-PD1/PD-L1 with chemotherapy, anti-CTLA4, epidermal growth factor receptor (EGFR) pathway inhibitors, transforming growth factor β (TGF-β) inhibitors or indoleamine-2,3-dioxygenase (IDO) inhibitors, respectively.42,43,44,45,46 It has been confirmed that cytotoxic T cell infiltration, antigen processing and presentation, PD-1/PD-L1 expression, and neo-antigen formation are critical for successful cancer immunotherapy. In this study, we reveal that BET inhibition triggers effective antitumor immunity via activating CD8+ T cells and enhances antigen processing in HNSCC. As expected, BET inhibition with the small molecule inhibitor JQ1 significantly increased the immunogenicity of tumor cells and enhanced the sensitivity of anti-PD-1 therapy in HNSCC. Taken together, these results serve as the basis for the combinatorial application of BET inhibitors with immunotherapy for HNSCC treatment. Considering the universal role of BRD4 in pan-cancer, combining BET inhibition with ICB may reinvigorate antitumor immunity as a widely accepted approach in cancer immunotherapy.
Materials and methods
Cell culture
HNSCC cell lines SCC1, HN6, CAL27, 293 T, and B16/F10 cells were used in this study. SCC1 cells were obtained from the University of Michigan and HN6 cells were obtained from Wayne State University. CAL27, 293 T, and B16/F10 were obtained from the American Type Culture Collection. HNSCC cell lines, 293 T, and B16/F10 cells were cultured in DMEM (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco) and incubated at 37°C in a humidified incubator with 5% carbon dioxide.
sgRNA and siRNA transfection
To generate the virus, the target plasmid and packing plasmid (ΔH8.2 and VSVG) were co-transfected into 293 T cells by lipo2000 transfection reagent. After 72 h, viral supernatants were collected and used to infect HNSCC cell lines supplemented with polybrene (2 μg/mL). The infected cells were selected with puromycin. For siRNA experiments, cells were plated into 6-well plates and transfected with Lipofectamine RNAiMax (Invitrogen, USA) according to the manufacturer’s instructions. The efficiency of transfection was confirmed by RT-qPCR and western blot. Sequences for human sgBRD4, mouse sgBrd4, human MHC class I siRNA (targeting the common sequence for HLA-A, HLA-B and HLA-C), humanG9a siRNA, and mouse H2-K siRNA are listed in Table S1.
4NQO-induced HNSCC model and treatment modality
C57BL/6 mice were purchased from Vital River Laboratories (China). Mice were housed under standard conditions in the animal facility of Sun Yat-sen University (SYSU), and all of the procedures were performed according to the SYSU Animal Research Committee-approved protocol. We chose a sample size of at least 6 mice per group based on our previous experience and publications.47,48,49,50,51 The induction of HNSCC was performed as previously described by using the 4NQO (TCI [USA] N0250-5 g) added in the drinking water.47,51 After 22 weeks of 4NQO induction, mice were treated with a series of drugs as indicated. For JQ1 treatment, mice were randomly divided into 2 groups and treated with vehicle control and JQ1 (50 mg/kg). Tumor sizes were assessed after JQ1 treatment for 4 weeks and immune profiling was performed after 10-day treatment. In the anti-CD8 blocking experiment, the mice were randomly divided into 4 groups and treated with vehicle control, JQ1 (50 mg/kg), anti-CD8 (InVivoMAb anti-mouse CD8a BE0061, BioXCell, 100 μg per mouse), and JQ1 combined with anti-CD8 for 4 weeks. In the JQ1 and anti-PD-1 combination treatment experiment, the mice were randomly divided into 4 groups and treated with vehicle control, JQ1 (50 mg/kg), anti-PD-1 (InVivoMAb anti-mouse PD-1, RMP1-14, BioXCell, 200 μg per mouse), or JQ1 plus anti-PD-1 for 4 weeks. JQ1 was administrated via intraperitoneal injection 5 days per week. Anti-PD-1 and anti-CD8 were administrated via intraperitoneal injection twice per week. Then, mice were sacrificed for the following experiments. Tongues were carefully dissected, and the lesion area was assessed as previously described.47 Invasive depth was measured in the H&E-stained sections as previously described.52
B16 syngeneic model
B16/F10 cells, 0.5 × 106, with genetic depletion of Brd4 and control cells were subcutaneously inoculated into C57BL/6 mice. Mice were treated with 200 μg anti-PD-1 antibodies or IgG (200 μg per mice, intraperitoneally, every 3 days). The tumors were measured every second day. Tumor volumes were calculated using the following equation: (length × width2)/2. After tumor collection, the tumors were weighed and the samples were used for flow cytometry analysis.
T cell cytotoxicity assays in vitro
The in vitro T cell killing activity was measured using a cleavage caspase-3 assay. T cells were isolated from peripheral blood mononuclear cells (PMBCs) using the Pan T Cell Isolation Kit (Miltenyi Biotech [Germany], no. 130-096-535) and frozen in FBS containing 10% dimethyl sulfoxide. For experiments, the 24-well plates were coated with Ultra-LEAF purified anti-human CD3 (Biolegend [USA], no. 300331) and Ultra-LEAF purified anti-human CD28 (Biolegend, no. 302933) in PBS overnight at 4°C and washed with ice-cold PBS twice. The isolated T cells were plated into 24-well plates to activate for at least 72 h in RPMI medium supplemented with 10% FBS, 1X MEM Non-Essential Amino Acids (Thermo Fisher [USA], no. 11140050), 1 mM sodium pyruvate (Thermo Fisher, no. 11360070), 100 U/mL penicillin, 100 μg/mL streptomycin, and 100 IU/mL human interleukin-2 (IL-2) (PeproTech [USA], no. 200-02-50). Tumor cells were treated with BRD4 inhibition for 24 h with or without IFN-γ exposure in advance. The prepared tumor cells and activated T cells were co-cultured at a 1:10 ratio for 6–8 h at 37°C. For anti-PD-1 treatment in vitro, HNSCC cells with or without BRD4 KO and activated T cells were co-cultured in the presence or absence of anti-PD-1 antibody. The cells were stained with allophycocyanin (APC)-conjugated anti-human CD3 antibody for 30 min on ice in the dark and washed with PBS plus 2% FBS. Cells were then fixed and permeabilized before staining with fluorescein isothiocyanate (FITC)-conjugated anti-human/mouse-cleaved caspase-3 antibody (BD [USA], no. 559341) for 30 min on ice and washed with PBS plus 2% FBS. The percentages of cleaved caspase-3+ tumor cells were analyzed by flow cytometry.
OT-1 T-cell cytotoxicity assays
OT-1 T cells isolated from the spleen of OT-1 TCR transgenic mice were cultured in a 75-cm2 flask and incubated with 1–2 ng/mL OVA 257–264 (Sigma [USA], cat. no. 138831-86-4) and 10 ng/mL IL-2 to expand CD8+ OT-1 T cells for 72 h in RPMI medium supplemented with 10% FBS, 20 nM HEPES (Gibco, 15630080), 1 mM sodium pyruvate (Thermo Fisher, no. 11360070), 2 nM L-glutamine (Gibco, 25030081), 0.05 nM 2-mercaptoethanol (Gibco, 21985023), 100 U/mL penicillin, and 100 μg/mL streptomycin. Successful enrichment of a CD8+ T cell population was confirmed by flow cytometry staining for PerCP/Cyanine5.5 anti-mouse CD8a antibody (Biolegend, no. 155013). B16 cells were pretreated as indicated BRD4 inhibitor, Brd4 siRNAs and H2-k siRNAs for 48 h. Subsequently, B16 cells were pulsed with 1–2 μg/mL of OVA 257–264 in an incubator for 2 h, which binds to cell surface MHC class I. The prepared B16 cells and CD8+ T cells were seeded into 96-well round bottom plates at a 1:1 ratio for 3 h at 37°C. The cells were stained with PerCP/Cyanine5.5 anti-mouse CD8a antibody for 30 min on ice in the dark and washed with PBS plus 2% FBS. Cells were then fixed and permeabilized before staining with FITC-conjugated anti-human/mouse cleaved caspase-3 antibody (BD, no. 559341) for 30 min on ice and washed with PBS plus 2% FBS. The percentages of cleaved caspase-3+ tumor cells were analyzed by flow cytometry.
Western blotting
Cells were harvested with 1× Cell RIPA Buffer (Cell Signaling Technology [USA], no. 9806 s) with a cocktail of protease inhibitors (Sigma, P7626-5g). Cell lysates were collected by centrifugation at 12,000 rpm for 15 min at 4°C. All protein samples were denatured with 6× SDS buffer and boiled at 100°C for 10 min and then separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Roche, no. 3010040001). The membranes were blocked for 1 h with 5% non-fat milk and then were incubated overnight at 4°C with primary antibody, including BRD4 (Abcam [UK], ab243862), HLA-ABC (Abcam, ab70328), β-tubulin (Cell Signaling Technologies [USA], no. 2148s). The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature and exposed by a western blotting detection kit (Merck Millipore [USA], WBKLS0500).
RT-qPCR
Total RNA was prepared with RNA-quick purification kit (EScience [USA], RN001), and RNA concentration was measured by NanoDrop. cDNA synthesis was performed with HiScript II One Step RT-PCR Kit (Vazyme [China], P611-01), per the manufacturers’ instructions. qRT PCR was conducted using SYBR Green master mix. All of the samples were assayed in triplicate. Relative expression levels were determined with the ΔCt method and normalized to TUBB4A. The qPCR primer sequences are listed in Table S2.
Flow cytometry
Cells were harvested and collected by centrifuge at 1,500 rpm for 5 min. For molecular staining of cell surfaces, resuspended cells were directly incubated with related antibody and protected from lights on ice for 30 min and then washed with staining buffer. For intracellular molecular staining, the BD cell fixation and permeabilization were used for the fixation of cells and the Perm/Wash Buffer was used to wash the cells and to dilute the antibodies for staining (BD, no. 554714). The supernatant was aspirated, and the cells were resuspended in 200 μL staining buffer for flow cytometric analysis. For immune cell populations in peripheral blood and spleens of mice, mice were sacrificed after drug administration to harvest peripheral blood and spleens. Spleen were dissociated into single-cell suspension and lysed with Red Blood Cell Lysis Buffer (BioLegend [USA], no. 420301), then stained with antibody dye. The peripheral blood was directly lysed with Red Blood Cell Lysis Buffer and stained with antibody dye. All of the flow cytometry analysis was performed with Navios and Gallios (Beckman Coulter [USA]) and the data were analyzed with FlowJo and CytExpert software. All of the antibodies used for flow cytometry analysis are anti-mouse FVS700 APC-700 (BD, no. 564997), anti-mouse CD45 Percp-cy5.5 (BioLegend, no. 103132), anti-mouse CD4 PE-cy7 (BioLegend, no. 100422), anti-mouse CD3 Alexa Fluor 488 (BioLegend, no. 100210), anti-mouse CD8 BV786 (BioLegend, no. 563332), anti-mouse NK1.1 BV510 (BioLegend, no. 108738), anti-mouse GranzymeB Efluor450 (Invitrogen, no. 48-8898-82), anti-mouse IFN-γ APC-cy7 (BioLegend, no. 505850), anti-mouse CD11b BV605 (Biolegend, no. 101257), anti-mouse F4/80 BV650 (Biolegend, no. 123149), and anti-mouse Gr-1 PE/Dazzle 594 (Biolegend, no. 108452).
Immunohistochemistry
Sections were dewaxed with xylene and graded ethanol, and antigen repair was performed by a pressure cooker in sodium citrate pH 6.0 for 5 min. We used 3% hydrogen peroxide to block tissue endogenous peroxidase activity for 15 min. Furthermore, the sections were incubated with primary antibody at 4°C overnight and incubated with secondary antibody (ZS, China, PV-6001) at 37°C for 30 min and counterstained with hematoxylin, dehydrated, and mounted with neutral gum. Immunostaining slides were recorded with an Olympus BX51 Research System Microscope with 10× and 20× UPlanApo air objective lenses (Olympus America). The primary antibodies include BRD4 (Abcam, ab243862), MHC class I (Abcam, ab134189), MHC class I (SAB, USA, no. 48250), CD8 (Abcam, ab93278), CD8 (Cell Signaling Technology, #85336).
scRNA-seq
Primary murine HNSCC samples were harvested from 4NQO-induced mice treated with JQ1 or control for 10 days. To obtain enough cells for scRNA-seq, HNSCC samples from 3 mice were collected in each group. Then, single-cell suspension was performed as described above51 and processed by using the 10x Genomics platform. Sequencing libraries were prepared by using Single Cell 3′ Reagent Kits version 3 (10x Genomics) and converted using the MGIEasy Universal Library Conversion Kit, following the MGISEQ-2000 platform.
Cellranger (version 3.1.0) was used to generate a raw gene expression matrix and then processed by using the Seurat R package (R Foundation for Statistical Computing, Austria). For quality filtering, cells with <201 or >5,999 expressed genes or >10% mitochondria genes were removed. Doublets were filtered out by using the DoubletFinder R package (version 2.0.3)53 according to default parameters. For dimensionality reduction, all of the samples were performed and merged by using the SCTransform R package (version 0.3.2). Principal-component analysis was used to summarize the variably expressed genes and then the UMAP function was used to further summarize the principal components.
To identify cell types, cells were clustered by using the FindClusters function after computing a shared nearest neighbor with Louvain algorithm. Then, the clusters were annotated by using the SingleR package (version 1.4.1) and the well-known marker genes. In total, 7 clusters were identified in our murine HNSCC samples, including cancer cells (Epcam, Krt5, Krt6a, Krt13, Krt15, and Krt17), myeloid cells (Lyz2, Cd14, and S100a9), fibroblasts (Col1a1, Col1a2, Lum, and Dcn), neurons (Plp1 and Chl1), myofibroblasts (Rgs5, Myl9, Acta2, and Mylk), endothelial cells (Pecam1, Cdh5, and Eng), and lymphoid cells (Cd3d, Cd3 g, and Cd2). To further dissect immune cells, 6 different clusters were determined by repeating dimensionality reduction and unsupervised reclustering. We identified neutrophils (S100a8, S100a9, and Cd14), DCs (Cst3, Ccl17, and Ccl22), αβ T cells (Trac and Trbc2), δγ T cells (Trdc and Tcrg-C1), macrophage (Apoe, C1qa, C1qb, and C1qc), mast cells (Mcpt1, Mcpt2, Mcpt4, and Tpsb2). During reclustering in immune cells, we also observed small clusters co-expressing keratinocyte markers and immune cell markers, which were inferred to be doublets and removed for the further analysis.54 To obtain a more comprehensive profile of T cells status, we split all T cells into 9 clusters, including Treg (Foxp3 and Ctla4), naive-like T (Klf2, Tcf7, and Klf3), γδ T_1 (Il17a, Il17f, Trdc, and Tcrg-C1), Ifng+ Hsp+ T (Hspa1a, Hspa1b, Dnajb1, Ifng, and Fos), CD8+ cytotoxic T (Nkg7, Klrd1, Ccl4, Ccl3, Cd8b1, and Cd8a), CD4+ T effector (Tnfsf8, Tnfsf11, Bhlhe40, Cd4, and Pdcd1), γδ T_2 (Trdc, Tcrg-C1, Fos, and Junb), Xcl1+ T (Xcl1, Ccl5, Klrb1b1, and Arl4d), and ILC2 (Gata3 and Kit).
RNA-seq
Genome-wide transcriptional profiling was performed in the SCC1 cell line treated with JQ1 or iBET-151 in the presence or absence of IFN-γ for 24 h. Differentially expressed genes were identified using the edgeR program. Genes showing altered expression with p < 0.05 and more than 1.5-fold changes were considered as differentially expressed. A gene list that was significantly increased upon the JQ1 and iBET-151 treatment at the presence and absence of IFN-γ was obtained via Venn diagram. The gene list of upregulated genes was input into IPA software (Qiagen) to assess changes in enrichment signaling pathways as previously described.49 A p value of <0.05 was considered statistically significant.
ChIP assay
For each ChIP reaction mixture, approximately 1 × 106 cells were used. Cells were treated with 10 mM dimethyl 3,30-dithiobispropionimidate-HCl (DTBP) (Thermo Fisher, no. 20665) in PBS for 10 min and then cross-linked with 1% formaldehyde at 37°C for 10 min. Total cell lysates were sonicated to generate 200- to 400-bp DNA fragments. Chromatin complexes were immunoprecipitated with the following antibodies: anti-BRD4 (Abcam, ab243862), anti-G9a (Cell Signaling Technology, 3306S), anti-H3k9me1 (Abcam, ab9045), anti-H3K9me2 (Abcam, ab1220), and anti-H3K9me3 (Abcam, ab8898). The precipitated DNA samples were measured by qPCR. Data are expressed as the percentage of input DNA. The ChIP-PCR primer sequences are listed in Table S2.
TCGA data analysis
The mRNA expression of BRD4 was evaluated based on Starbase 3.0 from SYSU. The correlation of BRD4 with IFNG, CD8A, CD8B, GZMA, GZMB, HLA-A, HLA-B, and HLA-C gene expressions were evaluated based on TCGA-HNSCC datasets by using LinkedOmics (http://www.linkedomics.org/) online database. The correlation of BRD4 with tumor-infiltrating immune cells were analyzed based on the TIMER 2.0 online database (http://timer.cistrome.org).55,56,57
Statistical analysis
Statistical analyses were carried out using GraphPad Prism (version 8.0, www.graphpad.com), and data were statistically determined by Student’s t test or ANOVA. All reported p values were 2-tailed and p < 0.05 was considered statistically significant. Each in vitro experiment was repeated at least 3 times.
Data availability
The accession numbers for the RNA-seq and scRNA-seq reported in this paper are NCBI GEO: GSE164155 and GSE207504. All other relevant data are available from the corresponding authors upon request.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (82073265 and 81572661) and Guangdong Financial Fund for High-Caliber Hospital Construction (174-2018-XMZC-0001-03-0125/D-14).
Author contributions
C.W. and J.L. conceived and designed the study. M.Z., G.W., Z.M., G.X., W.W., Z.H., Y.W., X.X., and R.G.H. developed the methodology and performed the in vitro and in vivo experiments. C.W., J.L., and M.Z. wrote the manuscript. G.W., C.Y., J.H., X.L., and D.C. aided in experimental design, interpretation of results, writing, and figure design. All of the authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.07.022.
Contributor Information
Jiong Li, Email: jli29@vcu.edu.
Cheng Wang, Email: wangch75@mail.sysu.edu.cn.
Supplemental information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Hedberg M.L., Goh G., Chiosea S.I., Bauman J.E., Freilino M.L., Zeng Y., Wang L., Diergaarde B.B., Gooding W.E., Lui V.W.Y., et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Invest. 2016;126:169–180. doi: 10.1172/JCI82066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Economopoulou P., Agelaki S., Perisanidis C., Giotakis E.I., Psyrri A. The promise of immunotherapy in head and neck squamous cell carcinoma. Ann. Oncol. 2016;27:1675–1685. doi: 10.1093/annonc/mdw226. [DOI] [PubMed] [Google Scholar]
- 4.Chen D.S., Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 5.Wolchok J.D., Neyns B., Linette G., Negrier S., Lutzky J., Thomas L., Waterfield W., Schadendorf D., Smylie M., Guthrie T., Jr., et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164. doi: 10.1016/s1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 6.Topalian S.L., Hodi F.S., Brahmer J.R., Gettinger S.N., Smith D.C., McDermott D.F., Powderly J.D., Carvajal R.D., Sosman J.A., Atkins M.B., et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipson E.J., Forde P.M., Hammers H.J., Emens L.A., Taube J.M., Topalian S.L. Antagonists of PD-1 and PD-L1 in cancer treatment. Semin. Oncol. 2015;42:587–600. doi: 10.1053/j.seminoncol.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling D.C., Bakkenist C.J., Ferris R.L., Clump D.A. Role of immunotherapy in head and neck cancer. Semin. Radiat. Oncol. 2018;28:12–16. doi: 10.1016/j.semradonc.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Page D.B., Postow M.A., Callahan M.K., Allison J.P., Wolchok J.D. Immune modulation in cancer with antibodies. Annu. Rev. Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- 10.Shao Q., Kannan A., Lin Z., Stack B.C., Jr., Suen J.Y., Gao L. BET protein inhibitor JQ1 attenuates Myc-amplified MCC tumor growth in vivo. Cancer Res. 2014;74:7090–7102. doi: 10.1158/0008-5472.CAN-14-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong J., Li J., Li Y., Ma Z., Yu Y., Wang C.Y. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat. Commun. 2021;12:3974. doi: 10.1038/s41467-021-24137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adeegbe D.O., Liu S., Hattersley M.M., Bowden M., Zhou C.W., Li S., Vlahos R., Grondine M., Dolgalev I., Ivanova E.V., et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in kras-mutant non–small cell lung cancer. Cancer Immunol. Res. 2018;6:1234–1245. doi: 10.1158/2326-6066.Cir-18-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogg S.J., Vervoort S.J., Deswal S., Ott C.J., Li J., Cluse L.A., Beavis P.A., Darcy P.K., Martin B.P., Spencer A., et al. BET-bromodomain inhibitors engage the host immune System and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18:2162–2174. doi: 10.1016/j.celrep.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu H., Bengsch F., Svoronos N., Rutkowski M.R., Bitler B.G., Allegrezza M.J., Yokoyama Y., Kossenkov A.V., Bradner J.E., Conejo-Garcia J.R., Zhang R. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16:2829–2837. doi: 10.1016/j.celrep.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson M.A., Prinjha R.K., Dittmann A., Giotopoulos G., Bantscheff M., Chan W.I., Robson S.C., Chung C.W., Hopf C., Savitski M.M., et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Y., Fang K., Wang Y., Lu N., Sun H., Zhang C. Single-cell analysis reveals the origins and intrahepatic development of liver-resident IFN-γ-producing γδ T cells. Cell. Mol. Immunol. 2021;18:954–968. doi: 10.1038/s41423-021-00656-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaeger N., Gamini R., Cella M., Schettini J.L., Bugatti M., Zhao S., Rosadini C.V., Esaulova E., Di Luccia B., Kinnett B., et al. Single-cell analyses of Crohn's disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun. 2021;12:1921. doi: 10.1038/s41467-021-22164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C., Zhu B., Son Y.M., Wang Z., Jiang L., Xiang M., Ye Z., Beckermann K.E., Wu Y., Jenkins J.W., et al. The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8(+) T cell fitness and functionality. Immunity. 2019;51:491–507.e7. doi: 10.1016/j.immuni.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sallin M.A., Kauffman K.D., Riou C., Du Bruyn E., Foreman T.W., Sakai S., Hoft S.G., Myers T.G., Gardina P.J., Sher A., et al. Host resistance to pulmonary Mycobacterium tuberculosis infection requires CD153 expression. Nat. Microbiol. 2018;3:1198–1205. doi: 10.1038/s41564-018-0231-6. [DOI] [PubMed] [Google Scholar]
- 20.Mei Y., Xiao W., Hu H., Lu G., Chen L., Sun Z., Lü M., Ma W., Jiang T., Gao Y., et al. Single-cell analyses reveal suppressive tumor microenvironment of human colorectal cancer. Clin. Transl. Med. 2021;11:e422. doi: 10.1002/ctm2.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kroczek R.A., Henn V. The role of XCR1 and its ligand XCL1 in antigen cross-presentation by murine and human dendritic cells. Front. Immunol. 2012;3:14. doi: 10.3389/fimmu.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donati B., Lorenzini E., Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol. Cancer. 2018;17:164. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao H., Li L., Yang D., Zeng L., Yewei X., Yu B., Liao G., Chen J. Recent progress in histone methyltransferase (G9a) inhibitors as anticancer agents. Eur. J. Med. Chem. 2019;179:537–546. doi: 10.1016/j.ejmech.2019.06.072. [DOI] [PubMed] [Google Scholar]
- 24.Wei S.C., Levine J.H., Cogdill A.P., Zhao Y., Anang N.A.A.S., Andrews M.C., Sharma P., Wang J., Wargo J.A., Pe'er D., Allison J.P. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170:1120–1133.e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakamaki J.I., Wilkinson S., Hahn M., Tasdemir N., O'Prey J., Clark W., Hedley A., Nixon C., Long J.S., New M., et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell. 2017;66:517–532.e9. doi: 10.1016/j.molcel.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faivre E.J., McDaniel K.F., Albert D.H., Mantena S.R., Plotnik J.P., Wilcox D., Zhang L., Bui M.H., Sheppard G.S., Wang L., et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578:306–310. doi: 10.1038/s41586-020-1930-8. [DOI] [PubMed] [Google Scholar]
- 27.Leonard B., Brand T.M., O'Keefe R.A., Lee E.D., Zeng Y., Kemmer J.D., Li H., Grandis J.R., Bhola N.E. BET inhibition overcomes receptor tyrosine kinase-mediated cetuximab resistance in HNSCC. Cancer Res. 2018;78:4331–4343. doi: 10.1158/0008-5472.CAN-18-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W., Ge H., Jiang Y., Huang R., Wu Y., Wang D., Guo S., Li S., Wang Y., Jiang H., Cheng J. Combinational therapeutic targeting of BRD4 and CDK7 synergistically induces anticancer effects in head and neck squamous cell carcinoma. Cancer Lett. 2020;469:510–523. doi: 10.1016/j.canlet.2019.11.027. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y., Wang Y., Diao P., Zhang W., Li J., Ge H., Song Y., Li Z., Wang D., Liu L., et al. Therapeutic targeting of BRD4 in head neck squamous cell carcinoma. Theranostics. 2019;9:1777–1793. doi: 10.7150/thno.31581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mao W., Ghasemzadeh A., Freeman Z.T., Obradovic A., Chaimowitz M.G., Nirschl T.R., McKiernan E., Yegnasubramanian S., Drake C.G. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J. Immunother. Cancer. 2019;7:277. doi: 10.1186/s40425-019-0758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y.R., Ouyang S.S., Chen Y.L., Li P., Xu H.W., Zhu S.L. BRD4/8/9 are prognostic biomarkers and associated with immune infiltrates in hepatocellular carcinoma. Aging (Albany NY) 2020;12:17541–17567. doi: 10.18632/aging.103768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin M., Guo Y., Hu R., Cai W.L., Li Y., Pei S., Sun H., Peng C., Li J., Ye R., et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 2020;11:1833. doi: 10.1038/s41467-020-15290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wellinger L.C., Hogg S.J., Newman D.M., Friess T., Geiss D., Michie J., Ramsbottom K.M., Bacac M., Fauti T., Marbach D., et al. BET inhibition enhances TNF-mediated antitumor immunity. Cancer Immunol. Res. 2022;10:87–107. doi: 10.1158/2326-6066.CIR-21-0224. [DOI] [PubMed] [Google Scholar]
- 34.Tian C.Q., Chen L., Chen H.D., Huan X.J., Hu J.P., Shen J.K., Xiong B., Wang Y.Q., Miao Z.H. Inhibition of the BET family reduces its new target gene Ido1 expression and the production of L-kynurenine. Cell Death Dis. 2019;10:557. doi: 10.1038/s41419-019-1793-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lampen M.H., van Hall T. Strategies to counteract MHC-I defects in tumors. Curr. Opin. Immunol. 2011;23:293–298. doi: 10.1016/j.coi.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Sokol L., Koelzer V.H., Rau T.T., Karamitopoulou E., Zlobec I., Lugli A. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015;13:279. doi: 10.1186/s12967-015-0647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnsen A.K., Templeton D.J., Sy M., Harding C.V. Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J. Immunol. 1999;163:4224–4231. [PubMed] [Google Scholar]
- 38.Li H., Xiong H.G., Xiao Y., Yang Q.C., Yang S.C., Tang H.C., Zhang W.F., Sun Z.J. Long non-coding RNA LINC02195 as a regulator of MHC I molecules and favorable prognostic marker for head and neck squamous cell carcinoma. Front. Oncol. 2020;10:615. doi: 10.3389/fonc.2020.00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kokkali S., Ntokou A., Drizou M., Perdikari K., Makaronis P., Katsarou E., Koufopoulos N., Tzovaras A., Ardavanis A. Nivolumab in patients with rare head and neck carcinomas: a single center's experience. Oral Oncol. 2020;101:104359. doi: 10.1016/j.oraloncology.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Pai S.I., Faivre S., Licitra L., Machiels J.P., Vermorken J.B., Bruzzi P., Gruenwald V., Giglio R.E., Leemans C.R., Seiwert T.Y., Soulieres D. Comparative analysis of the phase III clinical trials of anti-PD1 monotherapy in head and neck squamous cell carcinoma patients (CheckMate 141 and KEYNOTE 040) J. Immunother. Cancer. 2019;7:96. doi: 10.1186/s40425-019-0578-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Y., Lee N.Y. JAVELIN Head and Neck 100: a Phase III trial of avelumab and chemoradiation for locally advanced head and neck cancer. Future Oncol. 2019;15:687–694. doi: 10.2217/fon-2018-0405. [DOI] [PubMed] [Google Scholar]
- 42.Bauml J., Seiwert T.Y., Pfister D.G., Worden F., Liu S.V., Gilbert J., Saba N.F., Weiss J., Wirth L., Sukari A., et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. J. Clin. Oncol. 2017;35:1542–1549. doi: 10.1200/jco.2016.70.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siu L.L., Even C., Mesía R., Remenar E., Daste A., Delord J.P., Krauss J., Saba N.F., Nabell L., Ready N.E., et al. Safety and efficacy of durvalumab with or without tremelimumab in patients with PD-L1-low/negative recurrent or metastatic HNSCC: the phase 2 CONDOR randomized clinical trial. JAMA Oncol. 2019;5:195–203. doi: 10.1001/jamaoncol.2018.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu F., Weigel K.J., Zhou H., Wang X.J. Paradoxical roles of TGF-β signaling in suppressing and promoting squamous cell carcinoma. Acta Biochim. Biophys. Sin. 2018;50:98–105. doi: 10.1093/abbs/gmx127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell T.C., Hamid O., Smith D.C., Bauer T.M., Wasser J.S., Olszanski A.J., Luke J.J., Balmanoukian A.S., Schmidt E.V., Zhao Y., et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037) J. Clin. Oncol. 2018;36:3223–3230. doi: 10.1200/jco.2018.78.9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burtness B., Harrington K.J., Greil R., Soulières D., Tahara M., de Castro G., Jr., Psyrri A., Basté N., Neupane P., Bratland Å., et al. KEYNOTE-048 Investigators Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet (London, England) 2019;394:1915–1928. doi: 10.1016/s0140-6736(19)32591-7. [DOI] [PubMed] [Google Scholar]
- 47.Chen D., Wu M., Li Y., Chang I., Yuan Q., Ekimyan-Salvo M., Deng P., Yu B., Yu Y., Dong J., et al. Targeting BMI1(+) cancer stem cells overcomes chemoresistance and inhibits metastases in squamous cell carcinoma. CelL Stem Cell. 2017;20:621–634.e6. doi: 10.1016/j.stem.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia L., Zhang W., Wang C.Y. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. 2020;27:238–253.e6. doi: 10.1016/j.stem.2020.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhuang Z., Yu P., Xie N., Wu Y., Liu H., Zhang M., Tao Y., Wang W., Yin H., Zou B., et al. MicroRNA-204-5p is a tumor suppressor and potential therapeutic target in head and neck squamous cell carcinoma. Theranostics. 2020;10:1433–1453. doi: 10.7150/thno.38507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang M., Hoyle R.G., Ma Z., Sun B., Cai W., Cai H., Xie N., Zhang Y., Hou J., Liu X., et al. FOSL1 promotes metastasis of head and neck squamous cell carcinoma through super-enhancer-driven transcription program. Mol. Ther. 2021;29:2583–2600. doi: 10.1016/j.ymthe.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang C., Li Y., Jia L., Kim J.K., Li J., Deng P., Zhang W., Krebsbach P.H., Wang C.Y. CD276 expression enables squamous cell carcinoma stem cells to evade immune surveillance. Cell Stem Cell. 2021;28:1597–1613.e7. doi: 10.1016/j.stem.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie N., Wang C., Liu X., Li R., Hou J., Chen X., Huang H. Tumor budding correlates with occult cervical lymph node metastasis and poor prognosis in clinical early-stage tongue squamous cell carcinoma. J. Oral Pathol. Med. 2015;44:266–272. doi: 10.1111/jop.12242. [DOI] [PubMed] [Google Scholar]
- 53.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ji A.L., Rubin A.J., Thrane K., Jiang S., Reynolds D.L., Meyers R.M., Guo M.G., George B.M., Mollbrink A., Bergenstråhle J., et al. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell. 2020;182:497–514.e22. doi: 10.1016/j.cell.2020.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li T., Fu J., Zeng Z., Cohen D., Li J., Chen Q., Li B., Liu X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48:W509–W514. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li T., Fan J., Wang B., Traugh N., Chen Q., Liu J.S., Li B., Liu X.S. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77:e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li B., Severson E., Pignon J.C., Zhao H., Li T., Novak J., Jiang P., Shen H., Aster J.C., Rodig S., et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the RNA-seq and scRNA-seq reported in this paper are NCBI GEO: GSE164155 and GSE207504. All other relevant data are available from the corresponding authors upon request.