

Abstract
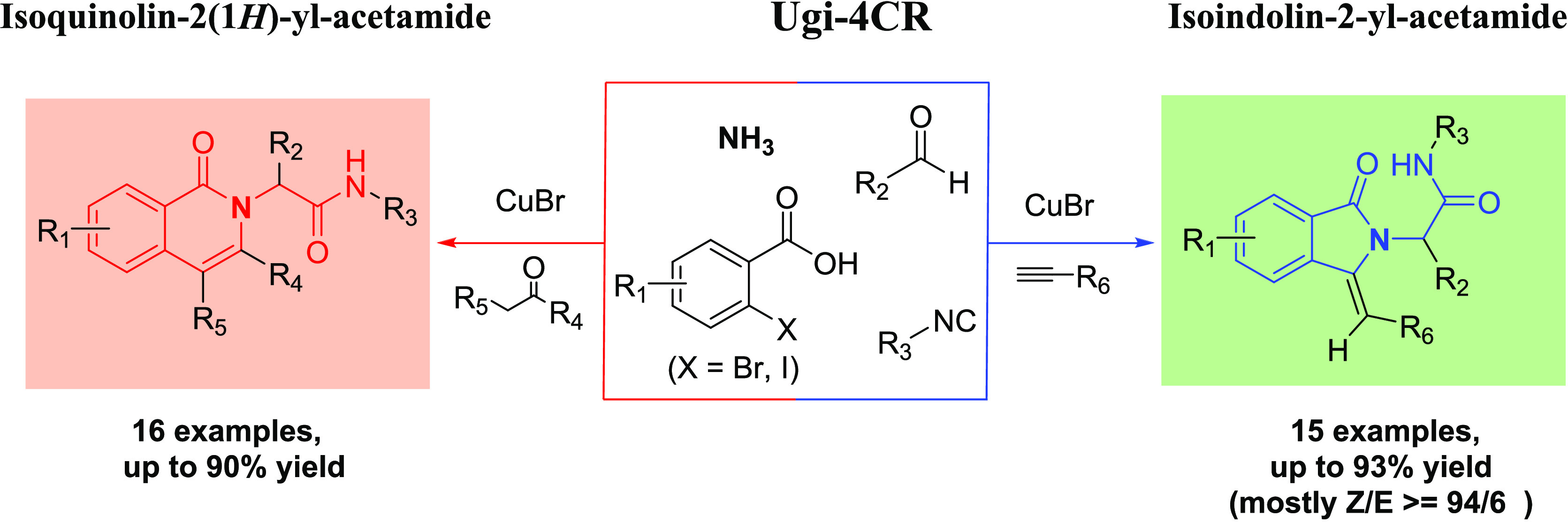
We achieved a divergent synthesis of isoquinolin-2(1H)-yl-acetamides (16 examples, up to 90% yields) and regioselective isoindolin-2-yl-acetamides (14 examples, up to 93% yields) in moderate to good yields by reacting various substituted ethanones or terminal alkynes with Ugi-4CR intermediates via an ammonia-Ugi-4CR/Copper(I)-catalyzed annulation sequence reaction. The same intermediate thus gives 2D distant but 3D closely related scaffolds, which can be of high interest in exploiting chemistry space on a receptor. The scopes and limitations of these efficient sequence reactions are described, as well as gram-scale synthesis.
Introduction
Multicomponent reactions (MCRs) and post transformations of MCR products have become progressively popular, which has made them some of the most successful methods leading to the rapid generation of small-molecule library of high structural diversity and molecular complexity.1 The Ugi reaction, one of the best-known MCRs, with the advantage of atom economy and environmental benefit, could typically afford a linear bis amide backbone.2 However, linear bis amides often have issues with stability, solubility, and distribution–metabolism–pharmacokinetic (DMPK) and are not a preferred scaffold in medicinal chemistry.3 Therefore, Ugi-4CR and its post-amide-cyclization reactions are widely used in medicinal chemistry research due to the diversity of scope and ability to improve the metabolic stability of final products.4,5 Isoquinolin-2(1H)-yl-acetamide and isoindolin-2-yl-acetamide have attracted increasing attention due to their possible diverse biological activities. The isoquinolin-2(1H)-yl-acetamide scaffold, for example, was found in P2X7 inhibitors I, proteasome inhibitors II or TLR agonists III (Figure 1A).6−8 Likewise, the isoindolin-2-yl-acetamide also appears in the FDA-proved anticancer drug Lenalidomide,9 as well as the antimicrobial compound IV or the EGFR inhibitor EAI045 (Figure 1B).10−12
Figure 1.
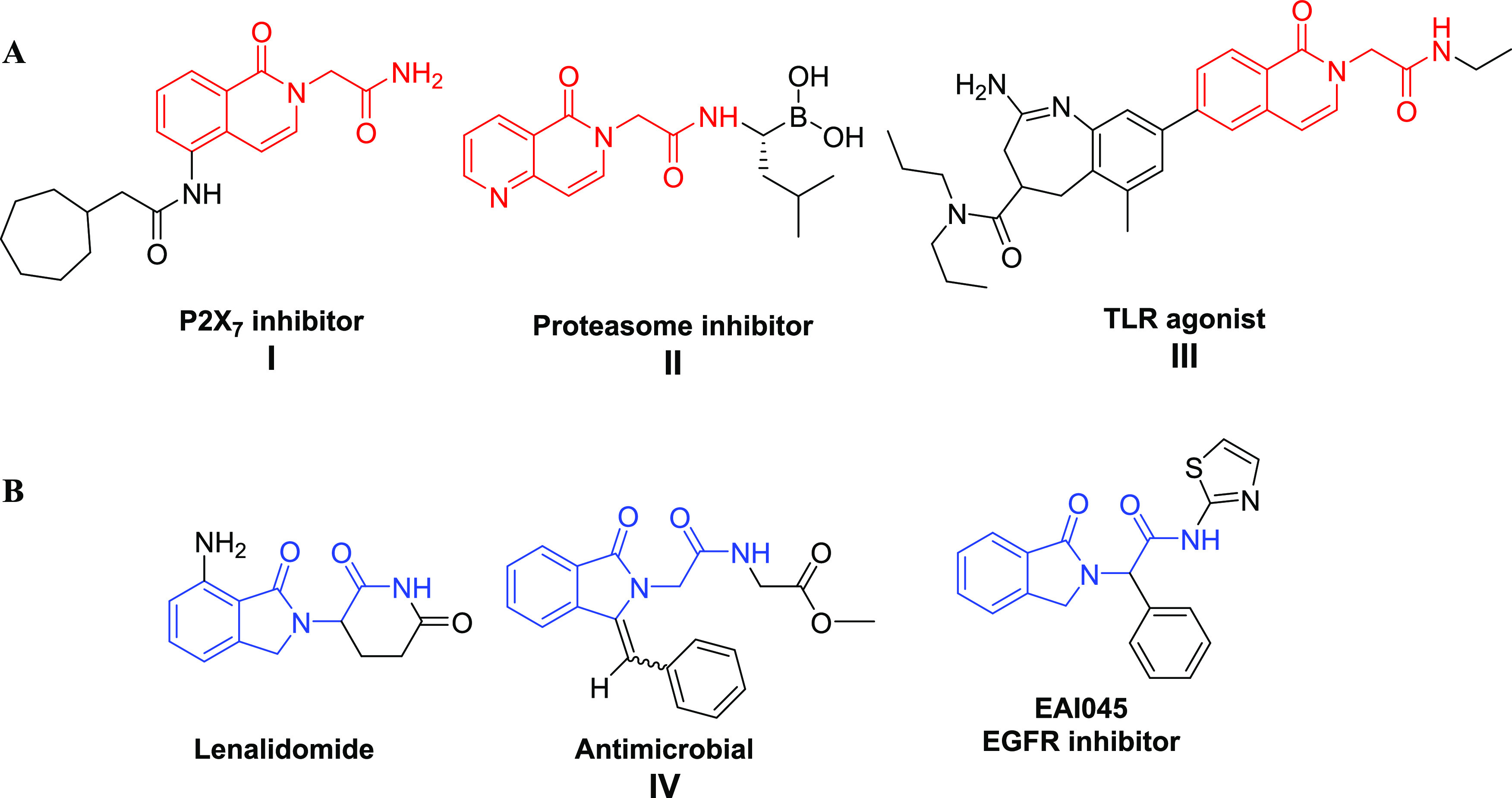
Representative bioactive molecules with isoquinolin-2(1H)-yl-acetamide scaffold (A) and isoindolin-2-yl-acetamide skeleton (B).
Not surprisingly, new and superior ways to construct the isoquinolin-2(1H)-yl-acetamide scaffold or isoindolin-2-yl-acetamide skeleton are in high demand. Yang’s group published a Ugi-4CR/Pd-catalyzed intramolecular arylation reaction to efficiently afford the tricyclic isoquinolin-2(1H)-yl-acetamides.13
Recently, our group reported a two-step synthesis of privileged tetracyclic isoquinolin-2(1H)-yl-acetamide scaffold via an ammonia-Ugi-4CR/copper-catalyzed annulation sequence (Scheme 1A).14 As for the isoindolin-2-yl-acetamides (Scheme 1B), Ibrahim synthesized a series of (1-benzylidene-3-oxoisoindolin-2-yl)-acetamide derivatives from 3-benzalphthalide and the corresponding amino acids.10 Most recently, Li’s group developed an efficient protocol to construct a DNA-encoded, isoindolin-2-yl-acetamide-based chemical library with potential lenalidomide-like pharmacological properties.15 However, to date, no article has achieved the fast synthesis of both isoquinolin-2(1H)-yl-acetamide and isoindolin-2-yl-acetamide derivatives from a common Ugi MCR-based precursor.
Scheme 1. Strategies of Isoquinolin-2(1H)-yl-acetamide and Isoindolin-2-yl-acetamide.
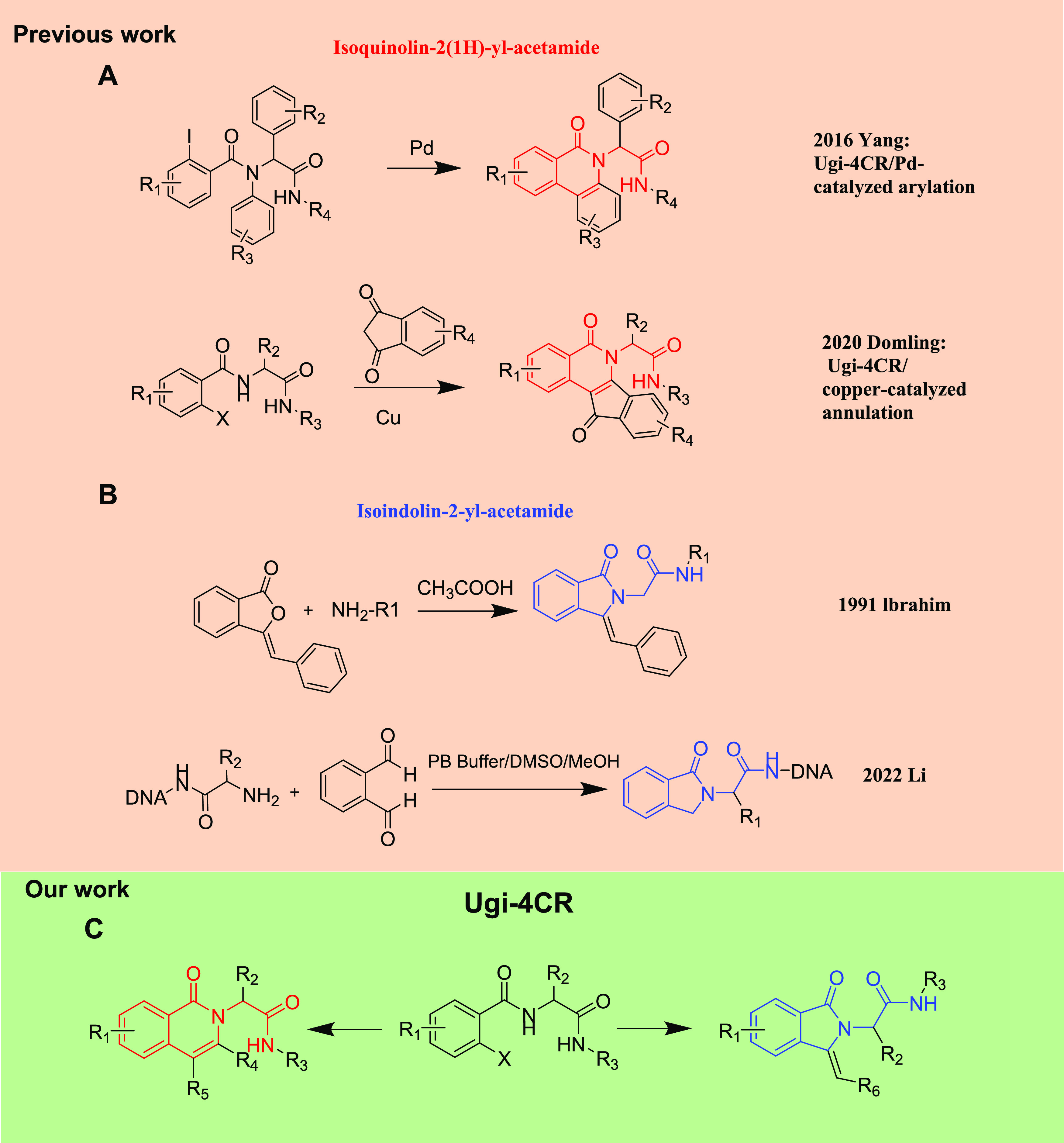
Based on previous works on copper-catalyzed cyclization reactions16 and our ongoing experience in MCR chemistry and post-MCR modifications,17 we hypothesized that two different polycyclic heterocycles could be accessed by a Ugi/Cu-catalyzed annulation sequence reaction between Ugi adducts and different corresponding reagents. Hence, we report herein the synthesis of either isoquinolin-2(1H)-yl-acetamides or isoindolin-2-yl-acetamides in moderate to good yields by adopting Cu(I)-catalyzed C–C coupling/annulation reaction of the C(sp2)–I/Br bond of Ugi-4CR adducts with substituted ethanones or terminal alkynes, respectively (Scheme 1C).
Results and Discussion
First, we synthesized 11 Ugi adducts in 27–58% yields via Ugi-ammonia-4CR based on our previous work (Scheme 2).4,14
Scheme 2. Ugi Adducts in This Work,,,

Reaction conditions: carboxylic acid (2 mmol), 25% ammonia solution (2.4 mmol), aldehyde (2 mmol), isocyanide (2 mmol), TFE (2 mL), 60 °C, overnight.
Yield refers to the purified products.
Published Ugi adducts from our previous articles.
New synthesized Ugi adducts in this work.
Then, the Ugi adduct 1a and acetophenone 2a were selected for the model copper-catalyzed cyclization reaction for the synthesis of isoquinolin-2(1H)-yl-acetamide 3a. The target compound 3a could be achieved in 62% yield by reacting 1a with 1.5 equimolar 2a in DMSO at 80 °C for 16 h under N2 in the presence of 10 mol % CuCl and 2 equivalent Cs2CO3 (Table 1, entry 1). Further optimization was done by variation of the nature of the Cu catalyst, base, solvent, reaction time and temperature (Table 1). CuCl2, CuSO4, and Cu2O all failed to improve yield, affording 3a in 41, 42, and 39%, respectively (entries 2–4). Comparable to CuCl, CuBr2 and CuI could achieve 63 and 61% yield (entries 5 and 6). When replacing CuCl with CuBr, the yield of 3a slightly increased to 67% (entry 7). K2CO3 and Na2CO3 failed to give better yield compared to Cs2CO3, only trace products were obtained (entries 8 and 9). Further attempts to change the solvent DMSO to MeCN, dioxane, MeOH, toluene, THF and DMF didn’t achieve any yield improvements (entries 10–15). To our delight, increasing the temperature from 80 to 90 or 100 °C provided higher yields of 79% and 78%, respectively (entries 16 and 17). Furthermore, decreasing the reaction time significantly reduced the yield (entries 18–20), and likewise, using microwave irradiation to promote the reaction was not advantageous, resulting in a lower yield (33%, entry 21). Therefore, the optimal reaction condition is: Ugi intermediate 1a (0.3 mmol), acetophenone 2a (0.45 mmol), 10 mol % CuBr, and 2.0 equiv of Cs2CO3 in DMSO (2 mL) at 90 °C for 16 h (entry 16). At the same time, we also screened the reaction condition for the synthesis of isoindolin-2-yl-acetamide 5a and found the best condition as follows: Ugi intermediate 1a (0.3 mmol), phenylethyne 4a (0.45 mmol), 20 mol % CuBr, and 2.0 equiv of K2CO3 in PEG (2 mL) at 100 °C for 2 h (Table S2, entry 13).
Table 1. Optimization of Reaction Conditions.

| entry | catalyst (10 mol %) | base | solvent | time (h) | temp (°C) | yield 3a (%) |
|---|---|---|---|---|---|---|
| 1 | CuCl | Cs2CO3 | DMSO(dry) | 16 | 80 | 62a |
| 2 | CuCl2 | Cs2CO3 | DMSO(dry) | 16 | 80 | 41b |
| 3 | CuSO4 | Cs2CO3 | DMSO(dry) | 16 | 80 | 42b |
| 4 | Cu2O | Cs2CO3 | DMSO(dry) | 16 | 80 | 39b |
| 5 | CuBr2 | Cs2CO3 | DMSO(dry) | 16 | 80 | 63b |
| 6 | CuI | Cs2CO3 | DMSO(dry) | 16 | 80 | 61b |
| 7 | CuBr | Cs2CO3 | DMSO(dry) | 16 | 80 | 67b |
| 8 | CuBr | K2CO3 | DMSO(dry) | 16 | 80 | trace |
| 9 | CuBr | Na2CO3 | DMSO(dry) | 16 | 80 | trace |
| 10 | CuBr | Cs2CO3 | MeCN | 16 | 80 | 8b |
| 11 | CuBr | Cs2CO3 | dioxane | 16 | 80 | 49b |
| 12 | CuBr | Cs2CO3 | MeOH | 16 | 80 | 16b |
| 13 | CuBr | Cs2CO3 | toluene | 16 | 80 | trace |
| 14 | CuBr | Cs2CO3 | THF | 16 | 80 | trace |
| 15 | CuBr | Cs2CO3 | DMF | 16 | 80 | 30b |
| 16 | CuBr | Cs2CO3 | DMSO(dry) | 16 | 90 | 79a |
| 17 | CuBr | Cs2CO3 | DMSO(dry) | 16 | 100 | 78a |
| 18 | CuBr | Cs2CO3 | DMSO(dry) | 4 | 80 | 37b |
| 19 | CuBr | Cs2CO3 | DMSO(dry) | 8 | 80 | 43b |
| 20 | CuBr | Cs2CO3 | DMSO(dry) | 12 | 80 | 55b |
| 21c | CuBr | Cs2CO3 | DMSO(dry) | 2 | 80 | 30b |
Reaction conditions: 1a (0.3 mmol), 2a (0.45 mmol), catalyst (10 mmol %), base (0.6 mmol), solvent (2 mL), isolated yields.
Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), catalyst (10 mmol %), base (0.2 mmol), solvent (1 mL), HPLC yields.
Microwave.
With optimized conditions in hand, we then investigated the substrate scope and limitations toward the corresponding isoquinolin-2(1H)-yl-acetamides 3a–3p (Scheme 3) and isoindolin-2-yl-acetamides 5a–5o (Scheme 4). First, we summarized the scope for the synthesis of isoquinolin-2(1H)-yl-acetamide derivatives. Overall, paraformaldehyde-based Ugi starting materials were tolerated in the domino reactions to efficiently give the desired products (3a–3p). Various substituted ethanones performed well with tert-butyl isocyanide-derived Ugi adducts in the cyclization reaction to give corresponding products (3a–3j) in moderate to good yields (53–90%). Aryl-substituted ethanones (acetophenones) proceeded smoothly to afford the isoquinolin-2(1H)-yl-acetamide derivatives in good yield (3a–3e). Among them, acetophenone gave optimal yield (3a, 79%), weak electron-withdrawing groups such as 4-Br resulted in slightly lower yield (3b, 70%), likewise, double-substituted ethanones like 1-phenyl-2-methyl-ethanone (3c, 62%) or 1,2-diphenyl-ethanone (3e, 70%) also gave decreased yields. 1-Aceto-naphthone gave 65% yields (3d).
Scheme 3. Substrate Scope for the Synthesis of Isoquinolin-2(1H)-yl-acetamides,

Reaction conditions: 1 (0.3 mmol), 2 (0.45 mmol), Cs2CO3 (0.6 mmol), CuBr (0.03 mmol), DMSO (2 mL), 90 °C, 16 h.
Yield refers to the purified products through a single step.
Scheme 4. Substrate Scope for the Synthesis of Isoindolin-2-yl-acetamides,,

Reaction conditions: 1 (0.3 mmol), 4 (0.45 mmol), K2CO3 (0.6 mmol), CuBr (0.06 mmol), PEG-400 (2 mL), 100 °C, 2 h.
Yield refers to the purified products through a single step.
E/Z was calculated based on 1H NMR.
When cyclohexanone and cyclopentanone were employed, the desired compounds could be obtained in 75% (3f) and 53% (3g) yields, comparable to the yields achieved by the above aromatic ethanones. Interestingly, acetone worked very well, providing the target compound (3h) in 90% yield. Noteworthy, acetaldehyde was also tolerated in this reaction, yielding 60% of 3i. The substituted Ugi intermediate 4-Br-2-iodobenzamide worked also well and resulted in the formation of 3j (53% yield). When Ugi adducts formed by tert-octyl-isocyanide were applied to the annulation reaction, the final products (3k–3o) were obtained in moderate to good yields, whereas the cyclohexyl isocyanide-based Ugi substrate provided compound 3p in 48% yield. We also observed limitations of the reaction (Table S4, 3q and 3r). 4-F-acetophenone yielded only trace amounts of product 3q, possibly due to lower acetyl activity in the structure. In addition, 4-NO2-2-iodobenzamide even couldn’t undergo the reaction, probably because it is difficult to achieve oxidative addition with acetophenone. The structures of 3e and 3i were unambiguously elucidated by X-ray.
Then, the scope for constructing the isoindolin-2-yl-acetamides was investigated (Scheme 4). Different terminal alkynes could be successfully employed in the reaction and produce the desired target compounds (5a–5d) with excellent regioselectivity (Z/E ≥ 94/6), aryl alkynes such as phenylethyne (5a), 4-Br-phenylethyne (5b), 2-pyridylethyne (5c) gave good yields (81–93%), while alkyl alkyne like trimethylsilylacetylene (5d) resulted in 15% yield. 5-Methoxy-2-bromobenzamide and ethyl aldehyde originated Ugi adducts were also tolerated, providing compounds 5e (Z/E = 95/5, 90%) and 5f (Z/E = 88/12, 72%) with good regioselectivity.
Aside from Ugi intermediates based on tert-butyl isocyanide, the Ugi adducts generated from other alkyl isonitriles like tert-octyl-isocyanide and cyclohexyl isocyanide afforded final products 5g–5m in moderate to excellent yields.
Gratifyingly, Ugi synthons from two aryl nitriles, phenylethyl isocyanide and 2-methoxyphenyl isocyanide could also result in corresponding isoindolin-2-yl-acetamides (5n–5o) in good yield, thus widely broadening the scope of the reaction. It should be noted that when the Ugi product from cyclohexyl isonitrile reacted with 2-pyridineacetylene, we could obtain a product (5k, Z/E = 51/49) with almost equal amounts of Z and E forms in 73% yield, which was further purified to give 5l (Z/E = 96/4) and 5m (Z/E = 9/91) with better regioselectivity. The 2D NMR correlations were done to assign the structure of 5a which was subsequently unambiguously determined by X-ray (Figure S5).
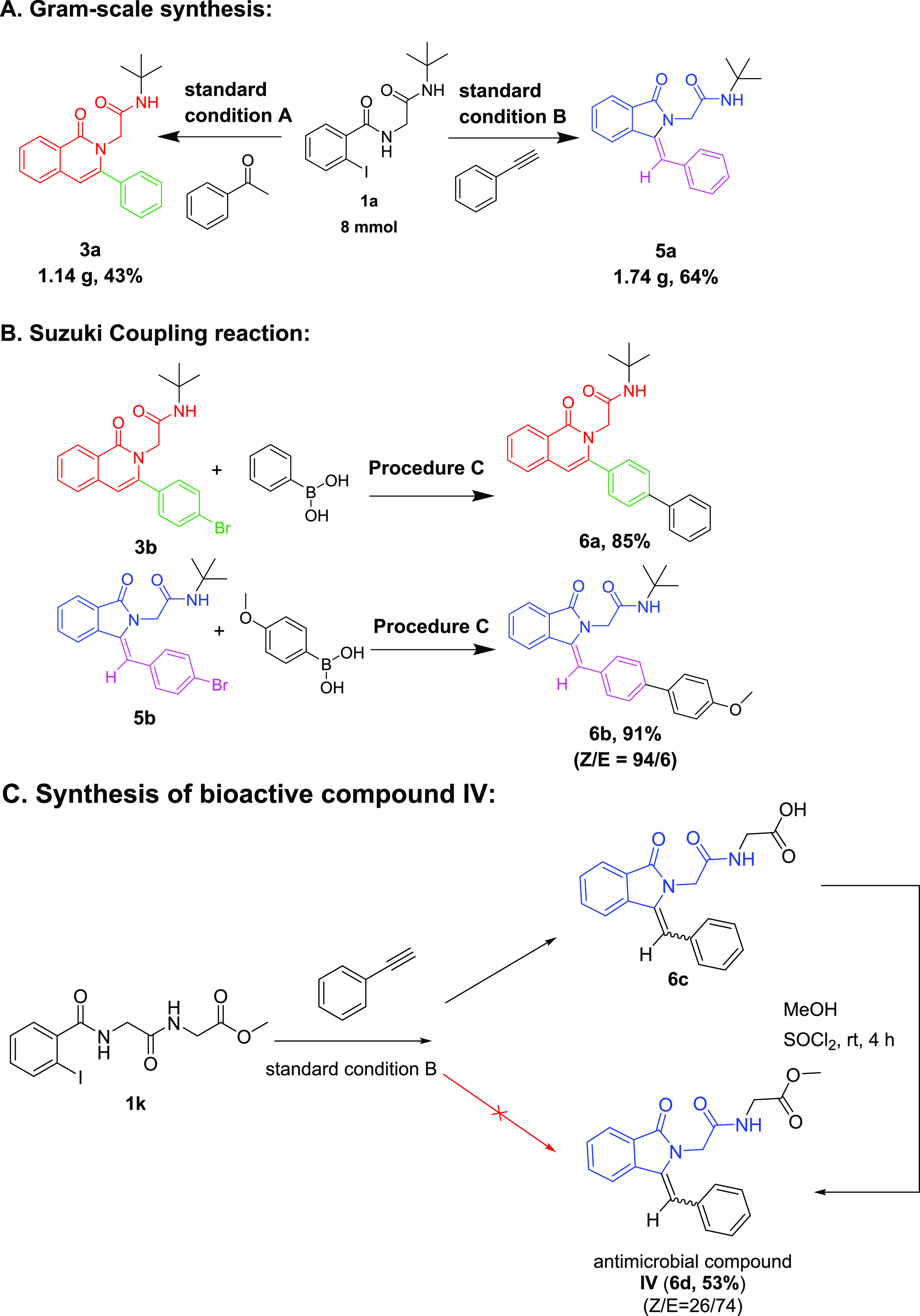
To support the preparative usefulness of our method, gram-scale experiments were carried out for the synthesis of isoquinolin-2(1H)-yl-acetamide 3a and isoindolin-2-yl-acetamide 5a in moderate yields (Scheme 5A). Two CuBr-promoted cyclization reactions of Ugi-4CR-based 2-iodobenzamide 1a with acetophenone and phenylethyne were conducted on an 8 mmol scale, producing 3a (1,14 g) and 5a (1,74g) in 43% and 64% yield, respectively. To further evaluate the potential of the above-described two scaffolds, we performed the Suzuki coupling late-stage functionalization. 3b and 5b were coupled with phenylboronic acid and 4-methoxyphenyl boronic acid separately, giving the corresponding products 6a and 6b in good yields via Pd-catalyzed Suzuki reaction (Scheme 5B). We also applied our method to synthesize antibacterial compound IV (Scheme 5C); however, instead of obtaining the normal cyclization product IV directly, we obtained the carboxylic acid 6c, which may be due to the similar hydrolysis of the methyl ester occurring in the process as in our previous work.4 Then, we reacted 6c with MeOH and SOCl2 to realize product IV (6d, Z/E = 26/74) in 53% yield.
Scheme 5. Gram-Scale Reaction, Suzuki Coupling Functionalization, and Bioactive Compound Synthesis.
The herein-reported two complementary syntheses of two related scaffolds are based on a common Ugi-4CR intermediate. Switching between two related scaffolds is often applied in medicinal chemistry and sometimes called “scaffold hopping”. It is of high importance during lead optimization as the two related scaffolds might bind similarly into the same receptor site but might have different pharmacokinetic/pharmacodynamic (PKPD) parameters. Clearly, the two scaffolds align well in 3D (Figure 2) and are therefore potential bioisosteres.
Figure 2.
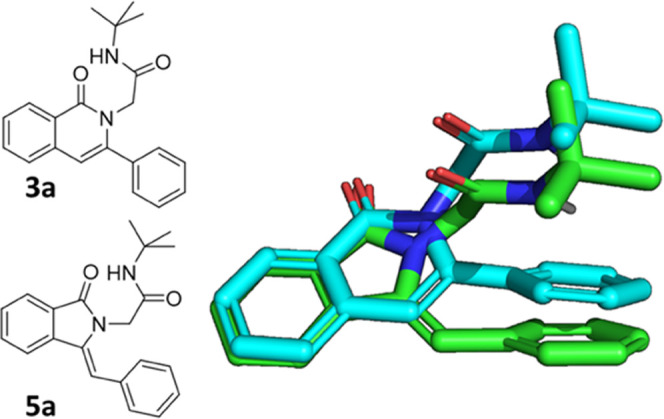
Alignment of energy-minimized isoquinolinone 3a and isoindolinone 5a supporting the similar 3D shape of the two scaffolds.
Conclusions
In summary, we developed an efficient Ugi-4CR/copper(I) catalytic system for the synthesis of two different bioactive potential scaffolds: isoquinolin-2(1H)-yl-acetamide and isoindolin-2-yl-acetamide, with the advantages of atom economy, good yields and absence of ligands. Additionally, product diversity can be achieved not only through the Ugi starting materials aldehydes, isocyanides, and 2-halogene benzoic acids but also by different substituted ethanones and terminal alkynes. The proposed two different reaction mechanisms were also discussed in the Supporting Information. Having access to two different scaffolds from a common MCR precursor potentially facilitates structure–activity relationship (SAR) enormously, while allowing optimization of “druglike” properties through scaffold hopping.
Experimental Section
General Information
Nuclear magnetic resonance spectra were recorded on a Bruker Avance 500 spectrometer. Chemical shifts for 1H NMR were reported relative to TMS (δ 0 ppm) or internal solvent peak (CDCl3 δ 7.26 ppm, CD3OD δ 3.31 ppm or D2O δ 4.79 ppm) and coupling constants were in hertz (Hz). The following abbreviations were used for spin multiplicity: s = singlet, d = doublet, t = triplet, dt = double triplet, ddd = doublet of double doublet, m = multiplet, and br = broad. Chemical shifts for 13C NMR reported in ppm relative to the solvent peak (CDCl3 δ 77.23 ppm, DMSO δ 39.52 ppm, CD3OD δ 49.00 ppm). Flash chromatography was performed on a Grace Reveleris X2 using Grace Reveleris Silica columns (12 g) and a gradient of petroleum ether/ethyl acetate (0–100%) or dichloromethane/methanol (0–20%) was applied. Thin layer chromatography was performed on Fluka precoated silica gel plates (0.20 mm thick, particle size 25 μm). All isocyanides were made in-house via the Ugi procedure.18 Benzoic acids, ethanones (2), terminal alkynes (4), and other reagents were purchased from Sigma Aldrich, ABCR, Acros, Fluorochem, AK Scientific, Combiblocks, or A2B and were used without further purification. Mass spectra were measured on a Waters Investigator Supercritical Fluid Chromatograph with a 3100 MS Detector (ESI) using a solvent system of methanol and CO2 on a Viridis silica gel column (4.6 × 250 mm2, 5 μm particle size) and reported as (m/z). High-resolution mass spectra (HRMS) were recorded using an LTQ-Orbitrap-XL (Thermo Fisher Scientific; ESI pos. mode) at a resolution of 60000@m/z400. Melting points were obtained on a melting point apparatus and were uncorrected. Yields given refer to chromatographically purified compounds unless otherwise stated. Compounds 1a, 1d, 1f, and 1j were all prepared following our reported literature4,14 (Table S3).
General Experimental Procedure and Characterization
General Procedure for Ugi-4CR Products
To a stirred solution of the carboxylic acid (2 mmol, 1.0 equiv) in 2,2,2-trifluoroethanol (2 mL) in a 5 mL vial, 0.3 mL of 25% ammonia solution (2.4 mmol, 1.2 equiv) was added. Aldehyde (2 mmol, 1.0 equiv) and isocyanide (2 mmol, 1.0 equiv) were then introduced into the mixture, the vial was capped, and then the reaction mixture was placed in a heated metal block and stirred at 60 °C overnight. After the completion of the reaction, the solvent was removed in vacuo and the crude products were purified by column chromatography to give the desired products 1b, 1c, 1e, and 1g–1i (Table S3).
General Procedure A
A sealed tube was charged with Ugi adduct 1 (0.2/0.3 mmol, 1.0 equiv), substituted ethanones (0.3/0.45 mmol, 1.5 equiv), CS2CO3 (0.4/0.6 mmol, 2.0 equiv), and the CuBr (0.02/0.03 mmol, 0.1 equiv). DMSO (1/2 mL) was added, and the mixture was stirred under N2 at 90 °C for 16 h. After the reaction, saturated aqueous NaCl (10 mL) and EtOAc (10 mL) were added successively to the cooled reaction mixture. The organic phase was separated, and the aqueous phase was further extracted with EtOAc (3 × 10 mL). Then, the combined organic layers were dried over anhydrous Na2SO4 and concentrated. The residues were purified by column chromatography on silica gel to afford the compounds 3a–3p.
General Procedure B
A sealed tube was charged with Ugi adduct 1 (0.2/0.3 mmol, 1.0 equiv), terminal alkynes (0.3/0.45 mmol, 1.5 equiv), K2CO3(0.4/0.6 mmol, 2.0 equiv), and the CuBr(0.04/0.06 mmol, 0.2 equiv). PEG-400 (1/2 mL) was added, and the mixture was stirred under N2 at 100 °C for 2 h. After the reaction, the solvent was removed in vacuo and the residues were purified by column chromatography on silica gel to afford the compounds 5a–5n.
Procedure C
Compound 3b or 5b (0.15 mmol, 1.0 equiv) and the corresponding phenylboronic acid (0.225 mmol, 1.5 equiv) were placed in a 10 mL tube, and toluene/ethanol (v/v = 5:1) (3 mL) and sat. NaHCO3 (3 mL) were added. The tube was flushed with N2 for 10 min, then Pd(dppf)Cl2 (0.015 mmol, 0.1 equiv) was added, and the tube was sealed. The mixture was allowed to react at 90 °C in an oil bath for 12 h. Then, the reaction mixture was cooled to room temperature and treated with H2O and extracted with EtOAc. The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After the removal of EtOAc, the residues were purified by silica gel column chromatography to afford Suzuki coupling products 6a and 6b.
4-Bromo-N-(2-(tert-butylamino)-2-oxoethyl)-2-iodobenzamide (1b)
It was synthesized according to the procedure of Ugi-4CR reaction on a 2 mmol scale to afford 1b (236 mg, 27%) as a white solid. mp: 181–182 °C. Rf = 0.59 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 1.9 Hz, 1H), 7.48 (dd, J = 1.9, 8.2 Hz, 1H), 7.24 (d, J = 8.2 Hz, 1H), 7.18 (t, J = 5.3 Hz, 1H), 6.46 (s, 1H), 4.02 (d, J = 5.0 Hz, 2H), 1.34 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.9, 167.5, 142.2, 140.1, 131.4, 129.5, 124.7, 93.3, 51.8, 44.6, 28.9. HRMS (ESI) m/z: [M + H]+ calcd for C13H17BrIN2O2, 438.9518, found, 438.9510.
2-Iodo-N-(2-oxo-2-((2,4,4-trimethylpentan-2-yl)amino)ethyl)benzamide (1c)
It was synthesized according to the procedure of Ugi-4CR reaction on a 2 mmol scale to afford 1c (285 mg, 38%) as a white solid. mp: 158–160 °C. Rf = 0.38 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.87 (dd, J = 1.1, 8.0 Hz, 1H), 7.44–7.32 (m, 2H), 7.11 (td, J = 1.9, 7.5 Hz, 1H), 6.81 (t, J = 5.1 Hz, 1H), 6.13 (s, 1H), 4.05 (d, J = 5.2 Hz, 2H), 1.76 (s, 2H), 1.42 (s, 6H), 1.00 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.7, 167.3, 141.3, 140.2, 131.6, 128.5, 128.3, 92.7, 55.9, 51.4, 44.7, 31.8, 31.6, 29.4. HRMS (ESI) m/z: [M + H]+ calcd for C17H26IN2O2, 417.1039, found, 417.1021.
N-(1-(tert-Butylamino)-1-oxopropan-2-yl)-2-iodobenzamide (1e)
It was synthesized according to the procedure of Ugi-4CR reaction on a 2 mmol scale to afford 1e (285 mg, 38%) as a brown oil. Rf = 0.29 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.93–7.75 (m, 1H), 7.37–7.32 (m, 2H), 7.08 (ddd, J = 3.3, 5.8, 7.9 Hz, 1H), 6.69 (d, J = 7.7 Hz, 1H), 6.40 (s, 1H), 4.61 (dd, J = 6.9, 7.7 Hz, 1H), 1.46 (d, J = 6.9 Hz, 3H), 1.35 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 171.0, 169.1, 141.6, 140.1, 131.4, 128.4, 128.3, 92.6, 51.6, 50.0, 28.9, 18.5. HRMS (ESI) m/z: [M + H]+ calcd for C14H20IN2O2, 375.0570, found, 375.0557.
2-Bromo-N-(2-(tert-butylamino)-2-oxoethyl)-5-methoxybenzamide (1g)
It was synthesized according to the procedure of Ugi-4CR reaction on a 2 mmol scale to afford 1g (287 mg, 42%) as a white solid. mp: 153–155 °C. Rf = 0.30 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.45 (dd, J = 0.9, 8.8 Hz, 1H), 7.23 (t, J = 5.1 Hz, 1H), 7.05 (dd, J = 1.0, 3.1 Hz, 1H), 6.83 (ddd, J = 0.9, 3.2, 8.8 Hz, 1H), 6.55 (s, 1H), 4.08 (d, J = 5.2 Hz, 2H), 3.79 (d, J = 0.9 Hz, 3H), 1.37 (d, J = 1.1 Hz, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.8, 167.6, 158.9, 137.7, 134.3, 118.0, 114.6, 109.6, 55.7, 51.7, 44.5, 28.8. HRMS (ESI) m/z: [M + H]+ calcd for C14H20BrN2O3, 343.0657, found, 343.0645.
2-Iodo-N-(2-oxo-2-(phenethylamino)ethyl)benzamide (1h)
It was synthesized according to the procedure of Ugi-4CR reaction on a 2 mmol scale to afford 1h (318 mg, 39%) as a white solid. mp: 167–169 °C. Rf = 0.44 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.87–7.83 (m, 1H), 7.38–7.34 (m, 2H), 7.30–7.26 (m, 2H), 7.23–7.16 (m, 3H), 7.11 (ddd, J = 3.1, 6.1, 7.9 Hz, 1H), 6.80 (t, J = 5.3 Hz, 1H), 6.63 (t, J = 5.9 Hz, 1H), 4.10 (d, J = 5.2 Hz, 2H), 3.55 (td, J = 5.8, 7.2 Hz, 2H), 2.83 (t, J = 7.2 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.7, 168.4, 141.2, 140.1, 138.7, 131.6, 128.9, 128.8, 128.5, 128.3, 126.7, 92.6, 43.8, 40.9, 35.7. HRMS (ESI) m/z: [M + H]+ calcd for C17H18IN2O2, 409.0413, found, 409.0400.
2-Iodo-N-(2-((2-methoxyphenyl)amino)-2-oxoethyl)benzamide (1i)
It was synthesized according to the procedure of Ugi-4CR reaction on a 2 mmol scale to afford 1i (238 mg, 29%) as a white solid. 181–183 °C. Rf = 0.54 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.30 (dd, J = 1.7, 8.0 Hz, 1H), 8.26 (s, 1H), 7.89 (dd, J = 1.1, 8.0 Hz, 1H), 7.47 (dd, J = 1.9, 7.6 Hz, 1H), 7.40 (td, J = 1.3, 7.6 Hz, 1H), 7.13 (td, J = 1.8, 7.6 Hz, 1H), 7.08 (td, J = 1.7, 7.9 Hz, 1H), 6.96 (td, J = 1.4, 7.9 Hz, 1H), 6.92–6.87 (m, 1H), 6.73 (s, 1H), 4.33 (d, J = 5.2 Hz, 2H), 3.89 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.6, 166.2, 148.0, 141.3, 140.1, 131.5, 128.5, 128.2, 126.9, 124.4, 121.1, 120.0, 110.1, 92.5, 55.8, 44.5. HRMS (ESI) m/z: [M + H]+ calcd for C16H16IN2O3, 411.0206, found, 411.0190.
Methyl (2-iodobenzoyl)glycylglycinate (1j)
It was synthesized according to the procedure of Ugi-4CR reaction on a 2 mmol scale to afford 1j (293 mg, 39%) as a white solid. 100–102 °C. Rf = 0.24 (3% MeOH/DCM). 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.0 Hz, 1H), 7.39 (dd, J = 7.6, 1.8 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.30 (d, J = 6.4 Hz, 1H), 7.15–7.02 (m, 2H), 4.22 (d, J = 5.3 Hz, 2H), 4.04 (d, J = 5.5 Hz, 2H), 3.71 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.1, 169.9, 169.1, 141.2, 140.0, 131.5, 128.5, 128.3, 92.6, 52.5, 43.6. HRMS (ESI) m/z: [M + H]+ calcd for C12H14IN2O4I, 376.9998, found, 376.9988.
N-(tert-Butyl)-2-(1-oxo-3-phenylisoquinolin-2(1H)-yl)acetamide (3a)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3a (79 mg, 79%) as a yellow solid. mp: 203–205 °C. Rf = 0.48 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 8.0 Hz, 1H), 7.68–7.62 (m, 1H), 7.51–7.43 (m, 7H), 6.48 (s, 1H), 5.92 (s, 1H), 4.43 (s, 2H), 1.31 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.0, 163.5, 143.9, 136.7, 135.7, 132.8, 129.4, 129.3, 128.7, 128.2, 126.9, 126.1, 124.8, 108.2, 51.5, 50.4, 28.8. HRMS (ESI) m/z: [M + H]+ calcd for C21H23O2N2, 335.1754, found, 335.1746.
2-(3-(4-Bromophenyl)-1-oxoisoquinolin-2(1H)-yl)-N-(tert-butyl)acetamide (3b)
It was synthesized according to procedure A on a 0.2 mmol scale to yield final compound 3b (58 mg, 70%) as a white solid. mp: 214-216 °C. Rf = 0.31 (25% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.43 (dd, J = 8.1, 1.3 Hz, 1H), 7.67 (td, J = 7.4, 1.5 Hz, 1H), 7.59 (d, J = 8.5 Hz, 2H), 7.54–7.47 (m, 2H), 7.38 (d, J = 8.4 Hz, 2H), 6.46 (s, 1H), 5.87 (s, 1H), 4.38 (s, 2H), 1.33 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.9, 163.5, 142.8, 136.6, 134.6, 133.0, 132.0, 131.2, 128.3, 127.2, 126.2, 125.0, 123.9, 108.4, 51.7, 50.6, 28.8. HRMS (ESI) m/z: [M + H]+ calcd for C21H22O2N2Br, 413.0859, found, 413.0858.
N-(tert-Butyl)-2-(4-methyl-1-oxo-3-phenylisoquinolin-2(1H)-yl)acetamide (3c)
It was synthesized according to procedure A on a 0.3 mmol scale to yield the final compound 3c (65 mg, 62%) as a white solid. mp: 178-180 °C. Rf = 0.29 (33% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.52 (dt, J = 8.0, 1.3 Hz, 1H), 7.77–7.67 (m, 2H), 7.54 (ddd, J = 8.2, 6.4, 1.9 Hz, 1H), 7.47 (dd, J = 5.0, 1.9 Hz, 3H), 7.32–7.28 (m, 2H), 5.65 (s, 1H), 4.34 (s, 2H), 2.02 (s, 3H), 1.28 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.9, 162.8, 140.0, 137.6, 135.1, 132.8, 130.0, 129.1, 129.0, 128.5, 126.8, 125.2, 123.5, 111.3, 51.5, 50.5, 28.9, 28.8, 15.1. HRMS (ESI) m/z: [M + H]+ calcd for C22H25O2N2, 349.1911, found, 349.1910. [M + Na]+ calcd for C22H24O2N2Na, 371.1731, found, 371.1729.
N-(tert-Butyl)-2-(3-(naphthalen-1-yl)-1-oxoisoquinolin-2(1H)-yl)acetamide (3d)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3d (75 mg, 65%) as a white solid. mp: 217–219 °C. Rf = 0.43 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.51 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 7.1 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.63–7.58 (m, 2H), 7.54 (dd, J = 13.4, 8.1 Hz, 4H), 7.46 (t, J = 6.9 Hz, 1H), 6.59 (s, 1H), 5.46 (s, 1H), 1.61 (s, 2H), 1.21 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.5, 163.5, 141.5, 136.8, 133.4, 132.9, 132.4, 132.0, 129.9, 128.8, 128.7, 128.4, 127.4, 127.1, 126.6, 126.2, 125.5, 125.1, 109.1, 51.5, 49.6, 28.7. HRMS (ESI) m/z: [M + H]+ calcd for C25H25O2N2, 385.1911, found, 385.1913.
N-(tert-Butyl)-2-(1-oxo-3,4-diphenylisoquinolin-2(1H)-yl)acetamide (3e)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3e (86 mg, 70%) as a white solid. mp: 209–211 °C. Rf = 0.19(20% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.53 (dd, J = 8.0, 1.7 Hz, 1H), 7.52 (dddd, J = 27.0, 8.2, 7.1, 1.5 Hz, 2H), 7.22–7.12 (m, 9H), 7.11–7.05 (m, 2H), 5.73 (s, 1H), 4.43 (s, 2H), 1.30 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.8, 162.9, 141.2, 137.6, 136.5, 134.4, 132.6, 131.6, 130.6, 128.6, 128.2, 128.2, 128.0, 127.0, 126.8, 125.7, 124.9, 119.6, 51.6, 50.4, 28.8. HRMS (ESI) m/z: [M + H]+ calcd for C27H27O2N2, 411.2054,found, 411.2059.
N-(tert-Butyl)-2-(6-oxo-2,3,4,6-tetrahydrophenanthridin-5(1H)-yl)acetamide (3f)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3f (70 mg, 75%) as a white solid. m.p.: 204–206 °C. Rf = 0.52(20% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.43 (dd, J = 8.1, 1.7 Hz, 1H), 7.67 (dd, J = 6.8, 1.4 Hz, 1H), 7.62 (dd, J = 8.3, 1.3 Hz, 1H), 7.45 (s, 1H), 6.57 (s, 1H), 4.67 (s, 2H), 2.82–2.74 (m, 4H), 1.86 (ddd, J = 13.2, 4.7, 3.0 Hz, 4H), 1.30 (s, 9H).; 13C{1H} NMR (126 MHz, CDCl3) δ 167.8, 163.2, 137.3, 137.2, 132.7, 128.3, 126.2, 124.2, 121.8, 111.8, 51.5, 48.8, 28.8, 27.6, 24.4, 22.8, 22.0. HRMS (ESI) m/z: [M + H]+ calcd for C19H25O2N2, 313.1911, found, 313.1911.
N-(tert-Butyl)-2-(5-oxo-1,2,3,5-tetrahydro-4H-cyclopenta[c]isoquinolin-4-yl)acetamide (3g)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3g (47 mg, 53%) as a white solid. mp: 227–229 °C. Rf = 0.36 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 8.1 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.49–7.36 (m, 2H), 6.53 (s, 1H), 4.57 (s, 2H), 3.14–3.03 (m, 2H), 2.97 (t, J = 7.4 Hz, 2H), 2.22 (q, J = 7.5 Hz, 2H), 1.30 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.2, 163.7, 142.6, 135.5, 132.7, 128.8, 125.8, 124.3, 123.1, 116.0, 51.5, 50.8, 32.6, 29.0, 28.8, 21.5. HRMS (ESI) m/z: [M + H]+ calcd for C18H23O2N2, 299.1754, found, 299.1754.
N-(tert-Butyl)-2-(3-methyl-1-oxoisoquinolin-2(1H)-yl)acetamide (3h)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3h (73 mg, 90%) as a white solid. mp: 176-178 °C. Rf = 0.3 (25% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.43–8.19 (m, 1H), 7.61 (td, J = 7.6, 1.6 Hz, 1H), 7.46–7.39 (m, 2H), 6.41 (s, 1H), 6.36 (d, J = 5.4 Hz, 1H), 4.67 (s, 2H), 2.48 (d, J = 0.9 Hz, 3H), 1.31 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.4, 163.8, 139.6, 137.1, 132.8, 128.1, 126.3, 125.4, 124.2, 106.7, 51.6, 49.4, 28.8, 20.9. HRMS (ESI) m/z: [M + H]+ calcd for C16H21O2N2, 273.1598; found, 273.1598. [M + Na]+ calcd for C16H20O2N2Na, 295.1417, found, 295.1414.
N-(tert-Butyl)-2-(1-oxoisoquinolin-2(1H)-yl)acetamide (3i)
It was synthesized according to procedure A on a 0.2 mmol scale to yield final compound 3i (31 mg, 60%) as a white solid. mp: 202-204 °C. Rf = 0.12 (25% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.42 (dd, J = 8.1, 1.3 Hz, 1H), 7.66 (ddd, J = 8.4, 7.1, 1.4 Hz, 1H), 7.54 (dd, J = 8.0, 1.6 Hz, 1H), 7.50 (ddd, J = 8.2, 7.0, 1.3 Hz, 1H), 7.17 (d, J = 7.4 Hz, 1H), 6.56 (d, J = 7.3 Hz, 1H), 6.44 (s, 1H), 4.52 (s, 2H), 1.31 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.9, 162.7, 137.4, 132.7, 132.1, 128.0, 127.2, 126.3, 125.9, 107.0, 54.2, 51.7, 28.8. HRMS (ESI) m/z: [M + H]+ calcd for C15H19O2N2, 259.1441, found, 259.1441.
2-(6-Bromo-1-oxo-3-phenylisoquinolin-2(1H)-yl)-N-(tert-butyl)acetamide (3j)
It was synthesized according to procedure A on a 0.2 mmol scale to yield final compound 3j (44 mg, 53%) as a white solid. mp: 215-217 °C. Rf = 0.36 (20% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) 8.28 (d, J = 8.6 Hz, 1H), 7.58 (s, 1H), 7.58 (ddd, J = 8.6, 2.0, 0.9 Hz, 1H), 7.48–7.43 (m, 5H), 6.38 (s, 1H), 5.65 (s, 1H), 4.39 (s, 2H), 1.32 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.6, 163.0, 145.3, 138.1, 135.3, 130.14, 130.09, 129.5, 129.3, 128.8, 128.5, 127.9, 123.5, 106.9, 51.7, 50.3, 28.8. HRMS (ESI) m/z: [M + H]+ calcd for C21H22O2N2Br, 413.0859, found, 413.0858.
2-(1-Oxo-3-phenylisoquinolin-2(1H)-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (3k)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3k (54 mg, 46%) as a white solid. mp: 203–206 °C. Rf = 0.26 (20% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.51–8.32 (m, 1H), 7.64 (ddd, J = 8.2, 7.1, 1.4 Hz, 1H), 7.53–7.40 (m, 7H), 6.48 (s, 1H), 6.04 (s, 1H), 4.44 (s, 2H), 1.67 (s, 2H), 1.36 (s, 6H), 0.89 (s, 9H).; 13C{1H} NMR (126 MHz, CDCl3) δ 166.6, 163.4, 143.9, 136.6, 135.6, 132.8, 129.5, 129.3, 128.6, 128.1, 126.9, 126.1, 124.8, 108.3, 55.5, 51.8, 50.5, 31.6, 31.5, 29.1. HRMS (ESI) m/z: [M + H]+ calcd for C25H31O2N2, 391.2380, found, 391.2377.
2-(1-Oxo-3,4-diphenylisoquinolin-2(1H)-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (3l)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3l (63 mg, 45%) as a white solid. mp: 184-186 °C. Rf = 0.68 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.53 (dd, J = 8.0, 1.8 Hz, 1H), 7.52 (dddd, J = 25.5, 8.4, 7.1, 1.5 Hz, 2H), 7.21–7.12 (m, 9H), 7.09–7.06 (m, 2H), 5.80 (s, 1H), 4.42 (s, 2H), 1.66 (s, 2H), 1.36 (s, 6H), 0.90 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.4, 162.8, 141.2, 137.6, 136.5, 134.4, 132.6, 131.6, 130.6, 128.6, 128.2, 128.0, 127.0, 126.9, 125.6, 124.9, 120.0, 55.5, 52.0, 50.6, 31.7, 31.5, 29.1. HRMS (ESI) m/z: [M + H]+ calcd for C31H35O2N2, 467.2693, found, 467.2689.
2-(3-(Naphthalen-1-yl)-1-oxoisoquinolin-2(1H)-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (3m)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3m (86 mg, 65%) as a white solid. mp: 218–220 °C. Rf = 0.63 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.55–8.39 (m, 1H), 7.95 (dd, J = 20.3, 7.7 Hz, 2H), 7.69 (d, J = 1.6 Hz, 1H), 7.62–7.57 (m, 2H), 7.57–7.49 (m, 4H), 7.46 (s, 1H), 6.60 (s, 1H), 5.62 (s, 1H), 4.80 (d, J = 15.3 Hz, 1H), 3.76 (d, J = 15.6 Hz, 1H), 1.62 (s, 2H), 1.27 (d, J = 3.0 Hz, 6H), 0.83 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.1, 163.4, 141.6, 136.8, 133.5, 132.9, 132.4, 132.0, 130.0, 128.8, 128.7, 128.3, 127.4, 127.1, 126.6, 126.2, 125.5, 125.2, 125.0, 109.1, 55.4, 51.9, 49.8, 31.6, 31.4, 29.0 (d, J = 6.0 Hz). HRMS (ESI) m/z: [M + H]+ calcd for C29H33O2N2, 441.2537, found, 441.2534.
2-(5-Oxo-1,2,3,5-tetrahydro-4H-cyclopenta[c]isoquinolin-4-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (3n)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3n (54 mg, 51%) as a white solid. mp: 164–166 °C. Rf = 0.22 (20% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.40 (dd, J = 8.1, 1.7 Hz, 1H), 7.72–7.60 (m, 1H), 7.50–7.39 (m, 2H), 6.57 (s, 1H), 4.58 (s, 2H), 3.11 (t, J = 7.9 Hz, 1H), 2.99 (t, J = 7.4 Hz, 1H), 2.22 (p, J = 7.4 Hz, 2H), 1.66 (s, 2H), 1.36 (s, 6H), 0.85 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.9, 163.8, 142.5, 135.5, 132.8, 128.8, 125.9, 124.4, 123.2, 116.2, 77.4, 77.2, 76.9, 55.4, 51.7, 51.3, 32.6, 31.6, 31.3, 29.2, 29.1, 21.7. HRMS (ESI) m/z: [M + H]+ calcd for C22H31O2N2, 355.2380, found, 355.2377.
2-(3-Methyl-1-oxoisoquinolin-2(1H)-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (3o)
It was synthesized according to procedure A on a 0.2 mmol scale to yield final compound 3o (47 mg, 72%) as a yellow oil. mp: 145–147 °C. Rf = 0.68 (5% Acetone/DCM). 1H NMR (500 MHz, CDCl3) δ 8.35 (dd, J = 8.0, 1.3 Hz, 1H), 7.62 (ddd, J = 8.2, 7.1, 1.3 Hz, 1H), 7.45–7.40 (m, 2H), 6.56 (s, 1H), 6.41 (s, 1H), 4.67 (s, 2H), 2.50 (d, J = 0.9 Hz, 3H), 1.66 (s, 2H), 1.36 (s, 6H), 0.84 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.0, 163.8, 139.5, 136.9, 132.8, 128.1, 126.3, 125.4, 124.1, 106.8, 55.4, 51.7, 49.8, 31.6, 31.3, 29.2, 21.0. HRMS (ESI) m/z: [M + H]+ calcd for C20H29O2N2, 329.2224, found, 329.2220.
N-Cyclohexyl-2-(1-oxo-3,4-diphenylisoquinolin-2(1H)-yl)acetamide (3p)
It was synthesized according to procedure A on a 0.3 mmol scale to yield final compound 3p (63 mg, 48%) as a white solid. mp: 149–151 °C. Rf = 0.28 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.54 (dd, J = 8.0, 1.8 Hz, 1H), 7.54 (ddd, J = 19.5, 8.0, 1.4 Hz, 2H), 7.23–7.13 (m, 9H), 7.09–7.06 (m, 2H), 5.80 (d, J = 7.7 Hz, 1H), 4.45 (s, 2H), 3.73 (dddd, J = 14.7, 11.8, 8.0, 4.0 Hz, 1H), 1.91–1.88 (m, 1H), 1.67–1.61 (m, 3H), 1.32–1.28 (m, 2H), 1.15–1.09 (m, 3H), 0.88–0.85 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 166.9, 162.9, 141.2, 137.6, 136.5, 134.3, 132.7, 131.6, 130.6, 128.7, 128.24, 128.18, 128.1, 127.03, 126.95, 125.7, 124.9, 119.7, 50.3, 48.6, 33.1, 25.6, 24.9. HRMS (ESI) m/z: [M + H]+ calcd for C29H29O2N2, 437.2224, found, 437.2222.
(Z)-2-(1-Benzylidene-3-oxoisoindolin-2-yl)-N-(tert-butyl)acetamide (5a)
It was synthesized according to procedure B on a 0.3 mmol scale to yield final compound 5a (93 mg, 93%) as a white solid. mp: 174–176 °C. Rf = 0.40 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.86 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.61 (td, J = 1.4, 7.6 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.39–7.31 (m, 5H), 6.81 (s, 1H), 5.05 (s, 1H), 4.16 (s, 2H), 1.22 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.3, 166.0, 138.5, 135.1, 134.4, 132.4, 129.7, 129.2, 128.5, 127.82, 127.78, 123.7, 119.7, 107.1, 51.4, 46.2, 28.8, 28.7. HRMS (ESI) m/z: [M + H]+ calcd for C21H23O2N2, 335.1754, found, 335.1744.
(Z)-2-(1-(4-Bromobenzylidene)-3-oxoisoindolin-2-yl)-N-(tert-butyl)acetamide (5b)
It was synthesized according to procedure B on a 0.2 mmol scale to yield final compound 5b (67 mg, 81%) as a white solid. mp: 184–186 °C. Rf = 0.25 (20% EA/PE). 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 7.6 Hz, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.61 (t, J = 7.5 Hz, 1H), 7.53–7.47 (m, 3H), 7.22 (d, J = 8.0 Hz, 2H), 6.68 (s, 1H), 5.11 (s, 1H), 4.16 (s, 2H), 1.24 (s, 9H).13C{1H} NMR (126 MHz, CDCl3) δ 169.3, 165.8, 138.4, 135.7, 133.4, 132.6, 131.7, 131.4, 129.5, 127.8, 123.8, 121.9, 119.8, 105.5, 51.6, 46.2, 28.7. HRMS (ESI) m/z: [M + H]+ calcd for C21H22O2N2Br, 413.0859, found, 413.0857.
(Z)-N-(tert-Butyl)-2-(1-oxo-3-(pyridin-2-ylmethylene)isoindolin-2-yl)acetamide (5c)
It was synthesized according to procedure B on a 0.2 mmol scale to yield final compound 5c (56 mg, 83%) as a white solid. mp: 212–214 °C. Rf = 0.26 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.64–8.61 (m, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.79 (d, J = 6.9 Hz, 1H), 7.68 (td, J = 1.9, 7.7 Hz, 1H), 7.63 (t, J = 7.6 Hz, 1H), 7.55–7.50 (m, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.17 (dd, J = 4.9, 7.6 Hz, 1H), 6.69 (s, 1H), 5.31 (s, 1H), 4.78 (s, 2H), 1.17 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.3, 167.2, 153.5, 149.3, 139.0, 137.0, 136.6, 132.6, 129.7, 127.8, 125.6, 123.9, 121.9, 119.8, 106.1, 51.2, 47.8, 28.7. HRMS (ESI) m/z: [M + H]+ calcd for C20H22O2N3, 336.1707, found, 336.1708.
(Z)-N-(tert-Butyl)-2-(1-oxo-3-((trimethylsilyl)methylene)isoindolin-2-yl)acetamide (5d)
It was synthesized according to procedure B on a 0.3 mmol scale to yield final compound 5d (15 mg, 15%) as a yellow oil. Rf = 0.28 (EA/PE/DCM = 1:2:1). 1H NMR (500 MHz, CDCl3) δ 8.35 (t, J = 5.1 Hz, 1H), 8.08–8.00 (m, 1H), 7.59–7.51 (m, 1H), 7.47–7.37 (m, 2H), 6.14 (s, 1H), 4.09 (d, J = 5.2 Hz, 2H), 1.36 (s, 9H), 0.28 (s, 9H).13C{1H} NMR (126 MHz, CDCl3) δ 167.6, 166.4, 135.0, 134.1, 130.9, 130.2, 129.1, 120.0, 103.0, 102.7, 51.6, 44.9, 28.9. HRMS (ESI) m/z: [M + H]+ calcd for C16H25ON5Si, 331,1823, found, 331,1823.
(Z)-2-(1-Benzylidene-5-methoxy-3-oxoisoindolin-2-yl)-N-(tert-butyl)acetamide (5e)
It was synthesized according to procedure B on a 0.3 mmol scale to yield final compound 5e (98 mg, 90%) as a white solid. mp: 174–176 °C. Rf = 0.58 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.4 Hz, 1H), 7.39–7.34 (m, 2H), 7.34–7.30 (m, 4H), 7.17 (dd, J = 2.4, 8.5 Hz, 1H), 6.69 (s, 1H), 5.00 (s, 1H), 4.15 (s, 2H), 3.89 (s, 3H), 1.23 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.3, 166.1, 161.1, 135.0, 134.6, 131.3, 129.7, 129.3, 128.5, 127.7, 121.2, 121.1, 106.1, 105.9, 55.9, 51.5, 46.4, 28.7. HRMS (ESI) m/z: [M + H]+ calcd for C22H25O3N2, 365.1784, found, 365.1781.
(Z)-2-(1-Benzylidene-3-oxoisoindolin-2-yl)-N-(tert-butyl)propenamide (5f)
It was synthesized according to procedure B on a 0.3 mmol scale to yield final compound 5f (75 mg, 72%) as a white solid. mp: 166–168 °C. Rf = 0.56 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.64 (t, J = 7.6 Hz, 1H), 7.52 (t, J = 7.5 Hz, 1H), 7.38–7.31 (m, 5H), 6.82 (s, 1H), 6.27 (s, 1H), 4.34 (q, J = 7.2 Hz, 1H), 1.45 (d, J = 7.1 Hz, 3H), 1.36 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.9, 169.6, 139.1, 135.4, 134.0, 132.6, 129.38, 129.36, 128.8, 128.2, 123.4, 119.6, 113.8, 107.7, 55.9, 51.3, 28.8, 15.0. HRMS (ESI) m/z: [M + H]+ calcd for C22H25O2N2, 349.1911, found, 349.1909.
(Z)-2-(1-Benzylidene-3-oxoisoindolin-2-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (5g)
It was synthesized according to procedure B on a 0.2 mmol scale to yield final compound 5g (69 mg, 88%) as a white oil. Rf = 0.15 (1% MeOH/DCM). 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 7.9 Hz, 1H), 7.62 (t, J = 6.9 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.40–7.32 (m, 5H), 6.82 (s, 1H), 5.18 (s, 1H), 4.16 (s, 2H), 1.57 (s, 2H), 1.31 (s, 6H), 0.89 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.3, 165.8, 138.5, 135.0, 134.2, 132.5, 129.7, 129.3, 128.6, 127.9, 127.8, 123.7, 119.7, 107.3, 55.5, 52.4, 46.5, 31.6, 31.5, 28.7. HRMS (ESI) m/z: [M + H]+ calcd for C25H31O2N2, 391.2380, found, 391.2382.
(Z)-2-(1-Benzylidene-3-oxoisoindolin-2-yl)-N-cyclohexylacetamide (5h)
It was synthesized according to procedure B on a 0.2 mmol scale to yield final compound 5h (54 mg, 75%) as a white solid. mp: 206–208 °C. Rf = 0.15 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 7.6 Hz, 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.65 (td, J = 1.3, 7.6 Hz, 1H), 7.53 (td, J = 0.9, 7.4 Hz, 1H), 7.39–7.30 (m, 5H), 6.84 (s, 1H), 5.23 (d, J = 8.4 Hz, 1H), 4.20 (s, 2H), 3.64 (dtd, J = 4.0, 6.9, 10.9 Hz, 1H), 1.83 (dt, J = 4.2, 12.1 Hz, 2H), 1.69–1.63 (m, 2H), 1.57 (dt, J = 3.9, 12.9 Hz, 1H), 1.36–1.24 (m, 3H), 1.13–1.03 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.4, 166.1, 138.5, 135.0, 134.1, 132.6, 129.5, 129.4, 128.6, 128.0, 127.8, 123.8, 119.7, 107.5, 48.5, 46.1, 33.0, 25.6, 24.9. HRMS (ESI) m/z: [M + H]+ calcd for C23H25O2N2, 361.1911, found, 361.1911.
(Z)-N-Cyclohexyl-2-(1-(4-methylbenzylidene)-3-oxoisoindolin-2-yl)acetamide (5i)
It was synthesized according to procedure B on a 0.2 mmol scale to yield final compound 5i (102 mg, 91%) as a white solid. mp: 213–215 °C. Rf = 0.74 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.64 (td, J = 1.3, 7.6 Hz, 1H), 7.52 (td, J = 1.0, 7.5 Hz, 1H), 7.22–7.15 (m, 4H), 6.81 (s, 1H), 5.31 (d, J = 10.6 Hz, 1H), 4.22 (s, 2H), 3.72–3.62 (m, 1H), 2.37 (s, 3H), 1.84 (dd, J = 4.0, 12.5 Hz, 2H), 1.67 (t, J = 3.9 Hz, 1H), 1.58 (dt, J = 3.9, 13.1 Hz, 1H), 1.37–1.23 (m, 3H), 1.15–0.99 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.5, 166.3, 138.6, 137.9, 134.6, 132.6, 131.0, 129.4, 129.34, 129.26, 127.7, 123.7, 119.7, 107.8, 48.4, 46.2, 33.0, 25.6, 24.9, 21.5. HRMS (ESI) m/z: [M + H]+ calcd for C24H27O2N2, 375.2067, found, 375.2063.
(Z)-2-(1-(4-Bromobenzylidene)-3-oxoisoindolin-2-yl)-N-cyclohexylacetamide (5j)
It was synthesized according to procedure B on a 0.2 mmol scale to yield final compound 5j (110 mg, 84%) as a white solid. mp: 214–216 °C. Rf = 0.52 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.88 (dt, J = 1.0, 7.6 Hz, 1H), 7.77–7.74 (m, 1H), 7.64 (td, J = 1.3, 7.6 Hz, 1H), 7.53 (td, J = 1.0, 7.5 Hz, 1H), 7.49 (d, J = 8.4 Hz, 2H), 7.22–7.19 (m, 2H), 6.70 (s, 1H), 5.28 (d, J = 8.2 Hz, 1H), 4.19 (s, 2H), 3.65 (tdt, J = 4.0, 7.9, 10.9 Hz, 1H), 1.84 (dt, J = 3.9, 12.3 Hz, 2H), 1.66 (dt, J = 3.9, 14.2 Hz, 2H), 1.58 (dt, J = 3.8, 12.9 Hz, 1H), 1.31 (ddt, J = 4.2, 13.2, 14.8 Hz, 2H), 1.15–1.00 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.4, 165.9, 138.4, 135.6, 133.2, 132.7, 131.7, 131.2, 129.6, 127.7, 123.8, 122.1, 119.7, 105.9, 48.6, 46.0, 33.0, 25.6, 24.9. HRMS (ESI) m/z: [M + H]+ calcd for C23H24O2N2Br, 439.1016, found, 439.1014.
N-Cyclohexyl-2-(1-oxo-3-(pyridin-2-ylmethylene)isoindolin-2-yl)acetamide (Z:E=51:49) (5k)
It was synthesized according to procedure B on a 0.3 mmol scale to yield final compound 5k (79 mg, 73%) as a white solid. mp: 224-224 °C. Rf = 0.24 (50% EtOAc/petroleum ether). (Z) 1H NMR (500 MHz, CDCl3) δ 8.60 (d, J = 5.5 Hz, 1H), 7.91–7.85 (m, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.70–7.60 (m, 2H), 7.54 (t, J = 7.5 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.26–7.23 (m, 1H), 7.16 (dd, J = 7.7, 4.7 Hz, 1H), 6.69 (s, 1H), 5.53 (d, J = 8.4 Hz, 1H), 4.77 (s, 2H), 3.66 (tdt, J = 11.3, 7.7, 3.9 Hz, 1H), 1.65–1.52 (m, 5H), 1.34–1.26 (m, 2H), 1.11–0.92 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.4, 167.4, 153.2, 149.3, 139.0, 136.8, 136.6, 132.7, 129.8, 127.7, 125.5, 123.9, 122.0, 119.8, 106.4, 48.2, 47.8, 33.0, 25.6, 24.9. (E) 1H NMR (500 MHz, CDCl3) δ 8.73 (d, J = 5.0 Hz, 1H), 8.57–8.52 (m, 1H), 7.95–7.85 (m, 2H), 7.74 (td, J = 7.7, 2.0 Hz, 1H), 7.57–7.51 (m, 2H), 7.39 (d, J = 8.0 Hz, 1H), 6.53 (s, 1H), 5.88 (d, J = 8.5 Hz, 1H), 4.55 (s, 2H), 3.78 (tdt, J = 11.8, 8.0, 4.0 Hz, 1H), 1.85–1.74 (m, 2H), 1.65–1.52 (m, 3H), 1.34–1.26 (m, 2H), 1.11–0.92 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.2, 166.6, 154.0, 149.4, 138.4, 136.8, 135.0, 132.9, 130.3, 129.8, 126.2, 125.9, 123.5, 122.5, 111.7, 48.6, 44.6, 33.0, 25.4, 24.9. HRMS (ESI) m/z: [M + H]+ calcd for C22H24O2N3, 362.1863, found, 362.1862.
(Z)-N-Cyclohexyl-2-(1-oxo-3-(pyridin-2-ylmethylene)isoindolin-2-yl)acetamide (5l)
It was purified from 5k on a 0.2 mmol scale to yield final compound 5l (24 mg, 33%) as a white solid. mp: 232-234 °C. Rf = 0.40 (25% EtOAc/DCM). 1H NMR (500 MHz, CDCl3) δ 8.60 (d, J = 3.7 Hz, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.70–7.61 (m, 2H), 7.54 (t, J = 7.5 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.16 (dd, J = 7.6, 4.9 Hz, 1H), 6.69 (s, 1H), 5.52 (d, J = 8.2 Hz, 1H), 4.78 (s, 2H), 3.66 (tdt, J = 11.3, 8.1, 4.0 Hz, 1H), 1.76 (d, J = 4.1 Hz, 2H), 1.64–1.49 (m, 3H), 1.33–1.20 (m, 2H), 1.06 (tt, J = 12.4, 3.5 Hz, 1H), 1.02–0.91 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.4, 167.4, 153.2, 149.4, 139.0, 136.9, 136.6, 132.7, 129.8, 127.7, 125.5, 123.9, 122.0, 119.8, 106.4, 48.2, 47.8, 33.0, 25.6, 24.9. HRMS (ESI) m/z: [M + H]+ calcd for C22H24O2N3, 362.1863, found, 362.1862.
(E)-N-Cyclohexyl-2-(1-oxo-3-(pyridin-2-ylmethylene)isoindolin-2-yl)acetamide (5m)
It was purified from 5k on a 0.2 mmol scale to yield final compound 5m (22 mg, 30%) as a white solid. mp: 228-230 °C. Rf = 0.29 (25% EtOAc/DCM). 1H NMR (500 MHz, CDCl3) δ 8.74 (d, J = 3.9 Hz, 1H), 8.58 (dd, J = 6.2, 1.5 Hz, 1H), 7.95–7.85 (m, 1H), 7.75 (td, J = 7.7, 1.9 Hz, 1H), 7.61–7.50 (m, 2H), 7.41 (d, J = 8.0 Hz, 1H), 6.54 (s, 1H), 5.82 (d, J = 8.0 Hz, 1H), 5.30 (s, 1H), 4.56 (s, 2H), 3.78 (tdt, J = 10.7, 8.1, 4.1 Hz, 1H), 1.86 (dd, J = 12.5, 3.5 Hz, 2H), 1.69–1.61 (m, 2H), 1.55 (d, J = 3.9 Hz, 1H), 1.35–1.26 (m, 2H), 1.14–1.01 (m, 3H).13C{1H} NMR (126 MHz, CDCl3) δ 167.2, 166.7, 154.0, 149.5, 138.4, 136.9, 135.0, 132.9, 130.4, 129.7, 126.2, 125.9, 123.6, 122.5, 111.7, 48.7, 44.7, 33.0, 25.5, 24.9. HRMS (ESI) m/z: [M + H]+ calcd for C22H24O2N3, 362.1863, found, 362.1862.
(Z)-2-(1-Benzylidene-3-oxoisoindolin-2-yl)-N-phenethylacetamide (5n)
It was synthesized according to procedure B on a 0.2 mmol scale to yield final compound 5n (61 mg, 80%) as a white solid. mp: 180–182 °C. Rf = 0.76 (20% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 7.6 Hz, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.65 (td, J = 1.4, 7.6 Hz, 1H), 7.53 (td, J = 0.8, 7.6 Hz, 1H), 7.36–7.28 (m, 3H), 7.24–7.14 (m, 5H), 7.08 (d, J = 6.5 Hz, 2H), 6.80 (s, 1H), 5.41 (t, J = 5.8 Hz, 1H), 4.21 (s, 2H), 3.36–3.30 (m, 2H), 2.70 (t, J = 7.1 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.0, 167.0, 138.7, 138.3, 134.8, 134.0, 132.6, 129.5, 129.4, 128.8, 128.7, 128.5, 128.0, 127.7, 126.6, 123.7, 119.7, 107.5, 45.7, 40.8, 35.5. HRMS (ESI) m/z: [M + H]+ calcd for C25H23O2N2, 383.1754, found, 383.1755.
(Z)-2-(1-(4-Bromobenzylidene)-3-oxoisoindolin-2-yl)-N-(2-methoxyphenyl)acetamide (5o)
It was synthesized according to procedure B on a 0.3 mmol scale to yield final compound 5o (123 mg, 89%) as a white solid. mp: 185–186 °C. Rf = 0.50 (30% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.21 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.45 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.03 (t, J = 7.8 Hz, 1H), 6.91 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.73 (s, 1H), 4.41 (s, 2H), 3.81 (d, J = 1.0 Hz, 3H).13C{1H} NMR (126 MHz, CDCl3) δ 169.0, 164.6, 147.6, 138.2, 135.6, 133.3, 132.7, 131.6, 131.2, 129.6, 127.9, 127.1, 124.0, 123.9, 122.1, 121.1, 119.8, 119.5, 109.9, 105.8, 56.0, 46.3. HRMS (ESI) m/z: [M + H]+ calcd for C24H20O3N2Br, 463.0652, found, 463.0652.
2-(3-([1,1′-Biphenyl]-4-yl)-1-oxoisoquinolin-2(1H)-yl)-N-(tert-butyl)acetamide (6a)
It was synthesized according to procedure C on a 0.15 mmol scale to yield final compound 6a (52 mg, 85%) as a white solid. mp: 194–196 °C. Rf = 0.62 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 8.45 (d, J = 8.0 Hz, 1H), 7.69–7.62 (m, 5H), 7.57–7.46 (m, 6H), 7.39 (t, J = 7.3 Hz, 1H), 6.54 (s, 1H), 5.88 (s, 1H), 4.48 (s, 2H), 1.34 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.1, 163.6, 143.7, 142.2, 140.3, 136.7, 134.6, 132.9, 129.9, 129.1, 128.2, 127.9, 127.4, 127.3, 126.9, 126.1, 124.9, 108.4, 51.6, 50.6, 28.8. HRMS (ESI) m/z: [M + H]+ calcd for C27H27O2N2, 411,2067, found, 411,2062.
(Z)-N-(tert-Butyl)-2-(1-((4′-methoxy-[1,1′-biphenyl]-4-yl)methylene)-3-oxoisoindolin-2-yl)acetamide(6b)
It was synthesized according to procedure C on a 0.15 mmol scale to yield final compound 6b (60 mg, 91%) as a light yellow oil. Rf = 0.76 (50% EtOAc/petroleum ether). 1H NMR (500 MHz, CDCl3) δ 7.89 (dd, J = 7.6, 0.9 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.67–7.44 (m, 7H), 7.39 (d, J = 8.0 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 6.82 (s, 1H), 5.05 (s, 1H), 4.26 (s, 2H), 3.86 (s, 3H), 1.23 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.4, 166.1, 159.6, 140.2, 138.6, 135.2, 132.9, 132.7, 132.5, 130.2, 129.2, 127.8, 126.6, 123.7, 119.7, 114.5, 106.9, 55.5, 51.5, 46.4. HRMS (ESI) m/z: [M + H]+ calcd for C28H29O3N2, 441,2173, found, 441,2172.
Methyl(2-(1-benzylidene-3-oxoisoindolin-2-yl)acetyl) glycinate (Z/E = 26:74) (6d)
It was synthesized according to procedure C on a 0.5 mmol scale to yield carboxylic acid 6c, which was suspended in methanol and cooled to 0 °C. Then, SOCl2 was added dropwise and the mixture was further stirred for 4 h. After the reaction, the solvent was removed under reduced pressure and the crude product was extracted with water and DCM, the combined organic layer was dried over Na2SO4, the solvent was removed, and the crude was purified to give final compound 3d (92 mg, 53%). White solid. mp: 149–150 °C. Rf = 0.16 (50% EtOAc/petroleum ether). (Z) 1H NMR (500 MHz, CDCl3) δ 7.86 (t, J = 7.3 Hz, 3H), 7.77 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.6 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.40–7.36 (m, 3H), 6.86 (s, 1H), 5.97 (t, J = 5.2 Hz, 1H), 4.28 (s, 2H), 3.90 (d, J = 5.1 Hz, 2H), 3.73 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.1, 169.3, 167.4, 138.4, 134.9, 134.4, 134.1, 132.4, 129.4, 128.5, 128.1, 123.8, 123.7, 119.8, 107.7, 52.5, 45.7, 41.3. (E) 1H NMR (500 MHz, CDCl3) δ 7.44 (dt, J = 13.3, 7.6 Hz, 5H), 7.36–7.30 (m, 4H), 6.64 (s, 1H), 6.63–6.60 (m, 1H), 4.61 (s, 2H), 4.06 (d, J = 5.5 Hz, 2H), 3.71 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.9, 168.0, 166.9, 135.8, 135.3, 134.7, 132.7, 129.7, 129.6, 128.8, 128.2, 127.7, 123.7, 123.5, 112.4, 52.5, 43.9, 41.2. HRMS (ESI) m/z: [M + H]+ calcd for C20H19O4N2, 351,1345, found, 351,1332.
Acknowledgments
The authors thank Marcel de Vries and W.H.C. Huibers (University of Groningen) for their help in HRMS analysis. X.L., Q.W., and Q.Z. acknowledge the China Scholarship Council for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c01905.
Experimental procedure and 1H and 13C{1H} NMR spectra
for all compounds with the X-ray crystallographic data for 3e, 3i, 5a, 1H-13C HMBC NMR spectra for 3a and 5a (PDF)
Author Contributions
§ X.L. and Q.W. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Ruijter E.; Scheffelaar R.; Orru R. V. Multicomponent reaction design in the quest for molecular complexity and diversity. Angew. Chem., Int. Ed. 2011, 50, 6234–6246. 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]; b Xu Z.; De Moliner F.; Cappelli A. P.; Hulme C. Ugi/Aldol Sequence: Expeditious Entry to Several Families of Densely Substituted Nitrogen Heterocycles. Angew. Chem., Int. Ed. 2012, 51, 8037–8040. 10.1002/anie.201202575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreye O.; Türünç O.; Sehlinger A.; Rackwitz J.; Meier M. A. Structurally diverse polyamides obtained from monomers derived via the Ugi multicomponent reaction. Chem.-Eur. J. 2012, 18, 5767–5776. 10.1002/chem.201103341. [DOI] [PubMed] [Google Scholar]
- Brown M. L.; Aaron W.; Austin R. J.; Chong A.; Huang T.; Jiang B.; Kaizerman J. A.; Lee G.; Lucas B. S.; McMinn D. L.; et al. Discovery of amide replacements that improve activity and metabolic stability of a bis-amide smoothened antagonist hit. Bioorg. Med. Chem. Lett. 2011, 21, 5206–5209. 10.1016/j.bmcl.2011.07.052. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Mgimpatsang K. C.; Li X.; Domling A. Isoquinolone-4-Carboxylic Acids by Ammonia-Ugi-4CR and Copper-Catalyzed Domino Reaction. J. Org. Chem. 2021, 86, 9771–9780. 10.1021/acs.joc.1c01170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Basso A.; Banfi L.; Riva R. A marriage of convenience: combining the power of isocyanide-based multicomponent reactions with the versatility of (hetero) norbornene chemistry. Eur. J. Org. Chem. 2010, 2010, 1831–1841. 10.1002/ejoc.200901438. [DOI] [Google Scholar]; b Ugi I.; Werner B.; Dömling A. The Chemistry of Isocyanides, their MultiComponent Reactions and their Libraries. Molecules. 2003, 8, 53–66. 10.3390/80100053. [DOI] [Google Scholar]; c Quan B.-X.; Shuai H.; Xia A.-J.; Hou Y.; Zeng R.; Liu X.-L.; Lin G.-F.; Qiao J.-X.; Li W.-P.; Wang F.-L.; et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol. 2022, 7, 716–725. 10.1038/s41564-022-01119-7. [DOI] [PubMed] [Google Scholar]; d Casak S. J.; Pradhan S.; Fashoyin-Aje L. A.; Ren Y.; Shen Y. L.; Xu Y.; Chow E. C.; Xiong Y.; Zirklelbach J. F.; Liu J. FDA Approval Summary: Ivosidenib for the treatment of patients with advanced unresectable or metastatic, chemotherapy refractory cholangiocarcinoma with an IDH1 mutationFDA Approval: Ivosidenib in IDH1-mutated CCA. Clin. Cancer Res. 2022, 28, OF1–OF5. 10.1158/1078-0432.CCR-21-4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M. G.; Kincaid J.; Fang Y.; He J.; Cao Y.; Kaub C.; Gowlugari S.; Wang Z.. Bicycloheteroaryl Compounds as p2x7 Modulators and Uses Thereof. US20070225324A1, 2007.
- Troiano V.; Scarbaci K.; Ettari R.; Micale N.; Cerchia C.; Pinto A.; Schirmeister T.; Novellino E.; Grasso S.; Lavecchia A.; Zappalà M. Optimization of peptidomimetic boronates bearing a P3 bicyclic scaffold as proteasome inhibitors. Eur. J. Med. Chem. 2014, 83, 1–14. 10.1016/j.ejmech.2014.06.017. [DOI] [PubMed] [Google Scholar]
- Gao D. X.; Wang Y. X.; Chen S. J.; Yang H. P.. Benzazepine Derivative, Preparation Method, Pharmaceutical Composition and Use Thereof. WO2017190669A2017.
- a Armoiry X.; Aulagner G.; Facon T. Lenalidomide in the treatment of multiple myeloma: a review. J. Clin. Pharm. Ther. 2008, 33, 219–226. 10.1111/j.1365-2710.2008.00920.x. [DOI] [PubMed] [Google Scholar]; b Li S.; Gill N.; Lentzsch S. Recent advances of IMiDs in cancer therapy. Curr. Opin. Oncol. 2010, 22, 579–585. 10.1097/CCO.0b013e32833d752c. [DOI] [PubMed] [Google Scholar]; c Kotla V.; Goel S.; Nischal S.; Heuck C.; Vivek K.; Das B.; Verma A. Mechanism of action of lenalidomide in hematological malignancies. J. Hematol. Oncol. 2009, 2, 36. 10.1186/1756-8722-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yang B.; Yu R.-l.; Chi X.-h.; Lu X.-c. Lenalidomide Treatment for Multiple Myeloma: Systematic Review and Meta-Analysis of Randomized Controlled Trials. PLoS One 2013, 8, e64354 10.1371/journal.pone.0064354. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bennett C. L.; Angelotta C.; Yarnold P. R.; Evens A. M.; Zonder J. A.; Raisch D. W.; Richardson P. Thalidomide- and Lenalidomide-Associated Thromboembolism Among Patients With Cancer. JAMA 2006, 296, 2555–2560. 10.1001/jama.296.21.2558-c. [DOI] [PubMed] [Google Scholar]
- Ibrahim T. M. Synthesis of biologically active 3-benzalphthalide derivatives. Arch. Pharm. Res. 1991, 14, 342–345. 10.1007/BF02876881. [DOI] [Google Scholar]
- Wang S.; Song Y.; Liu D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. 10.1016/j.canlet.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Yun C.-H.; Park E.; Ercan D.; Manuia M.; Juarez J.; Xu C.; Rhee K.; Chen T.; Zhang H.; et al. Overcoming EGFR (T790M) and EGFR (C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.; Xiang Z.; Luo T.; Lu K.; Xu Z.; Chen J.; Yang Z. Synthesis of functionalized quinolines via Ugi and Pd-catalyzed intramolecular arylation reactions. J. Comb. Chem. 2006, 8, 696–704. 10.1021/cc060066b. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Tuinhof J.; Mgimpatsang K. C.; Kurpiewska K.; Kalinowska-Tluscik J.; Dömling A. Copper-Catalyzed Modular Assembly of Polyheterocycles. J. Org. Chem. 2020, 85, 9915–9927. 10.1021/acs.joc.0c01238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Q.; Fang X.; Liu C.; Zhang G.; Fan X.; Li Y.; Li Y. DNA-Compatible ortho-Phthalaldehyde (OPA)-Mediated 2-Substituted Isoindole Core Formation and Applications. J. Org. Chem. 2022, 87, 2551–2558. 10.1021/acs.joc.1c02496. [DOI] [PubMed] [Google Scholar]
- a Wang F.; Liu H.; Fu H.; Jiang Y.; Zhao Y. An efficient one-pot copper-catalyzed approach to isoquinolin-1 (2H)-one derivatives. Org. Lett. 2009, 11, 2469–2472. 10.1021/ol900847t. [DOI] [PubMed] [Google Scholar]; b Li L.; Wang M.; Zhang X.; Jiang Y.; Ma D. Assembly of substituted 3-methyleneisoindolin-1-ones via a CuI/L-proline-catalyzed domino reaction process of 2-bromobenzamides and terminal alkynes. Org. Lett. 2009, 11, 1309–1312. 10.1021/ol9000922. [DOI] [PubMed] [Google Scholar]; c Liu C.-C.; Parthasarathy K.; Cheng C.-H. Synthesis of highly substituted isoquinolone derivatives by nickel-catalyzed annulation of 2-halobenzamides with alkynes. Org. Lett. 2010, 12, 3518–3521. 10.1021/ol101371c. [DOI] [PubMed] [Google Scholar]; d Kavala V.; Wang C.-C.; Barange D. K.; Kuo C.-W.; Lei P.-M.; Yao C.-F. Synthesis of isocoumarin derivatives via the copper-catalyzed tandem sequential cyclization of 2-halo-N-phenyl benzamides and acyclic 1, 3-diketones. J. Org. Chem. 2012, 77, 5022–5029. 10.1021/jo300501j. [DOI] [PubMed] [Google Scholar]; e Shi Y.; Zhu X.; Mao H.; Hu H.; Zhu C.; Cheng Y. Synthesis of Functionalized Isoquinolin-1 (2H)-ones by Copper-Catalyzed α-Arylation of Ketones with 2-Halobenzamides. Chem.–Eur. J. 2013, 19, 11553–11557. 10.1002/chem.201301621. [DOI] [PubMed] [Google Scholar]; f GangadharaáChary R.; VaraáPrasad K.; ShivaáKumar K. A simple access to N-(un) substituted isoquinolin-1 (2 H)-ones: unusual formation of regioisomeric isoquinolin-1 (4 H)-ones. ChemComm. 2014, 50, 6797–6800. [DOI] [PubMed] [Google Scholar]; g Huang C.-Y.; Kavala V.; Kuo C.-W.; Konala A.; Yang T.-H.; Yao C.-F. Synthesis of biologically active indenoisoquinoline derivatives via a one-pot copper (II)-catalyzed tandem reaction. J. Org. Chem. 2017, 82, 1961–1968. 10.1021/acs.joc.6b02814. [DOI] [PubMed] [Google Scholar]; h Yu X.; Chen K.; Guo S.; Shi P.; Song C.; Zhu J. Direct access to cobaltacycles via C–H activation: N-Chloroamide-enabled room-temperature synthesis of heterocycles. Org. Lett. 2017, 19, 5348–5351. 10.1021/acs.orglett.7b02632. [DOI] [PubMed] [Google Scholar]
- a Zheng Q.; Kurpiewska K.; Dömling A. SNAr Isocyanide Diversification. Eur. J. Org. Chem. 2022, 2022, e202101023 10.1002/ejoc.202101023. [DOI] [Google Scholar]; b Lei X.; Thomaidi M.; Angeli G. K.; Dömling A.; Neochoritis C. G. Fluorene-based multicomponent reactions. Synlett. 2022, 33, 155–160. 10.1055/a-1471-9080. [DOI] [Google Scholar]; c Sutanto F.; Shaabani S.; Oerlemans R.; Eris D.; Patil P.; Hadian M.; Wang M.; Sharpe M. E.; Groves M. R.; Dömling A. Combining High-Throughput Synthesis and High-Throughput Protein Crystallography for Accelerated Hit Identification. Angew. Chem., Int. Ed. 2021, 60, 18231–18239. 10.1002/anie.202105584. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang Q.; Mgimpatsang K. C.; Konstantinidou M.; Shishkina S. V.; Dömling A. 1,3,4-Oxadiazoles by Ugi-Tetrazole and Huisgen Reaction. Org. Lett. 2019, 21, 7320–7323. 10.1021/acs.orglett.9b02614. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wang Q.; Osipyan A.; Konstantinidou M.; Butera R.; Mgimpatsang K. C.; Shishkina S. V.; Dömling A. Pd-Catalyzed de Novo Assembly of Diversely Substituted Indole-Fused Polyheterocycles. J. Org. Chem. 2019, 84, 12148–12156. 10.1021/acs.joc.9b01258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil P.; Ahmadian-Moghaddam M.; Dömling A. Isocyanide 2.0. Green Chem. 2020, 22, 6902–6911. 10.1039/D0GC02722G. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.