

Abstract
Stimulator of interferon genes (STING) is an essential signaling protein that is located on the endoplasmic reticulum (ER) and triggers the production of type I interferons (IFN) and proinflammatory cytokines in response to pathogenic DNA. Aberrant activation of STING is linked to autoimmune diseases. The mechanisms underlying homeostatic regulation of STING are unclear. Here, we report that UNC13D, which is associated with familial hemophagocytic lymphohistiocytosis (FHL3), is a negative regulator of the STING‐mediated innate immune response. UNC13D colocalizes with STING on the ER and inhibits STING oligomerization. Cellular knockdown and knockout of UNC13D promote the production of interferon‐β (IFN‐β) induced by DNA viruses, but not RNA viruses. Moreover, UNC13D deficiency also increases the basal level of proinflammatory cytokines. These effects are diminished by an inhibitor of STING signaling. Furthermore, the domains involved in the UNC13D/STING interaction on both proteins are mapped. Our findings provide insight into the regulatory mechanism of STING, the previously unknown cellular function of UNC13D and the potential pathogenesis of FHL3.
Keywords: IFN‐β, innate immunity, proinflammatory cytokines, STING, UNC13D
Subject Categories: Membranes & Trafficking; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
STING signaling is essential for the production of type I interferons and proinflammatory cytokines in response to pathogenic DNA. UNC13D, which is associated with familial hemophagocytic lymphohistiocytosis, is a negative regulator of STING‐mediated innate immune responses.
Introduction
For mammals, the innate immune system serves as the first line of defense protecting the host from pathogens and the effects of damaged cells. Some pattern recognition receptors (PRRs) sense pathogenic RNA or DNA to activate transcription factors, including interferon (IFN) regulatory factor 3 (IRF3) and nuclear factor‐κB (NF‐κB), which coordinately regulate the production of type I IFNs and proinflammatory cytokines (Takeuchi & Akira, 2010). Type I IFNs inhibit microbial replication by inducing the expression of interferon‐stimulated genes (ISGs; Darnell et al, 1994). IFNs and proinflammatory cytokines also activate subsequent adaptive immune responses (Vallabhapurapu & Karin, 2009).
Cyclic guanosine monophosphate (GMP)‐adenosine monophosphate (AMP) (cGAMP) synthase (cGAS) recognizes aberrant cytosolic DNA released from pathogens or damaged mitochondria and synthesizes the second‐messenger cGAMP (Sun et al, 2013; Wu et al, 2013). cGAMP activates the endoplasmic reticulum (ER)‐anchored protein STING (stimulator of interferon genes, also named MITA, MPYS, ERIS) (Ishikawa & Barber, 2008; Jin et al, 2008; Ishikawa et al, 2009; Sun et al, 2009; Xu et al, 2013), after which the STING dimer undergoes a conformational change, which leads to the formation of STING tetramers and oligomers through side‐by‐side packing (Shang et al, 2019). Once activated, STING oligomers translocate from the ER via the ER‐Golgi intermediate compartment (ERGIC), enter the Golgi via membrane trafficking and aggregate into polymers on the Golgi (Saitoh et al, 2009; Xu et al, 2013). STING polymers serve as scaffolds that recruit and activate TANK‐binding kinase 1 (TBK1) and the IκB kinase (IKK) complex, leading to phosphorylation of STING, followed by phosphorylation of the transcription factors IRF3 and NF‐κB (p65/p50), which induce the expression of type I IFNs and proinflammatory cytokines (Ergun et al, 2019; Shang et al, 2019; Zhang et al, 2019; Zhao et al, 2019).
STING hyperactivity is responsible for autoimmune syndromes such as STING‐associated vasculopathy with onset in infancy (SAVI; Liu et al, 2014). Given the clinical importance of STING in human diseases, there is an imperative need to uncover the mechanisms governing its activity. Many regulatory mechanisms have been found to control the activity of STING by regulating STING auto‐repression, degradation, trafficking, etc. In addition, STING undergoes post‐translational modifications such as ubiquitination, phosphorylation, palmitoylation and sumoylation (Eaglesham & Kranzusch, 2020; Hu & Shu, 2020). However, the mechanisms underlying homeostatic regulation of STING on the ER membrane remain unclear.
UNC13D (also known as Munc13‐4) belongs to the Munc13 (mammalian homolog of Caenorhabditis elegans uncoordinated gene 13) family, which participates in membrane fusion and synaptic transmission (Brenner, 1974; Palfreyman & Jorgensen, 2017). UNC13 family members display distinct patterns of expression; UNC13A and UNC13C are expressed specifically in nerve tissues, while UNC13B and UNC13D are widely expressed (Augustin et al, 1999; Koch et al, 2000; Dittman, 2019). Human UNC13D is essential for cytolytic granule exocytosis of lymphocytes. Mutations in the human UNC13D gene are associated with familial hemophagocytic lymphohistiocytosis 3 (FHL3), a syndrome characterized by hyperinflammatory responses (Feldmann et al, 2003). In comparison with healthy people, FHL3 patients have markedly lower UNC13D levels (Santoro et al, 2006; Amirifar et al, 2021). Recent studies suggest that genetic defects in UNC13D may also contribute to the development of acquired hemophagocytic lymphohistiocytosis (HLH), which is usually associated with viral infections (Janka, 2012; Miao et al, 2019). Previous studies of UNC13D function have mainly focused on hematopoietic cells, but UNC13D is also expressed in many nonhematopoietic cells and tissues, where its functions are unknown.
In this study, we identify FHL3‐associated protein UNC13D as a negative regulator of STING‐mediated innate immune signaling. UNC13D interacts with STING and inhibits oligomerization of STING on the ER in the presence or absence of pathogenic DNA, thus attenuating subsequent signaling and reducing the expression of type I IFNs and proinflammatory cytokines. These findings provide new insight into the mechanisms controlling STING activity and provide new directions for the development of treatments for FHL3 patients.
Results
UNC13D negatively regulates cytosolic DNA‐triggered innate immune signaling
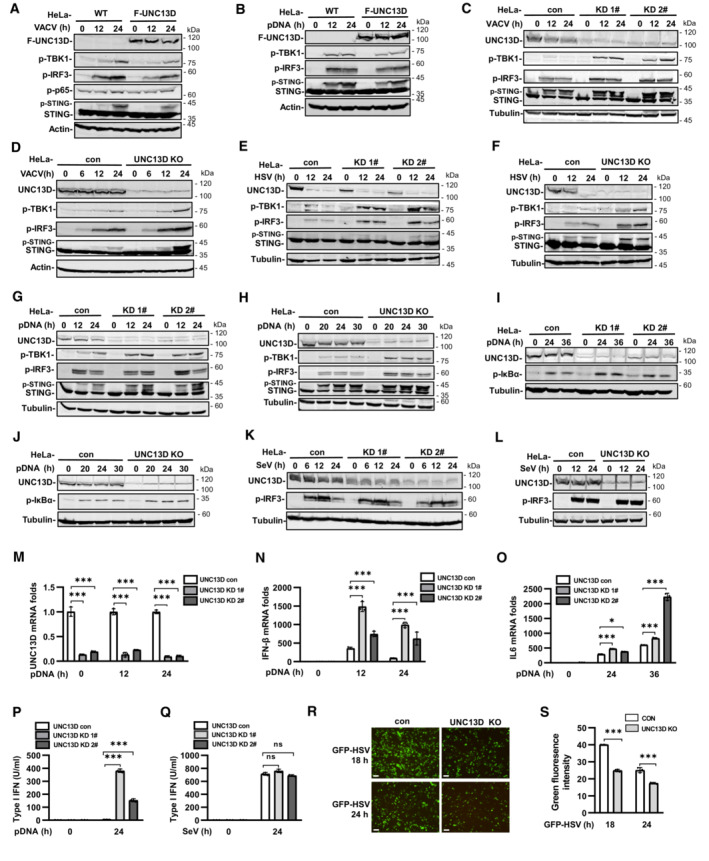
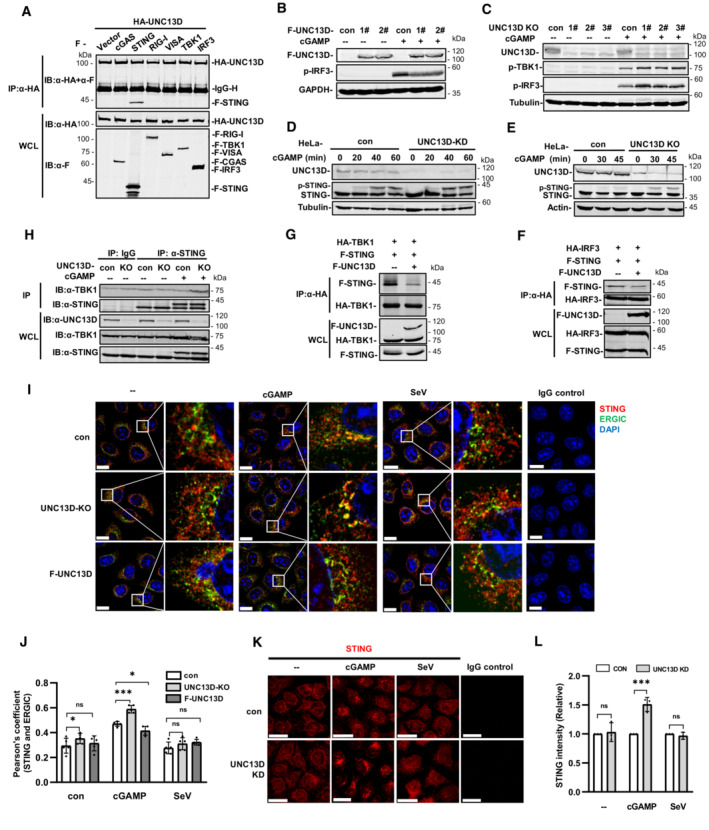
We performed yeast two‐hybrid assays with the STING C‐terminal cytoplasmic domain to identify proteins that may play roles in STING regulation. We screened a human fetal kidney and leukocyte cDNA mixed library and obtained 66 independent positive clones. One of these clones encoded FHL3‐associated protein UNC13D, dysfunction of which leads to hyperinflammatory responses. Since the interaction of UNC13D with STING has not been reported, we performed experiments to gain insight into the role of UNC13D in innate immune signaling. We generated HeLa cells stably expressing FLAG‐tagged UNC13D. Overexpression of UNC13D reduced the phosphorylation of IRF3, TBK1 and STING induced by infection with vaccinia virus (VACV), a DNA virus (Figs 1A and EV1A) and plasmid DNA transfection (Figs 1B and EV1B). In addition, overexpression of UNC13D reduced phosphorylation of p65, a marker of NF‐κB signaling activation, in response to VACV (Figs 1A and EV1A).
Figure 1. UNC13D negatively regulates cytosolic DNA‐triggered innate immune signaling.
-
A–LHeLa cells stably expressing FLAG‐tagged UNC13D (F‐UNC13D) (A and B), UNC13D stable knockdown (KD) (C, E, G, I and K) or knockout (KO) (D, F, H, J and L) HeLa cells, and their control cells were infected with VACV (A, C and D), HSV (E and F), or SeV (K and L), or they were transfected with plasmid DNA (pDNA) (1 μg/ml) (B, G–J) for the indicated time, before immunoblotting with anti‐Flag, ‐UNC13D, ‐p‐TBK1, ‐p‐IRF3, ‐STING, ‐p‐P65, ‐p‐IκB‐α, ‐actin or ‐tubulin antibody.
-
M–QUNC13D stable knockdown (KD) HeLa cells and their control cells were transfected with plasmid DNA (1 μg/ml) (M–P) or infected with SeV (Q) for the indicated time. UNC13D, IFN‐β and IL6 mRNA levels were measured by qPCR (M–O). The secretion of IFN‐β was measured by bioassay (P and Q). n = 3 technical replicates.
-
R, SUNC13D knockout (KO) HeLa cells and their control cells were infected with GFP‐HSV for 18 or 24 h. GFP expression was visualized (left) and the green fluorescence intensity was determined by ImageJ. Scale bar: 100 μm. n = 3 biological replicates.
Data information: Statistically significant differences were determined using ANOVA. Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; ***P < 0.001.
Source data are available online for this figure.
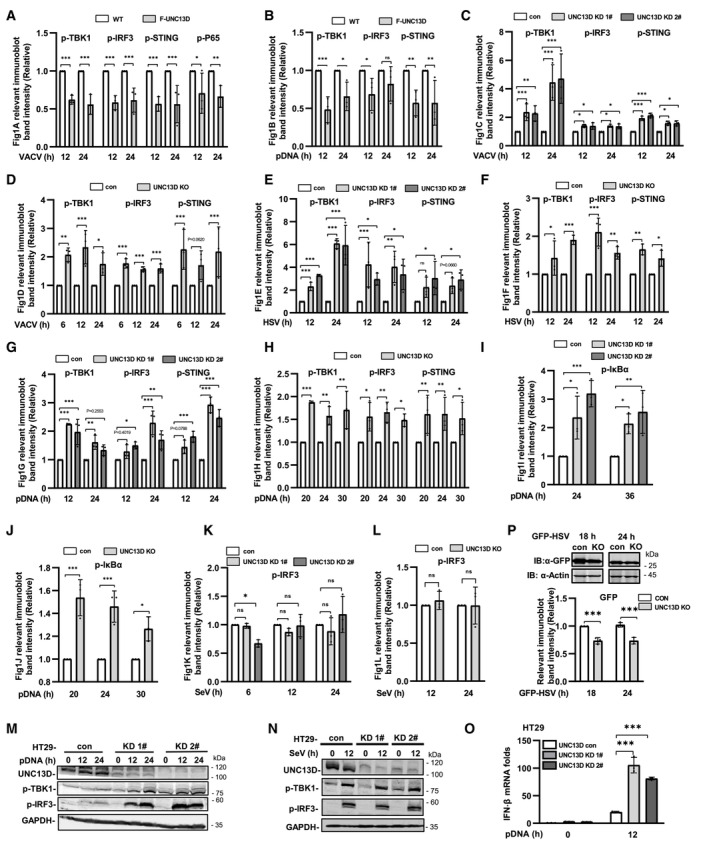
Figure EV1. UNC13D inhibits signaling induced by cytoplasmic DNA, but not RNA.
-
A–LQuantification of the relative expression of relevant proteins in Fig 1A–L. n = 3 biological replicates.
-
M–OUNC13D stable knockdown (KD) HT29 cells and their control cells were transfected with plasmid DNA (1 μg/ml) (M and O) or infected with SeV (N) for the indicated time. Protein levels were detected by immunoblotting with anti‐UNC13D, ‐p‐TBK1, ‐p‐IRF3 or ‐GAPDH antibody (M and N). IFN‐β mRNA levels were measured by qPCR (O). n = 3 technical replicates.
-
PUNC13D knockout (KO) HeLa cells and their control cells were infected with GFP‐HSV for 18 or 24 h. GFP expression was examined via whole cell lysis (WCL) with immunoblotting (IB). The relevant band intensities were quantified using ImageJ. n = 3 biological replicates.
Data information: Statistically significant differences were determined using ANOVA. Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Source data are available online for this figure.
To verify the functions of endogenous UNC13D, we generated stable UNC13D knockdown HeLa cells using shRNA. In addition, HeLa knockout cell lines were constructed by CRISPR‐Cas9 gene editing to further characterize the role of UNC13D. Knocking down or knocking out UNC13D in HeLa cells significantly elevated the phosphorylation of IRF3, TBK1 and STING following infection with DNA viruses VACV and herpes simplex virus (HSV), as well as after plasmid DNA transfection (Figs 1C–H and EV1C–H). In addition, we observed enhancement of IκBα phosphorylation, indicating that NF‐κB signaling was activated in the absence of UNC13D (Figs 1I and J, and EV1I and J). We also explored whether UNC13D performed the same functions in response to RNA viruses. However, there was no significant change in the level of p‐IRF3 induced by Sendai virus (SeV) in UNC13D knockdown and knockout HeLa cells (Figs 1K and L, and EV1K and L). We also generated stable UNC13D knockdown HT29 cells. Similar to the results in HeLa cells, knocking down UNC13D significantly enhanced phosphorylation of IRF3 and TBK1 in response to plasmid DNA transfection (Fig EV1M), but not SeV infection (Fig EV1N).
We also observed increased levels of IFN‐β and IL‐6 mRNA in UNC13D knockdown HeLa cells after transfection with plasmid DNA for different periods of time by quantitative real‐time PCR (qPCR) analysis (Fig 1M–O). In addition, the mRNA level of IFN‐β was also increased significantly in UNC13D knockdown HT29 cells upon transfection with plasmid DNA (Fig EV1O). Moreover, bioassays suggested that secretion of IFN‐β induced by plasmid DNA, but not SeV, was also promoted in UNC13D knockdown cells (Fig 1P and Q). Finally, we showed that knocking out UNC13D inhibited replication of GFP‐HSV in HeLa cells (Figs 1R and S, and EV1P). Taken together, these results suggest that UNC13D negatively regulates innate immune signaling triggered by cytosolic DNA, but not that induced by RNA.
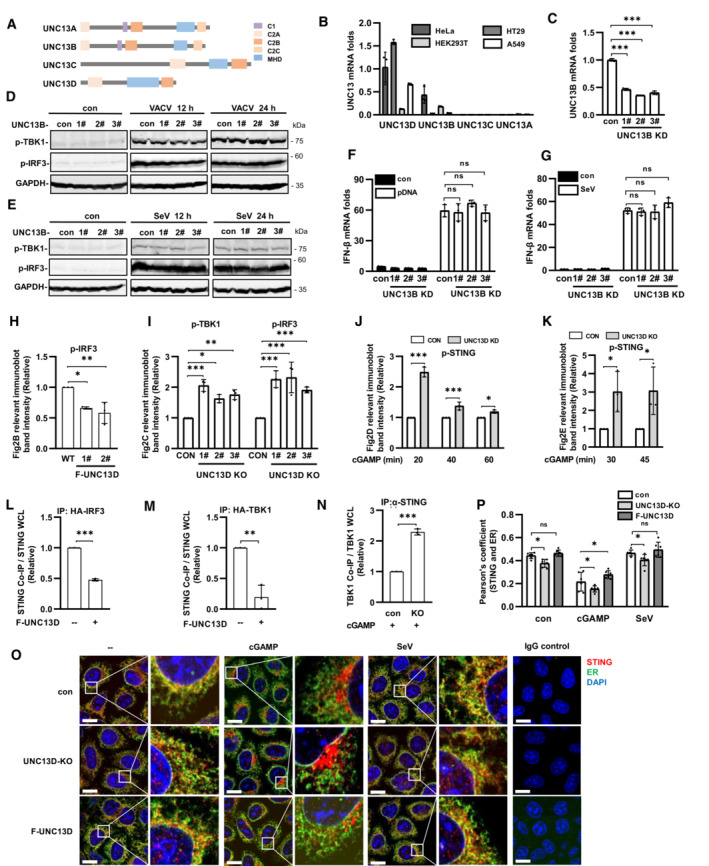
We also explored the functions of other members of the UNC13 family (Fig EV2A). The mRNA transcripts encoding UNC13A and UNC13C were detected at extremely low levels in HeLa, HT29, A549 and HEK293T cells, while mRNA transcripts encoding UNC13B and UNC13D were readily detected in HeLa and HT29 cells (Fig EV2B). We constructed UNC13B knockdown HeLa cells using shRNA (Fig EV2C) and found that the phosphorylation levels of IRF3 and TBK1 were changed only slightly after stimulation with VACV or SeV in the absence of UNC13B (Fig EV2D and E). In addition, the mRNA level of IFN‐β showed no significant changes following transfection with plasmid DNA or SeV infection (Fig EV2F and G). These results suggested that UNC13B had no significant role in the induction of IFN‐β expression by DNA or RNA viruses.
Figure EV2. UNC13B has no effect on interferon‐β induced by viruses, UNC13D inhibits STING signaling.
-
ADiagram of UNC13 family members.
-
BqPCR analysis of UNC13 mRNA levels in wild‐type HeLa, HT29, HEK293T and A549 cells. n = 3 technical replicates.
-
CqPCR analysis of UNC13B mRNA levels in UNC13B stable knockdown (KD) HeLa cells. n = 3 technical replicates.
-
D–GUNC13B stable knockdown (KD) HeLa cells and control HeLa cells were infected with VACV (D) or SeV (E and G), or transfected with plasmid DNA (F) (1 μg/ml), for 12 h or for the indicated time. Protein levels were detected by immunoblotting with anti‐p‐TBK1, ‐p‐IRF3 or ‐GAPDH antibody (D and E). IFN‐β mRNA levels were measured by qPCR (F and G). n = 3 technical replicates.
-
H–NQuantification of the relative expression of relevant proteins in Fig 2B–H. n = 3 biological replicates.
-
O, PUNC13D knockout (KO) HeLa cells and HeLa cells stably expressing FLAG‐tagged UNC13D (F‐UNC13D) and control HeLa cells were treated with cGAMP (100 nM) for 45 min or SeV for 12 h. Confocal microscopy was used to determine the localization of endogenous STING (red), ER marker Calnexin (green) and nuclei (blue) (O). Scale bars: 15 μm. The colocalization of STING and calnexin was analyzed using ImageJ (P). n = 6 biological replicates.
Data information: Statistically significant differences were determined using Student's t‐test (L–N) or ANOVA (C, F–K, P). Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Source data are available online for this figure.
UNC13D inhibits STING signaling
Yeast two‐hybrid assays revealed the interaction between STING and UNC13D. Therefore, in order to verify this interaction in mammalian cells, we carried out co‐immunoprecipitation with plasmids containing hemagglutinin (HA)‐tagged UNC13D and FLAG‐tagged signaling molecules. We found that STING, but not cGAS, Retinoic acid‐inducible gene I (RIG‐I), virus‐induced signaling adaptor (VISA), TBK1, or IRF3, specifically co‐precipitated with UNC13D (Fig 2A). Furthermore, upon exposure to STING ligand 2′,3′‐cGAMP, cells stably overexpressing UNC13D had lower p‐IRF3 levels, whereas UNC13D knockout cells showed higher levels of p‐TBK1 and p‐IRF3 (Figs 2B and C, and EV2H and I). Moreover, stimulation with 2′,3′‐cGAMP promoted STING phosphorylation in UNC13D‐deficient cells (Figs 2D and E, and EV2J and K). These results confirm that UNC13D interacts with STING and inhibits STING‐mediated innate immune signaling.
Figure 2. UNC13D inhibits STING signaling.
-
AHEK293T cells were transfected with the indicated plasmids (5 μg each) for 24 h. Whole cell lysates (WCL) were examined, and cell lysates were subjected to immunoprecipitation (IP) with anti‐HA antibodies, followed by immunoblotting (IB) with anti‐HA and ‐Flag antibody.
-
B–EHeLa cells stably expressing FLAG‐tagged UNC13D (F‐UNC13D) (B), UNC13D stable knockdown (KD) (D) or knockout (KO) (C and E) HeLa cells, and their control cells were stimulated with cGAMP (100 nM) for 30 min (B and C) or for the indicated time (D and E) before immunoblotting with anti‐Flag, ‐UNC13D, ‐p‐TBK1, ‐p‐IRF3, ‐STING, ‐GAPDH, ‐actin or ‐tubulin antibody.
-
F–HHEK293T cells were transfected with the indicated plasmids (5 μg each) for 24 h (F and G). UNC13D knockout (KO) and control HeLa cells were stimulated with cGAMP (100 nM) for 30 min (H). Whole cell lysates (WCL) were examined, and cell lysates were immunoprecipitated (IP) with anti‐HA antibodies (F and G), IgG and anti‐STING antibodies (H), followed by immunoblotting (IB) with anti‐HA, ‐Flag antibody or the indicated antibodies.
-
I, JUNC13D knockout (KO) HeLa cells and HeLa cells stably expressing FLAG‐tagged UNC13D (F‐UNC13D) and control HeLa cells were treated with cGAMP (100 nM) for 30 min or SeV for 12 h. Confocal microscopy was used to determine the localization of endogenous STING (red), ERGIC marker ERGIC‐53 (green) and nuclei (blue). Scale bars: 15 μm (I). The colocalization of STING and ERGIC was analyzed using ImageJ (J). n = 5 biological replicates.
-
K, LUNC13D stable knockdown (KD) HeLa cells and control HeLa cells were treated with cGAMP (100 nM) for 30 min or SeV for 12 h. The aggregation of endogenous STING (red) was examined via confocal microscopy (K). Scale bar: 25 μm. The red fluorescence intensity was determined by ImageJ (L). n = 3 biological replicates.
Data information: Statistically significant differences were determined using ANOVA. Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; ***P < 0.001.
Source data are available online for this figure.
Previous results demonstrated that UNC13D impaired phosphorylation of IRF3, TBK1 and STING induced by pathogenic DNA or cGAMP stimulation. Therefore, we investigated whether UNC13D inhibits recruitment of TBK1 and IRF3 to STING. The immunoprecipitation results showed that overexpression of UNC13D inhibited the binding of IRF3 and TBK1 to STING (Figs 2F and G, and EV2L and M). We also found that UNC13D deficiency enhanced the binding of endogenous TBK1 to STING during stimulation with cGAMP (Figs 2H and EV2N).
Trafficking of STING from the ER to the Golgi via the ERGIC is crucial for STING activation (Ishikawa et al, 2009; Saitoh et al, 2009). We also conducted immunofluorescence experiments in UNC13D knockout and control HeLa cells to investigate whether UNC13D prevented STING trafficking. By utilizing ERGIC marker (ERGIC‐53) or ER marker (calnexin) co‐staining, we showed that cGAMP induced translocation of STING from the ER to the ERGIC in control HeLa cells, and STING trafficking was not observed upon SeV infection (Figs 2I and J, and EV2O and P). In comparison with their WT counterparts, UNC13D‐deficient cells showed significantly more STING accumulation in the ERGIC (Fig 2I and J) and less STING distribution on the ER (Fig EV2O and P) following cGAMP exposure. Conversely, STING translocation to the ERGIC was inhibited (Fig 2I and J), and ER retention was promoted (Fig EV2O and P) in HeLa cells that stably overexpressed UNC13D. In addition, immunofluorescence experiments showed that knocking down UNC13D promoted the formation of STING aggregation puncta induced by cGAMP. Control cells infected with SeV showed no noticeable change in STING aggregation (Fig 2K and L).
Taken together, these data suggest that UNC13D suppresses STING aggregation, STING translocation, and recruitment of IRF3 and TBK1 in response to cGAMP, indicating that UNC13D functions upstream of STING signaling.
UNC13D inhibits STING oligomerization on the endoplasmic reticulum
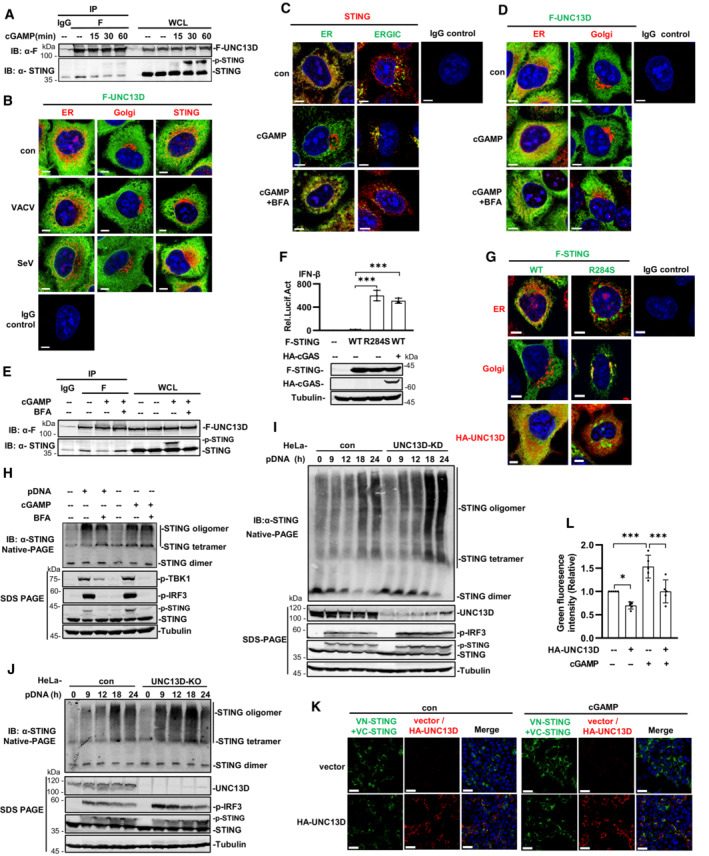
The results described above suggested that UNC13D inhibited the early steps of STING activation. To corroborate our findings, we carried out co‐immunoprecipitation using HeLa cells that stably expressed FLAG‐tagged UNC13D. We observed that UNC13D interacted with STING in the absence of stimulation. As the stimulation time with cGAMP was prolonged, the interaction between UNC13D and STING ceased gradually (Figs 3A and EV3A). To verify these observations, we explored the distributions of FLAG‐tagged UNC13D and endogenous STING in HeLa cells infected with a DNA virus. In the absence of stimulation, UNC13D was partly localized on the ER and colocalized with endogenous STING. Upon VACV infection, UNC13D was still located on the ER and did not colocalize with aggregated STING on the Golgi (Figs 3B and EV3B).
Figure 3. UNC13D inhibits STING oligomerization on the endoplasmic reticulum.
-
AUNC13D knockout HeLa cells stably expressing FLAG‐tagged UNC13D were stimulated with cGAMP (100 nM) or left unstimulated for the indicated time. Whole cell lysates (WCL) were examined, and cell lysates were immunoprecipitated (IP) with IgG and anti‐Flag antibodies, followed by immunoblotting (IB) with the indicated antibodies.
-
BUNC13D knockout HeLa cells stably expressing FLAG‐tagged UNC13D were infected with VACV or SeV for 12 h. The localization of FLAG‐UNC13D (green), ER marker calnexin (red), Golgi marker GM130 (red), endogenous STING (red) and nuclei (blue) was examined via confocal microscopy. Scale bar: 6 μm.
-
C–EWild‐type HeLa cells (C) and UNC13D knockout HeLa cells stably expressing FLAG‐tagged UNC13D (D and E) were pretreated with BFA (20 μM) or left untreated for 2 h, followed by stimulation with cGAMP (100 nM) (or left unstimulated) for 30 min. Localization of endogenous STING (red), FLAG‐UNC13D (green), ER marker calnexin (red/green), ERGIC maker ERGIC‐53 (green), Golgi marker GM130 (red) and nuclei (blue) was examined via confocal microscopy. Scale bar: 6 μm (C and D). Whole cell lysates (WCL) were examined, and cell lysates were immunoprecipitated (IP) with IgG and anti‐Flag antibodies, followed by immunoblotting (IB) with the indicated antibodies (E).
-
FHEK293T cells were transfected for 24 h with IFN‐β‐luciferase reporter plasmid, pRL‐TK and the indicated expression plasmids, followed by dual‐luciferase reporter assays. The expression of plasmids was examined by western blotting with anti‐Flag, ‐HA or ‐tubulin antibody. n = 3 technical replicates.
-
GSTING knockout HeLa cells were transfected with FLAG‐tagged STING WT/R284S (1 μg), HA‐tagged UNC13D (1 μg) or vector (1 μg) for 24 h. The localization of FLAG‐STING WT or R284S (green), endogenous ER marker calnexin (red), Golgi marker GM130 (red), HA‐UNC13D (red) and nuclei (blue) was examined via confocal microscopy. Scale bar: 5 μm.
-
HWild‐type HeLa cells were pretreated with BFA (20 μM) or left untreated for 2 h before stimulation with plasmid DNA (1 μg/ml) for 18 h or cGAMP (100 nM) for 30 min (or left unstimulated). Cell lysates were separated by native (top) or SDS (bottom) PAGE and analyzed by immunoblotting (IB) with anti‐p‐TBK1, ‐p‐IRF3, ‐STING or ‐tubulin antibody.
-
I, JUNC13D stable knockdown (KD) (I) and knockout (KO) (J) HeLa cells and control cells were transfected with plasmid DNA (pDNA) (1 μg/ml) for the indicated time. Cell lysates were separated by native (top) or SDS (bottom) PAGE and analyzed by immunoblotting (IB) with anti‐UNC13D, ‐p‐TBK1, ‐p‐IRF3, ‐STING or ‐tubulin antibody.
-
K, LHEK293T cells were transfected with HA‐tagged UNC13D, VN/VC‐STING or vector for 24 h, followed by stimulation with cGAMP (100 nM) (or left unstimulated) for 30 min. Nucleus marker DAPI (blue), VN/VC‐STING (green) and HA‐UNC13D (red) were examined via confocal microscopy (K). Scale bar: 25 μm. The green fluorescence intensity was determined by ImageJ (L). n = 5 biological replicates.
Data information: Statistically significant differences were determined using ANOVA. Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; ***P < 0.001.
Source data are available online for this figure.
Figure EV3. UNC13D impairs STING oligomerization.
-
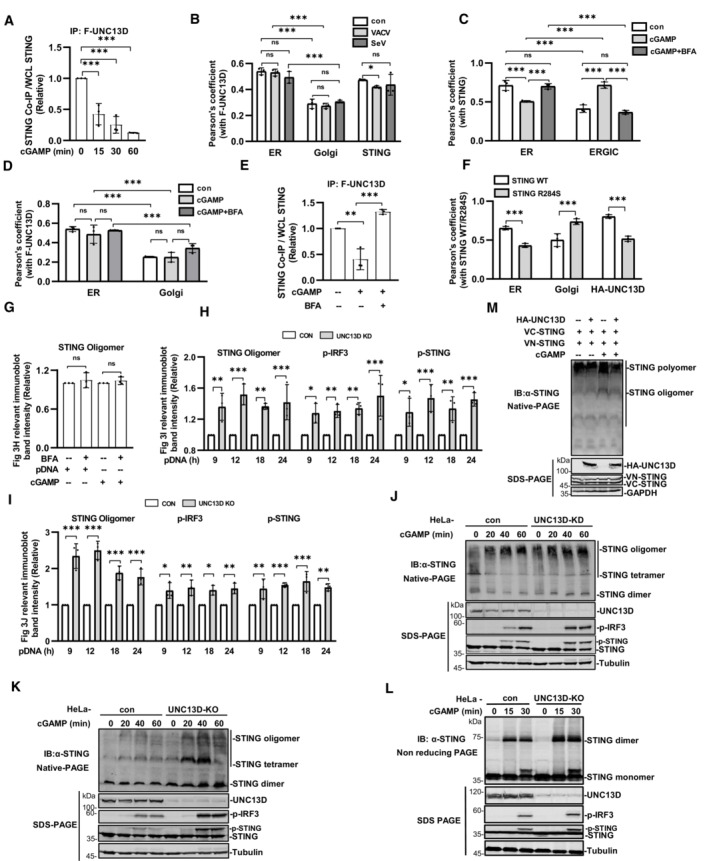
A–F(A, E) Quantification of the relative expression of STING from co‐immunoprecipitation (IP) results shown in Fig 3A and E. n = 3 biological replicates. (B–D, F) Quantification of the colocalization of relevant proteins in Fig 3B–D and F. n = 3 biological replicates.
-
G–IQuantification of the relative expression of relevant proteins in Fig 3H–J. n = 3 biological replicates.
-
J–LUNC13D stable knockdown (KD) (J) or knockout (KO) (K and L) HeLa cells and control HeLa cells were stimulated with cGAMP for the indicated time. Cell lysates were separated by native (top) (J and K), non‐reducing (top) (L) or SDS (bottom) PAGE and analyzed by immunoblotting (IB) with anti‐UNC13D, ‐p‐IRF3, ‐STING or ‐tubulin antibody.
-
MHEK293T cells were transfected with HA‐tagged UNC13D, VN/VC‐STING or vector for 24 h, followed by stimulation with cGAMP (100 nM) (or unstimulated) for 30 min. Cell lysates were separated by native (top) or SDS (bottom) PAGE and analyzed by immunoblotting (IB) with anti‐HA, ‐STING or ‐GAPDH antibody.
Data information: Statistically significant differences were determined using ANOVA. Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Source data are available online for this figure.
The translocation of STING from the ER to the Golgi via the ERGIC depends on membrane vesicle transport. Brefeldin A (BFA) was reported to disrupt membrane trafficking and inhibit STING translocation, thus blocking downstream signaling (Lippincott‐Schwartz et al, 1990; Konno et al, 2013; Ergun et al, 2019). The immunofluorescence results showed that STING did not translocate following pretreatment with BFA, and it remained localized on the ER upon cGAMP stimulation (Figs 3C and EV3C). Similarly, UNC13D remained on the ER in response to BFA pretreatment and cGAMP stimulation (Figs 3D and EV3D). Consistently, co‐immunoprecipitation results showed that localizing STING on the ER by BFA treatment restored the interaction between UNC13D and STING after cGAMP stimulation (Figs 3E and EV3E). These results indicate that UNC13D interacts with STING on the ER in the presence or absence of stimuli, and it does not interact with STING that has translocated in response to stimuli.
STING‐associated vasculopathy with on‐set in infancy (SAVI) is an autoimmune disease caused by STING hyperactivity. SAVI‐associated STING alterations promote constitutive TBK1 and IRF3 activation and type I IFN production without the requirement for STING agonists (Liu et al, 2014; Melki et al, 2017). We constructed STING with point mutation R284S, as it is expressed by SAVI patients, and we determined that STING R284S is constitutively active without stimulation using an IFN‐β reporter assay (Fig 3F). Not surprisingly, immunofluorescence experiments showed that, in contrast to ER‐localized wild‐type STING, R284S was localized on the Golgi as aggregation puncta in the absence of stimuli. Moreover, co‐expressed UNC13D did not colocalize with STING R284S on the Golgi, and R284S aggregation was not affected by the presence of UNC13D (Figs 3G and EV3F). These data also indicate that UNC13D negatively regulates STING localized on the ER, but not in the Golgi.
Several studies have revealed that STING undergoes ligand‐dependent oligomerization on the ER (Ergun et al, 2019; Yu et al, 2021). We verified that BFA blocks translocation of STING (Fig 3C) and phosphorylation of IRF3, TBK1 and STING induced by cGAMP or plasmid DNA (Fig 3H). However, native PAGE experiments showed that BFA did not affect STING oligomerization induced by cGAMP or plasmid DNA (Figs 3H and EV3G), indicating that oligomerization of STING takes place on the ER. We also examined whether STING oligomerization was regulated by UNC13D using native PAGE. As the stimulus time was prolonged, the abundance of tetramers and oligomers of STING increased under the native condition. In comparison with control cells, cells in which UNC13D was knocked down or knocked out showed enhanced STING oligomerization upon stimulation with plasmid DNA or cGAMP (Figs 3I and J, and EV3H–K). Meanwhile, similar to our previous results, SDS‐PAGE results showed that IRF3 phosphorylation induced by plasmid DNA and cGAMP was markedly increased in UNC13D‐deficient cells (Figs 3I and J, and EV3H–K). Non‐denaturing SDS‐PAGE can depolymerize the STING polymer and preserve the dimer (Ergun et al, 2019; Sing et al, 2022). We have also performed non‐reducing SDS‐PAGE to detect oligomerization of STING. In comparison with their corresponding control cells, UNC13D‐knockout cells showed significantly more STING dimers, indicating that more polymers were formed upon cGAMP stimulation (Fig EV3L). This result is in line with the results obtained from the native PAGE experiments. Taken together, these results reveal that UNC13D negatively regulates STING signaling by attenuating STING oligomerization on the ER.
We further carried out Venus‐based bimolecular fluorescence complementation (BiFC) to confirm that UNC13D regulates STING polymerization in live cells. The BiFC assay is based on the reconstitution of a fluorescent protein in vivo, and it allows protein–protein association to be visualized directly in living cells (Kodama & Hu, 2012; Miller et al, 2015). We constructed two plasmids, VN‐STING and VC‐STING, in which a component of fluorescent Venus protein (VN or VC) was fused to the N terminal of STING. Expression of either the VN‐STING or VC‐STING alone did not produce fluorescence, while co‐expression of both fusion proteins resulted in green fluorescence. Stimulation with cGAMP enhanced the Venus fluorescence signal (Fig 3K and L). We found that the green fluorescence was apparently reduced after overexpression of UNC13D with co‐expression of both fusion proteins in the presence or absence of cGAMP stimulation (Fig 3K and L). Native PAGE using co‐expressed STING‐Venus fusion proteins revealed the presence of prominent polymers, which were increased in abundance upon cGAMP stimulation, while UNC13D overexpression reduced their abundance (Fig EV3M), and this change was correlated with the change in the fluorescence signal. These results indicate that UNC13D suppressed the formation of STING oligomers and polymers.
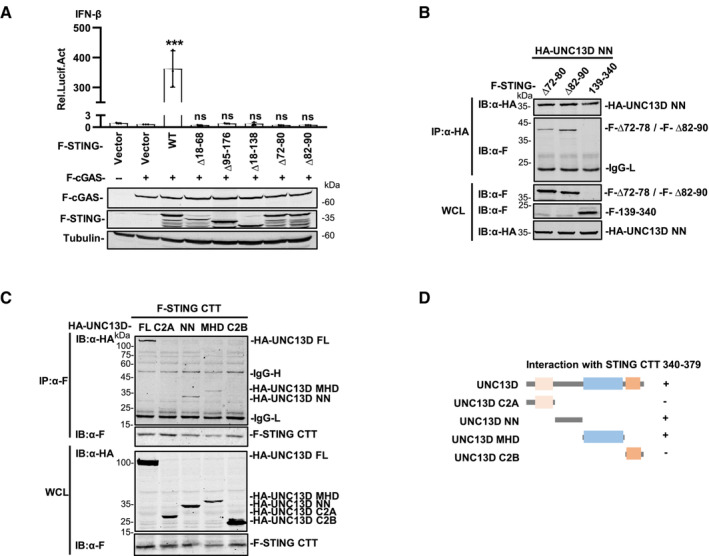
UNC13D interacts with the TM2‐TM3 linker and the CTD of STING
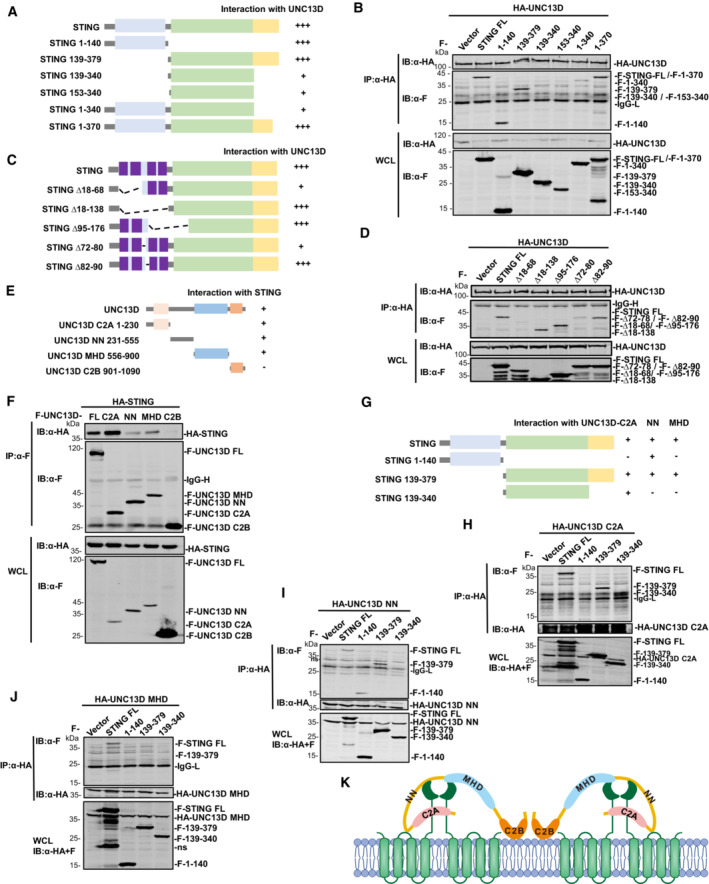
STING is an ER membrane protein that contains four transmembrane helices followed by a cytoplasmic ligand binding domain and a signaling domain (Shang et al, 2019). To uncover the domain(s) of STING involved in interaction with UNC13D, we generated a series of truncations of STING: STING N‐terminal transmembrane domains (TMD) (aa 1–140), STING C‐terminal domain (CTD) (aa 139–379), STING ligand binding domain (LBD) (aa 139–340, aa 153–340), STING lacking the C‐terminal tail (aa 1–340) and STING lacking its nine C‐terminal residues (aa 1–370) (Fig 4A). The results of co‐immunoprecipitation experiments with full length (FL) UNC13D showed that the STING TMD (aa 1–140) and the truncations containing the STING C‐terminal tail (CTT) (aa 340–379) interacted strongly with UNC13D (Fig 4A and B). In contrast, the binding between the STING LBD (aa 139–340, aa 153–340) and UNC13D was much weaker (Fig 4A and B), indicating that UNC13D mostly associates with the TMD and CTT of STING.
Figure 4. UNC13D interacts with the TM linker and CTD of STING.
-
A–JDiagram of domain mapping for UNC13D‐STING truncation interactions (A), UNC13D‐STING TMD truncation interactions (C), STING‐UNC13D truncation interactions (E) and STING truncation‐UNC13D truncation interactions (G). Truncated forms of UNC13D are indicated as follows: C2A (aa 1–230), NN (aa 231–555), MHD (aa 556–900) and C2B (aa 901–1,090) (E). (B, D, F, H–J) HEK293T cells were transfected with the indicated plasmids (5 μg each) for 24 h. Whole cell lysates (WCL) were examined, and cell lysates were immunoprecipitated (IP) with the indicated antibodies, followed by immunoblotting (IB) with anti‐HA or Flag antibody.
-
KModel of domain mapping of the STING‐UNC13D interaction.
Source data are available online for this figure.
The results described above demonstrated that UNC13D lacks a transmembrane domain and interacts with the STING TMD. Therefore, we conducted experiments to determine the residues through which these two proteins interact on the ER. STING contains four transmembrane helices, and the linker between transmembrane helix 2 and 3 (TM2 and TM3) (aa 70–91), which is exposed to the cytoplasm, contains two short helices. To determine whether UNC13D binds with the linker between TM2 and TM3 of STING, we measured the activity of mutant STING proteins lacking the TM1/TM2 helices (∆ aa 18–68), the TM3/TM4 helices (∆ aa 95–176), all four TM helices (∆ aa 18–138), or the two short helices of the TM2/TM3 linker (STING ∆ aa 72–80, ∆ aa 82–90). Luciferase assays showed that the IFN‐β promoter was not activated by any of the tested mutants (Fig EV4A). Co‐immunoprecipitation experiments showed that the interaction between UNC13D and the STING mutant lacking the TM1/TM2 helices (∆ aa 18–68) or the first short helix of the TM2/TM3 linker (aa 72–80) was much weaker (Fig 4C and D). These results suggest that UNC13D associates with the first short helix in the STING TM2‐TM3 linker region that is exposed to cytoplasm.
Figure EV4. Domain mapping of the STING‐UNC13D interaction.
-
AHEK293T cells were transfected for 24 h with the IFN‐β‐luciferase reporter plasmid, pRL‐TK and the other indicated plasmids, followed by dual‐luciferase reporter assays. The expression of plasmids was examined by western blotting with anti‐Flag or ‐tubulin antibody. n = 3 technical replicates.
-
B, CHEK293T cells were transfected with the indicated plasmids (5 μg each) for 24 h. Whole cell lysates (WCL) were examined, and cell lysates were immunoprecipitated (IP) with the indicated antibodies, followed by immunoblotting (IB) with anti‐Flag or ‐HA antibody.
-
DDiagram of domain mapping for STING C‐terminal tail (CTT) (aa 340–379)‐UNC13D truncation interactions.
Data information: Statistically significant differences were determined using ANOVA. Data are shown as the mean ± s.d. ns, not significant, P > 0.05; ***P < 0.001.
Source data are available online for this figure.
To further investigate the functional mechanism of UNC13D, we also mapped the UNC13D region(s) that interacted with STING. UNC13D contains two C2 domains (C2A and C2B) that are separated by Munc13 homology domain (MHD). The C2B domain has been reported to be involved in calcium‐dependent phospholipid binding (Feldmann et al, 2003; Palfreyman & Jorgensen, 2017), suggesting that UNC13D may be a peripheral membrane protein. The MHD contains a typical coiled‐coil structure for protein interaction (Feldmann et al, 2003). In addition, UNC13D contains an unnamed domain between the C2A domain and the MHD, which we designated as the NN (unnamed) domain. Based on the structure of UNC13D, we constructed truncated forms of UNC13D as follows: C2A (aa 1–230), NN (aa 231–555), MHD (aa 556–900) and C2B (aa 901–1,090) (Fig 4E). Co‐immunoprecipitation experiments indicated that the C2A domain, NN domain and MHD of UNC13D, but not its C2B domain, interacted with STING (Fig 4E and F), suggesting that the C2B domain is not necessary for binding with STING.
To identify the precise regions of UNC13D and STING responsible for their interaction, we performed co‐immunoprecipitation assays using their respective truncation plasmids (Fig 4G). We observed that the UNC13D C2A truncation interacted with the CTD (aa 139–379) and LBD (aa 139–340) of STING (Fig 4H). The UNC13D NN truncation interacted with the TMD (aa 1–140) and CTD (aa 139–379) of STING, but failed to interact with the STING LBD (Fig 4I). We also detected the interaction between NN and TMD mutants of STING. Similar to full‐length UNC13D, the binding of NN with the STING mutant lacking the first short helix of the TM2/TM3 linker (aa 72–80) was weaker than that of the mutant lacking the second short helix (aa 82–90) (Fig EV4B). Taken together with the results described above, we speculated that UNC13D NN may associate with the TM2‐TM3 linker of the STING TMD and the STING CTT (aa 340–379). Furthermore, the UNC13D MHD truncation specifically interacted with the STING CTD (aa 139–379), but not with the STING LBD or TMD (Fig 4J).
To address the direct interaction between the STING CTT and the NN/MHD domains of UNC13D, we constructed a plasmid expressing the STING CTT (340–379) and performed co‐IP. The binding of STING CTT with the FL and NN domains of UNC13D was clear, whereas binding with the MHD of UNC13D was weak, and there was no detected binding with the C2A or C2B domains of UNC13D (Fig EV4C and D). These results confirmed the interaction between the STING CTT and the NN domain of UNC13D. In general, domain mapping assays revealed that UNC13D associated with STING mainly via interactions between the C2A domain of UNC13D and the STING LBD, as well as between the NN domain of UNC13D and the STING TMD/CTT (Figs 4H and I, and EV4B–D).
Guided by the results described above, we proposed a model in which different domains of UNC13D interact with the TM2/TM3 linker, LBD and CTT of STING. The STING TMD and CTT play important roles in STING oligomerization (Ergun et al, 2019; Shang et al, 2019). We propose that UNC13D acts like a lid to prevent the interaction between STING dimers, thus inhibiting the formation of STING tetramers and oligomers on the ER membrane (Fig 4K).
STING inhibitor H‐151 mitigates the overexpression of proinflammatory factors induced by UNC13D dysfunction
FHL3 is known to be associated with UNC13D dysfunction (Feldmann et al, 2003). With the appropriate triggers, the development of acquired HLH may also be related to UNC13D genetic defects (Miao et al, 2019). FHL3 and HLH patients present with multisystem inflammation and produce higher levels of cytokines such as IFNs, interleukin (IL) 6 and tumor necrosis factor (TNF) α (Henter et al, 1991; Schneider, 2002; Filipovich, 2008).
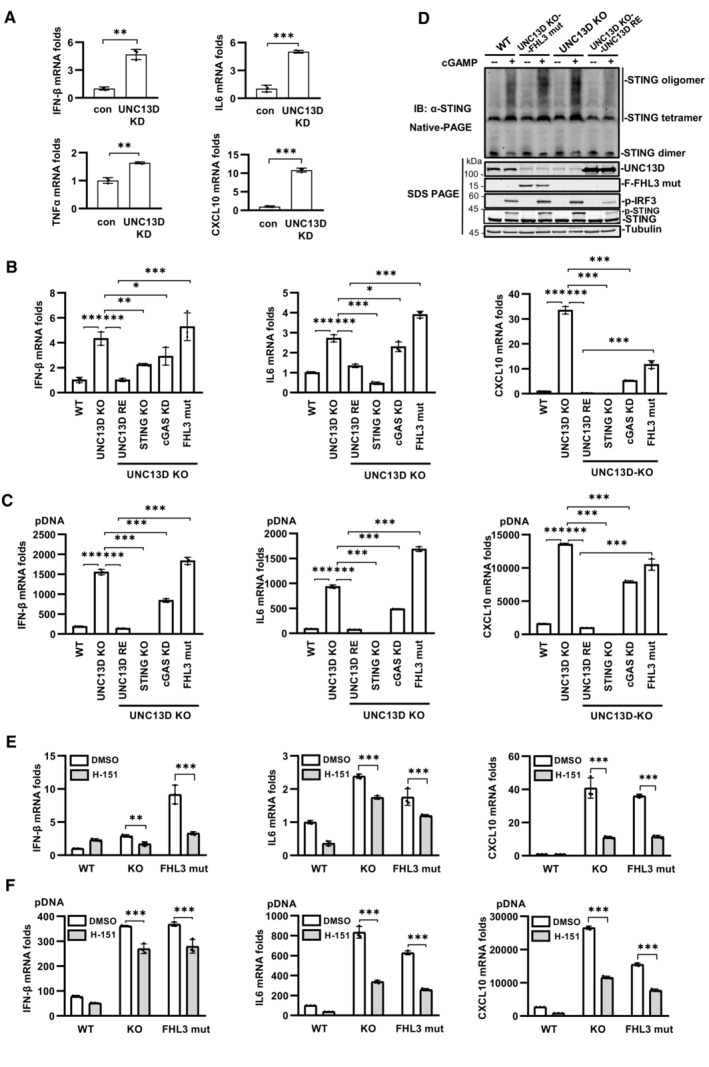
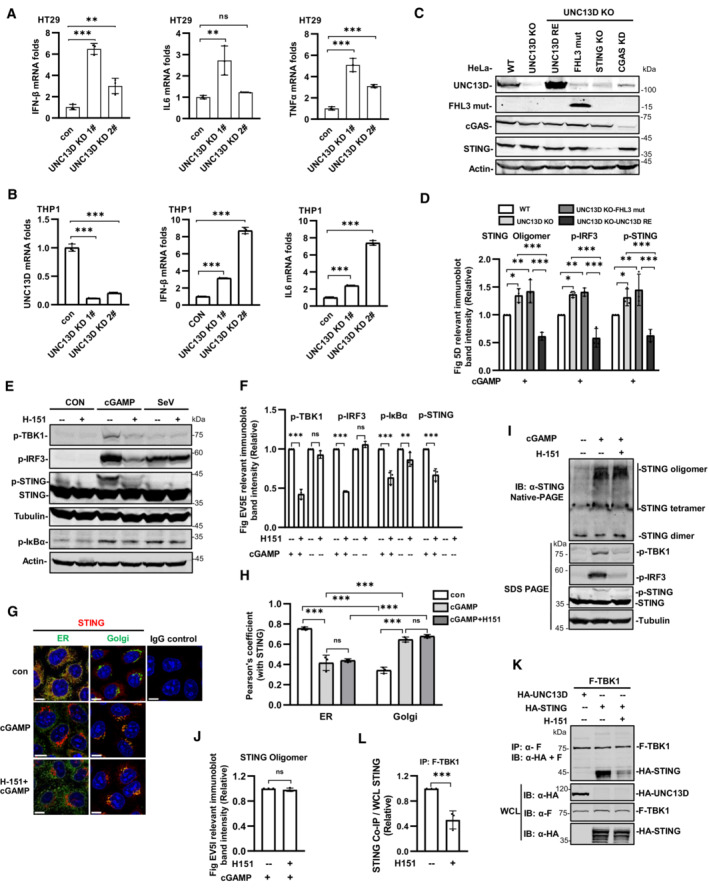
We found that the basal levels of the mRNA transcripts encoding IFN‐β, IL6, TNFα and CXCL10 were notably increased in UNC13D knockdown HeLa cells in comparison with those of control cells (Fig 5A). In addition, higher levels of the mRNA transcripts encoding IFN‐β, IL6 and TNFα were observed in unstimulated UNC13D knockdown HT29 cells (Fig EV5A). Measurements of the basal levels of cytokines in the context of UNC13D dysfunction using human macrophage cell line THP1 revealed increased levels of IFN‐β and IL6 in unstimulated UNC13D knockdown THP1 cells (Fig EV5B). Similar to the knockdown cells, the basal levels of IFN‐β, IL6 and CXCL10 were also notably increased in unstimulated UNC13D knockout HeLa cells (Fig 5B). In addition, expression of wild‐type UNC13D restored the basal levels of cytokines in unstimulated UNC13D knockout HeLa cells (Fig 5B).
Figure 5. STING inhibitor H‐151 mitigates the overexpression of proinflammatory factors induced by UNC13D dysfunction.
-
AqPCR analysis of IFN‐β, IL6, TNFα and CXCL10 mRNA levels in UNC13D stable knockdown (KD) HeLa cells without stimulation. n = 3 technical replicates.
-
B, CUNC13D knockout HeLa cells (UNC13D KO), UNC13D knockout HeLa cells stably expressing FLAG‐tagged wild‐type UNC13D (UNC13D RE), UNC13D knockout HeLa cells with STING knockout (STING KO), UNC13D knockout HeLa cells with cGAS stable knockdown (cGAS KD), and UNC13D knockout HeLa cells stably expressing FHL3 mutation (FHL3 mut) were transfected with plasmid DNA (C) for 12 h, or not transfected (B). IFN‐β, IL6 and CXCL10 mRNA levels were measured by qPCR. n = 3 technical replicates.
-
DUNC13D knockout (KO) HeLa cells, UNC13D knockout HeLa cells stably expressing FLAG‐tagged wild‐type UNC13D (UNC13D RE) and FHL3 mutation (FHL3 mut), and control cells were stimulated with cGAMP (100 nM) (or left unstimulated) for 30 min. Cell lysates were separated by native (top) or SDS (bottom) PAGE and analyzed by immunoblotting (IB) with anti‐UNC13D, ‐Flag, ‐p‐IRF3, ‐STING or ‐tubulin antibody.
-
E, FUNC13D knockout (KO) cells, UNC13D knockout HeLa cells stably expressing UNC13D FHL3 mutants (FHL3 mut), and their control cells were pretreated with H‐151 (2 μM) or untreated for 2 h, followed by transfection with (F) plasmid DNA for 12 h, or not transfected (E). IFN‐β, CXCL10 and IL6 mRNA levels were measured by qPCR. n = 3 technical replicates.
Data information: Statistically significant differences were determined using Student's t‐test (A) and ANOVA (B, C, E and F). Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Source data are available online for this figure.
Figure EV5. UNC13D dysfunction enhances basal levels of IFN‐β, IL6 and TNFα, H‐151 impairs the recruitment of TBK1 to STING.
-
A, BqPCR analysis of IFN‐β, IL6, UNC13D or TNFα mRNA levels in UNC13D stable knockdown (KD) HT29 (A) and THP1 cells (B) without stimulation. n = 3 technical replicates.
-
CConstruction of UNC13D knockout HeLa cells stably expressing FLAG‐tagged wild‐type UNC13D (UNC13D RE) and FHL3 mutation (FHL3 mut), UNC13D knockout HeLa cells with STING knockout cells (STING KO) and UNC13D knockout HeLa cells with cGAS stable knockdown (cGAS KD) cells. Cell lysates were subjected to immunoblotting (IB) with anti‐UNC13D, ‐Flag, ‐cGAS, ‐STING or ‐actin antibody.
-
DQuantification of the relative expression of relevant proteins in Fig 5D. n = 3 biological replicates.
-
E, FWild‐type HeLa cells were pretreated with H‐151 (2 μM) for 2 h before stimulation with cGAMP (100 nM) for 30 min, or they were infected with SeV for 6 h, or they were untreated (left). Protein levels were detected by immunoblotting with anti‐p‐TBK1, ‐p‐IRF3, ‐STING, ‐p‐IκBα, ‐tubulin or ‐actin antibody (E). The relative expression of relevant proteins was analyzed using ImageJ (F). n = 3 biological replicates.
-
G, HWild‐type HeLa cells were pretreated with H‐151 (2 μM) for 2 h before stimulation with cGAMP (100 nM) for 30 min, or they were untreated. Cell localization of ER marker calnexin (green), Golgi marker GRASP65 (green), STING (red) and nuclei (blue) were examined via confocal microscopy. Scale bar: 10 μm (G). The colocalization of relevant proteins was analyzed using ImageJ (H). n = 3 biological replicates.
-
I, JWild‐type HeLa cells were pretreated with H‐151 (2 μM) for 2 h before stimulation with cGAMP (100 nM) for 30 min, or they were untreated (left). Cell lysates were separated by native (top) or SDS (bottom) PAGE and analyzed by immunoblotting (IB) with anti‐p‐TBK1, ‐p‐IRF3, ‐STING or ‐tubulin antibody (I). The relative expression of relevant proteins was analyzed using ImageJ (J). n = 3 biological replicates.
-
K, LHEK293T cells were pretreated with H‐151 (2 μM) or left untreated and transfected with the indicated plasmids (5 μg each) for 24 h. Whole cell lysates (WCL) were examined, and cell lysates were immunoprecipitated (IP) with IgG and anti‐FLAG antibodies, followed by immunoblotting (IB) with anti‐Flag or ‐HA antibody. Quantification of the relative expression of STING from co‐immunoprecipitation (IP) experiments (L). n = 3 biological replicates.
Data information: Statistically significant differences were determined using Student's t‐test (J and L) or ANOVA (A, B, D, F and H). Data are shown as the mean ± s.d. ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Source data are available online for this figure.
We knocked out STING in UNC13D KO HeLa cells, which revealed that permanent removal of STING clearly rescued the elevated expression of cytokines in the presence or absence of stimulation (Fig 5B and C), indicating that overexpression of proinflammatory factors is mediated by STING. We also attempted to knock out cGAS in UNC13D KO HeLa cells, but only cGAS knockdown cells were obtained. Knockdown of cGAS significantly decreased the elevated expression of cytokines under stimulated or non‐stimulated conditions, which indicated that cGAS plays a role in increasing the abundance of proinflammatory factors in UNC13D‐deficient cells (Fig 5B and C). These results suggest that basal STING activation in UNC13D‐deficient cells requires an endogenous ligand trigger such as mtDNA or nuclear DNA damage, which is recognized by cGAS.
To explore the potential link between STING signaling and FHL3 caused by UNC13D mutations, we constructed a UNC13D substitution/frameshift mutation (A59T, P147 FsX14) based on genetic data from FHL3 patients (Santoro et al, 2006), and we reconstituted the mutant UNC13D into UNC13D KO HeLa cells (Fig EV5C). In comparison with KO cells restored with wild‐type UNC13D, the cells with the mutant form of UNC13D (A59T, P147 FsX14) exhibited higher basal levels of IFN‐β, IL6 and CXCL10 in the absence of stimulation (Fig 5B), and cells with the mutant form of UNC13D also exhibited elevated expression of various cytokines upon treatment with plasmid DNA (Fig 5C). We have demonstrated that knockdown or knockout of UNC13D enhanced ligand‐induced STING activation by promoting STING oligomerization (Figs 3I and J, and EV3J and K). We performed native PAGE to measure STING oligomerization in cells with the disease‐relevant mutant A59T P147 FsX14. In comparison with control cells, the mutant cells showed enhanced STING oligomerization upon stimulation with cGAMP (Figs 5D and EV5D). These results demonstrated that the disease‐relevant mutant of UNC13D failed to inhibit STING oligomerization in response to stimuli.
Next, we conducted experiments to examine whether a STING inhibitor could mitigate the elevation in the levels of various cytokines induced by UNC13D dysfunction. Small molecule inhibitor H‐151 was reported to inhibit STING signaling by blocking palmitoylation of STING on the Golgi upon stimulation (Haag et al, 2018). We confirmed that H‐151 remarkably inhibited phosphorylation of IκBα, IRF3, TBK1 and STING induced by cGAMP, but not by RNA virus SeV (Fig EV5E and F). We also observed that H‐151 had no obvious effects on the oligomerization, translocation or polymerization of STING induced by cGAMP using native‐PAGE and immunofluorescence assays (Fig EV5G–J), suggesting that H‐151 acts downstream of STING signaling. We also found that H‐151 suppressed the binding of TBK1 to STING (Fig EV5K and L). Treating UNC13D knockout cells and the disease‐relevant mutant A59T P147 FsX14 cells with H‐151 effectively decreased the elevated basal level of IFN‐β, IL6 and CXCL10 in the presence or absence of stimuli (Fig 5E and F). Taken together, these results indicate that UNC13D dysfunction induces higher levels of IFN‐β and proinflammatory cytokines via STING, and a STING inhibitor could mitigate this phenomenon.
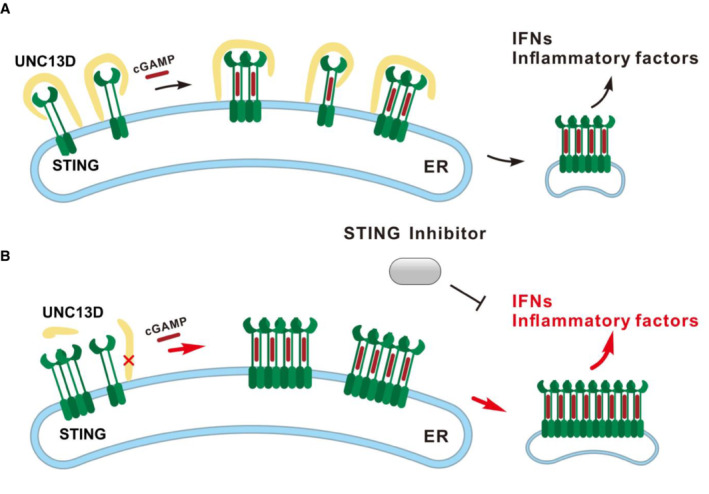
Based on the results described above, we proposed a functional model for UNC13D. In the resting and stimulated states, FHL3‐associated protein UNC13D negatively regulates STING oligomerization on the ER and maintains a homeostatic innate immune response (Fig 6A). UNC13D dysfunction may enhance STING oligomerization and facilitate subsequent downstream signaling events, leading to increased basal levels of inflammatory factors and elevated expression of cytokines upon pathogen invasion. Moreover, STING inhibitors can inhibit the excessive inflammatory response induced by UNC13D dysfunction (Fig 6B).
Figure 6. Model of UNC13D function.
-
AUNC13D interacts with STING in the presence or absence of cGAMP, and it acts like a lid to prevent the interaction between the STING dimer, thus impeding STING oligomerization on the ER membrane.
-
BUNC13D dysfunction may enhance STING oligomerization and facilitate subsequent downstream signaling events, resulting in increased basal levels of inflammatory factors and excessive cytokine expression upon pathogen invasion. A STING inhibitor can inhibit the hyperinflammatory response associated with UNC13D dysfunction.
Discussion
The cGAS‐STING pathway is essential for defense against infections and effective anti‐tumor immune responses (Li & Chen, 2018). Overactivation of STING has been observed in patients with many different autoimmune diseases, including systemic lupus erythematosus (SLE; Jeonghyun et al, 2012) and STING‐associated vasculopathy with on‐set in infancy (SAVI; Liu et al, 2014). As a central signaling component of the cytosolic DNA sensing pathway, STING is regulated precisely in a variety of ways to maintain an appropriate immune response. In this work, we showed that FHL3‐associated protein UNC13D functions as a negative regulator of STING in the cytosolic DNA‐triggered innate immune pathway by attenuating STING oligomerization on the ER. Furthermore, we found that UNC13D deficiency enhances phosphorylation of TBK1 and IRF3 induced by VACV, plasmid DNA and cGAMP. We also found that knocking down or knocking out UNC13D promoted IFN‐β production induced by plasmid DNA. Cells lacking UNC13D also showed higher activation of NF‐κB following treatment with cytosolic DNA. UNC13D specially binds with STING and does not affect its protein level. In addition, UNC13D has no significant effects on RNA virus‐induced responses, and UNC13D does not interact with RIG‐I or VISA, which function in the RNA virus‐triggered signaling pathway.
STING is localized on the ER, and it translocates from the ER to the Golgi when it is activated by cGAMP. Several ER‐localized proteins have been shown to regulate STING trafficking, including inactive rhomboid protein 2 (iRhom2; Luo et al, 2016), transmembrane emp24 protein transport domain containing 2 (TMED2; Sun et al, 2018) and the ER calcium sensor STIM1 (Srikanth et al, 2019). We found that UNC13D was present on the ER, but not in the Golgi, with or without stimulation, and UNC13D interacted with STING on the ER in the absence of stimuli. Upon stimulation with cGAMP, STING moved away from the ER, and the interaction between STING and UNC13D was weakened. BFA treatment blocked translocation of STING from the ER to the Golgi and restored the interaction between UNC13D and STING. These findings indicate that UNC13D negatively regulates STING on the ER. SAVI‐associated gain‐of‐function mutation STING R284S accumulated on the Golgi and did not colocalize with UNC13D, indicating that UNC13D may be unable to negatively regulate STING R284S.
High‐order oligomeric assemblies of activated receptors and adaptors have been observed in various innate immune signaling pathways (Yin et al, 2015). The oligomerization of STING is a necessary step for its activation. There is limited knowledge about the regulation of STING oligomerization on the ER. Recent studies reported that cGAMP induced the formation of ER‐associated oligomers or biocondensates of STING, and BFA treatment efficiently inhibited STING translocation, but not oligomerization or condensation (Ergun et al, 2019; Yu et al, 2021). We also found that preventing STING from translocating away from the ER via BFA treatment did not block cGAMP‐induced STING oligomerization. These findings suggest that cytosolic DNA‐induced STING oligomerization takes place on the ER before trafficking. We found that UNC13D deficiency promoted STING tetramerization and oligomerization upon stimulation with cytosolic DNA or cGAMP. Taken together with previous results, these findings demonstrate that UNC13D inhibited STING oligomerization on the ER. STING multimeric complexes serve as a critical scaffold for the recruitment of TBK1 and the IKK complex. Once STING oligomerization is inhibited, downstream signaling cannot be activated normally (Liu et al, 2015; Zhang et al, 2019). Accordingly, we observed that UNC13D inhibited the trafficking of STING from the ER to the ERGIC, recruitment of IRF3 and TBK1 by STING, and the production of type I interferons.
Accumulating evidence shows that various diseases are associated with abnormal STING accumulation and transportation in the absence of stimuli. Retrograde trafficking of STING from the Golgi to the ER is inhibited in coatomer protein subunit α (COPA) mutations, resulting in abnormal ligand‐independent activation of STING and COPA syndrome (Lepelley et al, 2020; Shum et al, 2020). In addition, STING is recruited to lysosomes for degradation with or without stimulation. Lysosomal membrane protein Niemann‐Pick type C1 (NPC1) deficiency blocks lysosomal degradation, enhancing STING signaling and resulting in the neurodegenerative disease known as Niemann‐Pick disease type C (Chu et al, 2021). A recent study showed that TOLLIP prevents lysosomal‐mediated degradation of STING protein to maintain immune homeostasis in the absence of stimuli (Pokatayev et al, 2020). These reports indicate that STING undergoes basal activation independent of ligands. We observed binding between UNC13D and STING in cells that were not treated with cGAMP, suggesting that UNC13D regulates STING in cells in the resting state. We posited that UNC13D acts as a homeostatic suppressor of STING on the ER, and UNC13D deficiency leads to a loss of control of STING oligomerization, resulting in a slightly increased level of STING signaling activation in the unstimulated state.
UNC13D mutations are associated with FHL3, and FHL3 patients may exhibit multisystemic inflammation (Feldmann et al, 2003; Chen et al, 2018). It has long been established that excessive cytokine production causes the clinical manifestations of FHL3 (Henter et al, 1991; Schneider, 2002). In FHL3 patients, UNC13D gene mutations are nearly always associated with a drastic decline in UNC13D protein abundance (Santoro et al, 2006; Meeths et al, 2011). In our system, knocking down or knocking out UNC13D mimicked the UNC13D expression status of FHL3 patients. Abnormally, low expression of UNC13D leads to higher basal levels of IFN‐β and proinflammatory cytokines in unstimulated cells, providing a mechanism for FHL3 mediated by UNC13D dysfunction. We demonstrated that STING inhibitor H‐151 functions downstream of STING oligomerization to suppress the binding of TBK1 with STING. We found that H151 efficiently inhibited the overactivation of STING signaling induced by UNC13D dysfunction, providing a promising direction for the development of new treatments for patients with FHL3.
Jinx mice with disruption of UNC13D are the first animal model of human FHL3. The phenotype of Jinx mice is conditional and depends on a specific infectious trigger. In comparison with WT homozygotes, Jinx homozygotes produce more IFN‐α/β, IL12 and IFN‐γ after viral inoculation, suggesting that FHL3 in humans could be caused by a similar infectious trigger (Crozat et al, 2007). Patients with a genetic predisposition for FHL3 may have a lower threshold for disease development upon viral infection. Consistent with this idea, UNC13D genetic defects also contribute to the development of acquired HLH, which has been shown to be triggered by viral infections, malignancies and even pregnancy (Jan‐Inge et al, 1993; Ménard et al, 2008; Janka, 2012; Wang et al, 2018; Miao et al, 2019). In comparison with WT cells, the human UNC13D dysfunction cell model that we constructed exhibited more excessive production of IFN‐β and IL6 upon simulation with plasmid DNA, and STING inhibitor H151 inhibited this elevation in cytokine production. These results demonstrate that our findings could have important therapeutic implications for patients with FHL3, HLH and other systemic inflammatory disorders.
In conclusion, our study showed that FHL3‐associated protein UNC13D negatively regulates cytosolic DNA‐triggered innate immune signaling by blocking STING oligomerization on the ER. These findings provide new insight into the manner in which regulation of cGAS‐STING controls innate immune responses, as well as the mechanisms underlying the pathogenesis of FHL and STING‐related immune disorders. It is important that future experiments test the physiological role of STING using primary FHL3 patient‐derived cells and FHL3 mouse models.
Materials and Methods
Antibodies and reagents
Mouse monoclonal antibodies against FLAG (Sigma Aldrich, F1804), HA (Sigma Aldrich, H9658), GFP‐Tag (Abmart, M20004), beta actin (Proteintech, 66009‐1‐Ig), GAPDH (Proteintech, 60004‐1‐lg), alpha tubulin (Proteintech, 66031‐1‐Ig), calnexin (Proteintech, 66903‐1), ERGIC53 (Santa Cruz Biotechnology, sc398893), GRASP65 (Santa Cruz Biotechnology, sc‐374423) and TBK1/NAK (Cell Signaling Technology, 51872S) were used in this study. Rabbit monoclonal antibodies against IRF3 (phospho S386) (Abcam, ab76493), TBK1/NAK (phospho S172) (Abcam, ab109272), Phospho‐NF‐κB p65 (Ser536) (Cell Signaling Technology, 3033), phospho‐IκBα (Ser32) (Cell Signaling Technology, 2859), calnexin (Cell Signaling Technology, 2679) and GM130 (Abcam, 52649) were used in this study. Rabbit polyclonal antibodies against TMEM173/STING (ABclonal, A3575; Proteintech, 19851‐1‐AP) and UNC13D (ABclonal, A13141) were used in this study. Alexa Fluor 488‐labeled goat anti‐mouse IgG (H+L) cross‐adsorbed secondary antibody (Thermo Fisher Scientific, A11001) and Alexa Fluor 594‐labeled donkey anti rabbit IgG (H+L) highly cross‐adsorbed secondary antibody (Thermo Fisher Scientific, A21207) were used in this study.
This study utilized 2′3′‐cGAMP (InvivoGen, tlrl‐nacga23m), Digitonin (SIGMA, 300410), Opti‐MEM®I (1*) + GlutaMAX™‐I (Gibco, 51985034), H‐151 (TOPSCIENCE, T5674), Brefeldin A (YEASEN, 50502ES03), LipoMax (SUNGEN, 32012), Liposomal transfection reagent (YEASEN, 40802ES03), Polyethylenimine (PEI) (polysciences, 24765), Puromycin (VWR LIFE SCIENCE, 97064‐280), qPCR SYBR green master mix (YEASEN, 11201ES08), the Dual‐Specific Luciferase Assay Kit (Promega, E1910), TriQuick reagent (Solarbio, R1100) and the Fast Mutagenesis System (Transgen, FM111‐01).
Plasmid constructs
Expression plasmids (HA‐ or FLAG‐tagged UNC13D and its truncation mutants, HA‐ or FLAG‐tagged STING and some of its mutants, and FLAG‐tagged RIG‐I and cGAS) were constructed using standard molecular biological techniques. Expression plasmids for FLAG‐ or HA‐tagged human VISA, TBK1 and IRF3, as well as the IFN‐β promoter luciferase reporter plasmid, were provided by Prof. HongBing Shu (Wuhan University, China). FLAG‐tagged human STING ∆18–138, ∆18–68 and ∆95–176 plasmids were provided by Prof. Zhengfan Jiang (Peking University, China). The vectors of the pcDNA3.1‐VN‐STING and pcDNA3.1‐VC‐STING expression plasmids were provided by Prof. ChuanMao Zhang (Peking University, China).
Transfection
HeLa and HT29 cells were transfected by LipoMax or Liposomal transfection reagent. HEK293T cells were transfected by polyetherimide. Transfections were performed following the manufacturer's instructions.
Cell lines and cultures
The cell lines used for the study were HEK293T, HeLa, Vero (Prof. HongBing Shu, Wuhan University, China), 2fTGH (Prof. ZhengFan Jiang, Peking University, China), HT29 and THP1 (Cell Resource Center, Institute of Basic Medicine, Chinese Academy of Medical Sciences/Peking Union Medical College, China). The HEK293T, HT29, HeLa, Vero and 2fTGH cell lines were cultured in DMEM (Gibco, USA) with 10% fetal bovine serum (GEMINI/PAN), 5 mg/ml penicillin and 10 mg/ml streptomycin at 37°C in 5% CO2. THP‐1 cells were cultured in RPMI‐1640 with 10% fetal bovine serum at 37°C in 5% CO2.
Virus amplification
Sendai virus (SeV), Herpes simplex virus‐1 (HSV‐1) wild‐type F strain and GFP‐HSV‐1 Kos strain (Prof. HongBing Shu, Wuhan University, China) and Vaccinia virus (VACV) Western Reserve strain (Prof. Min Fang, Chinese Academy of Medical Sciences, China) were obtained from the indicated sources. SeV was harvested using SPF chicken eggs. HSV‐1, GFP‐HSV‐1 and VACV were amplified by infecting Vero cells. Cells were collected 48 h after infection and re‐suspended in phosphate buffer, after which freezing (−80°C) and liquification (room temperature) were repeated three times to release the amplified virus.
Stimulation with 2′3′‐cGAMP
HeLa cells were incubated in reactions containing Opti‐MEM®I (1*) + GlutaMAXTM‐I, 10 mg/ml digitonin and 100 nM 2′3′‐cGAMP at 37°C for the indicated time before further experiments.
Generation of stable knock down or overexpression cells
To generate stable knockdown cells, short hairpin RNA (shRNA) lentiviruses expressing plasmids (Sigma) were transfected into HEK293T cells with three packaging plasmids. To generate stable overexpression cells, psin‐UNC13D‐puro plasmid or pSIN‐ FHL3 UNC13D mut (A59T, P147 FsX14)‐puro plasmid were transfected into HEK293T cells together with packing plasmids. After 12 h, cells were incubated with 15% fetal bovine serum medium for 36 h. The recombinant lentivirus‐containing medium was centrifuged to remove cell debris. Wild‐type HeLa cells, HT29 cells, and THP1 cells, as well as HeLa UNC13D knock out cells, were exposed to the indicated lentivirus supernatant in the presence of polybrene (8 mg/ml). The infected cells were selected with puromycin (1 mg/ml) for 48 h before further experiments. The shRNA sequences used in this study were as follows: human UNC13D 1#: GCTTTGCTACATGAACACCAA; human UNC13D 2#: GATCTTCCACAATACCCTCAA; human UNC13B 1#: CCGGACCCAGAACATTATCAT; human UNC13B 2#: CGAGTCCTATGAGTTGCAGAT; human UNC13B 3#: CCATTAAGAGAGAAGGACAAA; control (con): CGTGATCTTCACCGACAAGAT.
CRISPR/Cas9‐mediated genome editing
The target guide RNA was constructed into the lenti‐CRISPR vector to generate the indicated gene‐deficient cells. The lenti‐CRISPR‐UNC13D sgRNA plasmid and lenti‐CRISPR‐STING sgRNA plasmid were transfected into HeLa cells by Lipomax. After 12 h, cells were incubated with 15% fetal bovine serum medium for another 12 h. Cells were selected with 1 mg/ml puromycin for 1 week, and flow cytometry was used to separate individual cells. The knockout cells were identified by western blot and PCR. The control group consisted of monoclonal cells with no gene knocked out, and these control cells had activity similar to that of wild‐type cells. The guide RNA sequences were as follows: human UNC13D: CACCGTGCGCTGCCGAGAGGACCAG; human STING: CAGCTACTGGAGGACTGTGCGGG; human cGAS: CCGCCAGGAAGTCGGGATCCCGG.
RT‐PCR and quantitative RT‐PCR
Total RNA was isolated using TriQuick reagent (Solarbio) according to the manufacturer's instructions. Total RNA was converted into cDNA with oligo T primers and reverse transcriptase (Fermentas). PCR was performed with gene‐specific primer sets. Quantitative real‐time PCR was completed with SYBR green PCR Master Mix on an ABI7300 Detection System (Applied Biosystems, USA) or a LightCycle® 96 System (Roche, Germany), and gene expression levels were calculated by the accumulation index (2∆∆Ct). The primers for qRT‐PCR were as follows: human UNC13D: 5′‐AGTCTGTCCTGCCTGAGGAT‐3′ and 5′‐CAGGAGGCTGCTGAAGTTCT‐3′; human UNC13C: 5′‐TTCTGCACTCCAGGTGTGTG‐3′ and 5′‐CATGGCCACATCTCGGACAA‐3′; human UNC13B: 5′‐GATCCCGGGTGGAATGACGG‐3′ and 5′‐GTGACATGGCCGAGGATCG‐3′; human UNC13A: 5′‐AGCCGCACATTGAAGAGTGT‐3′ and 5′‐GGCTTGGCAATTTCACCCTG‐3′; human IFN‐β: 5′‐CCAACAAGTGTCTCCTCCAA‐3′ and 5′‐ATAGTCTCATTCCAGCCAGT‐3′; human CXCL10: 5′‐GTGGCATTCAAGGAGTACCTC‐3′ and 5′‐TGATGGCCTTCGATTCTGGATT‐3′; human TNFα: 5′‐TGCTTGTTCCTCAGCCTCTT‐3′ and 5′‐GGTTTGCTACAACATGGGCT‐3′; human IL6: 5′‐AGAGGCACTGGCAGAAAACAAC‐3′ and 5′‐AGGCAAGTCTCCTCATTGAATCC‐3′; human β‐actin: 5′‐ACGTGGACATCCGCAAAGAC‐3′ and 5′‐CAAGAAAGGGTGTAACGCAACTA‐3′.
Luciferase assay
HEK293T cells were cultured on 24‐well plates until they reached 70–80% confluence. Next, 50 ng firefly‐luciferase reporter plasmid and 50 ng of the expression plasmids were transfected together into HEK293T cells by polyetherimide, after which 50 ng Renilla‐luciferase reporter plasmid was transfected into every sample to normalize the transfection efficiency. Luciferase assays were performed using the Dual‐specific Luciferase Assay Kit (Promega) following the manufacturer's instructions.
Type I IFN bioassays
2fTGH cells that were stably transfected with type I IFN‐sensitive luciferase vectors were seeded into 96‐well plates and incubated with the supernatants from the indicated cells. After 4 h, 2fTGH cells were lysed and assayed using the Luciferase Reporter Assay System (Promega).
Native PAGE
Cells were treated with the indicated stimuli before they were lysed in lysis buffer (20 mM Tris, 150 mM NaCl, 1% Triton X‐100, 1 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulphonyl fluoride, pH 7.5) and solubilized by rocking for 30 min at 4°C. The cell lysates were centrifuged for 10 min at 15,000 rpm and supernatants were collected. Each lysate supernatant was mixed with 2× native sample buffer (125 mM Tris at pH 6.8, 20% glycerinum and 2.5% sodium deoxycholate). Next, the samples were electrophoresed for native PAGE. STING oligomers were detected using a standard western blot protocol. The final results were visualized using an Odyssey infrared imaging system (LICOR, USA).
Co‐immunoprecipitation and western blotting
HEK293T and HeLa cells were subjected to the indicated stimuli or transfected with the indicated plasmids before being lysed in Triton X‐100 lysis buffer or Nonidet P‐40 lysis buffer (20 mM Tris–HCl at pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100 or 1% Nonidet P‐40, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 1 mM phenylmethylsulphonyl fluoride). For every immunoprecipitation sample, 0.95 ml lysate was incubated with 30 μl Protein G Sepharose beads (GE Healthcare) and 1 μl of the indicated antibody at 4°C for 4 h. The Sepharose beads were washed gently with 1 ml lysis buffer three times. The precipitates were boiled for 10 min at 95°C and analyzed by SDS‐PAGE. The precipitates were examined by standard immunoblot procedures. The final results were visualized using an Odyssey infrared imaging system (LICOR, USA). The relevant immunoblot band intensities were quantified and normalized to their loading control using ImageJ.
Fluorescent confocal microscopy
Cells were cultured on slides, after which they were immobilized in absolute methanol for 20 min at −20°C and washed three times with PBS. Subsequently, the cells on the slides were incubated with 5% bovine serum albumin‐PBS for 30 min, incubated with 1% BSA‐PBS containing primary antibodies at 4°C overnight, and washed three times with PBS. Next, the cells were stained with Alexa Fluor 488‐labeled goat anti‐mouse IgG or Alexa Fluor 594‐labeled donkey anti‐rabbit IgG at 37°C for 1 h. Finally, the cells on the slides were washed three times with PBS. Nuclei were stained with 4,6‐diamidino‐2‐phenylindole (DAPI) at room temperature for 10 min. Images were captured by a confocal microscope (Dragonfly High Speed Spinning Disk Confocal Microscope, Andor, U.K.). The fluorescence intensity and the colocalization of relevant proteins were analyzed using ImageJ.
Bimolecular fluorescence complementation (BiFC)
Expression plasmids for pcDNA3.1‐VN‐STING and pcDNA3.1‐VC‐STING were constructed using standard molecular biological techniques. HEK293T cells were seeded on slides and transfected with the indicated plasmids. The cells were cultured at 37°C for 24 h, immobilized in methanol for 20 min at −20°C, and washed three times with PBS. Subsequently, nuclei were stained with 4,6‐diamidino‐2‐phenylindole (DAPI) at room temperature for 10 min and washed three times with PBS. Images were captured by a confocal microscope (Dragonfly High Speed Spinning Disk Confocal Microscope, Andor, U.K.).
Statistical analysis
Statistical analyses are described in each figure legend. The statistically significant differences were determined using Student's t‐test when comparing only two groups. Statistical analysis of more than two groups was performed using ANOVA. The graphs and statistical analysis were generated using GraphPad Prism 8. A dot plot is included with bar graphs to show the distribution of individual data points. All statistical results are shown as the mean ± s.d. (n ≥ 3). ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Author contributions
Pu Song: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Weiwei Yang: Validation; investigation. Karen F Lou: Investigation. Hao Dong: Investigation. Heng Zhang: Investigation. Beiming Wang: Investigation. Danying Chen: Conceptualization; supervision; funding acquisition; visualization; methodology; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We acknowledge Prof. HongBing Shu from Wuhan University, China, Prof. ZhengFan Jiang, Prof. Jianguo Chen, Prof. Junlin Teng and Prof. Chuanmao Zhang from Peking University, China, and Prof. Min Fang from the Chinese Academy of Sciences, China for plasmids, viruses, cell lines and instruments. We also acknowledge Ms. Liying Du, Dr. Siying Qin and Ming Du at the Core Facilities and Bioinformatics Core Facility, School of Life Sciences, Peking University, China for assistance with flow cytometry, immunofluorescence experiments and pattern diagram drawing. This work was supported by the National Natural Science Foundation of China (31870904).
EMBO reports (2022) 23: e55099
Contributor Information
Pu Song, Email: songpu17@pku.edu.cn.
Danying Chen, Email: dychen@pku.edu.cn.
Data availability
This study includes no data deposited in external repositories.
References
- Amirifar P, Ranjouri MR, Abolhassani H, Shad TM, Almasi‐Hashiani A, Azizi G, Moamer S, Aghamohammadi A, Yazdani R (2021) Clinical, immunological and genetic findings in patients with UNC13D deficiency (FHL3): a systematic review. Pediatr Allergy Immunol 32: 186–197 [DOI] [PubMed] [Google Scholar]
- Augustin I, Betz A, Herrmann C, Jo T, Brose N (1999) Differential expression of two novel Munc13 proteins in rat brain. Biochem J 337: 363–371 [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of caenorhabditis elegans. Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang F, Zhang Y, Teng W, Wang M, Nie D, Zhou X, Wang D, Zhao H, Zhu P et al (2018) Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet 94: 200–212 [DOI] [PubMed] [Google Scholar]
- Chu T, Tu X, Yang K, Wu J, Repa JJ, Yan N (2021) Tonic prime‐boost of STING signalling mediates Niemann–Pick disease type C. Nature 596: 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat K, Hoebe K, Ugolini S, Hong NA, Janssen E, Rutschmann S, Mudd S, Sovath S, Vivier E, Beutler B (2007) Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med 205: 737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J, Kerr I, Stark G (1994) Jak‐STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264: 1415–1421 [DOI] [PubMed] [Google Scholar]
- Dittman JS (2019) Unc13: a multifunctional synaptic marvel. Curr Opin Immunol 57: 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaglesham JB, Kranzusch PJ (2020) Conserved strategies for pathogen evasion of cGAS‐STING immunity. Curr Opin Immunol 66: 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergun SL, Fernandez D, Weiss TM, Li L (2019) STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 178: 290–301 [DOI] [PubMed] [Google Scholar]
- Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachée‐Chardin M, Chedeville G, Tamary H (2003) Munc13‐4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 115: 461–473 [DOI] [PubMed] [Google Scholar]
- Filipovich AH (2008) Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am 28: 293–313 [DOI] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R et al (2018) Targeting STING with covalent small‐molecule inhibitors. Nature 559: 269–273 [DOI] [PubMed] [Google Scholar]
- Henter JI, Elinder G, Sder O, Hansson M, Andersson B, Andersson U (1991) Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 78: 2918–2922 [PubMed] [Google Scholar]
- Hu MM, Shu HB (2020) Innate immune response to cytoplasmic DNA: mechanisms and diseases. Annu Rev Immunol 38: 79–98 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455: 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Zhe M, Barber GN (2009) STING regulates intracellular DNA‐mediated, type I interferon‐dependent innate immunity. Nature 461: 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan‐Inge H, Anneka E, Jan A, Gran E (1993) Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr 82: 369–372 [DOI] [PubMed] [Google Scholar]
- Janka GE (2012) Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med 166: 233–246 [DOI] [PubMed] [Google Scholar]
- Jeonghyun A, Delia G, Shinobu S, Barber GN (2012) STING manifests self DNA‐dependent inflammatory disease. Proc Natl Acad Sci USA 109: 19386–19391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC (2008) MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol 28: 5014–5026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch H, Hofmann K, Brose N (2000) Definition of Munc13‐homology‐domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J 349: 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama Y, Hu CD (2012) Bimolecular fluorescence complementation (BiFC): a 5‐year update and future perspectives. Biotechniques 53: 285–298 [DOI] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN (2013) Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155: 688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepelley A, Martin‐Niclos MJ, Bihan ML, Marsh JA, Frémond M‐L (2020) Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J Exp Med 217: e20200600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen ZJ (2018) The cGAS‐cGAMP‐STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215: 1287–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott‐Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD (1990) Microtubule‐dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell 60: 821–836 [DOI] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS et al (2014) Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371: 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV et al (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630 [DOI] [PubMed] [Google Scholar]
- Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, Shu HB (2016) iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol 17: 1057–1066 [DOI] [PubMed] [Google Scholar]
- Meeths M, Chiang SC, Wood SM, Entesarian M, Schlums H, Bang B, Nordenskjold E, Bjorklund C, Jakovljevic G, Jazbec J et al (2011) Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood 118: 5783–5793 [DOI] [PubMed] [Google Scholar]
- Melki I, Rose Y, Uggenti C, Eyck LV, Crow YJ (2017) Disease‐associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J Allergy Clin Immunol 140: 543–552 [DOI] [PubMed] [Google Scholar]
- Ménard F, Besson C, Rincé P, Lambotte O, Lazure T, Canioni D, Hermine O, Brousset P, Martin A, Gaulard P (2008) Hodgkin lymphoma‐associated hemophagocytic syndrome: a disorder strongly correlated with Epstein‐Barr virus. Clin Infect Dis 47: 531–534 [DOI] [PubMed] [Google Scholar]
- Miao Y, Zhu HY, Qiao C, Xia Y, Kong Y, Zou YX, Miao YQ, Chen X, Cao L, Wu W (2019) Pathogenic gene mutations or variants identified by targeted gene sequencing in adults with hemophagocytic lymphohistiocytosis. Front Immunol 10: 395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Kim Y, Huh WK, Park HO (2015) Bimolecular fluorescence complementation (BiFC) analysis: advances and recent applications for genome‐wide interaction studies. J Mol Biol 427: 2039–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfreyman MT, Jorgensen EM (2017) Unc13 aligns SNAREs and superprimes synaptic vesicles. Neuron 95: 473–475 [DOI] [PubMed] [Google Scholar]
- Pokatayev V, Yang K, Tu X, Dobbs N, Wu J, Kalb RG, Yan N (2020) Homeostatic regulation of STING protein at the resting state by stabilizer TOLLIP. Nat Immunol 21: 158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Akira S (2009) Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106: 20842–20846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro A, Cannella S, Bossi G, Gallo F, Trizzino A, Pende D, Dieli F, Bruno G, Stinchcombe JC, Micalizzi C et al (2006) Novel Munc13‐4 mutations in children and young adult patients with haemophagocytic lymphohistiocytosis. J Med Genet 43: 953–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E (2002) Hemophagocytic lymphohistiocytosis is associated with deficiencies of cellular cytolysis but normal expression of transcripts relevant to killer‐cell‐induced apoptosis. Blood 100: 2891–2898 [DOI] [PubMed] [Google Scholar]
- Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X (2019) Cryo‐EM structures of STING reveal its mechanism of activation by cyclic GMP‐AMP. Nature 567: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum AK, Deng Z, Chong Z, Law CS, Taguchi T (2020) A defect in COPI‐mediated transport of STING causes immune dysregulation in COPA syndrome. J Exp Med 217: e20201045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing RS, Vidhyasagar V, Yang S, Arna AB, Yadav M, Aggarwal A, Aguilera AN, Shinriki S, Bhanumathy KK, Pandey K et al (2022) DDX41 is required for cGAS‐STING activation against DNA virus infection. Cell Rep 39: 110856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Woo JS, Wu B, El‐Sherbiny YM, Leung J, Chupradit K, Rice L, Seo GJ, Calmettes G, Ramakrishna C et al (2019) The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol 20: 152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z (2009) ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 106: 8653–8658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MS, Zhang J, Jiang LQ, Pan YX, Tan JY, Yu F, Guo L, Yin L, Shen C, Shu HB et al (2018) TMED2 potentiates cellular IFN responses to DNA viruses by reinforcing MITA dimerization and facilitating its trafficking. Cell Rep 25: 3086–3098 [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M (2009) Regulation and function of NF‐kappaB transcription factors in the immune system. Annu Rev Immunol 27: 693–733 [DOI] [PubMed] [Google Scholar]
- Wang LY, Hu J, Ramsingh G, Theodory B, Yaghmour G (2018) A case of recurrent pregnancy‐induced adult onset familial hemophagocytic lymphohistiocytosis. World J Oncol 9: 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen Z (2013) Cyclic GMP‐AMP containing mixed phosphodiester linkages is an endogenous high‐affinity ligand for STING. Mol Cell 51: 226–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, Fu TM, Li J, Wu H (2015) Structural biology of innate immunity. Annu Rev Immunol 33: 393–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Zhang L, Shen J, Zhai Y, Jiang Z (2021) The STING phase‐separator suppresses innate immune signalling. Nat Cell Biol 23: 330–340 [DOI] [PubMed] [Google Scholar]
- Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ (2019) Structural basis of STING binding with and phosphorylation by TBK1. Nature 567: 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, Liu M, Lei Y, Gao X, Fu X et al (2019) A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569: 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]