

Abstract
The intestinal epithelium exhibits a rapid and efficient regenerative response to injury. Emerging evidence supports a model where plasticity of differentiated cells, particularly those in the secretory lineages, contributes to epithelial regeneration upon ablation of injury‐sensitive stem cells. However, such facultative stem cell activity is rare within secretory populations. Here, we ask whether specific functional properties predict facultative stem cell activity. We utilize in vivo labeling combined with ex vivo organoid formation assays to evaluate how cell age and autophagic state contribute to facultative stem cell activity within secretory lineages. Strikingly, we find that cell age (time elapsed since cell cycle exit) does not correlate with secretory cell plasticity. Instead, high autophagic vesicle content predicts plasticity and resistance to DNA damaging injury independently of cell lineage. Our findings indicate that autophagic status prior to injury serves as a lineage‐agnostic marker for the prospective identification of facultative stem cells.
Keywords: autophagy, facultative stem cell, organoid formation, paligenosis, regeneration
Subject Categories: Autophagy & Cell Death, Stem Cells & Regenerative Medicine
Facultative intestinal stem cells promote epithelial regeneration in response to injury, but the contributing cellular properties are unclear. This study shows that autophagic vesicle content positively correlates with plasticity and injury‐resistance in intestinal epithelial cells.
Introduction
The intestinal epithelium is a highly proliferative tissue, undergoing nearly complete turnover within 5 days. Turnover is driven by division of crypt base columnar stem cells (CBCs) marked by the Wnt target gene Lgr5. CBCs cycle rapidly at homeostasis, then commit either to the absorptive lineage to make enterocytes after transit‐amplification or to the secretory lineage (Barker et al, 2007; Clevers, 2013). Although CBCs are responsible for maintaining tissue turnover at homeostasis, they and their transit‐amplifying progeny are highly sensitive to DNA damage‐inducing injury, such as ionizing radiation (Yan et al, 2012; Tao et al, 2015). In contexts where CBCs are depleted, the intestinal epithelium can repopulate the vacant CBC niche through recruitment of facultative intestinal stem cells (f‐ISCs; Tian et al, 2011; Yan et al, 2012). Historically, f‐ISCs have been defined using reporter mice with insertion of CreER cassettes into the loci of genes including Bmi1, Hopx, or Lrig1 (as well as random integrant transgenics driven by the mTert promoter) which label heterogeneous populations of cells, some of which contribute to postinjury regeneration (Breault et al, 2008; Sangiorgi & Capecchi, 2008; Montgomery et al, 2011; Takeda et al, 2011; Powell et al, 2012; Yan et al, 2012). However, there is a varying degree of overlap between reporter‐marked populations, and the endogenous transcripts from these genes are broadly expressed throughout the crypt epithelium and are not specific to f‐ISCs (Li et al, 2014b).
Historically, f‐ISC activity has been studied within specific epithelial lineages. Accumulating evidence supports the secretory lineage as a major contributor to postinjury regeneration. Some studies specifically implicate secretory progenitor populations, such as those marked by Dll1‐ and Atoh1‐CreER reporter alleles, as the source of facultative stem cells (van Es et al, 2012; Tomic et al, 2018). Other studies suggest plasticity of more mature secretory lineage cells using markers of enteroendocrine cells (including Prox1‐CreER and Neurod1‐CreER) or Paneth cells (Lyz1‐CreER) (Yan et al, 2017; Schmitt et al, 2018; Sei et al, 2018; Yu et al, 2018; Jones et al, 2019). In agreement with this premise, studies utilizing Histone H2B pulse‐chase label retention systems demonstrated that quiescent label retaining cells in the intestinal epithelium are highly enriched in the enteroendocrine‐specific genes Chga and Chgb (Buczacki et al, 2013; Li et al, 2016). Buczacki et al (2013) demonstrate further using an H2B‐split‐Cre system that label retaining cells can behave as f‐ISCs in the face of tissue damage. Similarly, other studies show that cells marked by Bmi1‐GFP are largely composed of differentiated Chga+ enteroendocrine cells (Jadhav et al, 2017), and lineage tracing from a Bmi1‐CreER allele provides evidence for f‐ISC activity in what is presumed to be an analogous population to that marked by Bmi1‐GFP (Sangiorgi & Capecchi, 2008). Finally, recent evidence in Lgr5 Dtr‐GFP mice crossed with mice depleted in (i) secretory cells (via Atoh1−/−) or (ii) enterocytes (via Rbpj−/−) suggests that cells from either lineage can contribute to regeneration when Lgr5 cells are ablated with diphtheria toxin (Murata et al, 2020).
It remains unknown whether cells with f‐ISC capacity maintain the same degree of latent stem cell potential as they become increasingly committed to their fate, or whether additional functional characteristics confer facultative stem cell potential. A recent study by Sheahan et al (2021) suggested via lineage‐tracing experiments that epithelial regeneration following doxorubicin‐induced intestinal injury is mediated by immediate progenitors of CBCs, suggesting that cellular maturity may be inversely correlated with plasticity. Although current lineage tracing strategies have been indispensable for developing a deeper understanding of plasticity in the epithelium, there are limitations: (i) Not all labeled cells act as f‐ISCs, making it unclear which cellular properties confer plasticity, and (ii) although f‐ISC function has been observed across several lineages, inefficient Cre recombination together with infrequent tracing events makes it difficult to determine the extent to which each lineage contributes to regeneration. Furthermore, these studies collectively attempt to understand f‐ISC activity by employing lineage‐specific recombinases, with the underlying assumption that such stem cell activity should be a function of lineage. In this study, we aim to uncover cellular states that confer facultative stemness.
We demonstrated previously that calorie restriction, a nutritional regimen characterized by enhanced autophagic flux and decreased activity of the mammalian target of rapamycin complex 1 (mTORC1), is associated with increased frequency of f‐ISCs at homeostasis and a greater number of regenerative foci following 12 Gy gamma irradiation. Furthermore, pretreatment with the mTORC1 inhibitor Rapamycin, which can induce the autophagy pathway, decreased f‐ISC apoptosis following irradiation (Yousefi et al, 2018). Recent work in gastric chief and pancreatic acinar cells shows that these cells, through a process termed paligenosis, utilize autophagy to promote dedifferentiation and metaplastic gene expression following a high‐dose tamoxifen injury model (Willet et al, 2018). Collectively, these studies suggest that metabolic state at the time of injury may be a critical component for cell survival and plasticity to ensure tissue regeneration. In this study, we evaluate how cell age (as a proxy for lineage commitment) and autophagic status predict f‐ISC potential within the intestinal epithelium. We find that plasticity in long‐lived secretory cells is not altered as a function of cell age. Rather, we report that high autophagic vesicle content enables prospective identification of cells with f‐ISC potential regardless of lineage, correlating with plasticity and DNA damage resistance. Taken together, our data support a model in which autophagic status serves as a lineage‐agonistic marker enabling the prospective identification of epithelial cells with f‐ISC potential.
Results
Facultative intestinal stem cells are enriched for a secretory cell gene signature
Among the many Cre driver alleles used to identify f‐ISCs, Hopx‐CreER is one of the most robust and frequently used (Takeda et al, 2011; Li et al, 2014b; Wang et al, 2019) and largely overlaps with another frequently utilized marker, Bmi1‐CreER (Sangiorgi & Capecchi, 2008; Yan et al, 2012; Li et al, 2014b). Moreover, only Hopx‐CreER has been used to ablate genes important for f‐ISC activity with resulting decreases in regenerative capacity after injury (Yousefi et al, 2016). However, while the Hopx‐CreER and Bmi1‐CreER reporters efficiently label f‐ISCs, endogenous Hopx and Bmi1 transcripts can be found in all cells of the intestinal crypt, including those not marked by the targeted CreER reporters (Muñoz et al, 2012; Li et al, 2014b). This lack of concordance between CreER activity and mRNA distribution obfuscates f‐ISC identity. We therefore sought to more clearly define the f‐ISC population marked by Hopx‐CreER through transcriptome and proteome profiling of Hopx CreER ::R26 LSL‐tdTomato ‐marked cells.
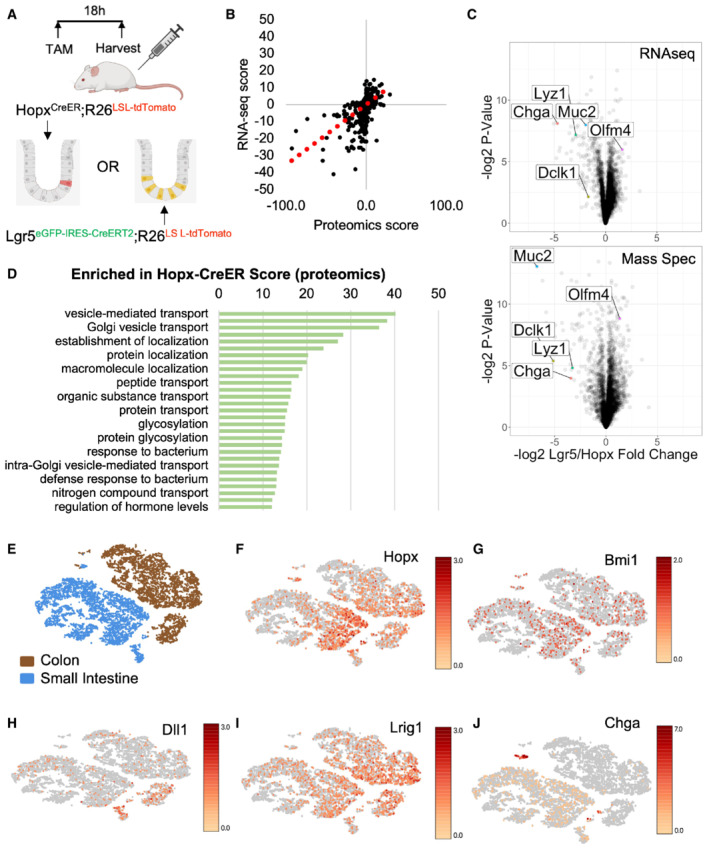
We evaluated tdTomato+ cells from Hopx CreER ::R26 LSL‐tdTomato mice along with CBCs marked by eGFP/tdTomato from Lgr5 eGFP‐IRES‐CreERT2 ::R26 LSL‐tdTomato mice (Fig 1A). Bulk RNA‐seq and mass spectrometry analysis revealed a high degree of concordance between enriched genes and proteins within the Hopx CreER ‐marked population, with almost no anticorrelated genes and proteins (R 2 = 0.286) (Fig 1B). Many of the top transcripts and proteins enriched in Hopx CreER ‐marked cells relative to CBCs were specific to secretory lineage cells including enteroendocrine, Paneth, tuft, and goblet cells (marked by Chga, Lyz1, Dclk1, and Muc2, respectively) (Fig 1C). This finding is consistent with the literature describing plasticity within the secretory lineages using Dll‐, Atoh1‐, Bmi1‐, Lyz1‐, Prox1‐, and Neurod1‐CreER reporter alleles and suggests that Hopx‐CreER marks f‐ISCs across multiple secretory branches. Pathway analysis of proteins enriched within the Hopx CreER ‐marked population revealed signatures related to vesicle‐mediated transport and protein and macromolecule localization, further consistent with secretory cell identity (Fig 1D). One of the most highly differentially expressed genes, also enriched in mass spectrometry data, was Chromogranin A (Chga), a marker of mature EECs (Rindi et al, 2004). Chga mRNA is expressed specifically within the EEC lineage and marks a more homogenous population of cells relative to known f‐ISC marker genes including Hopx, Bmi1, Dll1, and Lrig1 whose expression is not restricted to the secretory lineages (Fig 1E–J). Marker genes used to identify cell populations are displayed in Fig EV1. These findings underscore limitations of current putative f‐ISC reporter alleles, which lack concordance between cells with Cre activity and cells with endogenous expression of the Cre driver‐genes, and ultimately hinder the assignment of f‐ISC potential to specific cell types.
Figure 1. Facultative intestinal stem cells are enriched for secretory lineage markers, including Chromogranin A.
-
ASchema for tamoxifen injections to Hopx CreER ::Rosa26 LSL‐tdTomato and Lgr5 eGFP‐IRES‐CreERT2 ::R26 LSL‐tdTomato mice. Small intestines were processed for FACS 18 h after one tamoxifen dose and RNA and protein were extracted from collected cells for sequencing and mass spectrometry, respectively. N = 3 mice/group for transcriptomics and N = 4 mice/group for proteomics.
-
BConcordance between transcript and protein abundance as measured by the correlation between enrichment scores per gene [−log2 (P‐value) × fold change] for RNA and protein.
-
CVolcano plots of enriched transcripts/proteins by RNA‐seq and Mass Spectrometry in Hopx‐CreER labeled cells versus Lgr5‐CreER labeled cells.
-
DGene ontology analysis of proteins enriched in Hopx‐CreER labeled f‐ISCs. Score = −log2 (P‐value).
-
E–JtSNE plots of single‐cell RNA‐sequencing data from wild type small intestine and colon (E). tSNE plots colored by expression levels of common f‐ISC Cre‐drivers and Chga (F–J).
Figure EV1. Expression of marker genes in bulk small and large intestinal epithelium.
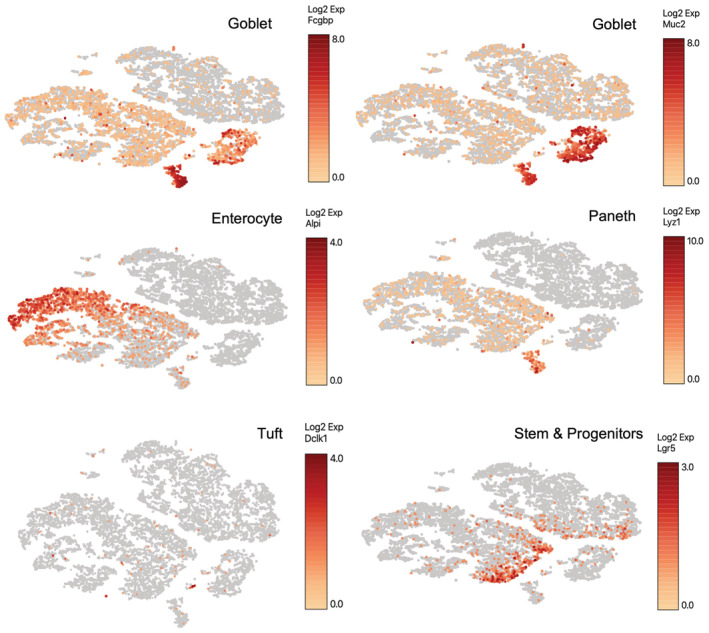
Marker genes used to identify cell clusters in the single‐cell RNAseq dataset included Fcgbp and Muc2 (Goblet cells), Alpi (enterocytes), Lyz1 (Paneth cells), Dclk1 (Tuft cells), and Lgr5 (Crypt base columnar stem cells).
A novel Chga CreER‐tdTomato allele marks EECs with f‐ISC activity
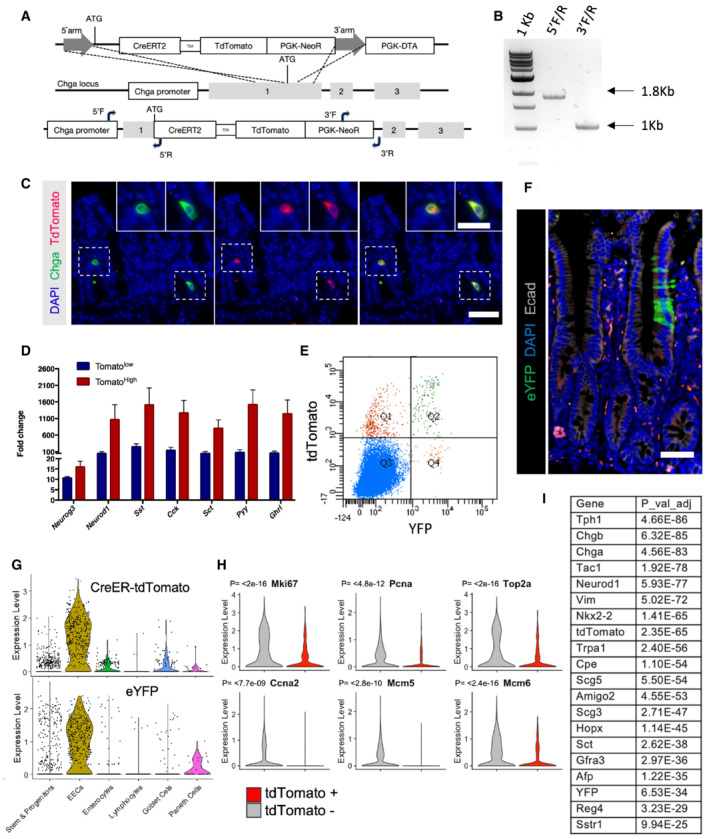
Cells of the secretory lineage and their common upstream progenitor have been implicated as f‐ISCs; however, it remains unclear to what degree their plasticity is related to their maturation. To address this question, we generated a Chga‐CreER‐2A‐tdTomato allele to mark mature enteroendocrine cells (Rindi et al, 2004) by inserting a CreER‐2A‐tdTomato reporter cassette into the endogenous Chga locus (Fig EV2A and B). We confirmed overlap of Chga protein and tdTomato reporter expression by immunofluorescence staining (Fig EV2C) and enrichment of EEC‐specific gene expression in FACs‐purified tdTomato+ cells (Fig EV2D).
Figure EV2. Generation and validation of a novel Chga‐CreER‐2A‐tdTomato allele.
-
ATargeting strategy to insert a CreERT2‐2A‐tdTomato sequence into the translational start site of the endogenous Chga locus.
-
BPCR to confirm proper targeting of the CreERT2‐2A‐tdTomato sequence, as demonstrated in (A). Expected 5′ fragment = 1.8 kb, expected 3′ fragment = 1 kb.
-
CImmunofluorescence staining of small intestine demonstrating overlap of Chga protein and tdTomato reporter in the same cells. Scale bar = 50 μm. Inset scale bar = 25 μm.
-
DCrypt enriched epithelial cell suspensions were generated from ChgaCreER‐2A‐tdTomato mice, sorted tdTomato+ cells, and qPCR was performed for transcription factors (Neurog3, Neurod1) and hormones (Sst, Cck, Sct, Pyy, Ghrl). Y‐axis is fold change relative to tdTomato negative cells. N = 3 technical replicates/group. Data are expressed as mean ± SEM.
-
EFlow cytometry analysis of ChgaCreER‐2A‐tdTomato::R26RLSL‐eYFP intestinal epithelial cells 48 h after five consecutive daily tamoxifen doses. Single‐cell RNA sequencing was performed on cells sorted from Q2 + Q4 and a bulk population (all quadrants).
-
FImmunofluorescence staining of small intestine from a ChgaCreER‐2A‐tdTomato::R26RLSL‐eYFP mouse 48 h after five tamoxifen doses. Scale bar = 50 μm.
-
GViolin plots demonstrating CreER‐tdTomato and eYFP expression by cell type.
-
HViolin plots of cell cycle related genes in tdTomato+ and tdTomato− populations. N = 1 mouse/group, tomato− = 1,907 cells/group, tomato+ = 429 cells/group. P‐value generated by two‐tailed Student's t‐test.
-
ITable of EEC related genes enriched in the tdTomato+ versus tdTomato− cell population.
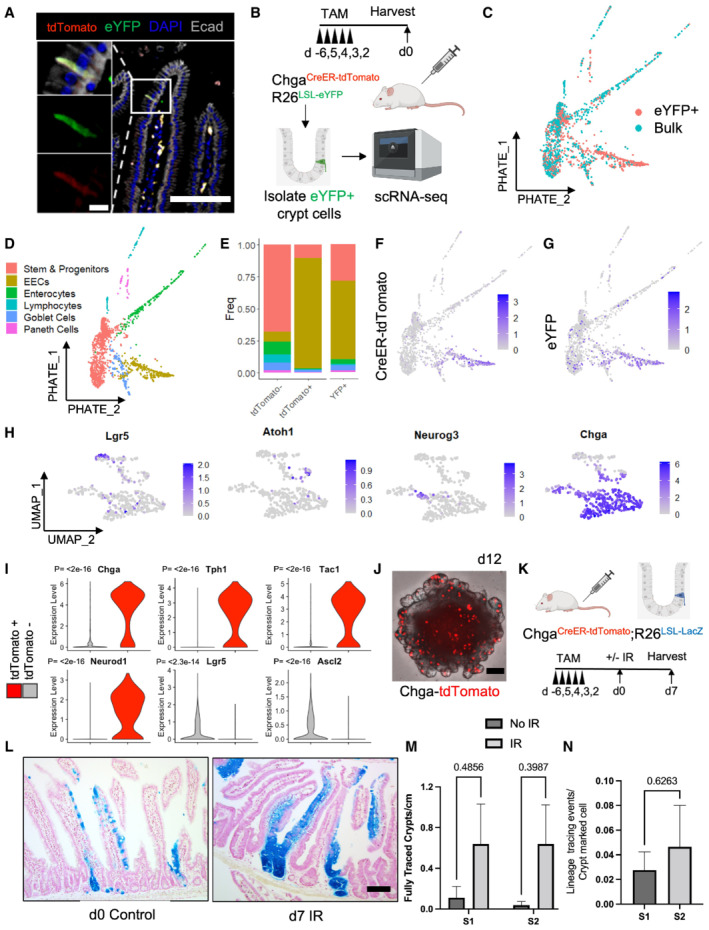
Given that prior studies demonstrated a dramatic disconnect between populations marked by putative f‐ISC Cre drivers, fluorescent reporters, and endogenous transcripts (Muñoz et al, 2012; Li et al, 2014b), we investigated which cells express the CreER‐2A‐tdTomato reporter transcript and which are marked by CreER activity (readout by activation of a R26‐lox‐stop‐lox‐eYFP reporter) by profiling intestinal epithelial cells from Chga CreER‐2A‐tdTomato ::R26 LSL‐eYFP mice. We confirmed overlap of the tdTomato and eYFP reporters histologically (Fig 2A) and performed single‐cell RNA sequencing (scRNAseq) on eYFP+ cells sorted from a crypt enriched epithelial cell suspension (Fig EV2E), as well as bulk epithelial cells for reference (Fig 2B and C). Crypt cells were profiled because they likely harbor the bulk of the regenerative potential due to their proximity to crypt base niche factors. We generated a PHATE map (Moon et al, 2019) to visualize lineage relationships between profiled cell types (Fig 2D). We probed for expression of both the CreER‐2A‐tdTomato and the eYFP transcripts in the scRNAseq data, allowing us to determine which cells underwent recombination (and their progeny). As expected, we detect the CreER‐2A‐tdTomato transcript primarily within the EEC lineage (Figs 2E–G and EV2G). Although eYFP transcripts were similarly most prominent within the EEC lineage, we also observed expression in a small number of stem, progenitor, and differentiated lineage cells (enterocytes, Paneth, and goblet cells), indicating either EEC dedifferentiation to a progenitor state at homeostasis (which can give rise to YFP‐traced progeny) (Fig EV2F), infrequent recombination occurring in non‐EEC cell types, or some combination of these possibilities (Figs 2E–G and EV2G). Looking within the tdTomato+ population, we observe that the reporter captures a spectrum from common secretory (Atoh1+) and EEC progenitors (Neurog3+) to mature EECs (Chga+) (Fig 2H). We also confirm the specificity of our Chga‐CreER‐tdTomato allele by demonstrating enrichment of EEC marker genes and the lack of robust CBC or TA gene expression (Figs 2I and EV2H and I).
Figure 2. A novel Chga CreER‐tdTomato allele marks EECs with f‐ISC activity.
-
AImmunofluorescence staining of small intestine demonstrating overlap of tdTomato and activated eYFP reporter. Scale bar = 100 μm. Inset scale bar = 10 μm.
-
BSchema for scRNAseq experiment. Bulk and eYFP+ cells were collected 48 h after five daily doses of 1 mg of tamoxifen.
-
CPHATE map projection of scRNAseq data color coded by sorted population (eYFP+ vs. bulk cells).
-
DPHATE map projection of eYFP and bulk cells demonstrating lineage relationships between intestinal cell types.
-
EHistogram of cell types that express tdTomato and eYFP.
-
FCreER‐tdTomato expression displayed on PHATE map.
-
GeYFP expression displayed on PHATE map.
-
HUMAP dimension reduction was performed on cells that express the tdTomato transcript, revealing a cluster of EECs (bottom Chga+ cluster) including some Neurog3+ EEC progenitors and a more immature secretory progenitor cluster (top, expressing Lgr5 and Atoh1).
-
IViolin plots of EEC (Chga, Tph1, Tac1, Neurod1) and CBC (Lgr5, Ascl2) marker genes in tdTomato+ and tdTomato− populations. N = 1 mouse/group, tomato− = 1,907 cells/group, tomato+ = 429 cells/group. P‐value generated by two‐tailed Student's t‐test.
-
JImage of an organoid formed from a single ChgaCreER‐tdTomato+ cell, 12‐day postplating. Scale bar = 100 μm.
-
KExperimental scheme for lineage tracing of Chga CreER ‐marked cells at homeostasis and postinjury. Five consecutive doses of 1 mg of tamoxifen were administered to Chga CreER‐2A‐tdTomato ::R26 LSL‐LacZ mice, followed by 12 Gy of gamma irradiation and tissue harvest 7 days later. N = 3 control mice, N = 4 IR mice.
-
LExample images of β‐Gal+ traced cells postirradiation and at homeostasis. Scale bar = 100 μm.
-
MThe number of fully traced β‐Gal+ crypt‐villus axes/cm in irradiated and nonirradiated mice. Lineage tracing events were quantified from small intestines divided into four equal segments (S1–4, with S1 being the most proximal), stained with X‐Gal, embedded in paraffin, cut into 5 μm thick sections, and counterstained with neutral red. Data are expressed as mean ± SEM. N = 3 mice/group, P‐value generated by multiple Student's t‐tests.
-
NThe number of β‐Gal+ fully traced crypt‐villus axes (i.e., lineage tracing events) normalized to the number of pre‐injury crypt marked β‐Gal+ cells (quantified from d0 control mice). S1 + S2 = the two most proximal intestinal segments. Data are expressed as mean ± SEM. N = 4 mice/group, P‐value generated by two‐tailed student's t‐test.
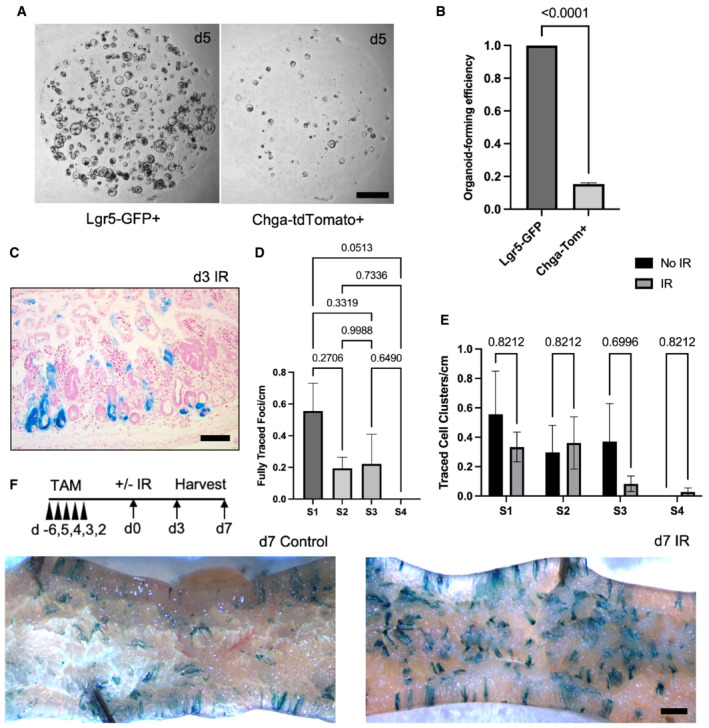
We next examined plasticity of EECs upon their exposure to crypt base stem cell niche factors in vitro. We sorted single Chga CreER‐2A‐tdTomato+ cells or Lgr5 eGFP‐High cells, embedded them in Matrigel and exposed them to media containing R‐spondin, Noggin, and EGF. We found that EECs form organoids at one‐fifth the rate of Lgr5 eGFP‐High CBCs (Fig EV3A and B). The spherical organoids on Day 5 developed into mature budding organoids by Day 12 and retained tdTomato+ fluorescence within the EEC population (Fig 2J). Together, these data indicate that EECs are plastic and can reprogram upon exposure to niche factors in vitro, although it remains unknown which tdTomato+ subset contributed most to the organoid formation.
Figure EV3. EECs contribute to tissue mass pre‐ and post‐DNA damaging injury.
-
ARepresentative images of enteroids grown from single sorted Lgr5‐GFPHigh or Chga‐tdTomato+ cells. Scale bar = 1 mm.
-
BOrganoid formation efficiency of cells plated in (E). N = 3 mice/genotype, N = 3 wells quantified per mouse. Data are expressed as mean ± SEM. P‐value generated by two‐tailed Student's t‐test.
-
CExample image of d3 small intestine stained with X‐Gal, embedded in paraffin, sectioned, and counterstained with neutral red. Scale bar = 100 μm.
-
DThe number of fully traced b‐Gal + clonal regenerative foci per cm of small intestine, measured on d3 postirradiation. Small intestines were divided into four equal segments, S1–4, with S1 being the most proximal and S4 the most distal. N = 3 mice. Data are expressed as mean ± SEM. P‐value generated by Ordinary one‐way ANOVA.
-
EThe number of b‐Gal + cell clusters per cm. These cell clusters were considered distinct events from clonally traced crypts. N = 3 mice/group. Data are expressed as mean ± SEM. P‐value generated by multiple student's t‐tests.
-
FWhole mount imaging of proximal jejunum from control and irradiated Chga CreER‐2A‐tdTomato ::R26 LSL‐LacZ mice. Scale bar = 1 mm.
Finally, we probed the degree of EEC lineage plasticity by performing lineage tracing experiments using Chga CreER‐2A‐tdTomato ::R26 LSL‐LacZ mice. Two days after tamoxifen injection, we exposed mice to 12 Gy of gamma irradiation and scored lineage tracing events 3‐ and 7‐day postinjury. Seven‐day postirradiation, we found frequent ribbons of continuous lineage traced cells emanating from the crypt base and extending into the villi (Fig 2K–M). At Day 3, we found numerous β‐Gal+ clonal regenerative foci, with more traced foci in the proximal segment of the jejunum (S1) as compared with more distal small intestinal segments (S2, S3 and S4) (Fig EV3C and D). In the absence of injury, we found rare clusters of β‐Gal+ lineage traced enterocytes, as well as labeled cells near the crypt base (Figs 2L and EV3E and F). We postulate that these tracing events emanate from rare progenitor cells marked by Chga CreER‐2A‐tdTomato (consistent with single‐cell transcriptomic data, Fig 2I), from marked EECs that dedifferentiate in the absence of injury to regain stem/progenitor activity, or a combination of these. We ultimately quantified the number of lineage tracing events relative to the number of β‐Gal+ crypt cells and found that only a fraction of marked EECs (approximately 2%) form lineage traced ribbons postinjury and thus act as f‐ISCs (Fig 2N). These data indicate that although EECs can contribute to intestinal epithelial regeneration after DNA damaging injury, there is considerable heterogeneity in this population with respect to their facultative stem cell potential.
Secretory cell plasticity is independent of cell age
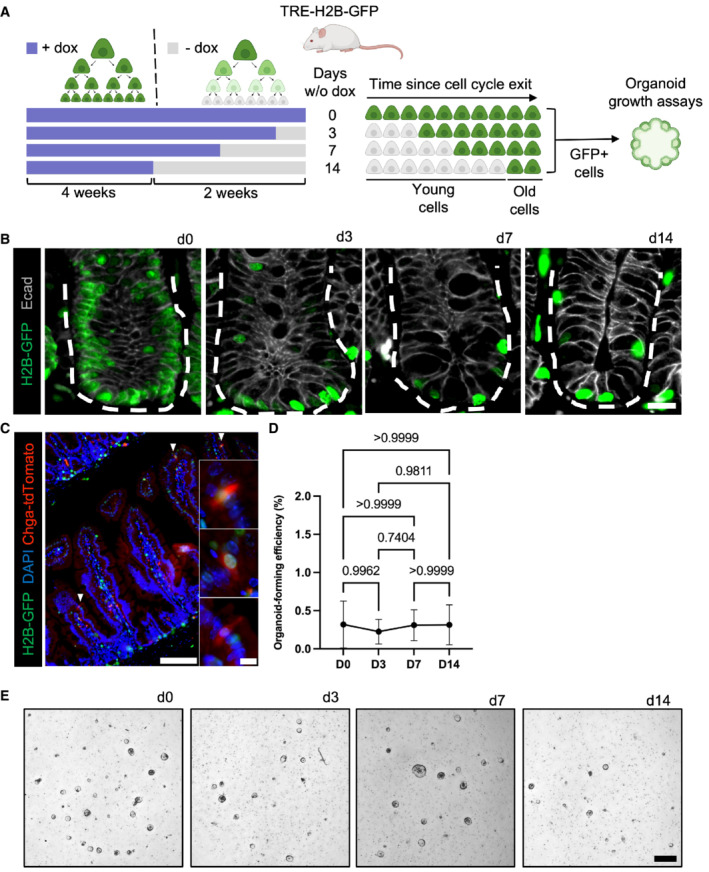
Given that only a small fraction of marked EECs exhibit f‐ISC potential, we sought to define properties that confer stemness to this plastic subset. A recent study using Lgr5 eGFP‐IRES‐CreERT2 ::R26 tdTomato mice demonstrated that recent progeny of Lgr5+ cells contribute the majority of cells to regeneration, suggesting that younger cells could be more plastic than older cells (Murata et al, 2020). Similarly, early CBC progeny upregulate DNA damage response genes in response to injury, which may confer survival and facilitate regeneration (Sheahan et al, 2021). We therefore hypothesized that younger secretory cells arising from Lgr5 eGFP+ CBCs may more readily re‐acquire stem cell activity relative to older secretory cells which may reach a point of terminal differentiation. To investigate this hypothesis, we utilized H2B‐GFP label retention to fractionate epithelial cells by elapsed time since cell cycle exit (Foudi et al, 2009). TRE‐H2B‐GFP mice harbor a doxycycline‐inducible green fluorescent histone 2B protein such that continuous doxycycline administration to mice loads chromatin of all dividing cells with GFP‐tagged H2B. Upon doxycycline withdrawal, the GFP is diluted in half with each round of cell division, and any cells that exit the cell cycle prior to dox withdrawal remain GFP+ (Figs 3A and EV4A and B). We and others previously demonstrated that short‐term (10 day) label retaining cells (LRCs) are a population of secretory cells, primarily EECs and Paneth cells that retain organoid forming capabilities (Buczacki et al, 2013; Li et al, 2016).
Figure 3. Epithelial plasticity does not correlate with cell age.
-
ADiagram depicting the use of TRE‐H2B‐GFP mice for label retention studies. TRE‐H2B‐GFP mice were given doxycycline continually for at least 4 weeks prior to variable withdrawal periods to generate label retaining cell populations of different ages. The GFP+ LRCs were then FACS‐purified, embedded in Matrigel, and exposed to niche growth factors (EGF, R‐Spondin, Noggin) for in vitro organoid formation assays.
-
BImmunofluorescence images of small intestines of TRE‐H2B‐GFP mice after the dox withdrawal periods outlined in panel (A). Crypts are outlined with dashed lines. Scale bar = 15 μm.
-
CImmunofluorescent image of a TRE‐H2B‐GFP Chga CreER‐2A‐tdTomato mouse after a 10 day dox withdrawal period, demonstrating that EECs and LRCs represent an overlapping population, with half of EECs retaining an H2B‐GFP label. Scale bar = 100 μm. Inset scale bar = 10 μm.
-
DOrganoid formation efficiency of different aged LRC populations. N = 3 mice/group, N = 3 wells quantified per mouse. Data are expressed as mean ± SEM. N = 3 mice/group, P‐value generated by Ordinary one‐way ANOVA.
-
EImages of organoids quantified in (D). Scale bar = 500 μm.
Figure EV4. TRE‐H2B‐GFP label retaining populations decrease over time.
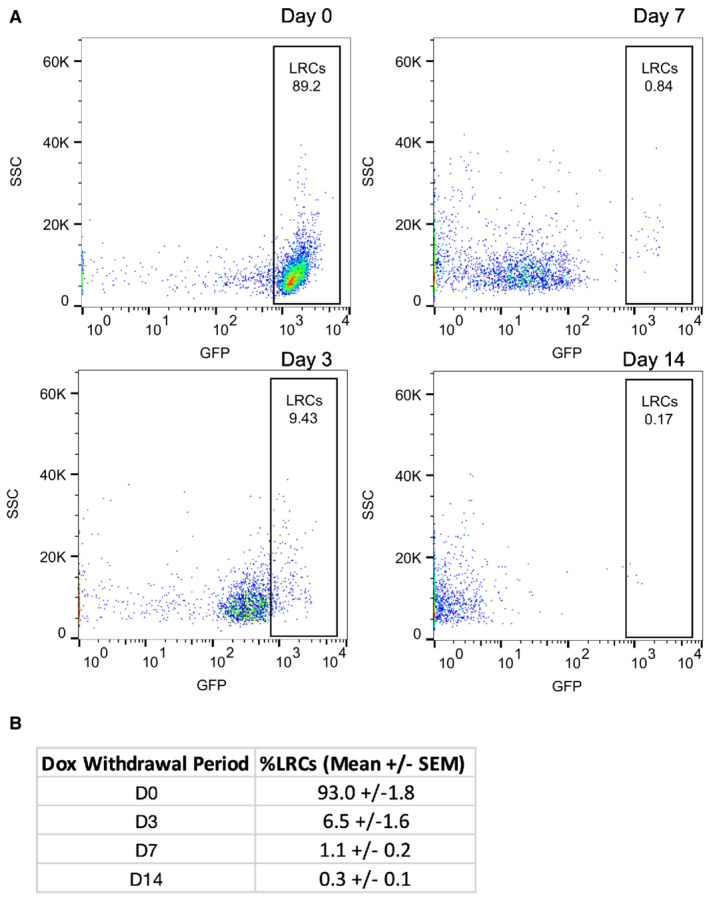
-
ARepresentative plots of the distribution of GFP‐positive cells from TRE‐H2B‐GFP mice after 0, 3, 7, and 14 day dox withdrawal periods.
-
BThe percentage of label retaining cells in each sample quantified from plots in (A). N = 3 mice per time point.
To determine whether younger LRCs exhibit plasticity that is progressively lost in older LRCs, we performed organoid formation assays on differentially aged LRC populations. We withdrew doxycycline for 0, 3, 7, or 14 days prior to isolating single GFP+ LRCs by FACS and embedding equal numbers of cells in Matrigel to assess organoid formation in response to niche growth factor exposure (Fig 3A). Histologically, we found the expected reduction in the number of LRCs for the different withdrawal periods (Fig 3B). Furthermore, we analyzed intestinal tissue from a Chga CreER‐2A‐tdTomato ::TRE‐H2B‐GFP mice and found that after a 10 day dox withdrawal, half of tdTomato+ EECs retained a GFP label (Fig 3C). These data were consistent with prior findings that 10‐day LRCs are approximately 50% EECs (Li et al, 2016). Surprisingly, there was no difference in organoid formation between younger and older GFP+ LRCs (Fig 3D and E). Taken together, these data support the conclusion that secretory cell plasticity is not correlated with cell age, and presumably, lineage commitment. Given that cell age could not explain why a subset of EECs participate in regeneration, but others do not, we next considered additional cell states that could underlie variable plasticity within the secretory lineage.
Autophagic vesicle content correlates with facultative intestinal stem cell capacity in vitro
Facultative stem cells must be able to both survive injury and re‐acquire stem cell function to ensure tissue regeneration. Emerging literature in other contexts suggests that autophagy may support both properties. Autophagy is a cellular self‐degradation mechanism that clears proteins, damaged organelles, and intracellular pathogens to regulate energy levels during stress or injury. Recent work evaluating cell dedifferentiation in both gastric chief and pancreatic acinar cells following metaplasia‐inducing injury showed that autophagy is required for metaplastic gene expression (Willet et al, 2018). Furthermore, autophagy is suppressed by activity of mTORC1, and in the intestine high mTORC1 activity renders f‐ISCs susceptible to DNA damage‐induced apoptosis (Yousefi et al, 2018). Additionally, mice with AhCre‐ or VillinCreER‐mediated deletion of key autophagy regulators, Atg5 or Atg7, respectively, exhibit increased cell death and DNA damage in the crypt base following ionizing irradiation (Asano et al, 2017; Trentesaux et al, 2020). These prior studies highlight a role for autophagy in regulating injury‐resistance and cellular plasticity, making autophagy a strong candidate marker for prospective identification of f‐ISCs.
We asked whether autophagic vesicle content could predict secretory cell plasticity in vitro. To measure autophagic vesicle content, we employed CytoID, an amphiphilic tracer dye that marks autophagic vesicles including early‐forming autophagosomes as well as late‐stage autolysosomes (Guo et al, 2015). We sorted the top (high) and bottom (low) 15% of CytoID‐stained intestinal epithelial cells from freshly isolated small intestinal crypts to evaluate the ability to segregate crypt cells based on autophagic state (Fig 4A and B; Fig EV6). We confirmed that CytoIDhigh cells have higher amounts of autophagic vesicles compared to CytoIDlow cells by sorting equal numbers of cells and performing a Western blot for LC3‐I/LC3‐II protein expression, where an increase in the phosphatidylethanolamine‐modified LC3‐II serves as a marker for increased autophagosomes (Mizushima & Yoshimori, 2007; Dikic & Elazar, 2018; Fig 4C). We further confirmed that CytoIDhigh cells exhibit pronounced autophagic puncta using ImageStream flow cytometry (Fig 4D). Together these data confirm that CytoID faithfully identifies cells with high autophagic vesicle content. We next investigated whether autophagic vesicle content correlates with the ability of single crypt cells to form organoids in vitro. We observed that CytoIDhigh cells form significantly more organoids than CytoIDlow cells, with an efficiency comparable to Lgr5+ CBCs (Fig 4E and F). Next, we inhibited autophagic flux via exposure to bafilomycin, an inhibitor of autophagosome‐lysosome fusion, and observed decreased organoid formation capacity from CytoIDhigh cells in a dose‐dependent manner (Mauvezin & Neufeld, 2015; Fig 4G and H). This finding suggests a functional requirement of autophagic flux for in vitro organoid formation. Taken together, these data support our hypothesis that high autophagic vesicle content can prospectively identify intestinal epithelial cells with f‐ISC capacity.
Figure 4. Intestinal epithelial cells with high autophagic vesicle content exhibit greater organoid‐formation capacity.
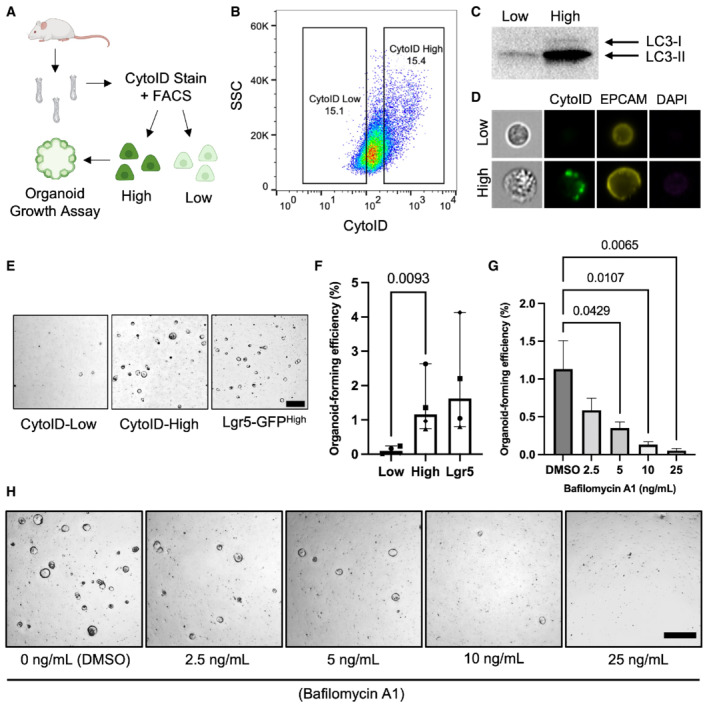
-
ASchema of experimental approach. Small intestinal crypts were isolated, processed into single cells, and stained with CytoID. The top and bottom 15% of CytoID‐stained cells were sorted and plated to evaluate organoid‐formation efficiencies.
-
BRepresentative flow plot demonstrating the CytoID staining pattern within intestinal epithelial cells.
-
CWestern blot demonstrating LC3 levels from equal numbers of sorted CytoIDlow and CytoIDhigh cells. 200,000 cells were loaded per lane.
-
DImages of sorted CytoIDlow and CytoIDhigh cells acquired using an imaging flow cytometer.
-
ERepresentative images of organoids formed from sorted CytoIDlow, CytoIDhigh, and Lgr5‐GFPhigh cells. Scale bar = 500 μm.
-
FQuantification of the organoid‐formation efficiency in (E). N = 4 mice/group, N = 3 wells quantified per mouse. P‐values displayed on graph. Data are expressed as mean ± SEM. P‐value generated by ratio paired two‐tailed t‐test.
-
GEffect of bafilomycin on organoid formation efficiency in CytoIDlow and CytoIDhigh cells. Cells were plated in media containing different concentrations of bafilomycin (0, 2.5, 5, 10, or 25 ng/ml) on day 0, media was changed on day 3 to media that did not contain bafilomycin and organoids were imaged on day 5 after plating. N = 4 mice/group, N = 4 wells quantified per mouse. Data are expressed as mean ± SEM. P‐value generated by Ordinary one‐way ANOVA.
-
HRepresentative images of organoids quantified in (G). Scale bar = 500 μm.
Figure EV6. Gating scheme for isolating CytoID low and high cell populations.
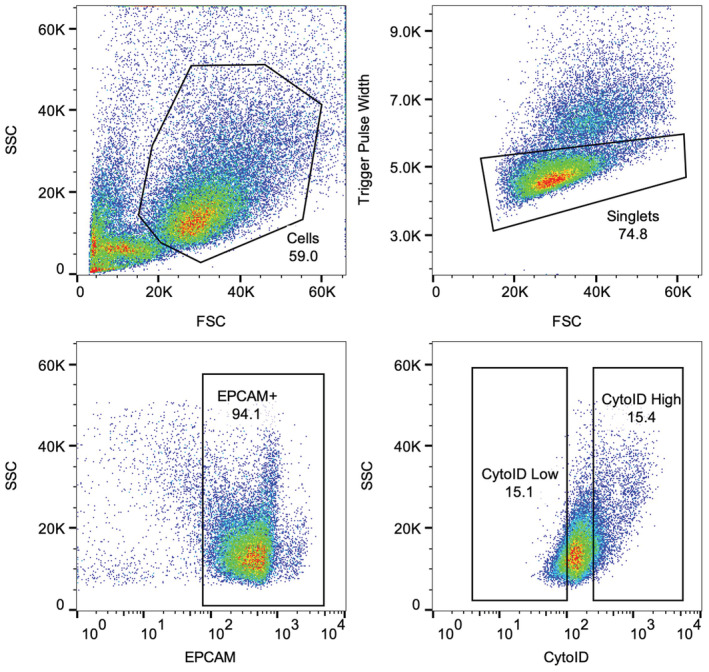
Representative plots of the gating scheme utilized to isolate CytoID low and high experiments for organoid‐formation efficiency and single‐cell sequencing experiments. Note that panel 4B is repeated in this figure to clearly demonstrate the gating strategy utilized for sorting CytoID low and high cells.
Single‐cell RNA sequencing analysis reveals autophagic epithelial cells are biased to the secretory lineages
To gain a detailed understanding of the spectrum of intestinal epithelial cell populations that have high autophagic vesicle content, we FACS‐purified CytoIDlow and CytoIDhigh cells and performed single‐cell RNA sequencing analysis (Fig 5A). We utilized UMAP dimension reduction and graph‐based clustering algorithms to partition the captured cell types and assigned cluster identity based on expression of known marker genes (Fig EV5A–C). We next performed PHATE map dimension reduction (Moon et al, 2019) and found the full spectrum of intestinal epithelial cell types represented within the CytoIDlow and CytoIDhigh populations, with an enrichment in goblet and Paneth cell lineages in the CytoIDhigh pool and no significant difference in EECs between groups. There were significantly more CBC stem and progenitor cells within the CytoIDlow population (Fig 5B–F). The low organoid formation efficiency of this population despite its substantial CBC content suggests that autophagy could also play a role in maintaining stemness of CBCs in a homeostatic context. Consistent with these observations, cell cycle analysis showed that CytoIDlow cells were more likely to be cycling, and CytoIDhigh cells more likely to be in G1 (Fig 5G and H). To confirm these cell population distributions histologically, we employed a CAG‐LC3‐GFP‐RFP reporter mouse expressing a dual‐fluorophore labeled LC3, with early autophagosomes labeled by both RFP and GFP and autolysosomes labeled with acid‐stable RFP only (Li et al, 2014a). We observed that epithelial cells containing RFP+ autophagic puncta were often positive for UEA‐1, a lectin that stains both Paneth and goblet cells. Consistent with the single‐cell transcriptomic data, cycling Ki67+ cells were mutually exclusive from cells containing RFP+ autophagic puncta (Fig 5I). GFP levels from this reporter allele were not detectable postfixation. In sum, these data indicate that the secretory lineages, particularly goblet and Paneth cells, are enriched in noncycling, autophagic cells.
Figure 5. Autophagy activity is enriched within noncycling cells of the secretory lineage.
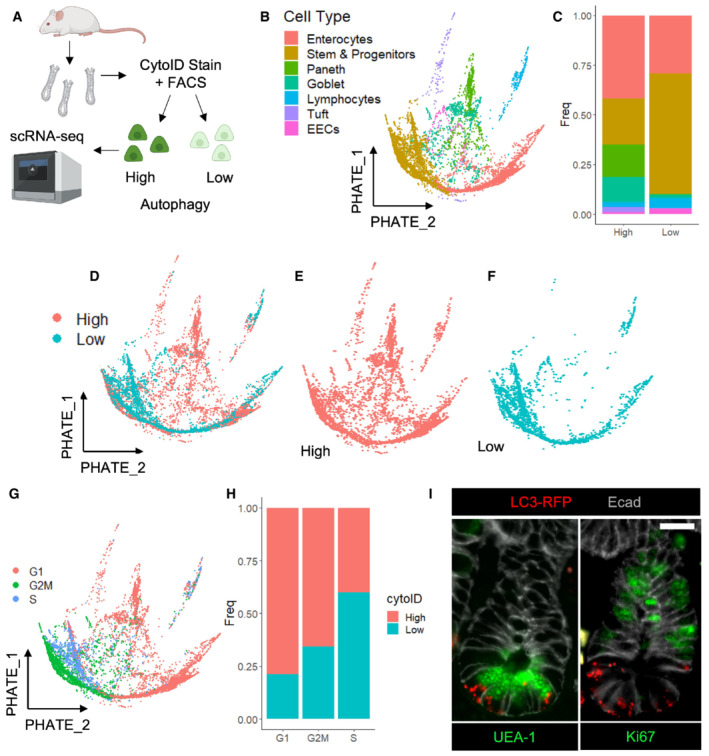
-
ACrypts were isolated from wildtype mice, dissociated into single cells, stained with CytoID and anti‐EpCam antibodies. Epcam+ CytoID high and low (brightest and dimmest 15%, respectively) cells were sorted out and processed for single‐cell RNA sequencing using 10× Chromium technology. Cells from N = 2 mice (one male, one female) were pooled, and mouse origin was demultiplexed using BioLegend Hashtag antibodies.
-
BPHATE map dimension reduction of all sequenced cells, colored by cell type.
-
CHistogram depicting the cell type make‐up of the CytoID high and low populations.
-
D–FPHATE maps with cells colored by autophagy status.
-
GCells were assigned cell cycle status based on expression of phase‐specific genes and projected onto a PHATE map.
-
HThe percentage of CytoID high versus low cells by cell cycle phase.
-
IImmunofluorescence images of crypts from CAG‐LC3‐GFP‐RFP mice stained with anti‐RFP, anti‐E‐cadherin, and either anti‐ki67 or UEA1 antibodies. Scale bar = 15 μm.
Figure EV5. Marker gene expression used to assign cell type identities to cell clusters.
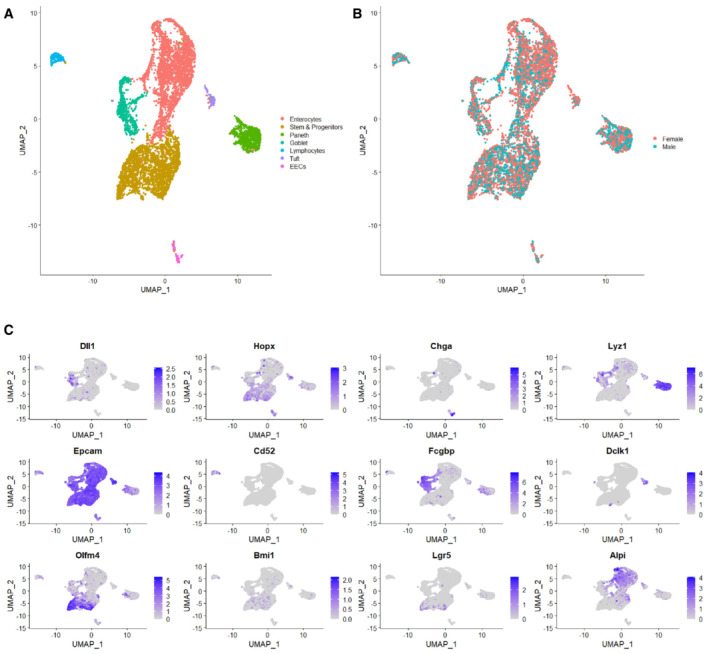
-
AUMAP plot of all sequenced cells with cell clusters colored by cell type identity.
-
BSequenced cells were a mix of N = 2 mice, one male and one female. UMAP of cells colored by sample origin.
-
CMarker genes used to identify cell type identities.
Autophagic vesicle content functions as a lineage agnostic marker of stem cell capacity and protects against DNA damaging injury
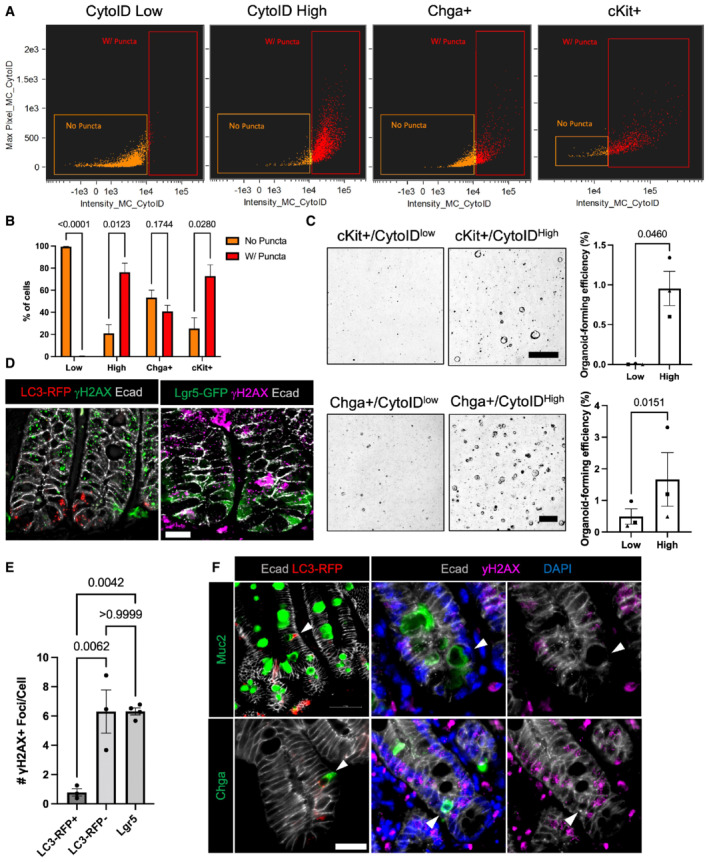
We next sought to confirm the distribution of autophagic cells within specific secretory lineages using imaging flow cytometry. We evaluated autophagic status of EEC and Paneth cell lineages by staining freshly isolated crypts with CytoID then sorting out tdTomato+ cells from Chga CreER‐tdTomato mice (Chga+) or cKit+ cells (a specific marker of Paneth cells within the epithelium; Rothenberg et al, 2012; Li et al, 2016) from wild‐type mice and analyzing the percentage of cells with and without CytoID puncta. Enteroendocrine cells exhibited a roughly equal distribution of cells with and without CytoID puncta, whereas most Paneth cells contained CytoID puncta (Fig 6A and B). These results are consistent with the cell type distributions observed in Fig 5C. We next asked how autophagic status relates to stem cell potential within secretory lineages. To address this, we sorted out the top (high) and bottom (low) 25% of autophagic cells within a c‐Kit+ Paneth cell population and evaluated their organoid‐formation efficiency. The higher CytoID% was chosen to increase cell yield of these rare populations. Here we demonstrate that CytoIDhigh Paneth cells form significantly more organoids than their CytoIDlow counterparts (Fig 6C). Published studies suggest that Paneth cell‐derived growth factors can contribute to stem cell potential in an ex vivo organoid culture setting (Sato et al, 2010). To exclude this possibility, we performed this experiment within Chga+ enteroendocrine cells. We similarly observed that CytoIDhigh EECs form organoids to a greater degree than CytoID low EECs (Fig 6C). These findings demonstrate that autophagic activity directly correlates with epithelial cell plasticity, irrespective of lineage.
Figure 6. Autophagic activity predicts stemness within the secretory lineages and protects against DNA damaging injury.
-
ARepresentative plots demonstrating the distribution of cells with and without CytoID puncta within sorted CytoID low, high, Chga+, and c‐Kit+ cells.
-
BQuantification of the percentage of cells with and without puncta within CytoID low, high, Chga+, and c‐Kit+ cells. N = 3 mice/group (biological replicates). Data are expressed as mean ± SEM. N = 3 mice/group, P‐value generated by multiple student's t‐tests.
-
CRepresentative images and quantification of organoid‐formation efficiency of CytoID low and high cKit+ and Chga+ cells. Scale bars = 500 μm. N = 3 mice/group (biological replicates), N = 4 wells quantified per mouse (technical replicates). P‐values displayed on graph. Data are expressed as mean ± SEM. P‐value generated by paired and ratio paired two‐tailed t‐test, respectively.
-
DImmunofluorescent images of crypts from CAG‐LC3‐GFP‐RFP and Lgr5‐GFP mice 6 h postirradiation. Sections were stained for γH2AX, E‐cadherin, and either LC3‐RFP or Lgr5‐GFP, respectively. Scale bar = 15 μm.
-
EQuantification of the number of γH2AX foci in cells with RFP+ puncta, without autophagic puncta, or in Lgr5+ CBCs. N = 3 mice/group, 40–80 cells quantified/mouse. P‐values displayed on graph. Data are expressed as mean ± SEM. P‐value generated by unpaired two‐tailed t‐test.
-
FImmunofluorescence staining of crypt base goblet cells (Muc2+) and EECs (Chga+) in CAG‐LC3‐GFP‐RFP reporter mice pre‐ (RFP stained) and 6 h postirradiation (γH2AX stained). Arrowheads point to goblet cells (top left) and EECs (bottom left) with high autophagic vesicle content (LC3‐RFP+). On the right panels, arrowheads point to goblet cells (top) and EECs (bottom) with fewer yH2AX foci. Scale bar = 25 μm.
Because f‐ISC activity requires a cell to not only re‐acquire stem cell activity upon exposure to niche factors, but first survive injury to the tissue, we next asked whether autophagic activity is protective against DNA damaging injury. We irradiated CAG‐LC3‐GFP‐RFP and Lgr5‐GFP mice with 12 Gy of ionizing irradiation and collected small intestines 6 h later to evaluate the DNA damage response. Immunofluorescence for γH2AX foci, an early DNA damage response marker, demonstrates that autophagic LC3‐RFP+ cells have significantly fewer γH2AX+ foci than either nonautophagic cells or Lgr5+ CBCs (Fig 6D and E) We next evaluated whether injury resistant crypt base cells include other secretory cells besides Paneth cells. We found that 24.4 ± 3.6% of EECs are located in the crypt base. Furthermore, we demonstrate the presence of EECs and goblet cells localized to the crypt base that are both high in autophagic vesicle content and negative for γH2AX+ foci (Fig 6F). Taken together, these data indicate that autophagic vesicle content is a prospective identifier of f‐ISCs and plays a role in supporting two essential properties of cells with facultative stem cell potential: (i) Allowing the f‐ISC to survive injury and (ii) promoting plasticity by enabling cells to reacquire CBC identity when exposed to niche factors.
Discussion
To act as a facultative stem cell, a cell must survive injury and reprogram to a CBC‐like state. We found that cells with high autophagic vesicle content are protected from DNA damaging injury and have enhanced organoid formation capacity, thus conferring both required properties for f‐ISC activity. These findings describe a functional state that correlates with f‐ISC capacity across intestinal cell lineages.
The primary limitation of using reserve ISC marker genes to identify f‐ISCs, including Hopx and Bmi1, is the discrepancy between cells marked by the mRNA versus CreER activity. In this study, we aimed to bring clarity to the composition of the Hopx‐CreER‐marked f‐ISCs by profiling their transcriptome and proteome and found that f‐ISCs are enriched in secretory‐specific genes such as Chga, Chgb, Lyz1, Dclk1, and Muc2 both at the mRNA and at the protein level. This finding is consistent with the literature describing plasticity within multiple branches of the secretory lineage (Yan et al, 2017; Schmitt et al, 2018; Sei et al, 2018; Yu et al, 2018). This emerging literature continues to ascribe regenerative capacity based on cell lineage identity, yet in these studies, only a small subset of cells in any of the secretory lineages examined function as facultative stem cells, prompting the question of whether additional attributes confer f‐ISC potential to this rare subset.
We initially postulated that the subset of cells which exhibit plasticity and subsequently mediate the regenerative response would primarily be less mature progenitors as opposed to older, more committed secretory cells. This is consistent with the study by (Jadhav et al, 2017) demonstrating that secretory progenitor cells in the epithelium retain similar chromatin accessibility signatures to Lgr5+ CBCs, suggesting that as secretory cells mature and differentiate they may lose the ability to re‐acquire CBC identity after reaching an epigenetic “point of no return.” Remarkably, we observed no difference in plasticity as a function of cell age, using in vitro organoid formation in response to niche cytokine exposure as a readout. One limitation of our approach is that this phenotype could be different in vivo. Furthermore, it is possible that in vivo, older cells may be less likely to contribute to regeneration given their higher position along the crypt‐villus axis away from the source of niche cytokines at the crypt base (Gehart et al, 2019).
A long‐standing question since the initial development of intestinal stem cell Cre alleles in Bmi1 and Lgr5 has been whether postinjury regeneration is driven by a dedicated pool of reserve stem cells, or by plasticity of differentiating cells downstream of the Lgr5+ CBC. Evidence has accumulated demonstrating the plasticity of both absorptive and secretory lineages post‐CBC ablation. Dedifferentiation as a primary mechanism for regeneration is further supported by Murata et al (2020) and Sheahan et al (2021) who demonstrated that immediate CBC progeny restore the bulk of the ISC compartment after CBC ablation. These findings are not mutually exclusive from our data indicating that epithelial cell plasticity does not decline as a function of age, given that the robust contribution from younger cells in their studies could be explained by positioning of recent progeny near the crypt base niche factors as opposed to inherently more robust plasticity within young cells. We add to the body of evidence that EECs specifically are plastic postinjury using our novel Chga‐CreER‐2A‐tdTomato reporter. We demonstrate that this allele captures the full spectrum of EECs from progenitors to mature states, and that only a small fraction of these cells contributes to regeneration. One limitation of this reporter that should be noted is the presence of the neomycin resistance cassette in the allele, which could be associated with minimal transcriptional changes in the marked EEC population.
We evaluated autophagic state as a predictor of f‐ISC potential and found that cells with high autophagic vesicle content exhibit plasticity in vitro, are protected from injury in vivo, and are biased toward noncycling secretory lineages and against cycling stem and absorptive progenitor cells. A recent study demonstrated that organoid‐formation efficiency is inversely correlated with Lgr5‐GFP expression in the small intestine, lending support to our findings that cellular plasticity in vitro is enhanced within differentiated secretory cells compared with CBCs and Lgr5‐expressing progenitors (Azkanaz et al, 2022). The requirement for autophagy in intestinal regeneration has been demonstrated previously using pan‐epithelial genetic ablation (Asano et al, 2017; Trentesaux et al, 2020), although these studies did not dissect the contribution of autophagy prior to injury versus a requirement for injury‐induced autophagy. Ultimately, our data are in line with Willet et al's (2018) description of paligenosis—the process in which mature cells reprogram to a stem cell state by utilizing autophagy to repurpose mature cell machinery and subsequently activate mTOR to enter the cell cycle. In support of intestinal paligenosis, we and others have reported that autophagy is increased in the intestine following irradiation (Chatterji et al, 2019). In addition, our prior studies found that calorie‐restricted mice have increased regenerative ability, where calorie restriction reduces mTORC1 activity, a known repressor of autophagy (Yousefi et al, 2018), thereby increasing the pool of injury‐resistant f‐ISCs capable of epithelial regeneration. Similar observations are made with short‐term fasting (Richmond et al, 2015). One avenue for future investigation will be to prospectively mark cells with high autophagy in vivo and trace their contribution to regeneration postinjury, although genetic tools to execute such an analysis do not yet exist (e.g., the ability to lineage trace specifically from autophagic cells). In addition, it will be interesting to understand why some cells exhibit seemingly stochastic autophagy states in the epithelium. Indeed, the role of autophagy in the epithelium during homeostasis is poorly understood.
In summary, our study demonstrates a previously unappreciated cell state that confers plasticity and survival of multiple epithelial lineages, enabling differentiated cells to act as facultative stem cells. Since autophagy is intimately tied to a cell's environment, including nutrient fluctuations and external stressors, it is likely that these factors may influence how well or poorly an individual's epithelium can regenerate based on the number of cells that exist in a high autophagy state at a given time. It could therefore be useful clinically to induce a transient high autophagy state to both prevent DNA‐damaging injury and enhance intestinal regeneration, for example, in patients receiving chemotherapy and/or radiation therapy.
Materials and Methods
Animals
All mice used for these studies were between 8 and 12 weeks of age, fed ad libitum, and housed under standard ULAR conditions. The following mice were obtained from Jax Laboratories: Hopx‐CreER (#017606); Lgr5‐GFP (#008875); R26‐eYFP (#006148); R26‐tdTomato (#007909); R26‐LacZ (#003474); TRE‐H2B‐GFP (#016836); LC3‐GFP‐RFP (#027139). Chga‐CreER mice were generated in house, with the allele generation described below.
To activate CreERT2 based alleles, mice received 1 mg tamoxifen doses dissolved in corn oil via intraperitoneal injection. To activate Hopx‐CreER and Lgr5‐eGFP‐IRES‐creERT2 alleles, mice received one dose of tamoxifen followed mice cell harvest 18 h later. To activate the lox‐stop‐lox eYFP and LacZ reporters using the Chga‐creERT2 allele, mice received five consecutive daily doses of tamoxifen followed by tissue harvest or injury 48 h later. To induce GFP‐tagged H2B expression, 4‐week‐old mice were given 1 mg/ml doxycycline in their drinking water with 1% sucrose for 4 consecutive weeks prior to doxycycline withdrawal to generate label retaining cell populations. All irradiated mice were injured with 12 Gy of either X‐ray or gamma‐ray irradiation.
Chga mouse generation
Chga‐CreERT2‐2A‐tdTomato mice were derived by inserting a CreERT2‐2A‐tdTomato‐phosphoglycerate kinase I‐NeomycinR cassette into the endogenous Chga locus via homology‐directed repair in mouse V6.5 embryonic stem cells (129/sv × C57Bl/6 F1 hybrid male). The targeting vector was generated by In‐Fusion Cloning (Takara Bio Inc, Shiga, Japan), and arms of homology were amplified from genomic DNA of the V6.5 ES cells to be targeted. All components were verified by Sanger sequencing. Fifty‐six base pairs of the Chga coding sequence downstream of the ATG were deleted, rendering this reporter allele null for Chga protein expression. The linearized targeting vector was introduced by nucleofection (Amaxa; Lonza, Basel, Switzerland) into V6.5 cells, after which the cultures were subjected to G418 selection for 7 days and drug‐resistant colonies were subcloned and screened for proper insertion. Insertion was validated at both the 5′ and 3′ insertion sites using the following primers: 5′Forward: 5′‐GCTCTGAAGGATGCCAGTCA‐3′, 5′Reverse: 5′‐GCTAACCAGCGTTTTCGTTC‐3′, 3′Forward: 5′‐CTGAAGAACGAGATCAGCAGC‐3′, 3′Reverse: 5′‐ACCATAGCTTCTCCTGCCCTA‐3′. Several correctly targeted clones were identified, and one was injected into C57Bl/6 blastocysts, resulting in the generation of several high‐contribution male chimeras. Chimeras were backcrossed to C57Bl/6 females to establish the Chga‐CreERT2‐2A‐tdTomato strain. Chga‐CreERT2‐2A‐tdTomato mice subsequently were routinely genotyped using a 2‐primer polymerase chain reaction enabling detection of the Chga‐CreERT2‐2A‐tdTomato allele. Primer Chga forward: 5′‐TGTTACCACCACCGCTACTG‐3′ Chga reverse: 5′‐TAGCTGGCCCAAATGTTGCT‐3′.
RT–PCR
RNA was isolated from sorted cells using QIAGEN RNeasy mini plus kit. Two hundred nanogram total RNA was used for cDNA synthesis with SuperScript III First‐Strand Synthesis SuperMix (Life Technologies). Real‐time PCR was performed in a 10‐μl reaction volume using Fast SYBR Green Master Mix (Applied Biosystems), and the concentration of primer is 100–300 nM. The following primer sets used in the assay: Neurog3 (forward: 5′‐GCTGCTTGACACTGACCCTA‐3′; reverse: 5′‐ATGAGGCGCCATCCTAGTTC‐3′); Neurod1 (forward: 5′‐AACAACAGGAAGTGGAAACATGAC‐3′; reverse: 5′‐CATCTGTCCAGCTTGGGGGA‐3′); Sst (forward: 5′‐ACCCCAGACTCCGTCAGTT‐3′; reverse: 5′‐AAGTACTTGGCCAGTTCCTGTT‐3′); Cck (forward: 5′‐CCAATTTTTCCTGCCCGCAT‐3′; reverse: 5′‐AGAAGGAGCAGTCAAGCCAAA‐3′); Sct (forward: 5′‐GACCCCAAGACACTCAGACG‐3′; reverse: 5′‐TTTCTGTGTCCTGCGCTTCC‐3′); Pyy (forward: 5′‐AGGTCTCGGCCAGAAGGTTT‐3′; reverse: 5′‐CATGCAAGTGAAGTCGGTGT‐3′); Ghrl (forward: 5′‐CCCCAGGCATTCCAGGTCAT‐3′; reverse: 5′‐CAAACTGCAGATGGTGCCTG‐3′).
Bulk RNA‐seq
Hopx+ or Lgr5+ cells were FACS sorted (BD FACS Aria II SORP) from Hopx‐CreERT2::R26‐LSL‐tdTomato or Lgr5‐eGFP‐IRES‐CreERT2 mice into TRIzol LS (Thermo Fisher Scientific). After phase separation using the standard TRIzol LS protocol, the aqueous phase containing RNA was mixed with 70% ethanol and transferred to RNeasy MinElute spin columns (QIAGEN). The cleanup and further concentration of the RNA was made using the standard procedure of RNeasy MinElute Cleanup Kit.
The cDNA synthesis was carried out using SMART‐Seq v4 Ultra Low Input RNA Kit for Sequencing (Clontech), and the library was prepared using Nextera XT DNA Library Prep kit (Illumina) and sequenced using Illumina NextSeq 550. Sequencing of mRNA libraries generated 20–40 million high‐quality 75‐bp reads/sample. Raw sample data were mapped to the mouse reference transcriptome using kallisto (Bray et al, 2016), and data were analyzed in the statistical computing environment R. RUVseq (Risso et al, 2014) and edgeR (Robinson et al, 2009; McCarthy et al, 2012). RNA‐sequencing data have been submitted to GEO, accession number GSE205889.
LC–MS/MS and analysis
Samples from the 0.5 cm gel were digested with trypsin and analyzed the digests by LC–MS/MS on a Q Exactive HF mass spectrometer using a 4‐h gradient. MS/MS spectra generated from the LC–MS/MS runs were searched using full tryptic specificity against a UniProt mouse database (www.uniprot.org) using the MaxQuant 1.5.2.8 program. False discovery rates for protein and peptide identifications were set at 1%. Common contaminants and low‐confidence identifications (i.e., proteins identified by a single nonredundant (razor + unique) peptide) were removed. The abundance of a protein in a sample can be determined from the MS/MS count and intensity. MS/MS count refers to how many times peptides belonging to the protein were sequenced. Intensity is the sum of the peptide MS intensities for the protein. Quantitation based on intensity is considered more accurate than MS/MS count. In addition, the intensity values are normalized (LFQ Intensity) to take into account the differences in sample load. Fold changes are also calculated based on the LFQ intensity.
Proteomics and transcriptomics data were compared using the same data transformation, normalization, and statistics (Aguilan et al, 2020). Briefly, raw proteins/RNA values were log2 transformed (to obtain a Gaussian distribution) and normalized by the center of their data distribution. Missing values were imputed using random sampling of values corresponding to the lower end of the normal distribution for each sample, that is, imputed values were two standard deviations negative compared to the center of the normal distribution. The P‐value was calculated using a two‐tails heteroscedastic t‐test. Gene Ontology enrichment was performed using the software GOrilla (Eden et al, 2009).
Immunofluorescence and lacZ staining
The first 2 cm of duodenum was removed and the subsequent proximal 5–10 cm of jejunum was cut open lengthwise, swiss‐rolled, and fixed overnight in Zn formalin and then processed for paraffin embedding. Five micrometer sections from paraffin blocks was used for immunofluorescence staining with the following primary antibodies: tdTomato (dsRed Mouse: Takara Biosystems 632392, Rabbit: Takara Biosystems 632496), GFP (Abcam 6673), Chga (Immuno Star 20085), E‐Cadherin (Mouse: BD 610182, Rabbit: CST 3195), Ki67 (Abcam 15580), yH2AX (Millipore 05‐636), Muc2 (GeneTex GTX100664), or Cy2 conjugated UEA‐1 (Vector NC9290132). All secondary antibodies were used at a 1:600 dilution. For yH2AX immunofluorescence, Z‐stack images were captured with a 63× objective on a Leica DM500 Widefield microscope. Z‐stacks were then deconvoluted using Huygens Deconvolution software. The number of gH2AX foci were counted from maximum intensity deconvoluted projections by blinded investigators.
LacZ staining was performed as previously described (Barker & Clevers, 2010). The entire length of the small intestine was divided into four segments, labeled as S1, S2, S3, S4 with S1 being the most proximal and S4 being the most distal. Each segment was flushed with fixative and stained with X‐Gal (Sigma‐Aldrich 10703729001). After staining, intestines were inspected by whole mount imaging and traced villi were counted along the entire length of the intestine. After counting and imaging by whole mount, the same intestinal segments were swiss rolled, embedded in paraffin, sectioned, and stained with neutral red.
Isolation of small intestinal crypts and FACS analysis
Following mouse euthanasia, the gastrointestinal tract of the mice was dissected and the most proximal 15 cm of the small intestine was isolated in PBS. The tissue was briefly washed in fresh PBS and was subsequently splayed open and transferred to a tube containing 10 ml of 1× HBSS with 1 mM NAC. Following collection, the tissue was vortexed for 15 s followed by a 15 s rest on ice; this was performed repeatedly during a 2‐min period. The tissue was then transferred to a tube containing 10 ml of 1× HBSS with 1 mM NAC and 10 mM EDTA and was placed on a rotator in 4°C for 45 min. After the incubation period, the tissue was vortexed for 30 s followed by a 30‐s rest period on ice; this was performed repeatedly during a 3‐min period. After vortexing, the tissue digestion was filtered through a 70 μM filter and the flow‐through was centrifuged at 300 g for 3 min. To generate a single‐cell suspension, the cell pellet was resuspended in a single‐cell suspension buffer containing DNAse (35 μg/ml) and Liberase (20 μg/ml) and was incubated at 37°C for 20 min. Following digestion, the cells were washed in PBS and were resuspended in media containing the desired antibodies and were incubated at 37°C for 30 min. The following antibodies and dilutions were used: EpCam‐PE (1:500, Thermo Fisher Scientific Cat# 12‐5791‐82); EpCam‐APC (1:500, Thermo Fisher Scientific Cat# 17‐5791‐82); cKit‐PE (1:500, Thermo Fisher Scientific Cat# 12‐1171‐82); CD24‐APC (1:500, BioLegend Cat# 101813); UEA‐1 (1:500, Vector Laboratories Cat# FL‐1061). Subsequently, the cells were washed in PBS and resuspended in FACS buffer (PBS with 4% FBS) prior to FACS analysis. The viability dyes DAPI and DRAQ7 were used to exclude dead cells. Cells were analyzed and sorted on the FACSJazz and FACSAria Fusion sorters. Data analysis was performed using FlowJo software.
Cyto‐ID autophagy detection kit staining procedure
Following the generation of a single‐cell suspension from isolated small intestinal crypts, the cells were washed in PBS and were resuspended in media containing the CytoID dye (1:1,000 dilution, ENZ‐KIT175) as well as additional desired antibodies (dilutions described above) and were incubated at 37°C for 30 min protected from light. After staining, the cells were once again washed in PBS and were resuspended in FACS buffer containing an appropriate viability dye for subsequent FACS analysis.
Small intestinal enteroid culture and analysis
Intestinal epithelial cells isolated by FACS were plated in Matrigel droplets at a density of 4,000 cells per 20 μl droplet. Matrigel droplets were overlaid with the following medium: advanced DMEM/F12 media containing 1× Glutamax, 10 mM Hepes buffer, 1× Antibiotic‐Antimycotic, titrated R‐spo1 and Noggin‐containing conditioned media, 1× N‐2 supplement, 1× B‐27 supplement, 5 μM CHIR99021, 1 mM NAC, 50 ng/ml mEGF, 5% Noggin/R‐spondin conditioned medium, and Y‐27632 (10 μM). Media was replaced the day after initial plating (Day 1), and every other day afterward. For bafilomycin experiments, bafilomycin was added to the organoid‐culture media at varying doses during initial plating and was maintained until the second media change on Day 3. All organoid experiments were imaged on Day 5 using a Keyence BZ‐X all‐in‐one fluorescence microscope to collect Z‐stack images of 3 × 3 focal planes that were subsequently stitched together to form one image that encompassed the entire Matrigel. The number of organoids per droplet were counted while making sure to only count each structure once. For H2B‐GFP organoid assays, one mouse from each time point (d0, d3, d7, and d14) was sacrificed for each experiment with three independent experiments performed. Comparing all time‐points on each experimental day allowed us to internally control for the variability in organoid formation efficiency between different sorts.
Western blotting
Cells were lysed with lysis buffer (Cell Lysis reagent (Cell Signaling #9803S), Halt Protease Inhibitor Cocktail (Life technologies #78430), 1 mM Sodium Orthovanadate, 10 μM Sodium Fluoride) on ice and spun at 9391G at 4°C for 10 min. Running buffer (final concentration 62.5 mM Tris–HCl pH 6.8, 2.5% SDS, 0.002% Bromophenol Blue, 5% B‐mercaptoethanol, and 10% glycerol) was added to supernatant and boiled for 6 min and loaded onto an SDS‐PAGE gel. Samples were run and transferred onto PVDF membrane using the Bio‐rad Trans‐blot Turbo system and then blocked in either 5% Milk or BSA in TBS‐T (20.7 mM Tris Base, 150.7 NaCl, 0.1% Tween‐20, pH 7.6) for 1 h at room temperature. Primary antibodies (LC3A/B (D3U4C) XP® Rabbit mAb CS#12741, 1:1,000) were diluted in 5% Milk or BSA in TBS‐T and incubated with gentle rocking at 4°C for 24 h. Blots were washed with TBS‐T and then incubated in appropriate secondary antibodies (Anti‐rabbit IgG, HRP‐linked Antibody CS#7074, 1:1,000) for 1 h at room temperature with rocking. Following washing, blots were incubated with luminol reagent (Santa Cruz Biotechnology sc‐2048) per manufacturer protocol and imaged using the ChemiDoc Imaging System.
Imaging flow cytometry analysis
For all imaging flow cytometry experiments, cells were previously sorted using FACS and were subsequently analyzed on an Amnis ImageStream imaging flow cytometer. Analysis was performed by trained staff at the University of Pennsylvania Flow Cytometry Core.
Single‐cell RNA‐seq
For ChgaCreER eYFP samples, single‐cell suspensions of intestinal epithelial cells were generated as described above. eYFP+ and bulk epithelial cells were incubated with anti‐mouse Hashtag antibodies (BioLegend Cat #155831, 155833, 155835, 155837) before sorting. For CytoID high and low samples, single‐cell suspensions of WT intestinal epithelium were stained with CytoID as detailed above and incubated with anti‐mouse Hashtag antibodies (BioLegend Cat #155839, 155841, 155843, 155845). CytoID high and low cells were then sorted and processed separately for sequencing library generation. Sequencing libraries were prepared from single‐cell suspensions using Chromium 10× V3.0 (eYFP samples) and V3.1 (CytoID samples) technology. Sorted cells were encapsulated using the Chromium Controller (10× Genomics) and the Chromium Single‐Cell 3′ Library & Gel Bead Kit (10× Genomics) following the standard manufacturer's protocols. In brief, cells loaded onto the Chromium controller were limited to 10,000–12,000 to reach a multiple rate no higher than 6%. All libraries were quantified using an Agilent Bioanalyzer and pooled for sequencing on an Illumina NovaSeq at Center for Applied Genomics (CAG) at Children's Hospital of Philadelphia. At least two technical replicates were run in parallel for each sample. Targeted median read depth is 50,000 reads per cell from total gene expression libraries and 10,000 reads per cell for hashtag barcode libraries. Cellranger (Version 3.1.0) was used to align reads to a custom reference transcriptome (GRCh38‐3.0.0 + transcripts for tdTomato and eYFP) (Zheng et al, 2017). Seurat (version 3.0.1) was used for standard QC, hashtag demultiplexing, doublet removal, log normalization, dimension reduction (TSNE, UMAP), and cell cycle analysis using standard workflows (Stuart et al, 2019). PHATE cell trajectory analysis was performed using the PhateR (version 1.0.7) package (Moon et al, 2019). ScRNA‐sequencing data have been submitted to GEO, accession number GSE205889.
Statistical methods
Data were analyzed using unpaired and paired, two‐tailed student's t‐tests, one‐way ANOVA, or two‐way ANOVA with post hoc tests and P‐values indicated in individual figures. Analyses were performed on data from a minimum of three experiments unless otherwise noted, in which case multiple fields are analyzed, and are presented as mean ± standard error of mean (SEM). Specific experimental replicates are described in each figure legend.
Author contributions
Nicolette M Johnson: Conceptualization; formal analysis; investigation; writing – original draft. Louis R Parham: Conceptualization; formal analysis; investigation; writing – original draft. Jeeyoon Na: Formal analysis; investigation. Keara E Monaghan: Formal analysis; investigation; project administration. Hannah M Kolev: Formal analysis; investigation. Alena Klochkova: Formal analysis; investigation. Melissa S Kim: Formal analysis; investigation. Charles H Danan: Formal analysis; investigation. Zvi Cramer: Formal analysis; investigation. Lauren A Simon: Formal analysis; investigation; project administration. Kaitlyn E Naughton: Formal analysis; investigation; project administration. Stephanie Adams‐Tzivelekidis: Formal analysis; investigation; project administration. Yuhua Tian: Formal analysis; investigation. Patrick A Williams: Formal analysis; investigation. N Adrian Leu: Formal analysis; investigation. Simone Sidoli: Formal analysis; investigation. Kelly A Whelan: Conceptualization; writing – review and editing. Ning Li: Conceptualization; writing – review and editing. Christopher J Lengner: Conceptualization; funding acquisition; writing – original draft; writing – review and editing. Kathryn E Hamilton: Conceptualization; funding acquisition; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
PDF+
Acknowledgements
The authors thank David Speicher of the Wistar Institute, Dr. Kai Tan and lab (Children's Hospital of Philadelphia, and the following core facilities: CHOP Flow Core, Penn Flow Core, Center for Molecular Studies in Digestive and Liver Diseases (NIH P30‐DK050306) Molecular Pathology and Imaging Core, Penn Vet Imaging Core (Gordon Ruthel)). This work was supported by the following: NIH F31AI150224 (NMJ), NIH F31‐DK124956 (LRP), NIH R01DK12115 (KAW), NIH R01‐DK106309 (CJL), the State of Pennsylvania Commonwealth Research Enhancement Foundation (CURE) Health Research Formula Fund (HRFF) (CJL), NIH R01‐DK124369 (KEH), and Children's Hospital of Philadelphia Institutional Development Funds and Gastrointestinal Epithelium Modeling Program (KEH). Simone Sidoli gratefully acknowledges the Leukemia Research Foundation (Hollis Brownstein New Investigator Research Grant), American Federation for Aging Research (Sagol Network GerOmics award), Relay Therapeutics, Merck, and the NIH Office of the Director (1S10OD030286‐01).
EMBO reports (2022) 23: e55209
Contributor Information
Christopher J Lengner, Email: lengner@vet.upenn.edu.
Kathryn E Hamilton, Email: hamiltonk1@chop.edu.
Data availability
RNA‐seq and single‐cell seq dat are submitted to GEO, accession number GSE205889: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205889.
References
- Aguilan JT, Kulej K, Sidoli S (2020) Guide for protein fold change and p‐value calculation for non‐experts in proteomics. Mol Omics 16: 573–582 [DOI] [PubMed] [Google Scholar]
- Asano J, Sato T, Ichinose S, Kajita M, Onai N, Shimizu S, Ohteki T (2017) Intrinsic autophagy is required for the maintenance of intestinal stem cells and for irradiation‐induced intestinal regeneration. Cell Rep 20: 1050–1060 [DOI] [PubMed] [Google Scholar]
- Azkanaz M, Corominas‐Murtra B, Ellenbroek SIJ, Bruens L, Webb AT, Laskaris D, Oost KC, Lafirenze SJA, Annusver K, Messal HA et al (2022) Retrograde movements determine effective stem cell numbers in the intestine. Nature 607: 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ et al (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007 [DOI] [PubMed] [Google Scholar]
- Barker N, Clevers H (2010) Lineage tracing in the intestinal epithelium. Curr Protoc Stem Cell Biol 13: 5A.4.1–5A.4.11 [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34(5): 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Breault DT, Min IM, Carlone DL, Farilla LG, Ambruzs DM, Henderson DE, Algra S, Montgomery RK, Wagers AJ, Hole N (2008) Generation of mTert‐GFP mice as a model to identify and study tissue progenitor cells. Proc Natl Acad Sci USA 105: 10420–10425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczacki SJA, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ (2013) Intestinal label‐retaining cells are secretory precursors expressing Lgr5. Nature 495: 65–69 [DOI] [PubMed] [Google Scholar]
- Chatterji P, Williams PA, Whelan KA, Samper FC, Andres SF, Simon LA, Parham LR, Mizuno R, Lundsmith ET, Lee DS et al (2019) Posttranscriptional regulation of colonic epithelial repair by RNA binding protein IMP1/IGF2BP1. EMBO Rep 22: e47074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2013) The intestinal crypt, a prototype stem cell compartment. Cell 154: 274–284 [DOI] [PubMed] [Google Scholar]
- Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364 [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, Hock H (2009) Analysis of histone 2B‐GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol 27: 84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart H, van Es JH, Hamer K, Beumer J, Kretzschmar K, Dekkers JF, Rios A, Clevers H (2019) Identification of enteroendocrine regulators by real‐time single‐cell differentiation mapping. Cell 176: 1158–1173 [DOI] [PubMed] [Google Scholar]
- Guo S, Liang Y, Murphy SF, Huang A, Shen H, Kelly DF, Sobrado P, Sheng Z (2015) A rapid and high content assay that measures cyto‐ID‐stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 11: 560–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav U, Saxena M, O'Neill NK, Saadatpour A, Yuan G‐C, Herbert Z, Murata K, Shivdasani RA (2017) Dynamic reorganization of chromatin accessibility signatures during dedifferentiation of secretory precursors into Lgr5+ intestinal stem cells. Cell Stem Cell 21: 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JC, Brindley CD, Elder NH, Myers MG, Rajala MW, Dekaney CM, McNamee EN, Frey MR, Shroyer NF, Dempsey PJ (2019) Cellular Plasticity of Defa4-Expressing Paneth Cells in Response to Notch Activation and Intestinal Injury. Cell Mol Gastroenterol Hepatol 7: 533–554. 10.1016/j.jcmgh.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang ZV, Hill JA, Lin F (2014a) New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol 25: 305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Yousefi M, Nakauka‐Ddamba A, Jain R, Tobias J, Epstein JA, Jensen ST, Lengner CJ (2014b) Single‐cell analysis of proxy reporter allele‐marked epithelial cells establishes intestinal stem cell hierarchy. Stem Cell Reports 3: 876–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Nakauka‐Ddamba A, Tobias J, Jensen ST, Lengner CJ (2016) Mouse label‐retaining cells are molecularly and functionally distinct from reserve intestinal stem cells. Gastroenterology 151: 298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Neufeld TP (2015) Bafilomycin A1 disrupts autophagic flux by inhibiting both V‐ATPase‐dependent acidification and ca‐P60A/SERCA‐dependent autophagosome‐lysosome fusion. Autophagy 11: 1437–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297. 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T (2007) How to interpret LC3 immunoblotting. Autophagy 3: 542–545 [DOI] [PubMed] [Google Scholar]
- Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk MEG, Henderson DE, Baffour‐Awuah NY, Ambruzs DM, Fogli LK, Algra S et al (2011) Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci USA 108: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, van den Elzen A, Hirn MJ, Coifman RR et al (2019) Visualizing structure and transitions in high‐dimensional biological data. Nat Biotechnol 37: 1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz J, Stange DE, Schepers AG, van de Wetering M, Koo B‐K, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S et al (2012) The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers: transcriptomic and proteomic signature of Lgr5+ stem cells. EMBO J 31: 3079–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata K, Jadhav U, Madha S, van Es J, Dean J, Cavazza A, Wucherpfennig K, Michor F, Clevers H, Shivdasani RA (2020) Ascl2‐dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell 26: 377–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE et al (2012) The pan‐ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149: 146–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond CA, Shah MS, Deary LT, Trotier DC, Thomas H, Ambruzs DM, Jiang L, Whiles BB, Rickner HD, Montgomery RK et al (2015) Dormant intestinal stem vcells are regulated by PTEN and nutritional status. Cell Rep 13: 2403–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindi G, Leiter AB, Kopin AS, Bordi C, Solcia E (2004) The “normal” endocrine cell of the gut: changing concepts and new evidences. Ann NY Acad Sci 1014: 1–12 [DOI] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, Dudoit S (2014) Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32: 896–902. 10.1038/nbt.2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2009) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg ME, Nusse Y, Kalisky T, Lee JJ, Dalerba P, Scheeren F, Lobo N, Kulkarni S, Sim S, Qian D et al (2012) Identification of a cKit+ colonic Crypt Base secretory cell that supports Lgr5+ stem cells in mice. Gastroenterology 142: 1195–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangiorgi E, Capecchi MR (2008) Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet 40: 915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H (2010) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418. 10.1038/nature09637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M, Schewe M, Sacchetti A, Feijtel D, van de Geer WS, Teeuwssen M, Sleddens HF, Joosten R, van Royen ME, van de Werken HJG et al (2018) Paneth cells respond to inflammation and contribute to tissue regeneration by acquiring stem‐like features through SCF/c‐kit signaling. Cell Rep 24: 2312–2328 [DOI] [PubMed] [Google Scholar]
- Sei Y, Feng J, Samsel L, White A, Zhao X, Yun S, Citrin D, McCoy JP, Sundaresan S, Hayes MM et al (2018) Mature enteroendocrine cells contribute to basal and pathological stem cell dynamics in the small intestine. Am J Physiol Gastrointest Liver Physiol 315: G495–G510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan BJ, Freeman AN, Keeley TM, Samuelson LC, Roper J, Hasapis S, Lee C‐L, Dekaney CM (2021) Epithelial regeneration after doxorubicin arises primarily from early progeny of active intestinal stem cells. Cell Mol Gastroenterol Hepatol 12: 119–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R (2019) Comprehensive integration of single‐cell data. Cell 177: 1888–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA (2011) Interconversion between intestinal stem cell populations in distinct niches. Science 334: 1420–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Tang D, Morita Y, Sperka T, Omrani O, Lechel A, Sakk V, Kraus J, Kestler HA, Kühl M et al (2015) Wnt activity and basal niche position sensitize intestinal stem and progenitor cells to dna damage. EMBO J 34: 624–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ (2011) A reserve stem cell population in small intestine renders Lgr5‐positive cells dispensable. Nature 478: 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomic G, Morrissey E, Kozar S, Ben‐Moshe S, Hoyle A, Azzarelli R, Kemp R, Chilamakuri CSR, Itzkovitz S, Philpott A et al (2018) Phospho‐regulation of ATOH1 is required for plasticity of secretory progenitors and tissue regeneration. Cell Stem Cell 23: 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trentesaux C, Fraudeau M, Pitasi CL, Lemarchand J, Jacques S, Duche A, Letourneur F, Naser E, Bailly K, Schmitt A et al (2020) Essential role for autophagy protein ATG7 in the maintenance of intestinal stem cell integrity. Proc Natl Acad Sci USA 117: 11136–11146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es JH, Sato T, van de Wetering M, Lyubimova A, Yee Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J et al (2012) Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 14: 1099–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chiang I‐L, Ohara TE, Fujii S, Cheng J, Muegge BD, Ver Heul A, Han ND, Lu Q, Xiong S et al (2019) Long‐term culture captures injury‐repair cycles of colonic stem cells. Cell 179: 1144–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willet SG, Lewis MA, Miao Z, Liu D, Radyk MD, Cunningham RL, Burclaff J, Sibbel G, Lo HG, Blanc V et al (2018) Regenerative proliferation of differentiated cells by mtorc 1‐dependent paligenosis. EMBO J 37: e98311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR et al (2012) The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA 109: 466–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Gevaert O, Zheng GXY, Anchang B, Probert CS, Larkin KA, Davies PS, Cheng Z, Kaddis JS, Han A et al (2017) Intestinal enteroendocrine lineage cells possess homeostatic and injury‐inducible stem cell activity. Cell Stem Cell 21: 78–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi M, Li N, Nakauka‐Ddamba A, Wang S, Davidow K, Schoenberger J, Yu Z, Jensen ST, Kharas MG, Lengner CJ (2016) Msi RNA‐binding proteins control reserve intestinal stem cell quiescence. J Cell Biol 13: 401–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi M, Nakauka‐Ddamba A, Berry CT, Li N, Schoenberger J, Simeonov KP, Cedeno RJ, Yu Z, Lengner CJ (2018) Calorie restriction governs intestinal epithelial regeneration through cell‐autonomous regulation of mTORC1 in reserve stem cells. Stem Cell Reports 10: 703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Tong K, Zhao Y, Balasubramanian I, Yap GS, Ferraris RP, Bonder EM, Verzi MP, Gao N (2018) Paneth cell multipotency induced by notch activation following injury. Cell Stem Cell 23: 46–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J et al (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049 [DOI] [PMC free article] [PubMed] [Google Scholar]