

Abstract
N6‐methyladenosine (m6A) and alternative polyadenylation (APA) are important regulators of gene expression in eukaryotes. Recently, it was found that m6A is closely related to APA. However, the molecular mechanism of this new APA regulation remains elusive. Here, we show that YTHDC1, a nuclear m6A reader, can suppress proximal APA sites and produce longer 3′ UTR transcripts by binding to their upstream m6A sites. YTHDC1 can directly interact with the 3′ end processing factor FIP1L1 and interfere with its ability to recruit CPSF4. Binding to the m6A sites can promote liquid–liquid phase separation of YTHDC1 and FIP1L1, which may play an important role in their interaction and APA regulation. Collectively, YTHDC1 as an m6A “reader” links m6A modification with pre‐mRNA 3′ end processing, providing a new mechanism for APA regulation.
Keywords: 3′ end processing factor, alternative polyadenylation, FIP1L1, m6A, YTHDC1
Subject Categories: RNA Biology
YTHDC1 inhibits proximal poly(A) sites by interfering with FIP1L1 – CPSF4 interaction, which results in mRNA transcripts with longer 3′ UTRs.
Introduction
N6‐methyladenosine (m6A), one of the most common modifications of eukaryotic mRNAs, is dynamically regulated by m6A “writers” (METTL3/14, WTAP, RBM15/15B, VIRMA and ZC3H13) and “erasers” (FTO, ALKBH5, and ALKBH3; Wang & He, 2014; Roundtree et al, 2017a; Shi et al, 2019). The dynamic regulation of m6A modification was found to be associated with diverse physiological processes, including cancers, cell development and differentiation, heart failure, viral infection and type 2 diabetes (Wang et al, 2020; Zhou et al, 2020; He & He, 2021). Mettl3‐mediated m6A modification and reader‐expedited mRNA decay also have fundamental functions in the innate immune response to infection and anti‐tumor immunity (Gao et al, 2020; Lu et al, 2020; Li et al, 2021).
Increasing evidence has shown that the biological functions of m6A are mediated by m6A “readers” (mainly YTHDF1–3 and YTHDC1‐2), through which m6A can regulate mRNA splicing, transportation, stability and translation. YTHDC1 recruits splicing factors to regulate mRNA alternative splicing and mediates the export of methylated mRNA from the nucleus to the cytoplasm (Xiao et al, 2016; Roundtree et al, 2017b). YTHDC2 is associated with the 5′‐3′ exoribonuclease XRN1, which affects the translation efficiency and mRNA abundance of its targets (Hsu et al, 2017; Wojtas et al, 2017). YTHDF2 selectively recognizes m6A sites on mRNAs and noncoding RNAs to regulate RNA degradation in a genome‐wide fashion (Batista et al, 2014). YTHDF1 and YTHDF3 work in concert to affect the translation of m6A‐containing mRNAs (Wang et al, 2015a; Shi et al, 2017).
Alternative polyadenylation (APA) is a widespread phenomenon in the transcription of eukaryotic genes. APA contributes to the complexity of the transcriptome by generating different mRNA isoforms with alternative 3′ ends and tunes the gene network by regulating mRNA stability, localization and translation (Fu et al, 2018; Yuan et al, 2019). APA is spatiotemporally regulated through the recognition of cis‐regulatory RNA motifs by the 3′ end processing machinery complex, whose precise recruitment is connected with transcription initiation, RNA Pol II elongation, exon splicing and other RNA binding proteins (Di Giammartino et al, 2011; Tian & Manley, 2017). In addition to these regulatory factors, m6A modification was recently found to be related to the choice of APA sites. Sequencing profiling of m6A revealed that m6A sites were enriched in 3′ UTRs and near stop codon regions (Meyer et al, 2012a) or the last exon (Ke et al, 2015). A negative correlation of m6A density and proximal APA usage was observed when comparing brain and liver tissues, and knockout of m6A methyltransferase METTL3 led to more genes switched to the proximal APA sites, suggesting the inhibitory effect of m6A on the usage of proximal APA sites (Ke et al, 2015). Consistently, knockout of the demethylase FTO increased the usage of distal APA sites (Bartosovic et al, 2017). In contrast, by sequencing intact m6A‐positive and m6A‐negative transcripts, Molinie et al (2016) observed a bias that methylated transcripts tended to use the proximal APA sites, resulting in a shortened 3′ UTR (Molinie et al, 2016). It was found that depletion of METTL3 and VIRMA induced 3′ UTR lengthening of several hundred mRNAs, and VIRMA could interact with the 3′ end processing machinery protein CPSF5 in an RNA‐dependent manner (Yue et al, 2018). YTHDC1 was also found to be related to APA during mouse oocyte development (Kasowitz et al, 2018). All these findings indicate that, to some extent, m6A is associated with APA; thus, it is necessary to study the relationship between m6A and APA as well as its underlying molecular mechanism in detail.
In this study, we observed shortened 3′ UTR in MCF‐7 and HEK293T cell lines with knockdown of YTHDC1. We also found that YTHDC1 could disrupt the interaction of the 3′ end processing factors of CPSF4 and FIP1L1 by competitively binding to FIP1L1 in an m6A‐dependent manner, through which YTHDC1 can inhibit the proximal usage of APA sites. This study describes a novel mechanism for APA regulation via m6A modification.
Results
YTHDC1 regulates alternative polyadenylation
Recent studies found that the nuclear reader YTHDC1 regulates alternative splicing (AS) by interacting with the splicing factors SRSF3 and SRSF10 (Xiao et al, 2016). Considering the similarity of AS and APA (Wang et al, 2015b; Tian & Manley, 2017), we deduced that YTHDC1 may regulate APA by interacting with the 3′ end processing factors and exert its function as a mediator of m6A modification and mRNA 3′ end processing. Therefore, we checked the STRING protein–protein interaction database and found the putative interaction between YTHDC1 and 3′ end processing factors (Fig EV1), indicating that YTHDC1 may take part in the regulation of APA by interacting with 3′ end processing factors. To investigate the role of YTHDC1 in the choice of global APA sites, we interfered YTHDC1 with two siRNAs in MCF‐7 and HEK293T cells (Fig 1A), respectively, and performed high‐throughput 3′ end sequencing with IVT‐SAPAS (Fu et al, 2015). We obtained an average of approximately 10 million reads and 181,574 poly(A) sites from knockdown samples that were uniquely mapped to the genome after filtering internal priming. The number of annotated poly(A) sites and genes is similar to previous studies (Fu et al, 2018), and details of each sample of the sequencing data are shown in Table EV1.
Figure EV1. The putative interaction between YTHDC1 and 3′ end processing factors analyzed with STRING protein–protein interaction database.
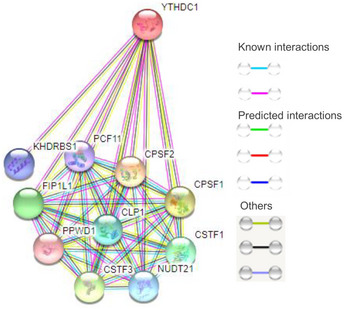
There are several 3′end processing factors (including CPSF, CFIm, CFIIm, CSTF) that may interact with YTHDC1, suggesting YTHDC1 as a potential candidate linking m6A to APA.
Figure 1. YTHDC1 regulates genome‐wide alternative polyadenylation.
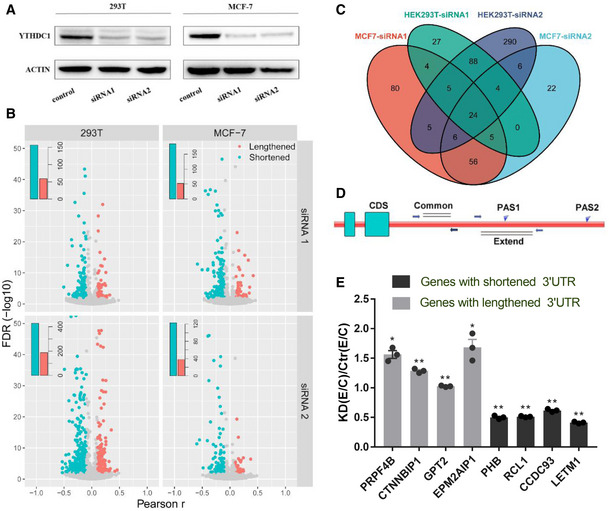
- Western blot analysis of YTHDC1 in HEK293T and MCF‐7 knockdown samples.
- Volcano plot of genes with significant switching to lengthened (red) or shortened (blue) 3′ UTRs according to test of linear trend alternative to independence in HEK293T (left panel) and MCF‐7 cells (right panel). Embedded bar plots show the number of significant genes. The x axis shows the Pearson correlation r from the linear trend alternative test, and the y axis shows the FDR.
- Venn diagram for genes with shortened 3′UTR after knockdown of YTHDC1 by different siRNA in MCF‐7 and HEK293T cells.
- Scheme of the validation of APA primer design. Common: total transcripts; Extend: long 3′ UTR transcripts.
- The qRT‐PCR validation of APA switching. Eight genes with lengthened and shortened 3′ UTRs in HEK293T and in MCF‐7 cells were chosen. PRPF4B, CTNNBIP1, PHB and RCL1 were selected from HEK293T cells, and rest of the genes are MCF‐7 cells. KD (Extended/Common)/Ctr (Extended/Common) < 1 indicates a shorter 3′ UTR in the knockdown sample, and vice versa. Data are presented as mean ± SEM of three biological replicates. *P < 0.05, **P < 0.01, the P values were obtained from unpaired two‐tailed Student's t‐test.
Source data are available online for this figure.
To evaluate the influence of YTHDC1 on genome‐wide APA regulation, we labeled the control sample as 0 and the knockdown sample as 1, and performed a test of linear trend alternative to independence (Agresti, 2003) to obtain the genes with significantly different 3′ UTR lengths between control and YTHDC1 knockdown samples as previously reported (Fu et al, 2011, 2015). A negative Pearson correlation r of the 3′UTR length and sample from the linear trend test means shortened 3′UTR in knockdown sample. In total, 464 and 202 genes significantly switched to the proximal and distal APA sites (FDR ≤ 0.01 and ¦r¦ > 0.1), respectively, after knockdown of YTHDC1 in HEK293T cells. Similarly, 217 and 68 genes with shortened and lengthened 3′ UTRs, respectively, were found in MCF‐7 cells (Fig 1B). The genes with shortened 3′ UTRs by two siRNAs in the same cell are highly overlapped (Fig 1C), suggesting that the APA regulation observed is not stochastic. Besides, the similar number of APA switching genes as previous studies (Yao et al, 2013; Li et al, 2015) also suggests that YTHDC1 is an important APA regulator.
To further display magnitude of length changes of genes with shortened 3′ UTRs, we compared 3′ UTR length between the control and YTHDC1 knockdown samples by calculating the standardized weighted average of the 3′ UTR length for each gene with shortening 3′ UTR (see Materials and Methods). Notched boxplot (Fig EV2) shows that the 3′ UTRs in the YTHDC1 knockdown are significantly shorter than those in control (P < 3.18 × 10−39 or even less). To validate the fidelity of the APA sequencing results, eight genes with significantly APA switching after knockdown of YTHDC1 in HEK293T and MCF‐7 cells were chosen to perform qRT‐PCR (Table EV2). For each gene, two pairs of primers were designed to quantify the expression of common and extended 3′ UTRs, and the relative ratio of transcripts represents trends in the choice of APA sites (Fig 1D). The qRT–PCR results showed a higher ratio of Extended/Common regions for those lengthening transcripts and a lower ratio for the shortened transcripts (Fig 1E), consistent with the proposed sequencing data (Fig EV3).
Figure EV2. Notched boxplot of weighted mean of 3′ UTR length.
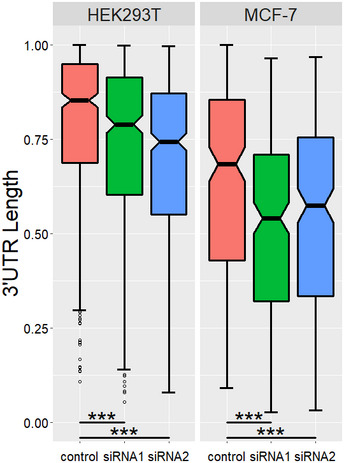
For each gene that undergoes 3′ UTR shortening, the length of each 3′ UTR isoform was normalized to the longest 3′UTR, and the weighted mean of 3′ UTR length was calculated. ***P < 3.18 × 10−39, P values were obtained with paired t‐test.
Figure EV3. IVT‐SAPAS sequencing reads distribution for genes validated with qRT‐PCR, related to Fig 1E.
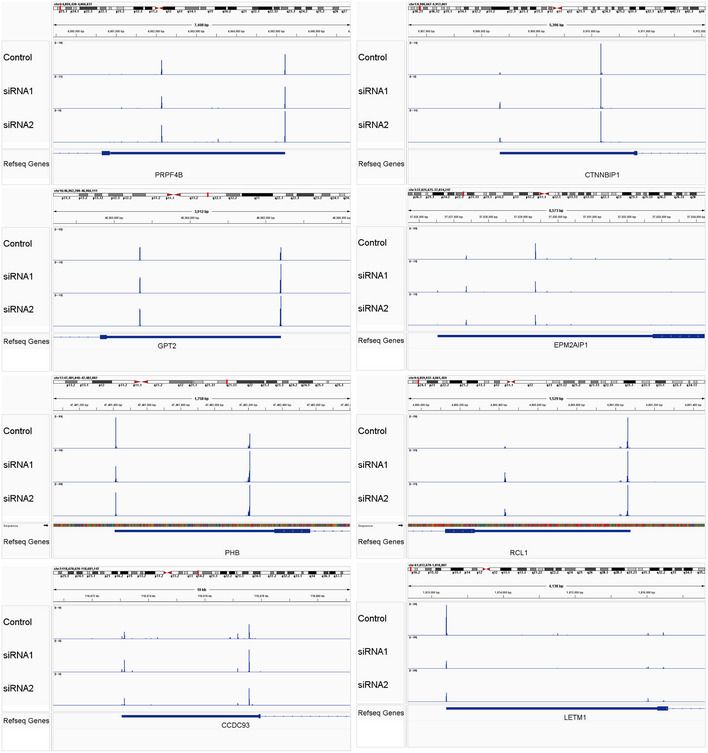
The x and y axes denote genome position of APA sites and sequencing reads number, respectively. The different rows represent the treatment.
Some effects of YTHDC1 knockdown on APA may be indirect effects mediated by expressional changes in m6A related and 3′ end processing factors. The qRT‐PCR results (Fig EV4A) showed that the mRNA expression levels of these related factors were not markedly altered after the knockdown of YTHDC1. We also analyzed the expression levels of genes with APA site switching after the knockdown of YTHDC1. There were few intersections between the genes with APA site switching and genes with differential expression levels (Fig EV4B), suggesting that these significant APA switched genes are unlikely to be regulated by mRNA degradation.
Figure EV4. The effects of YTHDC1 knockdown on APA is a direct regulation from YTHDC1.
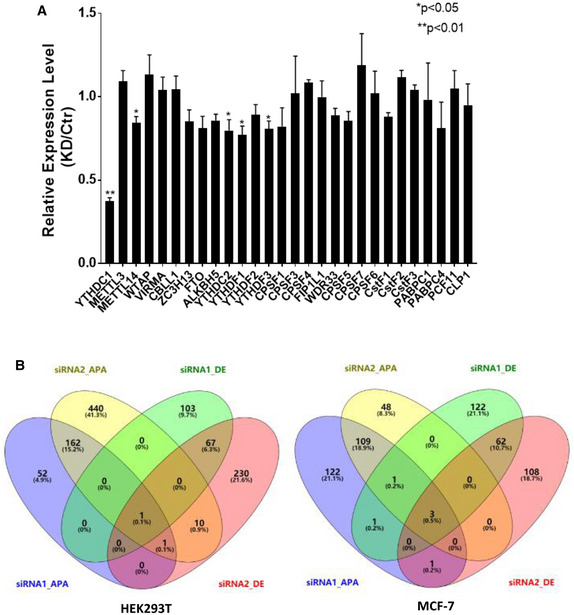
- qRT‐PCR analysis of m6A related factors and 3′ end processing factors after knockdown of YTHDC1 in HEK293T cells. Knockdown of YTHDC1 has little effect on mRNA expression of those factors, which may mediate indirect effect on APA. Data are presented as mean ± SEM of three biological replicates. *P < 0.05, **P < 0.01, the P values were obtained from unpaired two‐tailed Student's t‐test.
- Venn diagram of genes with differential expression levels and genes with APA sites switching after knockdown of YTHDC1 in HEK293T (left) and MCF7 (right). The siRNA‐APA denotes APA switching genes, siRNA‐DE denotes differential expression genes, and number denotes different siRNA. There are few intersections between genes with APA sites switching and genes with differential expression level, indicating that change of 3′UTR length was not caused by RNA degradation.
YTHDC1 inhibits the proximal APA sites in an m6A‐dependent manner
To clarify the effect of YTHDC1 on the choice of APA sites, we downloaded MeRIP‐Seq data (Meyer et al, 2012a; Data ref: Meyer et al, 2012b) and YTHDC1‐iCLIP data (Patil et al, 2016a; Data ref: Patil et al, 2016b) of HEK293T cells to identify YTHDC1 target and non‐target genes. We defined YTHDC1 target genes as those whose 3′ UTRs were covered by both YTHDC1‐iCLIP and MeRIP‐Seq data (see Materials and Methods). For the genes with multiple poly(A) sites covered with the proposed IVT‐SAPAS, we identified 1,373 YTHDC1 target genes and 4,516 non‐target genes. The target genes tended to be markedly more common in the list of genes with shortened 3′UTRs (P = 1.227 × 10−3 for siRNA1 and P = 3.725 × 10−4 for siRNA2 with Fisher's exact tests; Fig 2A). Next, with the Pearson correlation r value, which statistically characterizes changes in the 3′UTR length based on the test of linear trend alternative to independence (r < 0: shortened 3′ UTRs in knockdown of YTHDC1 sample, and vice versa), we used gene set enrichment analysis (GSEA) to identify the overall change of YTHDC1 target and non‐target genes (see Materials and Methods). The enrichment plots show that YTHDC1 target genes are significantly enriched at the bottom of the ranked list of r (Fig 2B, Enrichment Score = −0.30470, P value = 0), suggesting that YTHDC1 target genes are more prone to switching to the proximal APA sites than non‐target genes after the knockdown of YTHDC1.
Figure 2. YTHDC1 inhibits proximal APA sites in an m6A‐dependent manner.
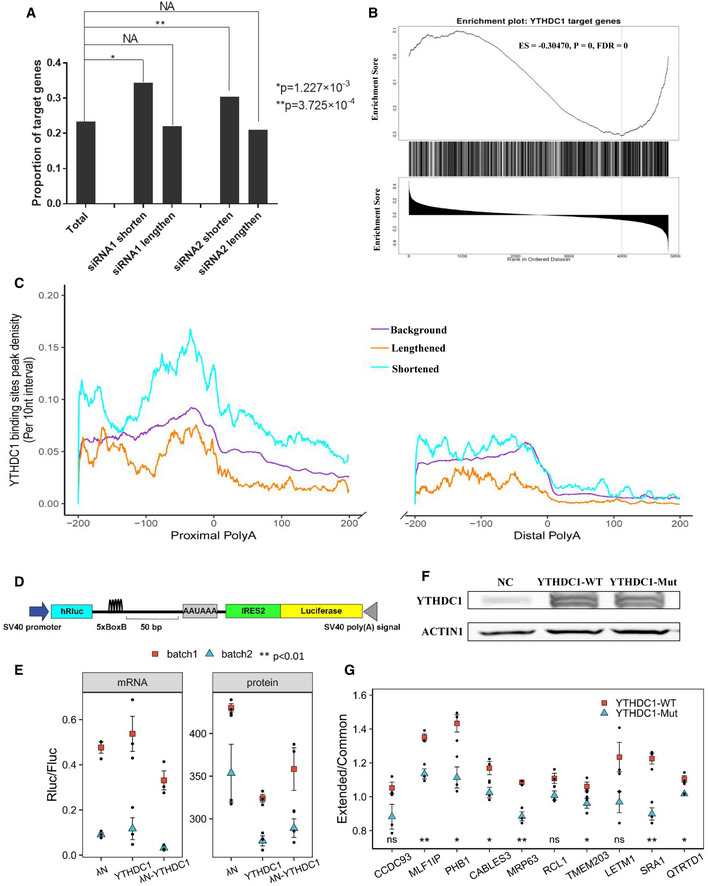
- Higher proportion of target genes in the list of genes with shortened 3′UTRs after knockdown of YTHDC1. The P values were obtained by Fisher's exact test. *P = 1.227 × 10−3, **P = 3.725 × 10−4, NA: no significance.
- Gene set enrichment analysis shows that YTHDC1 target genes are significantly enriched at the bottom of the list of genes ranked by Pearson correlation r (extracted from the linear trend test), suggesting that target genes tend to use shorter 3′ UTRs after YTHDC1 knockdown. ES: enrichment score; FDR: false discovery rate.
- Peak density distribution of YTHDC1 binding sites near different poly(A) sites. Genes with shortened 3′ UTR show a higher density of YTHDC1 binding peaks near proximal APA sites compared to background genes. The density of YTHDC1 binding site peak was calculated as the number of peak in a 10‐nt interval divided by the total number of mRNAs that contained this position.
- Schematic diagram of the bicistronic vector for the dual luciferase reporter assay. The PHB PAS and SV40 PAS were used as proximal and distal APA processing signals, respectively. The 5 × BoxB sequence was inserted 50 bp upstream of the PHB PAS. The ratio of Rluc/Fluc reflects the switch of APA sites.
- Scatter plot of protein and mRNA ratios of dual luciferase report assay with λN‐BoxB system. λN, YTHDC1 and λN‐YTHDC1 were co‐transfected with a bicistronic vector. Two batches of experiments with three biological repeats were performed. Both luciferase activity and mRNA levels were measured. To test the effect of YTHDC1 binding on the proximal APA site, a linear model with the co‐transfected genes and batch as independent variables was fitted. Data are presented as mean ± SEM of three biological replicates. **P < 0.01 with t‐test from linear model.
- Overexpression of YTHDC1‐WT and YTHDC1‐Mut (W377A, W428A) revealed by Western blot.
- Scatter plot of qRT‐PCR for APA validation. Mutation of the m6A binding site of YTHDC1 loses its inhibitory effect on proximal APA sites. Extended/Common ratios of 10 genes were obtained by qRT–PCR in HEK293T cells overexpressing YTHDC1‐WT and YTHDC1‐Mut. The ratios were normalized to those of control cells. Data are presented as mean ± SEM of three biological replicates. *P < 0.05, **P < 0.01, ns: no significance. The P values were obtained from unpaired two‐tailed Student's t‐test.
Source data are available online for this figure.
All of these results show that YTHDC1 promotes using distal APA sites. There are two possible mechanisms for this: (i) YTHDC1 binds to nearby distal APA sites and promotes its processing; and/or (ii) YTHDC1 binds to the proximal APA site and inhibits the usage of proximal APA sites. To identify the relevant mechanism(s), we explored the distribution of binding sites of YTHDC1 near the proximal and distal APA sites of target genes. However, there was a YTHDC1 binding peak ~50 bp upstream of all APA sites (Fig 2C) that is much higher at the proximal APA sites than at distal sites, which is consistent with the distribution of m6A sites (Ke et al, 2015). More interestingly, for the genes with a shortened 3′ UTR due to the knockdown of YTHDC1, their proximal but not distal APA sites showed a higher density of YTHDC1 binding peaks compared to background genes (Fig 2C). To further explicit the relationship between m6A and APA, we also analyzed distribution of m6A sites near the APA sites of genes with APA switching. Consistent with the result of YTHDC1, the genes with shortened 3′ UTR also show a higher m6A modification near of the proximal APA sites than the genes with lengthened 3′ UTR or background (Fig EV5A). Considering that m6A is enriched near stop codon regions (Meyer et al, 2012a), we also compared the distribution of m6A sites near the stop codon. However, the mRNA transcripts with shortened 3′ UTR does not show higher m6A modification level near stop codon than the mRNA with lengthened 3′ UTR or background (Fig EV5B). These results imply that YTHDC1 may regulate APA by binding to the nearby of the proximal APA sites via m6A and inhibiting the generation of a shortened 3′ UTR.
Figure EV5. Higher m6A levels in the proximal poly(A) sites correlates with genes with more proximal poly(A) sites usage after knockdown of YTHDC1.
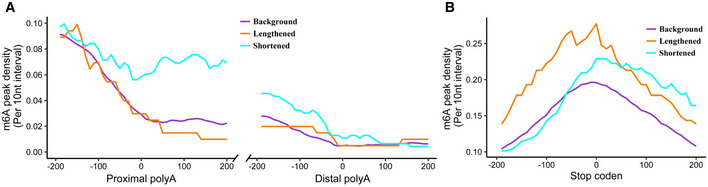
-
A, Bm6A peak density near APA sites and stop codon in (A and B), respectively. Genes with shortened 3′ UTR show a higher m6A modification near of the proximal APA sites compared with genes with lengthened 3′ UTR or background. But shortened 3′ UTR near of the stop codon did not show enriched m6A modification than mRNA with lengthened 3′ UTR or background. The density of m6A peak was calculated as the number of peak in a 10‐nt interval divided by the total number of mRNAs that contained this position.
Next, we confirmed the inhibitory effect of YTHDC1 on the proximal APA sites in HEK293T cells by a bicistronic dual luciferase report assay (Deng et al, 2018). We constructed a vector expressing a bicistronic mRNA with an internal ribosomal entry site 2 [IRES2] sequence, (Fig. 2D). Then, a proximal poly(A) site with PAS (AAUAAA) from a target gene (PHB) was inserted between the two luciferases, and a 5 × BoxB sequence was cloned 50 bp upstream of this poly(A) site (Table EV3). We evaluated the role of λN‐YTHDC1 in APA site processing by comparing the relative ratio of Rluc/Fluc. Two batches of experiments with three biological replicates were performed, and a dual luciferase assay demonstrated that both YTHDC1 and λN‐YTHDC1 could significantly reduce the protein ratios of Rluc/Fluc compared to λN (P < 0.01 with t‐test from linear model; Fig 2E, right), suggesting that YTHDC1 inhibits APA site processing. Considering that YTHDC1 regulates mRNA nuclear export and affects translation, which may interfere with the luciferase result, we also measured the mRNA expression levels with qRT–PCR and found a significantly reduced mRNA ratio of Rluc/Fluc in cells expressing λN‐YTHDC1 (P = 0.0315 with t‐test from linear model) but not YTHDC1 (P = 0.3311) compared to that in cells expressing λN (Fig 2E, left). All of these results suggest that YTHDC1 can inhibit the proximal APA sites by binding to upstream sites.
Two missense mutations (W377A and W428A) can completely abolish the m6A binding activity of YTHDC1 (Xu et al, 2014). To confirm that APA regulation by YTHDC1 requires its binding to m6A, we compared the effects of wild‐type and mutant YTHDC1. We overexpressed wild‐type (YTHDC1‐WT) and mutant YTHDC1 (YTHDC1‐Mut) in HEK293T cells (Fig 2F), and then measured Extended/Common ratios of 10 genes (significant shorter 3′UTR in our sequencing data) by qRT‐PCR. We firstly evaluated the effects of YTHDC1‐WT and YTHDC1‐Mut across genes by fitting a linear mixed model with the genes as a random effect using the “nlme” package (Bates et al, 2015) in R (R Core Team). Consistent with YTHDC1 knockdown, the overexpression of YTHDC1‐WT showed significantly higher ratio of Extended/Common than the control (P = 0 by t test from the linear mixed model), validating its effect on the suppression of the proximal APA sites. In contrast, YTHDC1‐Mut showed slightly lower ratio of extended/common than the control (P = 0.0184). We then directly compared the ratio for each gene, and seven genes showed significantly higher extended/common ratio with overexpression of YTHDC1‐WT than YTHDC1‐Mut (Fig 2G). The other three genes in YTHDC1‐Mut also showed a shorter 3′UTRs, although they were not statistically significant. To further test the role of m6A on YTHDC1 function, we also knocked down of METTL3 (Appendix Fig S1A) and measured APA switching of the above 10 YTHDC1 target genes with qRT‐PCR. Most of genes tend to use shorter 3′UTRs (Appendix Fig S1B), which is consistent with results of YTHDC1‐mut (Fig 2G). These results reveal that YTHDC1 plays a suppressive role on APA by binding the upstream of proximal APA sites in an m6A‐dependent manner.
YTHDC1 regulates APA co‐transcriptionally by preloading with the methyltransferase complex
Both m6A modification and 3′ end processing are known to be co‐transcriptionally regulated (Di Giammartino et al, 2011; Bentley, 2014; Huang et al, 2019). A challenge imposed on the regulation of APA by the m6A reader is how YTHDC1 effectively recognizes m6A and regulates 3′ end processing in a remarkably short time as transcription is so fast (~3.8 kb/min; Singh & Padgett, 2009; Veloso et al, 2014). Recent studies have indicated that the methyltransferase complex (MTC) deposits m6A co‐transcriptionally by interacting with RNA polymerase II (Huang et al, 2019). We propose that YTHDC1 is preloaded with MTC before m6A modification and then recognizes and binds with the m6A site immediately, which may be an effective way to coordinate the recognition of m6A by YTHDC1 and the regulation of 3′ end processing co‐transcriptionally. We performed Co‐IP to test the potential interaction between YTHDC1 and each MTC protein. Results showed that YTHDC1 was loaded in MTCs by interacting with WTAP but not other MTC protein in an RNA‐independent manner (Fig 3A–D). More interestingly, YTHDC1 mass spectrometry also identified RPB3, a core subassembly unit of RNA polymerase II (RNAP II), as a potential YTHDC1 interaction protein (peptide atlas access number: PASS01442). We also validated the interaction of YTHDC1 with RPB3 by co‐IP assay (Fig 3E). All of these results suggest that YTHDC1 can regulate APA co‐transcriptionally by preloading on MTC complexes.
Figure 3. YTHDC1 is pre‐loaded to RNA Pol II with MTC by interacting with WTAP.
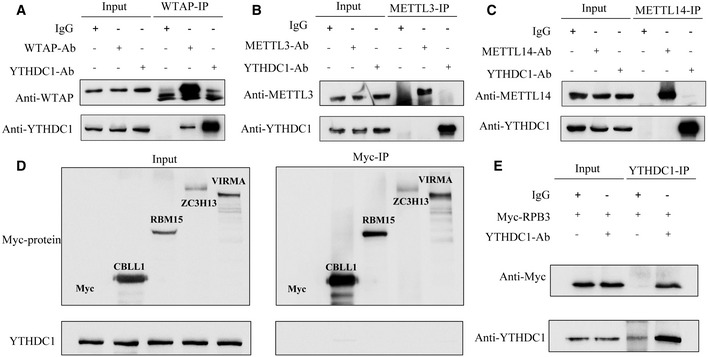
-
A–CThe Co‐IP results of endogenous WTAP, METTL3 and METTL14 with YTHDC1. The endogenous WTAP and YTHDC1 can be pulled down reciprocally by each other in the presence of RNase A.
-
DThe Co‐IP results of Myc‐CBLL1, Myc‐RBM15, Myc‐ZC3H13 and Myc‐VIRMA with Endogenous YTHDC1. None of them can pull down Endogenous YTHDC1 in the presence of RNase A.
-
EEndogenous YTHDC1 interacts with RPB3 in a RNA independent way.
Source data are available online for this figure.
YTHDC1 directly interacts with FIP1L1 in vitro and in vivo
To describe the mechanism of YTHDC1 on APA in more detail, we screened the interacting partners of YTHDC1 with co‐immunoprecipitation (Co‐IP) and mass spectrometry (see Materials and Methods). We overexpressed Flag‐YTHDC1 in HEK293T cells and performed co‐IP using FLAG and YTHDC1 as baits to exclude false‐positive proteins from nonspecific binding of antibodies (Appendix Fig S2), and then, the differential bands were analyzed with mass spectrometry. Consistent with a previous study (Xiao et al, 2016), many potential binding proteins are heterogeneous nuclear ribonucleoproteins and alternative splicing factors, such as HNRNPC, HNRNPM, SRSF3, SRSF10, PTBP1 and ELAVL1 (peptide atlas access number: PASS01442). This reproducibility suggests that the proposed mass spectrometry data are reliable and informative. Consistent with predictions, eight core 3′ end processing core factors were identified in our mass spectrometry assay, including core components of CPSF (CPSF2, CPSF3, FIP1L1), CSTF (CSTF3) and CFIm (NUDT21, CPSF6) complexes (Table EV4). To validate the interaction of YTHDC1 with these core 3′ end processing factors, co‐IP experiments were performed with Myc‐tagged 3′ end processing factors as baits (see Materials and Methods). The Co‐IP results show that the proteins Myc‐FIP1L1, Myc‐PABPC1 and Myc‐PABPC4 can pull down endogenous YTHDC1 in the presence of RNA (Fig 4A). To exclude the false interactions mediated by RNA, we repeated the Co‐IP experiments with the addition of RNase A. Results showed that only Myc‐FIP1L1, not Myc‐PABPC1 and Myc‐PABPC4, strongly interacted with YTHDC1 in an RNA‐independent manner (Fig 4B). We also detected the interaction of endogenous YTHDC1 and FIP1L1 using antibodies against YTHDC1 and FIP1L1. Consistently, both endogenous YTHDC1 and FIP1L1 can be pulled down by each other (Fig 4C). Next, we performed an in vitro pull‐down assay to investigate whether these two proteins can directly interact with each other. We expressed and purified FLAG‐YTHDC1 and Myc‐FIP1L1 proteins (Fig 4D). The Myc‐FIP1L11 protein was immobilized on anti‐Myc beads and then incubated with FLAG‐YTHDC1. Results showed that FLAG‐YTHDC1 can be pulled down by Myc‐FIP1L1 but not by Myc‐tag (Fig 4E), further supporting the direct interaction between YTHDC1 and FIP1L1.
Figure 4. YTHDC1 directly interacts with FIP1L1.
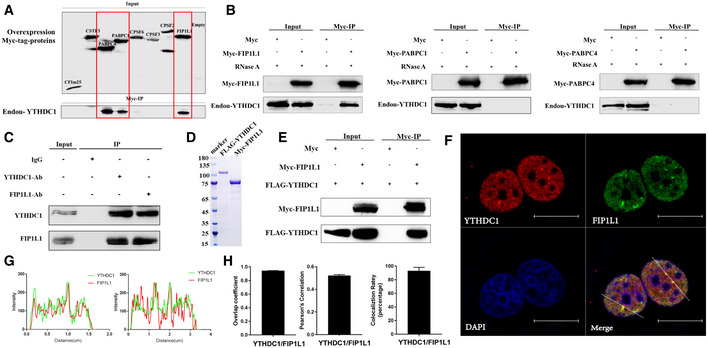
- Co‐IP assay using Myc‐tagged 3′ end processing factors to pull down endogenous YTHDC1 without the addition of RNase A. FIP1L1, PABPC1 and PABPC4 can pull down endogenous YTHDC1.
- Co‐IP assay using Myc‐FIP1L1, Myc‐PABPC1 and Myc‐PABPC4 to pull down endogenous YTHDC1 in the presence of RNase A.
- Co‐IP assay with antibodies against endogenous YTHDC1 and FIP1L1.
- Coomassie brilliant blue staining of purified different epitope‐tagged YTHDC1 and FIP1L1.
- An in vitro pull‐down assay using purified FLAG‐YTHDC1 and Myc‐FIP1L1 showed that FLAG‐YTHDC1 directly interacts with Myc‐FIP1L1.
- Endogenous YTHDC1 and FIP1L1 were stained with antibodies labeled with Alexa Fluor 488 and Alexa Fluor 568 fluorophores, respectively. Immunofluorescence results show that YTHDC1 and FIP1L1 have a high spatial association in the nucleus. Scale bars 15 μm.
- Scan graph analysis and nuclear staining density quantification of YTHDC1 and FIP1L1, indicating high co‐localization in the nucleus.
- Forty cells were randomly selected, and co‐localization was assessed by calculating the average overlap coefficient, Pearson's correlation and co‐localization rate according to Leica TCS SP8 microscope software. Data are presented as mean ± SD of 40 biological replicates.
Source data are available online for this figure.
The m6A reader YTHDC1 was found to localize in nuclear speckles, which are transcription active sites in the nucleus (Nayler et al, 2000). Given the interaction of YTHDC1 and FIP1L1 revealed by the above assays, we characterized the nature of the association between these two proteins in the nucleus. Then, we performed immunofluorescence assays with anti‐YTHDC1 and anti‐FIP1L1 antibodies coupled with Alexa Fluor 568 and 488, respectively (Fig 4F). Data from line scan graph analysis and nuclear staining density quantification demonstrated that YTHDC1 and FIP1L1 highly co‐localized in the nucleus (Fig 4G). To exclude the random diffusion of the two proteins in the nucleus, we randomly chose 40 cells, extracted the fluorescence intensity data of the nucleus and then calculated the overlap coefficient, Pearson's correlation coefficient and co‐localization rate. Results showed a strong correlation between red and green fluorescence (Fig 4H), suggesting the co‐localization of YTHDC1 and FIP1L1 proteins and potential for 3′ end processing.
The proline‐rich domain of FIP1L1 is necessary for the interaction with YTHDC1
To understand the mechanism of YTHDC1 and FIP1L1 in the regulation of APA, we dissected the interaction of YTHDC1 with FIP1L1 and mapped the region/domains involved in this interaction. We conducted FIP1L1 truncation analysis using immunoprecipitation assay, as shown in Fig 5A. By expressing a series of myc‐tagged FIP1L1 fragments in the HEK293T cell line and performing Co‐IP with an anti‐myc antibody, we found that the N‐terminal (aa 1–441) but not the C‐terminal (aa 442–589) fragment of FIP1L1 can pull down YTHDC1 (Fig 5B). Further truncated N‐terminal (N1, N2, N3) and C‐terminal (C1, C2, C3 and C4) fragments show that the fragment of FIP1L1 amino acids 351–426, which contains a proline‐rich domain (256–406 aa), plays an important role in the interaction with YTHDC1 (Fig 5B). To explore the role of the proline‐rich domain, we next mutated prolines to alanines (Fig 5A middle) and checked the interaction of mutant FIP1L1 with YTHDC1. Results showed that the mutation abrogates the interaction (Fig 5C), suggesting that the proline‐rich domain of FIP1L1 is necessary for the interaction with YTHDC1.
Figure 5. Proline‐rich domain of FIP1L1 is necessary for the interaction with YTHDC1.
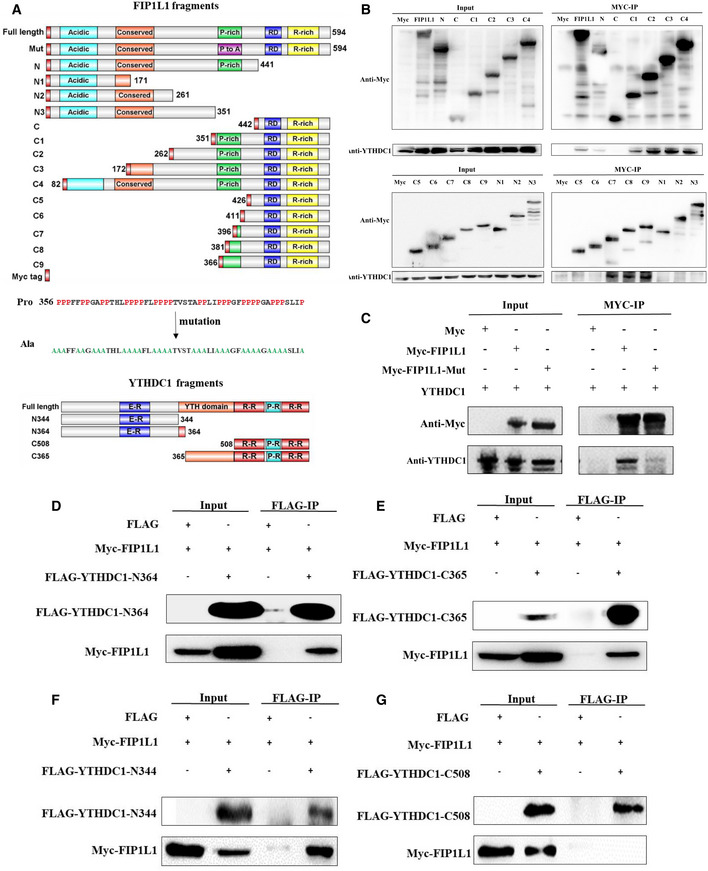
-
ASchematic diagram of the functional regions of YTHDC1 and FIP1L1 and fragment cloning. The prolines in the proline‐rich region (356–406) of FIP1L1 were mutated into alanines.
-
BCo‐IP assays using truncated FIP1L1 proteins revealed that the proline‐rich domain of FIP1L1 plays an important role in interacting with YTHDC1.
-
CMutation of FIP1L1 nearly abrogates the interaction between YTHDC1 and FIP1L1 compared with wild type.
-
D–GCo‐IP assays using different domains of YTHDC1 revealed that the N‐terminus (1–344 aa) and YTH domain (364–507 aa) of YTHDC1 interact with FIP1L1.
Source data are available online for this figure.
We also investigated the domains of YTHDC1 required for the interaction with FIP1L1 by cloning YTHDC1 fragments of N‐364 (1–364 aa) and C‐365 (365–727 aa; Fig 5A, bottom). Co‐IP results show that both N‐364 and C‐365 of YTHDC1 can pull down FIP1L1 (Fig 5D and E). Then, we mapped the YTHDC1 region/domains involved in this interaction by narrowing the fragments (Fig 5A bottom). The Co‐IP results showed that the N‐terminus (1–344 aa) and YTH domain (365–508 aa) of YTHDC1 but not the C‐terminus were responsible for the interactions with FIP1L1 (Fig 5F and G).
YTHDC1 and CPSF4 competitively bind to FIP1L1 and regulate the choice of APA sites
FIP1L1, as a core component of the CPSF complex, recruits other 3′ end processing factors and facilitates the usage of the proximal APA sites (Kaufmann et al, 2004; Lackford et al, 2014; Li et al, 2015). Specifically, FIP1L1 recognizes the upstream U‐rich RNA motif of the proximal APA sites and recruits CPSF4 and poly(A) polymerase (PAP) to promote APA site processing and produces shortened 3′UTR transcripts (Kaufmann et al, 2004). The C‐terminus of FIP1L1 contains an arginine‐rich domain mediating binding to the RNA U‐rich element, and the N‐terminus includes an acidic region and a conserved central domain interacting with PAP and CPSF4 (Fig 6A), respectively. Based on the interaction between YTHDC1 and FIP1L1, we wonder whether YTHDC1 will disrupt FIP1L1 recruitment of other 3′ end processing factors (CPSF4 and PAPOLA). Then, we co‐transfected HEK293T cells with Myc‐FIP1L1 and FLAG‐CPSF4 in combination with increased expression levels of FLAG‐YTHDC1 to test the inhibitory effect of YTHDC1 on the interaction between FIP1L1 and CPSF4. We found a markedly lower level of the interaction between FIP1L1 and CPSF4 when YTHDC1 expression was elevated (Fig 6B). We also investigated the inhibitory role of endogenous YTHDC1 on the interaction between FIP1L1 and CPSF4 by knocking down YTHDC1. Consistent with overexpression, knockdown of YTHDC1 enhanced the recruitment of CPSF4 by endogenous FIP1L1 (Fig 6C). However, we did not observe the effect of YTHDC1 on the interaction between FIP1L1 and PAPOLA (Fig 6D). These results indicate that YTHDC1 inhibits the usage of APA sites by disturbing the interaction of FIP1L1 with CPSF4, which is a key factor in recognizing poly(A) signals (AAUAAA or AUUAAA) and assembling 3′ end processing complexes (Shi et al, 2009; Chan et al, 2014; Clerici et al, 2018).
Figure 6. YTHDC1 and CPSF4 competitively bind to FIP1L1 and regulate the choice of APA sites.
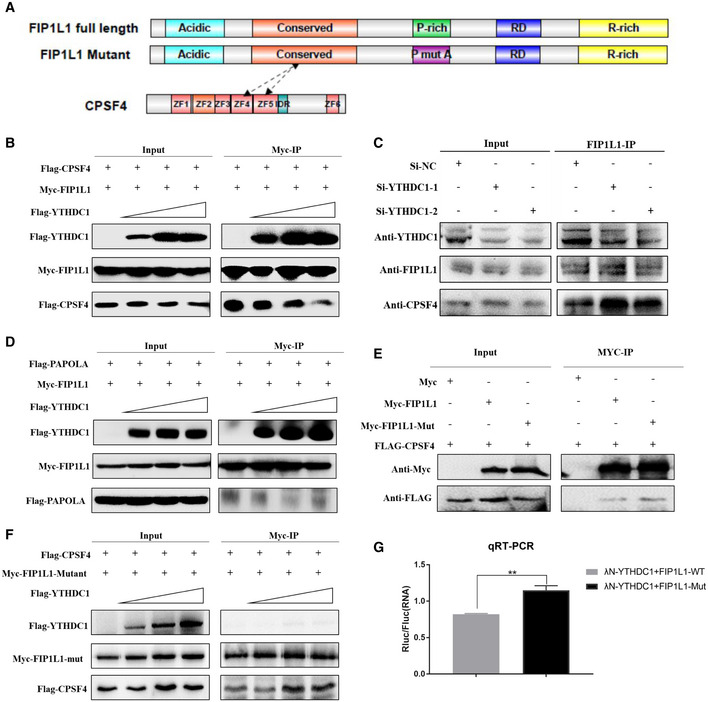
- Schematic diagram of functional regions of FIP1L1 and CPSF4.
- YTHDC1 inhibits the interaction between Myc‐FIP1L1 and FLAG‐CPSF4. A substantially suppressed level of interaction between Myc‐FIP1L1 and FLAG‐CPSF4 was observed when FLAG‐YTHDC1 input was increased.
- Knockdown of YTHDC1 enhances endogenous FIP1L1 recruitment to CPSF4, indicating that YTHDC1 plays an important role in interfering with the 3′ end processing complex interaction.
- YTHDC1 has little effect on the interaction between Myc‐FIP1L1 and FLAG‐PAPOLA. The interaction between Myc‐FIP1L1 and FLAG‐PAPOLA was not significantly affected when FLAG‐YTHDC1 was increased.
- Mutation of proline had little effect on the interaction between FIP1L1 and CPSF4 compared with the wild type.
- The mutation of prolines in FIP1L1 abrogated the inhibitory effect of YTHDC1 on the interaction between FIP1L1 and CPSF4.
- qRT–PCR validation of APA site switching in a bicistronic dual luciferase system. FIP1L1‐Mut could significantly increase the ratios of Rluc/Fluc compared to FIP1L1‐WT, indicating that YTHDC1 inhibits the use of proximal APA sites by interacting with the proline‐rich domain of FIP1L1. Data are presented as mean ± SEM of three biological replicates. **P = 8.77 × 10−4, the P values were obtained with unpaired two‐tailed Student's t‐test.
Source data are available online for this figure.
Given the role of the proline‐rich domain of FIP1L1 in the interaction with YTHDC1, we assume that mutation of this domain should be able to relieve the inhibitory effect of YTHDC1 on the interaction between FIP1L1 and CPSF4. Indeed, a co‐IP assay showed that the mutation of prolines in FIP1L1 had little effect on its interaction with CPSF4 (Fig 6E) but nearly abrogated the inhibitory effect of YTHDC1 on the interaction between FIP1L1 and CPSF4 (Fig 6F). This competitive binding assay implies that YTHDC1 regulates APA by interacting with the proline‐rich domain of FIP1L1, which disrupts the interaction between YTHDC1 and FIP1L1.
We have shown that YTHDC1 can suppress the proximal APA sites and interact with FIP1L1. To investigate whether the suppressive effects of YTHDC1 are mediated by its interaction with FIP1L1, we also compared the effects of wild‐type FIP1L1 (FIP1L1‐WT) and the proline mutant (FIP1L1‐Mut) on the regulation of APA. With bicistronic dual luciferase system (Fig 2D), we co‐expressed λN‐YTHDC1 with FIP1L1‐WT and FIP1L1‐Mut. The qRT–PCR showed that FIP1L1‐Mut significantly increased the ratios of Rluc/Fluc compared to FIP1L1‐WT (Fig 6G), indicating that the disruption of the interaction between YTHDC1 and FIP1L1 could abolish the inhibitory effect of YTHDC1 on APA site processing. Collectively, YTHDC1 regulates choice APA sites by interacting with FIP1L1, in which the proline‐rich domain plays an important role.
YTHDC1 forms nuclear condensates through LLPS and compartmentalizes FIP1L1 in an m6A‐dependent manner
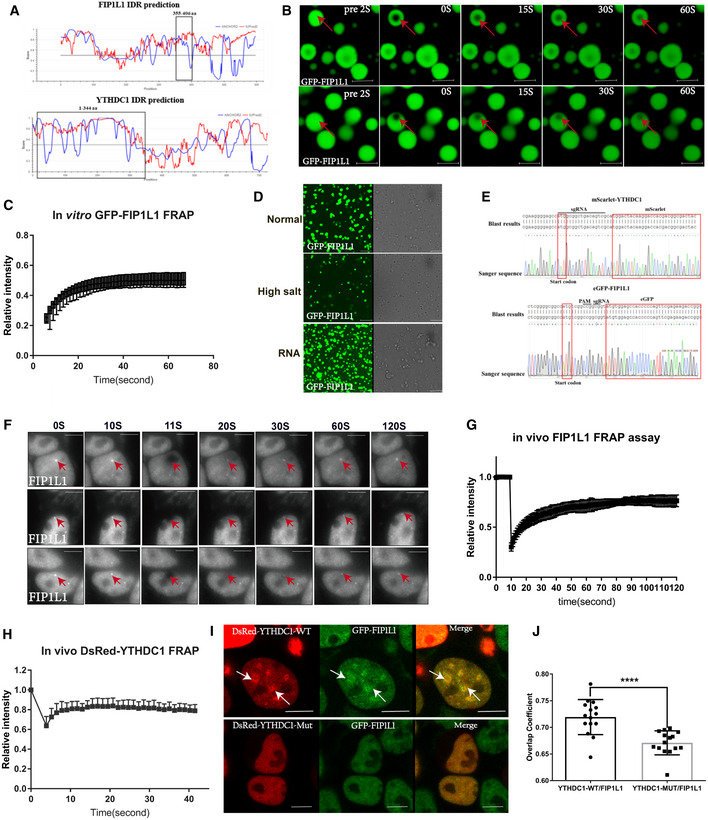
Recently, the m6A reader YTHDF1‐3 was reported to undergo liquid–liquid phase separation (LLPS) by binding to modified m6A RNA to regulate the stability and translation of transcripts (Ries et al, 2019; Fu & Zhuang, 2020), and YTHDC1 binds to m6A and forms nuclear condensates mediated by LLPS (Cheng et al, 2021). An immunofluorescence assay showed that YTHDC1 and FIP1L1 co‐localized and formed droplet‐like structures in the nucleus (Fig 4F), which is a physical characteristic of LLPS. LLPS proteins usually contain intrinsically disordered regions (IDRs) with low hydrophobicity and high net charge polypeptide segments. With IDRs score prediction tool IUPred2A (Meszaros et al, 2018), we found that the key domain mediating the interaction between YTHDC1 and FIP1L1 (Fig 7A black box) contained IDRs with high scores (score > 0.5). Indeed, purified FIP1L1 proteins could form liquid droplets in vitro (Fig 7B) and exhibit good dynamic liquid properties by fluorescence recovery after photobleaching (FRAP) assay (Fig 7C). Interestingly, these liquid droplets could be dissolved in a high salt buffer (Fig 7D middle), but promoted the incorporation of RNA (Fig 7D bottom). To investigate LLPS of FIP1L1 and YTHDC1 in vivo, we applied CRISPR/Cas9‐mediated precise integration into the target chromosome system (CRIS‐PITCh; Chen et al, 2020; Nakade et al, 2014) to knock in eGFP into FIP1L1 loci (see Materials and Methods). We obtained homozygous eGFP‐FIP1L1 knock‐in cell lines after flow cytometry sorting and Sanger sequencing validation (Fig 7E). Consistent with in vitro assay, endogenous GFP‐FIP1L1 formed nuclear condensates and displayed dynamic and liquid‐like properties in vivo (Fig 7F and G). We also obtained homozygous mScarlet‐YTHDC1 knock‐in cell lines (Fig 7E). However, endogenous mScarlet‐YTHDC1 proteins are too weak to perform FRAP assay. Then we overexpressed DsRed‐YTHDC1 to perform FRAP assay, and found that DsRed‐YTHDC1 also displayed dynamic and liquid‐like properties in vivo (Fig 7H).
Figure 7. YTHDC1 forms nuclear condensates through LLPS and compartmentalizes FIP1L1 in an m6A‐dependent manner.
-
ADiagram of the IDRs prediction of YTHDC1 and FIP1L1. The regions in the black box represent the interacting domain between YTHDC1 and FIP1L1 with a high disorder score.
-
BMicroscopy images of fluorescence recovery following partial photobleaching of GFP‐FIP1L1. Normal solutions containing 2.1 mg/ml GFP‐FIP1L1 were examined with a microscope at different time points. Scale bars: upper 3 μm, lower 2.98 μm.
-
CFRAP assay of GFP‐FIP1L1. Quantification of average relative fluorescence intensity and its initial rate of area of photobleaching across eight individual GFP‐FIP1L1 droplets. Data are presented as mean ± SD of eight biological replicates.
-
DThe GFP‐FIP1L1 phase separation assay was performed in normal buffer, high salt buffer and normal buffer with the addition of RNA. All these results indicated that the GFP‐FIP1L1 protein displayed dynamic and liquid‐like properties. Scale bars 15 μm.
-
ESanger sequencing was used to genotype eGFP‐FIP1L1 and mScarlet‐YTHDC1 homozygous cell lines.
-
FIn vivo FRAP assays for eGFP‐FIP1L1 and DsRed‐YTHDC1 were examined by time‐lapse phase‐contrast imaging. The arrows represent droplet of GFP‐FIP1L1. Nine cell‐independent experiments were selected for FRAP assays. Scale bars 10 μm.
-
G, HQuantification of average relative fluorescence intensity and its initial rate of area of photobleaching across 15 individual GFP‐FIP1L1 droplets in (G) and DsRed‐YTHDC1 in (H), respectively. Data are presented as mean ± SD of 15 biological replicates.
-
IGenome‐edited eGFP‐FIP1L1 HEK293T cells expressing Dsred‐YTHDC1‐WT and Dsred‐YTHDC1‐Mut were examined by confocal microscopy. DsRed‐YTHDC1‐WT forms LLPS to compartmentalize FIL1L1, but DsRed‐YTHDC1‐Mut diffuses in the nucleus without droplet structure. Droplet structures are indicated by arrows. Scale bars: upper 8.09 μm, lower 6.64 μm.
-
JCo‐localization of YTHDC1‐WT and YTHDC1‐Mut in (I) was assessed by calculating the average overlap coefficient according to Leica TCS SP8 microscope software. Data are presented as mean ± SEM of 15 cells. ****P = 6.46 × 10−4, the P values were obtained with unpaired two‐tailed Student's t‐test.
Source data are available online for this figure.
To test whether the function of YTHDC1 in APA regulation is related to phase separation, we overexpressed wild‐type YTHDC1 (DsRed‐YTHDC1‐WT) and mutant YTHDC1 (W377A, W428A; DsRed‐YTHDC1‐Mut) in HEK293T cells with eGFP‐FIP1L1 knock‐in. DsRed‐YTHDC1‐WT and eGFP‐FIP1L1 form micrometer‐sized and droplet‐like phase separation structures (Fig 7I top), while DsRed‐YTHDC1‐Mut and eGFP‐FIP1L1 nearly diffuse in the nucleus and cannot form droplets (Fig 7I bottom), suggesting that YTHDC1 can help LLPS of FIP1L1 in a m6A‐dependent manner. Overlap coefficient analysis also shows that the YTHDC1 mutant can reduce the co‐localization of YTHDC1 and FIP1L1 (Fig 7J). To test the role of m6A on YTHDC1 phase separation, we also knocked down of METTL3 (Appendix Fig S3A), and found that YTHDC1 condensates were significantly disrupted compared with control (Appendix Fig S3B and C). In summary, we provide both in vitro and in vivo data to demonstrate that YTHDC1 and FIP1L1 interacted by LLPS may play an important role in APA regulation.
Discussion
m6A and APA are two important post‐transcriptional mechanisms of gene regulation, and both have critical roles in various biological processes (Fu et al, 2011; Li et al, 2012; Chandola et al, 2015; Mayr, 2016). m6A was recently found to be related to the regulation of APA (Ke et al, 2015; Molinie et al, 2016; Bartosovic et al, 2017; Kasowitz et al, 2018; Yue et al, 2018). Here, we provide a molecular mechanism of m6A reader YTHDC1 to repress the proximal APA sites by interacting with FIP1L1 (Fig 8), which supports the previous finding that m6A inhibits the proximal 3′ end processing and prefers to distal APA sites (Ke et al, 2015; Bartosovic et al, 2017). m6A “readers” were found to mediate a variety of m6A effects (e.g., mRNA translocation, stability and splicing) by recruiting other effector proteins. Thus, it is reasonable to assume that m6A “readers” orchestrate 3′ end processing by collaborating with 3′ end processing factors. In this study, using APA sequencing, we found that the knockdown of YTHDC1, an m6A nuclear reader, promotes hundreds of YTHDC1 target gene switching to the proximal APA sites in an m6A‐dependent manner in HEK293T and MCF‐7 cells. We also identified that YTHDC1 and CPSF4 can competitively bind to FIP1L1, which disrupts the recruitment and assembly of the 3′ end machinery. In addition, we show that LLPS may take part in the interaction of YTHDC1 and FIP1L1. Based on these findings, we propose a model illustrating a novel mechanism for YTHDC1‐mediated APA regulation by m6A (Fig 8). YTHDC1 are pre‐loaded with MTC (Fig 3), which is associated with Pol II during transcription (Huang et al, 2019). When m6A modification is finished co‐transcriptionally by MTC, YTHDC1 subsequently binds to the modification sites upstream of the proximal APA sites. Then, YTHDC1 interacts with FIP1L1 by LLPS and disrupts the recruitment of CPSF4 and other CPSF factors, resulting in the suppression of the proximal APA sites and a lengthened 3′ UTR (Fig 8A). In contrast, in the absence of YTHDC1 binding due to the reduced level of YTHDC1 expression or m6A modification, the inhibition of the use of proximal APA sites was abrogated, resulting in a shortened 3′ UTR (Fig 8B).
Figure 8. Model of APA regulation by YTHDC1.
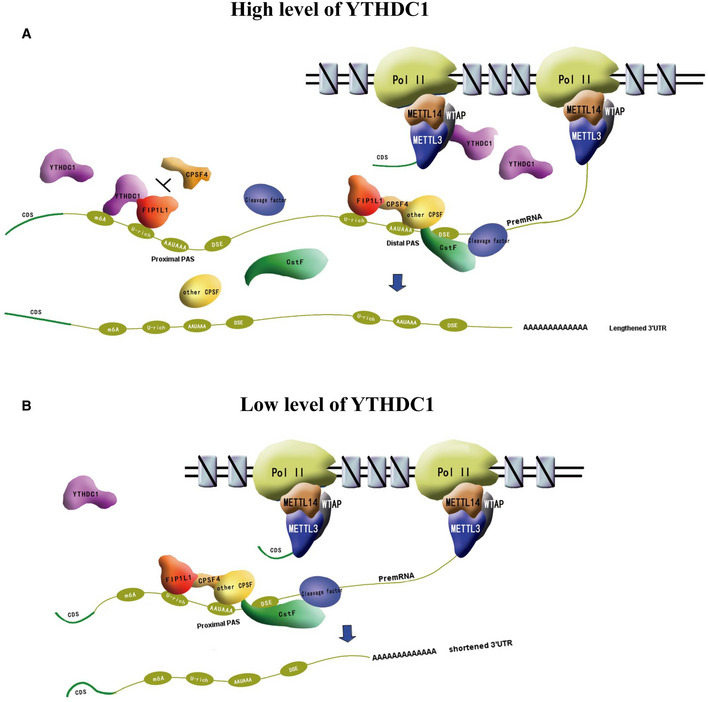
- YTHDC1 is associated with Pol II during transcription by interacting with MTC and binds to the modification sites upstream of proximal APA sites when m6A modification is finished. Then, YTHDC1 interacts with FIP1L1 and disrupts the recruitment of CPSF4 and other CPSF factors, resulting in the suppression of proximal APA sites and longer 3′ UTRs.
- This inhibition is abrogated at low levels of YTHDC1 or in the absence of m6A modification. FIP1L1 interacts with CPSF4, which recruits other CPSF factors and promotes the usage of proximal APA sites, resulting in shortened 3′ UTRs.
Recently, it was reported that YTHDC1 was involved in regulating the choice of APA sites during mouse oocyte development and could interact with CPSF6, suggesting that CPSF6 is a partner of YTHDC1 to regulate APA (Kasowitz et al, 2018). Consistent with this result, we also observed CSPF6 in our mass spectrometry (Table EV4 and peptide atlas access number: PASS01442). However, we found that this interaction was weak and had poor repeatability in the co‐IP assay (Fig 4A). In vitro pull‐down and immunofluorescence assays identified that YTHDC1 can directly interact with the FIP1L1 protein (Fig 4C and E), which suggests that the interaction between YTHDC1 and CPSF6 is likely mediated indirectly by other 3′ end processing factors, and FIP1L1 may be the key mediator connecting YTHDC1 and the 3′ end processing machinery. Indeed, in vitro pull‐down and competitive binding experiments revealed that YTHDC1 can interact with FIP1L1, and this interaction can disrupt the recruitment of CPSF4 and inhibit the use of the proximal APA sites (Fig 6). We also found that a proline‐rich domain of FIP1L1 plays an important role in the YTHDC1 regulation of APA (Figs 5 and 6G), which provides us with a potential target to develop new drugs for APA regulation by intervening the interaction between YTHDC1 and FIP1L1. Also, the role of the proline‐rich domain on LLPS of FIP1L1 and YTHDC1 should be addressed in future studies.
Both m6A and APA are known to regulate the stability, translational efficiency and localization of mRNA transcripts (Di Giammartino et al, 2011; Yue et al, 2015). There may be some cooperation between them to regulate the expression of mRNA transcripts. For example, some gene transcripts in the brain with higher m6A modification or YTHDC1 binding tend to use lengthened 3′ UTRs (Ke et al, 2015), and both long 3′ UTRs and m6A modifications are negative regulatory elements contributing to mRNA degradation (Sandberg et al, 2008; Nam et al, 2014). Previous studies typically evaluated the influence of m6A and APA on the regulation of gene expression separately, which may not see the whole picture. In this study, we highlight the complexity of m6A and APA in regulating gene expression and provide a mechanistic model for future research.
Interestingly, our APA analysis shows that only a subset of YTHDC1 target gene transcripts (about 11%) undergo a substantial shortening of the 3′ UTR and most of m6A transcripts are not significantly affected upon YTHDC1 deletion. The regulation effect of YTHDC1 on APA needs its binding to m6A sites nearby the proximal APA sites (Figs 2C and EV5A). Then it may only affect a subset of mRNAs with higher m6A level nearby the proximal APA sites but not all mRNAs with m6A modification. YTHDC1 inhibits proximal APA sites by interacting with FIP1L1, which preferentially binds to U‐rich sequences of transcripts (Kaufmann et al, 2004). This also limits a subset of genes to be regulated. Furthermore, most genes exhibit less than 50% methylation levels (Molinie et al, 2016), which can decrease the power of detection of significant APA switching too. All of these can explain that only a subset of genes with m6A switched to proximal APA sites by knockdown of YTHDC1, and the regulation effects of YTHDC1 is cell context‐dependent.
Both APA and m6A are involved in various biological processes, such as stem cell self‐renewal (Batista et al, 2014; Lackford et al, 2014), T‐cell differentiation (Li et al, 2017; Qiu et al, 2017), embryonic development (Li et al, 2012; Kasowitz et al, 2018) and carcinogenesis (Fu et al, 2011; Liu et al, 2018; Yuan et al, 2019). In this study, we found that YTHDC1, as an m6A “reader”, bridges m6A modification to pre‐mRNA 3′ end processing, which implies its relevance in the regulation of various biological functions. Thus, the crosstalk of m6A and APA on normal or pathological processes should be further studied thoroughly to uncover the complexity of gene transcriptional regulation.
Materials and Methods
Cell cultures, siRNA knockdown and plasmid transfection
Human HEK293T cells (ATCC, CRL‐11268) were cultured in DMEM supplemented with 10% FBS. The human breast cancer cell line MCF‐7 was cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, 11965092) supplemented with 10% fetal bovine serum (FBS; Gibco, 10100147) and 10 μg/ml human recombinant insulin (Sigma Aldrich, I9278). All media were supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco, 15140122). The siRNA oligos were designed and synthesized (RiboBio, Guangzhou), and the siRNA target sequences were as follows: control (RiboBio Co., LTD); YTHDC1‐siRNA1 sequence: GCAAGGAGTGTTATCTTAA; YTHDC1‐siRNA2 sequence: GGCGTCGACCAGAAGATTA. METTL3‐siRNA1: GCACTTGGATCTACGGAAT; METTL3‐siRNA2 sequence: CGACTACAGTAGCTGCCTT. Cells at ~30% density were transfected with siRNAs at a final concentration of 50 nM using Lipofectamine RNAiMAX Reagent (Invitrogen, 13778150) according to the manufacturer's recommendations. The plasmids transfected using Lipofectamine 3000 (Invitrogen, L3000015) following the manufacturer's instructions.
Antibodies and reagents
For immunoblotting, anti‐Myc (Sigma Aldrich, M4439), anti‐FLAG (Sigma Aldrich, F1804; Cell Signaling, 14793), anti‐HA (Cell Signaling, 3724), anti‐YTHDC1 (Abcam, ab122340), anti‐FIP1L1 (Novus, NB100‐74588), anti‐CPSF4 (Proteintech, 15023‐1‐AP), anti‐METTL3 (Cell Signaling, 86132), anti‐METTL14 (Cell Signaling, 51104) and anti‐WTAP (Cell Signaling, 56501) were used as primary antibodies. Anti‐mouse (Cell Signaling, 7076) and anti‐rabbit (Cell Signaling, 7074) HRP‐conjugated IgGs were used as secondary antibodies.
Preparation of the IVT‐SAPAS library, APA sequencing and analysis
HEK293T and MCF‐7 cells were transfected with YTHDC1 siRNAs for 48 h, and total RNA was used to prepare the IVT‐SAPAS library according to a previous report (Fu et al, 2015). Briefly, total RNA were extracted with Trizol (Invitrogen, 10296010) and digested with DNase I (Ambion, AM2222), and 1 μg total RNA was fragmented by heating at 95°C for 25 min. The first strand cDNA was transcribed by SuperScript III Reverse Transcriptase (Invitrogen, 18080044) with an anchored oligo d(T) primer (AAGCTTAGATATCTAATACGACTCACTATAGGGACCT‐ ACACGACGCTCTTCCGATC (T) 20VN) tagged with Illumina A adaptor and T7 promoter, and then the second strand was synthesized with PrimeScript™ Double Strand cDNA Synthesis Kit (Takara, 6111A). The second round of reverse transcription was performed with random primers (AGTTCAGACGTGCTCTTCCGATCTNNNNNN), and then cDNA was used as a template to perform PCR with 15 cycles to amplify the library. The P5 and P7 adapter were amplified by Platinum Taq DNA polymerase (Invitrogen, 15966025). Finally, 200–500 bp library fragments of the PCR product were purified by size selection with AMPure XP Beads (Beckman Coulter, A63881). The final pooled libraries were quantified and sequenced with Illumina HiSeq 2500 Sequencing System.
The reads were mapped to the human hg19 reference genomes using Bowtie and clustered into poly(A) sites as described previously (Fu et al, 2011, 2015). To detect the genes with significant 3′ UTR length changes, we performed the test of linear trend alternative to independence: a 2×C table was constructed with the number of reads of APA sites for each gene, with APA sites as columns (from the site with shortest 3′ UTR to that with longest) and the two samples as rows; then, Pearson correlation r and statistic were calculated to obtain the associated P values. A threshold of FDR ≤ 0.01 and ¦r¦ > 0.1 was used to identify significant genes. For expression level changes between two samples, we calculated the log2 ratio of normalized reads for each gene, and a threefold difference and FDR < 0.01 (Fisher's exact test) were defined as the threshold of differentially expressed genes. Considering average 3′ UTRs may be due to differential distribution of APA isoforms or mRNAs of different genes. To reduce the variance of 3′ UTR length across genes, we standardized the length by designating the longest 3′ UTR as 1.0 and calculated the weighted mean of 3′ UTR length with multiple APA sites for each gene. We fitted paired t test to test whether there is an overall 3′ UTR length difference between the control and YTHDC1 knockdown samples.
MeRIP, iCLIP and GSEA analysis
We downloaded (Meyer et al, 2012a; Data ref: Meyer et al, 2012b) and YTHDC1‐iCLIP data (Patil et al, 2016a; Data ref: Patil et al, 2016b) from the HEK293T cell line. Then, we counted the support reads mapped to the 3′ UTR of genes with UTR‐APA according to the proposed IVT‐SAPAS data. We defined YTHDC1 target genes as those whose support reads of both MeRIP and YTHDC1 were greater than 50, and MeRIP reads were twice as numerous as the negative control. With YTHDC1 iCLIP data (GSE78030), we counted YTHDC1 binding site and m6A peaks per 10 nt interval within a window 200 nt on each side of APA sites. Positional plots of YTHDC1 binding site and m6A peaks around APA sites were drawn with the ggplot2 package (ggplot2.org) in R software v3.3.3. The genes without APA site switching in the proposed IVT‐SAPAS data were chosen as the background genes.
We performed Fisher's exact test to examine the correlation between genes with significant 3′UTR length changes and target genes of YTHDC1. Then genes were ranked by the r values from the above tests of linear trend alternative to independence, and we performed Gene Set Enrichment Analysis (GSEA) to analyze the enrichment of YTHDC1 target genes in the list of genes with shortened 3′UTRs using the Gene Set Analysis Toolkit (http://www.webgestalt.org; Wang et al, 2017).
qRT‐PCR to validate APA sequencing validation experiments
HEK293T and MCF‐7 cells were harvested after siRNAs and plasmid transfection 48 h. Total RNA was extracted with TRIzol (Invitrogen, 10296010) according to the manufacturer's recommendations. cDNAs were synthesized with PrimeScript™ RT reagent Kit (TAKARA, RR037A) for next qRT‐PCR. The performance of YTHDC1 knockdown on mRNA level was appraised by 2–∆∆Ct method. For each gene, two pairs of primers were designed to quantify the expression of the common and extended regions of 3′UTR, respectively, and the ratio of Extended/Common was calculated. To evaluate the effects of the wild‐type and mutant comparing to the control, a linear mixed model was fitted with nlme package (Bates et al, 2015) in R (R Core Team), taking genes as random effect.
Genes cloning and construction of expression vectors
Total RNA extracted from HEK293T cells was used for cDNA synthesis by SuperScript® III Reverse Transcriptase (Invitrogen, 18080044) and an oligo (dT) primer. The obtained cDNA was used as a template to amplify the YTHDC1 and other 3′ end processing factors (Table EV3). YTHDC1 wild‐type, mutant with two missense mutations (W377A and W428A) and its different domains and CPSF4 were cloned into the pCMV‐FLAG tag expression vector (Clontech, 635688). FIP1L1 and its different domains fragments and 3′ end processing factors genes (Table EV3) were cloned into pCMV‐Myc tag expression vector (Clontech, 635689) with ClonExpress®Ultra One Step Cloning Kit (Vazyme, C115‐01). The cloning primers are shown in Table EV3.
Tethering assay and dual‐luciferase report assay
Bicistronic vectors were constructed as previously reported (Deng et al, 2018). A 5xBox B and polyadenylation signal (AAUAAA) sequence from PHB was synthesized (IGE BIOTECHNOLOGY., LTD) and subcloned downstream of the Renilla ORF by digestion with XhoI and NotI. The λN peptide sequence (MDAQTRRRERRAEKQAQWKAAN) was fused to the N‐terminus of YTHDC1 by subcloning into pCMV‐Myc (Clontech, 635689). The primer sequences for the tethering assay are shown in the Table EV3. The luciferase activity was measured by the GloMax Discover System (Promega, GM3000) according to the manufacturer's recommendations, and mRNA levels were measured by qRT–PCR. A linear model with the co‐transfected genes and batch as independent variables was fitted to test the effect of YTHDC1 binding on the proximal APA site.
Protein purification and in vitro pulldown
The HEK293T cells were harvested with lysis buffer (20 mM Tris–HCl pH 7.4, 500 mM NaCl, 1% NP‐40, 1 × cOmplete Protease Inhibitor Cocktail (Roche, 11873580001), 1 mg/ml RNase A (Sigma, 10109169001)) after being transfected with FLAG‐YTHDC1, Myc‐FIP1L1 and CMV‐Myc empty vector for 36 h. The cells were further lysed by sonication on ice using a Sonic Dismembrator (Toshiba; 15 cycles with 5 s pulse‐on and 10 s pulse‐off, 10% amplitude). The supernatant was cleared by centrifuging at 14,000 g for 10 min and filtered using 0.22 μm syringe filter (Millex) and the clear lysate was incubated with anti‐Flag M2 Affinity Gel (Sigma Aldrich, A2220) and EZviewTM Red Anti‐Myc Affinity Gel (Sigma Aldrich, E6654) by gently rotating for 1 h at room temperature. The beads were then washed twice with lysis buffer and twice with washing buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl) to exclude non‐specifically bound proteins. The FLAG‐YTHDC1 proteins were eluted by competing with 3 × Flag‐Peptide (Sigma Aldrich, F4799) and condensed using Pierce Protein Concentrators PES (Thermo Scientific, 88504). The purity of FLAG‐YTHDC1 proteins was checked by Commassie Blue Staining.
The Myc‐FIP1L1 and Myc tag immobilized on anti‐Myc beads were incubated with equal amounts of purified Flag‐YTHDC1 proteins, respectively, and gently rotated at 4°C for 3 h. The beads were then washed three times with washing buffer and eluted in 1 × protein loading buffer. The pulldown samples were analyzed on SDS‐PAGE gel, transferred onto PVDF membrane and immunoblotted with anti‐Flag or anti‐Myc antibodies as indicated.
CRIS‐PITCh system genome editing
In order to improve the efficiency of CRISPR/Cas9 genome editing, we applied with CRIS‐PITCH system to knock‐in GFP and DsRed in the FIP1L1 and YTHDC1 genome locus as previously reported (Nakade et al, 2014; Chen et al, 2020). Firstly, the linear DNA donor was generated from PCR and sgRNAs were designed on the CRISPR/Cas9 website (https://zlab.bio/guide‐design‐resources) and transcribed using the TranscriptAid T7 kit (Thermo Scientific, K0441). Then, the Cas9 RNPs were acquired by incubating 10 μg TrueCut Cas9 Protein v2 (Thermo Scientific, A36496) with 2 μg sgRNA at 37°C for 15 min. HEK293T cells (40–60% confluent) were harvested and centrifuged at 90 g for 10 min. Mix 4 μl of solution I (2 g ATP·disodium salt and 1.2 g MgCl2·6H2O in 10 ml) with 100 μl of solution II (6 g of KH2PO4, 0.6 g of NaHCO3, and 0.2 g of glucose in 500 ml) for electroporation solutions, and resuspended 10*6 cells together with 2 μg linear DNA donor. Electroporation of above cell mixtures and Cas9 RNPs by Lonza 4D‐Nucleofector (Lonza, AAF‐1003X) was performed under an optimized program (SOLUTION: Cell Line SE, PUSLE CODE: DS150). Four days after electroporation, GFP and mScarlet positive cells were sorted and seeded into 96‐well plates by Flow cytometry (BD FACSAria II). Finally, PCR and Sanger sequencing were performed for genotyping and screening of homozygous cell lines. The Immunofluorescence and western blot were done to verify the reliability of the genome editing. All sgRNA and PCR primers are shown in Table EV3.
The phase separation and FRAP assay
The in vitro phase separation of FIP1L1 was optimized according to previous studies (Alberti et al, 2018; Greig et al, 2020). Specifically, purified FIP1L1 was diluted to 2.1 mg/ml with a normal buffer (20 mM Tris–Hcl, 150 mM Nacl, 3% PEG6000), high salt buffer (20 mM Tris–Hcl, 600 mM Nacl, 3% PEG6000) and RNA Normal buffer (100 ng/μl HEK293T total RNA, 20 mM Tris–Hcl, 150 mM Nacl, 3% PEG6000), and left for 5 min at 37°C before imaging on an Leica microscope (Leica, TCS SP8 STED 3X) at 100× magnification. For in vitro FRAP assay, pre‐bleach frames from 10 droplets were taken before samples were bleached with 80% laser power twice at maximum speed following the recovery every 1 s for up to 120 s. For in vivo FRAP, GFP‐FIP1L1 cells were transiently transfected with and DsRed‐YTHDC1 plasmid in chamber slides for 24 h. Cells were incubated in a live‐cell imaging (37°C, 5% CO2) stage top incubator (Leica TCS SP8 STED). The nuclear condensates from 15 cells were taken at maximum acquisition speed for one‐way scanning before being bleached with 10% laser power for one frame, and then fluorescence intensity was recorded every 1 s for up to 120 s. All FRAP data were normalized according to LAS AF Lite software and recovery curve was calculated using GraphPad Prism 7 software.
Author contributions
Liutao Chen: Conceptualization; data curation; software; formal analysis; validation; visualization; writing – original draft; project administration. Yonggui Fu: Conceptualization; data curation; supervision; funding acquisition; project administration; writing – review and editing. Zhijie Hu: Software; validation. Ke Deng: Software; visualization. Zili Song: Data curation; software; visualization. Susu Liu: Validation; visualization. Mengxia Li: Software; validation. Xin Ou: Software; visualization. Runze Wu: Software; validation. Mian Liu: Validation. Rui Li: Software; visualization. Shuiying Gao: Validation. Lin Cheng: Validation. Shangwu Chen: Supervision; project administration. Anlong Xu: Conceptualization; resources; data curation; supervision; funding acquisition; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
PDF+
Acknowledgments
We would like to thank Dr. Runwen Yao (Lingling Chen lab, Shanghai Institute of Biochemistry and Cell Biology) for assisting with microscope imaging. This study was supported by the National Natural Science Foundation of China (No. 91942301 and 81430099 to A.X., No. 32000450 to L.C. and No. 31971332 to Y.F.), Guangdong Basic and Applied Basic Research Foundation (No. 2020A1515010293 to L.C.), and the Fundamental Research Funds for the Central Universities, Sun Yat‐sen University (No. 2021qntd26 to L.C).
EMBO reports (2022) 23: e54686
Footnotes
Correction added on 14 October 2022, after first online publication: The Acknowledgment section has been added.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The IVT‐SAPAS raw sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE198143 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE198143). YTHDC1 IP MS data have been submitted to the Peptideatlas database (http://www.peptideatlas.org/) under access number PASS01442 (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS01442).
References
- Agresti A (2003) Categorical data analysis. Hoboken, NJ: John Wiley & Sons; [Google Scholar]
- Alberti S, Saha S, Woodruff JB, Franzmann TM, Wang J, Hyman AA (2018) A User's guide for phase separation assays with purified proteins. J Mol Biol 430: 4806–4820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, Vanacova S (2017) N6‐methyladenosine demethylase FTO targets pre‐mRNAs and regulates alternative splicing and 3′‐end processing. Nucleic Acids Res 45: 11356–11370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Machler M, Bolker BM, Walker SC (2015) Fitting linear mixed‐effects models using lme4. J Stat Softw 67: 1–48 [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K et al (2014) M(6)a RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL (2014) Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Huppertz I, Yao C, Weng L, Moresco JJ, Yates JR III, Ule J, Manley JL, Shi Y (2014) CPSF30 and Wdr33 directly bind to AAUAAA in mammalian mRNA 3′ processing. Genes Dev 28: 2370–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandola U, Das R, Panda B (2015) Role of the N6‐methyladenosine RNA mark in gene regulation and its implications on development and disease. Brief Funct Genomics 14: 169–179 [DOI] [PubMed] [Google Scholar]
- Chen B, Deng S, Ge T, Ye M, Yu J, Lin S, Ma W, Songyang Z (2020) Live cell imaging and proteomic profiling of endogenous NEAT1 lncRNA by CRISPR/Cas9‐mediated knock‐in. Protein Cell 11: 641–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Xie W, Pickering BF, Chu KL, Savino AM, Yang X, Luo H, Nguyen DT, Mo S, Barin E et al (2021) N(6)‐Methyladenosine on mRNA facilitates a phase‐separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 39: 958–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Faini M, Muckenfuss LM, Aebersold R, Jinek M (2018) Structural basis of AAUAAA polyadenylation signal recognition by the human CPSF complex. Nat Struct Mol Biol 25: 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Zhang S, Gu S, Ni X, Zeng W, Li X (2018) Useful Bicistronic reporter system for studying poly(a) site‐defining cis elements and regulation of alternative polyadenylation. Int J Mol Sci 19: 279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, Manley JL (2011) Mechanisms and consequences of alternative polyadenylation. Mol Cell 43: 853–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sun Y, Li Y, Li J, Rao X, Chen C, Xu A (2011) Differential genome‐wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high‐throughput sequencing. Genome Res 21: 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Chen L, Chen C, Ge Y, Kang M, Song Z, Li J, Feng Y, Huo Z, He G et al (2018) Crosstalk between alternative polyadenylation and miRNAs in the regulation of protein translational efficiency. Genome Res 28: 1656–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Ge Y, Sun Y, Liang J, Wan L, Wu X, Xu A (2015) IVT‐SAPAS: low‐input and rapid method for sequencing alternative polyadenylation sites. PLoS ONE 10: e0145477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zhuang X (2020) M(6)A‐binding YTHDF proteins promote stress granule formation. Nat Chem Biol 16: 955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vasic R, Song Y, Teng R, Liu C, Gbyli R, Biancon G, Nelakanti R, Lobben K, Kudo E et al (2020) M(6)a modification prevents formation of endogenous double‐stranded RNAs and deleterious innate immune responses during hematopoietic development. Immunity 52: 1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig JA, Nguyen TA, Lee M, Holehouse AS, Posey AE, Pappu RV, Jedd G (2020) Arginine‐enriched mixed‐charge domains provide cohesion for nuclear speckle condensation. Mol Cell 77: 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He PC, He C (2021) M(6) a RNA methylation: From mechanisms to therapeutic potential. EMBO J 40: e105977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, Qi M, Lu Z, Shi H, Wang J et al (2017) Ythdc2 is an N(6)‐methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res 27: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F et al (2019) Histone H3 trimethylation at lysine 36 guides m(6)a RNA modification co‐transcriptionally. Nature 567: 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, Schultz RM, Wang PJ (2018) Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet 14: e1007412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann I, Martin G, Friedlein A, Langen H, Keller W (2004) Human Fip1 is a subunit of CPSF that binds to U‐rich RNA elements and stimulates poly(a) polymerase. EMBO J 23: 616–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker‐Scharff I, Moore MJ, Park CY et al (2015) A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev 29: 2037–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackford B, Yao C, Charles GM, Weng L, Zheng X, Choi EA, Xie X, Wan J, Xing Y, Freudenberg JM et al (2014) Fip1 regulates mRNA alternative polyadenylation to promote stem cell self‐renewal. EMBO J 33: 878–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhu L, Lu C, Wang C, Wang H, Jin H, Ma X, Cheng Z, Yu C, Wang S et al (2021) circNDUFB2 inhibits non‐small cell lung cancer progression via destabilizing IGF2BPs and activating anti‐tumor immunity. Nat Commun 12: 295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y et al (2017) M(6)a mRNA methylation controls T cell homeostasis by targeting the IL‐7/STAT5/SOCS pathways. Nature 548: 338–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL et al (2015) Systematic profiling of poly(a)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet 11: e1005166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun Y, Fu Y, Li M, Huang G, Zhang C, Liang J, Huang S, Shen G, Yuan S et al (2012) Dynamic landscape of tandem 3′ UTRs during zebrafish development. Genome Res 22: 1899–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZX, Li LM, Sun HL, Liu SM (2018) Link between m6A modification and cancers. Front Bioeng Biotechnol 6: 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Zhang Z, Xue M, Zhao BS, Harder O, Li A, Liang X, Gao TZ, Xu Y, Zhou J et al (2020) N(6)‐methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG‐I. Nat Microbiol 5: 584–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C (2016) Evolution and biological roles of alternative 3′UTRs. Trends Cell Biol 26: 227–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B, Erdos G, Dosztanyi Z (2018) IUPred2A: context‐dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46: W329–W337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012b) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore YV, Elemento O, Zumbo P, Mason CE, Jaffrey SR (2012a) Gene Expression Omnibus GSE29714 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE29714). [DATASET]
- Molinie B, Wang J, Lim KS, Hillebrand R, Lu ZX, Van Wittenberghe N, Howard BD, Daneshvar K, Mullen AC, Dedon P et al (2016) M(6)A‐LAIC‐seq reveals the census and complexity of the m(6)a epitranscriptome. Nat Methods 13: 692–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S, Tsubota T, Sakane Y, Kume S, Sakamoto N, Obara M, Daimon T, Sezutsu H, Yamamoto T, Sakuma T et al (2014) Microhomology‐mediated end‐joining‐dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun 5: 5560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JW, Rissland OS, Koppstein D, Abreu‐Goodger C, Jan CH, Agarwal V, Yildirim MA, Rodriguez A, Bartel DP (2014) Global analyses of the effect of different cellular contexts on microRNA targeting. Mol Cell 53: 1031–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayler O, Hartmann AM, Stamm S (2000) The ER repeat protein YT521‐B localizes to a novel subnuclear compartment. J Cell Biol 150: 949–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR (2016a) M(6)a RNA methylation promotes XIST‐mediated transcriptional repression. Nature 537: 369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil D, Chen C, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR (2016b) Gene Expression Omnibus GSE78030 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE78030). [DATASET]
- Qiu F, Fu Y, Lu C, Feng Y, Wang Q, Huo Z, Jia X, Chen C, Chen S, Xu A (2017) Small nuclear ribonucleoprotein polypeptide A‐mediated alternative polyadenylation of STAT5B during Th1 cell differentiation. J Immunol 199: 3106–3115 [DOI] [PubMed] [Google Scholar]
- Ries RJ, Zaccara S, Klein P, Olarerin‐George A, Namkoong S, Pickering BF, Patil DP, Kwak H, Lee JH, Jaffrey SR (2019) M(6)a enhances the phase separation potential of mRNA. Nature 571: 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, He C (2017a) Dynamic RNA modifications in gene expression regulation. Cell 169: 1187–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P et al (2017b) YTHDC1 mediates nuclear export of N(6)‐methyladenosine methylated mRNAs. Elife 6: e31311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB (2008) Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C (2017) YTHDF3 facilitates translation and decay of N(6)‐methyladenosine‐modified RNA. Cell Res 27: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wei J, He C (2019) Where, when, and how: context‐dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74: 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR III, Frank J, Manley JL (2009) Molecular architecture of the human pre‐mRNA 3′ processing complex. Mol Cell 33: 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA (2009) Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol 16: 1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Manley JL (2017) Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 18: 18–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veloso A, Kirkconnell KS, Magnuson B, Biewen B, Paulsen MT, Wilson TE, Ljungman M (2014) Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res 24: 896–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Vasaikar S, Shi Z, Greer M, Zhang B (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45: W130–W137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Kong S, Tao M, Ju S (2020) The potential role of RNA N6‐methyladenosine in cancer progression. Mol Cancer 19: 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, He C (2014) Dynamic RNA modifications in posttranscriptional regulation. Mol Cell 56: 5–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C (2015a) N(6)‐methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu J, Huang BO, Xu YM, Li J, Huang LF, Lin J, Zhang J, Min QH, Yang WM et al (2015b) Mechanism of alternative splicing and its regulation. Biomed Rep 3: 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS (2017) Regulation of m6A transcripts by the 3′→5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell 68: 374–387 [DOI] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY et al (2016) Nuclear m(6)a reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507–519 [DOI] [PubMed] [Google Scholar]
- Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, Min J (2014) Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10: 927–929 [DOI] [PubMed] [Google Scholar]
- Yao C, Choi EA, Weng L, Xie X, Wan J, Xing Y, Moresco JJ, Tu PG, Yates JR III, Shi Y (2013) Overlapping and distinct functions of CstF64 and CstF64tau in mammalian mRNA 3′ processing. RNA 19: 1781–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, Hankey W, Wagner EJ, Li W, Wang Q (2019) Alternative polyadenylation of mRNA and its role in cancer. Genes Dis 8: 61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H et al (2018) VIRMA mediates preferential m(6)a mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov 4: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, He C (2015) RNA N6‐methyladenosine methylation in post‐transcriptional gene expression regulation. Genes Dev 29: 1343–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Lv J, Yu H, Han J, Yang X, Feng D, Wu Q, Yuan B, Lu Q, Yang H (2020) Mechanism of RNA modification N6‐methyladenosine in human cancer. Mol Cancer 19: 104 [DOI] [PMC free article] [PubMed] [Google Scholar]