

Abstract
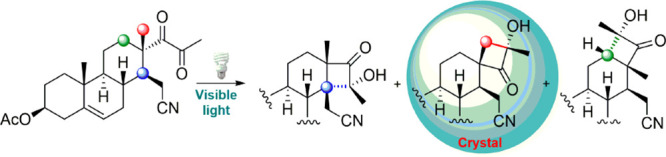
As shown by X-ray crystallography, crystals of 3β-acetoxy-16,17-seco-17,20-dioxopregn-5-ene-16-nitrile are dimorphic. The regioselectivity of the Norrish–Yang type II photocyclization under visible light of this steroidal 1,2-diketone, which bears primary, secondary, and tertiary nonequivalent abstractable γ-hydrogens, dramatically increases in the crystalline state of both polymorphs. X-ray crystallography and molecular mechanics calculations reveal crystal structure–solid state photochemistry relationships.
Norrish–Yang type II photocyclization is the almost exclusive photochemical reaction of 1,2-diketones (I, X = O) bearing abstractable γ-hydrogens after UV or visible light irradiation (Scheme 1).1,2 Thus, 1-hydroxy-2-cyclobutanones (III) are obtained from a highly regioselective 1,5-hydrogen atom transfer (1,5-HAT) reaction promoted by the excited carbonyl group and the subsequent Yang cyclization of the 1,4-biradical intermediate (II).3 These α-hydroxycyclobutanones readily undergo oxidative ring opening to form 4-oxo-acids (IV) in high yield.4 This tandem sequence (Norrish–Yang and oxidation) can be considered formally as a 1,3-stereocontrolled transference of an acyl group from the parent diketone5 and has been applied to the synthesis of a number of natural products with remarkable success.6
Scheme 1. Norrish Type II Reactions of Mono- and 1,2-Diketones.

HAT: Hydrogen atom transfer; RHAT: Reversal hydrogen atom transfer; ISC: intersystem crossing.
The competitive Norrish II photoelimination reaction is a highly unusual process in 1,2-diketones, and only two examples had been described in the literature until our recently published work.7
Although the Norrish-elimination is the preferred reaction pathway in monoketones (I, X = H2) where the 1,4-diradical (V) undergoes further cleavage to produce alkenes and enols (VI), both Norrish type II processes, exclusively promoted by UV irradiation, have been intensively investigated on this functional group.8 In many cases the Norrish–Yang photoreaction of monoketones has been utilized in the key step of natural products synthesis.2e,9 Due to the reversibility of the initial transfer reaction, the final photoproducts do not always proceed from the most stereoelectronically favored abstraction.10
It is well-known that the photochemical behavior of organic compounds changes considerably on going from solution to solid state.11 Thus, enhanced regioselectivity has been observed in the Norrish–Yang photocyclization of crystalline monoketones, with a significant improvement in the formation of cyclobutanols when solid state photochemistry was employed instead of conventional solution photochemistry.12 Even a direct correlation between the crystal structure and their reactivity in solid state has been established.10a,13 The photochemistry of crystalline polymorphic monoketones has also been the subjects of several studies.12e,14 In contrast, there is a paucity of information on the Norrish–Yang photocyclization of crystalline 1,2-diketones, and there do not appear to be studies on the influence of the geometry on the regio- and stereoselectivity of this process.15 Neither have we found information related to the photochemistry of polymorphic 1,2-diketones. A detailed study on the stereo-, chemo-, and diastereoselectivity of a solid-state Norrish–Yang cyclization of a related ketoamide has recently been published.15d
Herein we report on the results obtained from a comparative study in solution and in solid state on the photocyclization under visible light irradiation of the alkyl 1,2-diketone 1 that we had previously obtained from a pregnane derivative by using a methodology developed in our laboratory (Scheme 2).16 This molecule, which retains the conformational restrictions inherent in a steroid, has three different types of potentially abstractable γ-hydrogen atoms: the primary hydrogens of the 18-Me group (pregnane numbering), the secondary hydrogens at C-12, and the tertiary hydrogen atom at C-14. All these structural features provide the most suitable environment to study the regio- and stereoselectivity of the Norrish–Yang photocyclization. In addition, the fact that it is a dimorphic crystalline product renders it an ideal model to determine structure–reactivity relationships.
Scheme 2. Photocyclization of Alkyl 1,2-Diketone 1 in Solution by Irradiation.

With a Philips master PL electronic daylight lamp 23 W/865.
The photochemistry of diketone 1 was first investigated in deoxygenated benzene by irradiation under direct sunlight at room temperature for 3 h (Table 1, entry 1). After purification by silica gel chromatography α-hydroxycyclobutanones 2 (66%, from H-14 abstraction), 3 (20%, from H-18 abstraction), and 4 (6%, from H-12 abstraction) were obtained in good overall yield (Scheme 2). Remarkably, only one diastereoisomer of each α-hydroxycyclobutanone was formed.
Table 1. Photocyclization of Alkyl 1,2-Diketone 1 in Solution and Crystalline Statea.
| entry | medium | T (°C) | t (h) | 2/3/4b | (%)c |
|---|---|---|---|---|---|
| 1 | C6H6d | r.t. | 3 | 72/22/6 | 92 |
| 2 | C6D6 | 25 | 3 | 74/19/7 | 88 |
| 3 | CDCl3 | 25 | 2 | 78/20/2 | 81 |
| 4 | CDCl3 | –60 | 4 | 69/21/10 | 79 |
| 5 | CH3CN | 25 | 3.5 | 69/25/6 | 81 |
| 6 | tBuOH | 30 | 3.5 | 75/21/4 | 82 |
| 7 | Et2O | 25 | 2.5 | 77/17/4 | 83 |
| 8 | Et2O | –40 | 4.5 | 80/15/5 | 81 |
| 9 | Et2O | –90 | 4.5 | 83/13/4 | 84 |
| 10 | [BMIm]OTfe | 25 | 4 | 52/41/7 | 72 |
| 11 | 1A (crystals) | 25 | 9 | 0/100/0 | 92 |
| 12 | 1B (crystals) | 25 | 30 | 10/90/0 | 93 |
Irradiation with a Philips master PL electronic daylight lamp 23 W/865.
Ratio measured by 1H NMR spectroscopy.
Chromatographic isolated yield.
Irradiation with direct sunlight.
[BMIm]OTf: 1-butyl-3-metylimidazolium trifluoromethanesulfonate.
The structure and stereochemistry of the isomeric photoproducts 2–4 were established on the basis of 1D and 2D NMR experiments [see Figures S1 and S2 in the Supporting Information (SI)]. As observed, the carbon substitution pattern of the three cyclobutanones is quite different [2 (4 CH3, 7 CH2, 4 CH, 8 C); 3 (3 CH3, 8 CH2, 5 CH, 7 C); 4 (4 CH3, 6 CH2, 6 CH, 7 C)], and the 13C{1H} NMR spectra and their respective DEPT analyses provide sufficient information to easily distinguish them from one another.
The structure assigned to hydroxycyclobutanone 3 and the configuration of the new stereogenic center at C-20 were unambiguously confirmed by X-ray crystallographic analysis (see Figure S8 and Table S7 in the SI). Unfortunately, all attempts to obtain crystals suitable for X-ray analysis from the other α-hydroxycyclobutanones 2 and 4 failed completely. For both compounds NOE interactions between methyl hydrogens at C-21 and different hydrogens of the β-east side of the molecule are in concordance with the formation of hydroxycyclobutanones 2 and 4 as cis-fused rings, which allows us to assign tentatively the final configuration to the two new chiral centers (20R/14R and 20S/12S, respectively) generated in the cyclization (for more details, see Figure S1 in the SI).
Additionally we studied the photochemical behavior of diketone 1 in several common organic solvents by irradiation under visible light with a daylight lamp (Table 1, entries 2–10). Regardless of the reaction conditions, the same three hydroxycyclobutanones 2–4 were detected by 1H NMR, once again each as a single diastereomer.
The regioselectivity of the photocyclization reaction was not significantly affected by the polarity, protic or hydrogen bonding character of the solvents (entries 1–9).3c,17 Several experiments were even performed at lower temperatures, but no appreciable differences were detected (entries 3 and 4 or 7, 8, and 9). As it is known that the use of ionic liquids in radical reactions can affect the reactivity and dynamics of radicals,18 this photoreaction was carried out also in [BMIm]OTf.19 In this case, we observed a significant change in the photoproducts ratio although it is not synthetically useful (entry 10). The Norrish–Yang photocyclization reaction of 1,2-diketones, as far as we know, has not been previously described in ionic liquids.20
Then we moved on to study the photoreactivity of alkyl 1,2-diketone 1 in solid state. As determined by X-ray crystallography, this diketone can exist in two polymorphic forms: Polymorph 1A prepared by slow evaporation of a solution of acetone–n-hexane at rt for 60 h and polymorph 1B from a solution of EtOAc–n-hexane at 0 °C for 5 h (see Figures S6 and S7, and Table S6 in the SI).
Crystals of polymorph 1A, crushed between two Pyrex slides, were irradiated under visible light to provide exclusively 3. Under these conditions, none of the other five theoretical possible isomers were detected by 1H NMR analysis of the crude reaction residue (entry 11). Although a higher regioselectivity has generally been found on the Yang photocyclization of monoketones in crystalline state than in solution,12 such a dramatic increase as this had not previously been observed either in monoketones or 1,2-diketones. In the photocyclization of polymorph 1B, the formation of 3 predominates (90%), but a small amount of cyclobutanone 2 (10%) is also present in the reaction mixture (entry 12). The regio- and stereoselectivity observed in the high yielding photocyclization of polymorph 1A in the solid state encouraged us to investigate whether we are dealing with a crystal-to-crystal transformation. Initially we investigated whether a single-crystal-to-single-crystal (SCSC) process was possible, as has occasionally been observed in the Norrish–Yang photocyclization of monoketones.21 However, irradiation of large crystals of polymorph 1A caused cracking and breaking of the initial crystalline form into small fragments. Powder X-ray diffraction (PXRD) data confirm a microcrystalline structure for these fragments in excellent agreement with the single X-ray diffraction (SXRD) data of compound 3, indicative of the expected transformation (see Figures S9, S10, S13, and S14 in the SI). A similar result has been observed during the UV photocyclization of a monoketone derivative (2,4,6-triisopropylbenzophenone),21c and a possible explanation has been proposed. Since the photoreaction is initiated at the surface of the crystal, the strain generated inside causes the crack and break of the crystal lattice and the formation of microcrystals. In this sense, the reaction of polymorph 1A can be considered as a crystal-to-crystal transformation (see Figures S9 and S10 in the SI). The analogous reaction of polymorph 1B proceeded with crystal discoloration but without apparent change in their morphology to give an amorphous solid whose PXRD data show no evidence of a diffraction pattern (see Figures S11 and S12 in the SI).
To explain these photochemical results we must keep in mind that the viability of the initial 1,5-HAT reaction in Norrish type II processes is strongly dependent on the conformation of the TS (see Figure S3 in the SI). For this reason, in the restricted conformational mobility of the crystalline state the distance (d) between the excited carbonyl oxygen atom and the γ-hydrogen to be abstracted is of special importance and must be close to 2.72 Å (sum of the van der Waals radii).12e,13 In the three cases of Norrish Type II abstraction promoted by crystalline 1,2-diketones where d values have been calculated, these are in the range 2.4–2.69 Å.7a,15
A conformational analysis based in molecular mechanics calculations revealed that 1,2-diketone 1 adopts in solution two minimum energy staggered conformations by rotation around the C13–C17 single bond (Figure 1, see Tables S1, S2, and S3 in the SI for details).22 The two conformers are almost isoenergetic, where the global minimum syn-conformer A (Figure 1a, dihedral C18–C13–C17–O = 20°) is only about 0.5 kcal mol–1 more stable than the anti-conformer B (Figure 1b, dihedral C18–C13–C17–O = 141°) (see Figure S5 in the SI). Furthermore, the X-ray diffraction study established that both polymorphs 1A,B adopt in the crystalline state an anti-conformation (dihedral C18–C13–C17–O = 148.17(13)°, and 142.3(2)° respectively) very similar to that present in conformer B (see Figures S6 and S7, and Tables S4 and S5 in the SI).
Figure 1.

Solution conformational isomers of 1,2-diketone 1. (a) Conformer A (Dihedral C18–C13–C17–O = 20°). (b) Conformer B (Dihedral C18–C13–C17–O = 141°). X-ray structures are very similar to conformer B (Dihedral C18–C13–C17–O = 148.17(13)° for polymorph 1A and 142.3(2)° for polymorph 1B).
With regard to the photoreaction in solution, it appears clear that diketone 1 reacts through both conformations in equilibrium, which leaves all potentially abstractable γ-hydrogens involved (tertiary axial H14 and secondary axial H12α from conformer A and primary H18 and secondary equatorial H12β from conformer B) at a convenient distance (d = 2.4–2.7 Å) for the 1,5-HAT reaction to take place (see Tables S1 and S2 in the SI). The fact that 2 is the main photoproduct obtained in all cases suggests that conformer A is the preferred, as expected. The use of an ionic liquid as solvent seems to affect this conformational equilibrium, favoring a greater participation of conformer B. In any case, the final photocyclobutanones 2, 3, and 4 ratio does not follow a reactivity pattern determined by the expected stability of the radical intermediate (tertiary ≫ secondary > primary) and seems rather the result of a complex distribution of the three 1,4-biradicals between cyclization and the reversal hydrogen atom transfer reaction (RHAT).
On the basis of the crystalline geometrical parameters of polymorph 1A (see Table S4 in the SI), we could expect in solid state the initial abstraction of γ-hydrogens H18 (d = 2.5 Å) and H12β (d = 2.6 Å) (see Figure S6 in the SI). In the conformationally restricted crystal environment, the direct cyclization of the biradical generated from the H12β abstraction would lead to a highly energetic trans-fused cyclobutanone. In consequence the starting 1,2-diketone is regenerated from the RHAT reaction and the photochemical cyclization occurs exclusively from the H18 abstraction and the final formation of 3 as the sole product of the reaction. The loss of regioselectivity observed in the case of polymorph 1B may be rationalized in terms of fewer restrictions in the environment of the methyl 1,2-diketone group during the photochemical reaction allowing the rotation about C13–C17 single bond since, as previously commented, is a relatively low-energy process.
Finally, the diastereoselectivity of the reaction with the exclusive formation of a single isomer of the hydroxylmethyl grouping at C-20 can also be explained. In the crystalline conformation, the 1,4-biradical generated from the diastereotopic Si-face of the excited carbonyl rapidly collapses to give the 20S isomer of 3 exclusively. This feature that has been called retention of configuration at the carbonyl carbon is generally observed in the solid state photocyclization of monoketones.12d The formation of the 20R isomer would imply an attack by the Re-face of the carbonyl, which requires topochemically prohibited bond rotations in the crystal lattice.
The rapid collapsing of the 1,4-biradical can also be postulated as an explanation for the diastereoselectivity observed in solution during the formation of compounds 2 and 4. From the conformer A, the cyclization of the 14,20-biradical by the Re-face leads exclusively to 2 with a 20R stereochemistry while the Si-facial adduct 4 with a 20S configuration is obtained from the cyclization of the 12,20-biradical also with absolute diastereoselectivity.
In summary this work shows that Norrish–Yang photocyclization of alkyl 1,2-diketones with several nonequivalent abstractable γ-hydrogens can be rendered completely regioselective passing from the solution to crystalline state. The regio- and stereoselectivity of the photoreaction in the solid state seems to be governed by geometrical parameters in a manner similar to the photocyclization of monoketones.
Experimental Section
General Information
Melting points were determined with a hot-stage apparatus. Optical rotations were measured at the sodium line at ambient temperature in CHCl3 solutions. IR spectra were measured in CHCl3 solutions. 1H NMR spectra were determined at 400 or 500 MHz in CDCl3 or C6D6. Chemical shifts are reported in parts per million (ppm) and are calibrated to residual solvent peaks (CHCl3 7.26 ppm and C6H6 7.15 ppm). 13C{1H} NMR spectra were determined at 100.4 or 125.7 MHz in CDCl3. Chemical shifts are reported in parts per million (ppm) and are calibrated to residual solvent peaks (CHCl3 77.0 ppm). NMR peaks assignments and stereochemistries have been established using COSY, DEPT, HMBC, HSQC, and NOESY experiments. Low and high resolution mass spectra were recorded by using EI (70 eV). All single crystal diffraction measurements were performed at 123(2) K with a Oxford Diffraction Gemini instrument utilizing Cu radiation. Selected crystallographic parameters are given in the SI. PXRD was carried out using a Bruker D8 Advance diffractometer equipped with a LynxEye detector and monochromatic Cu Kα1 (λ = 1.54056 Å) radiation, operating in transmission capillary mode. Powdered samples were filled in 0.5 mm borosilicate glass capillaries, and data were initially collected over an angular range of 4 to 70° 2θ with a step size of 0.017°. Merck silica gel 60 PF (0.063–0.2 mm) was used for column chromatography. Circular layers of 1 mm of Merck silica gel 60 PF254 was used on a Chromatotron for centrifugally assisted chromatography. Elemental analyses were performed by the Microanalytical Service of the Institute. Commercially available reagents and solvents were analytical grade or were purified by standard procedures prior to use. The chloroform used in the photoreaction was purified by passing through a column of K2CO3 in the dark before use, but the utilization of strictly deoxygenated solvents does not seem to be necessary. The spray reagents for TLC analysis were conducted with 0.5% vanillin in H2SO4–EtOH (4:1) and further heating until development of color. The light source was a daylight Philips lamp (master PL electronic, 23 W/865) (for a description of the photoreaction equipment, see Figure S4 in the SI).
3β-Acetoxy-16,17-seco-17,20-dioxopregn-5-ene-16-nitrile (1)
Prepared as previously described by alkoxyl radical β-fragmentation of 3β-acetoxy-16β-azido-17α-hydroxy-preg-5-en-20-one.16 The product was isolated by silica gel column chromatography (benzene–EtOAc, 93:7) as bright yellow crystals (51%). [α]D −58.7 (c = 1.46, CHCl3). 1H NMR (400 MHz, CDCl3) δH 5.37 (m, 1H), 4.59 (m, 1H), 2.34 (s, 3H), 2.02 (s, 3H), 1.33 (s, 3H), 1.03 ppm (s, 3H). 13C{1H} NMR (100.4 MHz, CDCl3) δC 206.2 (C), 200.6 (C), 170.4 (C), 139.3 (C), 121.0 (CH), 118.7 (C), 73.4 (CH), 50.1 (C), 48.4 (CH), 41.9 (CH), 37.7 (CH2), 36.7 (C), 36.6 (CH2), 35.5 (CH2), 31.8 (CH), 31.3 (CH2), 27.5 (CH2), 26.8 (CH3), 21.3 (CH3), 19.2 (CH2), 19.1 (CH3), 18.0 (CH2), 14.3 ppm (CH3). IR (CHCl3) ν = 2248, 1722 cm–1. MS (EI, 70 eV) m/z (%) = 342 (1) [M – COCH3]+, 325 (11), 254 (100), 213 (10). HRMS (EI) m/z [M–COCH3]+ calcd for C21H28NO3 342.2069, found 342.2071. Anal. calcd for C23H31NO4: C, 71.66; H, 8.11; N, 3.63. Found: C, 71.84; H, 8.10; N, 3.63.
Polymorph 1A
Bright yellow crystals, mp 121–121.5 °C (slow evaporation of a solution of acetone–n-hexane at rt for 60 h); Crystal data and structure refinement: C23H31NO4, Mτ = 385.49, monoclinic, space group P21, a = 9.5769(1) Å, b = 9.8814(1) Å, c = 10.6257(1) Å, β = 93.098(1)°, V = 1004.074(17) Å3, Z = 2, ρcalcd = 1.275 Mg/m3, μ(CuKα) = 1.54180 Å, F(000) = 416, T = 123 (2) K, yellow crystal, 0.34 × 0.28 × 0.10 mm3, collected reflections 13014. The structure was solved by direct method, all hydrogen atoms were refined anisotropically using full-matrix least-squared based F2 to give R1 = 0.0316, wR2 = 0.0850 for 3655 independently observed reflections (|Fo| > 2σ(|Fo|)) and 257 parameters.
A small amount of polymorph 1A was ground to a fine powder and loaded into a 0.5 mm capillary, and powder X-ray diffraction (PXRD) data were collected. The data were in excellent agreement with the single crystal X-ray diffraction (SXRD) structure.
Polymorph 1B
Bright yellow crystals, mp 116.3–116.8 °C (slow evaporation of a solution of EtOAc–n-hexane at 0 °C for 5 h); Crystal data and structure refinement: C23H31NO4, Mτ = 385.49, monoclinic, space group P21, a = 7.6474(3) Å, b = 15.4448(5) Å, c = 9.2459(3) Å, β = 111.210(4)°, V = 1018.08(7) Å3, Z = 2, ρcalcd = 1.257 Mg/m3, μ(CuKα) = 1.54180 Å, F(000) = 416, T = 123 (2) K, yellow crystal, 0.30 × 0.25 × 0.05 mm3, collected reflections 4100. The structure was solved by direct method, all hydrogen atoms were refined anisotropically using full-matrix least-squared based F2 to give R1 = 0.0382, wR2 = 0.1028 for 3103 independently observed reflections (|Fo| > 2σ(|Fo|)) and 257 parameters.
A small amount of polymorph 1B was ground to a fine powder and loaded into a 0.5 mm capillary, and PXRD data were collected. The data were in excellent agreement with the SXRD structure.
Photocyclization of 3β-Acetoxy-16,17-seco-17,20-dioxopregn-5-ene-16-nitrile (1)
Method A (Solution, irradiation in C6H6): A deoxygenated solution of diketone 1 (50 mg, 0.13 mmol) in dry C6H6 (10 mL), placed in a Schlenk tube, was irradiated with sunlight at room temperature. The photoreaction was periodically monitored by TLC analysis showing complete disappearance of starting diketone in 3 h. The reaction mixture was concentrated under reduced pressure and the crude residue purified by silica gel Chromatotron chromatography (hexanes–EtOAc, 85:15) to give 2 (33 mg, 0.09 mmol, 66%), 3 (9.8 mg, 0.03 mmol, 20%), and 4 (3 mg, 0.008 mmol, 6%).
Method B (Solution, general procedure for irradiation in common organic solvents): A deoxygenated solution of diketone 1 (5 mg, 0.013 mmol) in the corresponding dry solvent (0.6 mL), placed in a resonance tube, was irradiated with a daylight-lamp (Philips master PL electronic, 23 W/865). Progress of the reaction was monitored until completion by TLC. The solvent, temperature, time of irradiation, and hydroxycyclobutanones ratio and yields as established from 1H NMR spectroscopy and chromatography are specified in Table 1.
Method C (Solution, irradiation in ionic liquid): The IL [BMIm][OTf] (1-butyl-3-metylimidazolium trifluoromethanesulfonate)18 (70 mg) was taken in a vial and a solution of diketone 1 (5 mg, 0.013 mmol) in dry CH2Cl2 was added. After evaporation of CH2Cl2 under reduced pressure the resulting oil was extended as a film over the vial wall and irradiated under nitrogen with a daylight-lamp (Philips master PL electronic, 23 W/865) at 25 °C. When the yellow color of the reaction mixture completely faded (3 h), it was extracted twice with Et2O and the IL residues were removed with ultrasound in a water bath. After evaporation of the solvent, the crude was analyzed by 1H NMR and purified by chromatography (hexanes–EtOAc, 80:20) to give (3.6 mg, 0.0094 mmol, 2/3/4, 52:41:7, 72%).
Method D (Solid-state, irradiation of single crystals): Crystals of polymorph 1A (12.8 mg, 0.0333 mmol) were placed in a resonance tube and irradiated (Philips master PL electronic, 23 W/865) at 25 °C for 9 h. The reaction proceeded with cracking and breaking of the initial single crystalline form to give pure 3 as a polycrystalline solid (11.8 mg, 0.0306 mmol, 92%), which was analyzed by 1H NMR and purified by chromatography (hexanes–EtOAc, 80:20).
Analogously, for crystals of polymorph 1B (11.5 mg, 0.0299 mmol) under irradiation (Philips master PL electronic, 23 W/865) at 25 °C for 30 h, the reaction proceeded with crystal discoloration but without an apparent change in their morphology to give an amorphous solid as a mixture of 2 and 3 (10.7 mg, 0.0278 mmol, 10:90, 93%), which was analyzed by 1H NMR and purified by chromatography (hexanes–EtOAc, 80:20).
Method E (Solid-state, irradiation of crushing crystals): The procedure consisted of crushing crystals of polymorph 1A (2 mg, 0.0052 mmol) between two Pyrex microscope slides, taping the plates together, sealing the resulting “sandwich” under nitrogen in a polyethylene bag, and irradiating the ensemble on both sides with a daylight-lamp (Philips master PL electronic, 23 W/865) at 25 °C for 3 h, until the yellow color faded. 1H NMR spectroscopy of the reaction crude showed complete conversion of diketone 1A exclusively into the hydroxycyclobutanone 3 (1.9 mg, 0.0049 mmol, 94%). This proceeded analogously for polymorph 1B (2.5 mg, 0.0065 mmol), which after irradiation (Philips master PL electronic, 23 W/865) at 25 °C for 10 h gave a mixture of hydroxycyclobutanones 2 and 3, which was purified by chromatography (hexanes–EtOAc, 80.20) to give (2.3 mg, 0.006 mmol, 10:90, 92%).
Compound 2: Crystalline solid, mp 176–178 °C (slow evaporation of a solution of n-hexane–EtOAc at 0 °C). [α]D +83 (c = 0.21, CHCl3). 1H NMR (500 MHz, CDCl3) δH 5.43 (m, 1H), 4.62 (m, 1H), 2.75 (d, J = 16.8 Hz, 1H), 2.31 (d, J = 16.8 Hz, 1H), 2.03 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H), 1.03 ppm (s, 3H). 1H NMR (500 MHz, C6D6) δH 5.28 (m, 1H), 4.73 (m, 1H), 2.45 (ddd, J = 13.1, 5.1, 2.5 Hz, 1H), 1.91 (d, J = 16.8 Hz, 1H), 1.74 (s, 3H), 1.13 (s, 3H), 0.84 (s, 3H), 0.76 ppm (s, 3H). 13C{1H} NMR (125.7 MHz, CDCl3) δC 214.3 (C), 170.8 (C), 139.5 (C), 121.8 (CH), 118.0 (C), 91.7 (C), 73.8 (CH), 62.7 (C), 45.5 (C), 44.9 (CH), 37.83 (C), 37.78 (CH2), 36.5 (CH2), 34.6 (CH), 32.0 (CH2), 27.5 (CH2), 27.4 (CH2), 23.3 (CH2), 21.4 (CH3), 20.1 (CH2), 19.9 (CH3), 18.1 (CH3), 16.9 ppm (CH3). IR (CHCl3) ν = 3581, 2248, 1775, 1725 cm–1. MS (EI; 70 eV) m/z (%) = 385 (<1) [M]+, 345 (3). HRMS (EI) m/z [M]+ calcd for C23H31NO4 385.2253, found 385.2228. Anal. calcd for C23H31NO4: C, 71.66; H, 8.11; N, 3.63. Found: C, 71.63; H, 8.13; N, 3.67.
Compound 3: Crystalline solid, mp 185–188 °C (slow evaporation of a solution of n-hexane–EtOAc at 0 °C). [α]D −65 (c = 0.77, CHCl3). 1H NMR (500 MHz, CDCl3) δH 5.38 (m, 1H), 4.60 (m, 1H), 2.72 (dd, J = 13.2, 1.6 Hz, 1H), 2.58 (dd, J = 18.0, 6.1 Hz, 1H), 2.18 (dd, J = 18.0, 2.7 Hz, 1H), 2.10 (d, J = 13.3 Hz, 1H), 2.03 (s, 3H), 1.45 (s, 3H), 1.01 ppm (s, 3H). 13C{1H} NMR (100.4 MHz, CDCl3) δC 216.5 (C), 170.5 (C), 139.6 (C), 120.8 (CH), 118.1 (C), 85.8 (C), 73.5 (CH), 65.9 (C), 48.5 (CH), 40.7 (CH), 37.7 (CH2), 36.8 (CH2), 36.7 (C), 36.6 (CH2), 34.9 (CH2), 32.2 (CH), 30.7 (CH2), 27.5 (CH2), 21.4 (CH3), 21.0 (CH3), 20.3 (CH2), 19.1 (CH3), 17.8 ppm (CH2). IR (CHCl3) ν = 3585, 2256, 1774, 1728 cm–1. MS (EI; 70 eV) m/z (%) = 385 (2) [M]+, 325 (26). HRMS (EI, 70 eV) m/z [M]+ calcd for C23H31NO4 385.2253, found 385.2278. Anal. calcd for C23H31NO4: C, 71.66; H, 8.11; N, 3.63. Found: C, 71.87; H, 8.14; N, 3.61. Crystal data and structure refinement for compound 3: C23H31NO4, Mτ = 385.49, orthorhombic, space group P212121, a = 7.7874(13), b = 9.0718(19), c = 28.882(6) Å, β = 90°, V = 2040.4(7) Å3, Z = 4, ρcalcd = 1.255 Mg/m3, μ(CuKα) = 1.54180 Å, F(000) = 832, T = 123 (2) K, colorless crystal, 0.28 × 0.20 × 0.05 mm3, collected reflections 10623. The structure was solved by direct method, all hydrogen atoms were refined anisotropically using full-matrix least-squared based F2 to give R1 = 0.0492, wR2 = 0.0856 for 3627 independently observed reflections (|Fo| > 2σ(|Fo|)) and 260 parameters.
PXRD data from the exposed (6500 K light for in excess of 30 h) powder sample of polymorph 1A showed excellent agreement with the SXRD structure of compound 3, indicative of the expected transformation. A single crystal of polymorph 1A transforms, upon the light irradiation (6500 K light for in excess of 30 h), into a microcrystalline powder with disintegration of the initial single crystalline form.
PXRD data from the exposed (6500 K light for in excess of 30 h) powder sample of polymorph 1B showed no crystalline diffraction, indicative of a loss of crystallinity. A single crystal of polymorph 1B exposed (6500 K light for in excess of 30 h) was remounted and scanned on the diffractometer but yielded no diffraction spots, despite looking essentially identical (in morphology) to the crystal before exposure.
Compound 4: Amorphous white solid, [α]D −34 (c = 0.2, CHCl3). 1H NMR (500 MHz, CDCl3) δH 5.37 (m, 1H), 4.59 (m, 1H), 3.04 (dd, J = 7.4, 1.6 Hz, 1H), 2.57 (dd, J = 17.6, 6.1 Hz, 1H), 2.51 (dd, J = 17.7, 3.1 Hz, 1H), 2.03 (s, 3H), 1.48 (s, 3H), 1.43 (s, 3H), 1.0 ppm (s, 3H). 1H NMR (500 MHz, C6D6) δH 5.09 (ddd, J = 4.9, 2.1, 2.1 Hz, 1H), 4.74 (dddd, J = 11.4, 11.4, 4.7, 4.7 Hz, 1H), 2.46 (ddd, J = 13.2, 5.0, 2.4 Hz, 1H), 2.00 (dd, J = 17.4, 3.4 Hz, 1H), 1.95 (dd, J = 17.6, 6.3 Hz, 1H), 1.75 (s, 3H), 0.98 (s, 3H), 0.91 (s, 3H), 0.66 ppm (s, 3H). 13C{1H} NMR (125.7 MHz, CDCl3) δC 209.9 (C), 170.5 (C), 139.1 (C), 121.0 (CH), 119.7 (C), 89.6 (C), 73.3 (CH), 57.5 (CH), 44.6 (CH), 41.0 (CH), 38.7 (C), 37.7 (CH2), 36.6 (C), 36.4 (CH2), 30.8 (CH2), 29.6 (CH), 27.5 (CH2), 21.4 (CH3), 20.3 (CH3), 19.2 (CH3), 18.3 (CH2), 17.5 (CH2), 15.6 ppm (CH3). IR (CHCl3) ν = 3586, 2246, 1775, 1727 cm–1. MS (EI; 70 eV) m/z (%) = 385 (<1) [M]+, 325 (22). HRMS (EI; 70 eV) m/z [M]+ calcd for C23H31NO4 385.2253, found 385.2226. Anal. calcd for C23H31NO4: C, 71.66; H, 8.11; N, 3.63. Found: C, 71.85; H, 8.22; N, 3.61.
Acknowledgments
Financial support by the Investigation Programs of the Ministerio de Economía y Competitividad (CTQ2010-18244) is acknowledged. D.A.-D. thanks the Ministerio de Economía y Competitividad for a fellowship. The authors acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c01855.
Most significant NOESY and HMBC correlations for compounds 2–4; geometrical parameters of conformers and polymorphs of compound 1; crystal data and structure refinement for compounds 1A, 1B, and 3; powder X-ray diffraction patterns before and after irradiation of polymorph 1A and 1B; copies of the 1H and 13C{1H} NMR spectra of all new compounds (PDF)
Author Present Address
∥ Chimie et Interdisciplinarité: Synthèse, Analyze, Modélisation, UMR CNRS 6230, UFR des Sciences et Techniques, Université de Nantes, 2 rue de la Houssiniere, BP 92208, 44322 Nantes Cedex 3, France
The authors declare no competing financial interest.
Supplementary Material
References
- a Yang N. C.; Yang D.-D. H. Photochemical Reactions of Ketones in Solution. J. Am. Chem. Soc. 1958, 80, 2913–2914. 10.1021/ja01544a092. [DOI] [Google Scholar]; b Urry W. H.; Trecker D. J. Photochemical Reactions of 1,2-Diketones. J. Am. Chem. Soc. 1962, 84, 118–120. 10.1021/ja00860a034. [DOI] [Google Scholar]
- For reviews on Norrish–Yang reaction, see:; a Wagner P. J.Abstraction of γ-Hydrogens by Excited Carbonyls. In Synthetic Organic Photochemistry (Molecular and Supramolecular Photochemistry); Griesbeck A. G., Mattay J., Eds.; Marcel Dekker: New York, 2005; Vol. 12, pp 11–39. 10.1201/9780203997369. [DOI] [Google Scholar]; b Wessig P.; Mühling O.. Abstraction of (γ ± n)-Hydrogen by Excited Carbonyls. In Synthetic Organic Photochemistry (Molecular and Supramolecular Photochemistry); Griesbeck A. G., Mattay J., Eds.; Marcel Dekker: New York, 2005; Vol. 12, pp 41–87. 10.1201/9780203997369. [DOI] [Google Scholar]; c Scheffer J. R.The Solid-State Ionic Chiral Auxiliary Approach to Asymmetric Induction in Photochemical Reactions. In Chiral Photochemistry (Molecular and Supramolecular Photochemistry); Ramamurthy V., Schanze K. S., Eds.; Marcel Dekker: New York, 2004; Vol. 11, pp 463–483. 10.1201/9780203026342. [DOI] [Google Scholar]; d Hoffmann N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]; e Bach T.; Hehn J. P. Photochemical Reactions as Key Steps in Natural Products Synthesis. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]; f Chen C. The Past, Present, and Future of the Yang Reaction. Org. Biomol. Chem. 2016, 14, 8641–8647. 10.1039/C6OB01214K. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kärkäs M. D.; Porco J. A.; Stephenson C. R. J. Photochemical Approaches to Complex Chemotypes: Applications in Natural Products Synthesis. Chem. Rev. 2016, 116, 9683–9747. 10.1021/acs.chemrev.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Ravelli D.; Protti S.; Fagnoni M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. 10.1021/acs.chemrev.5b00662. [DOI] [PubMed] [Google Scholar]
- a Hamer N. K. Photocyclization of α-Bromomethyl-1,2-diketones. Tetrahedron Lett. 1982, 23, 473–474. 10.1016/S0040-4039(00)86864-1. [DOI] [Google Scholar]; b Obayashi M.; Mizuta E.; Noguchi S. Photochemical Reaction of 21-Methyl-20, 21-diketosteroids: The Formation of New 3′,4′,16α,17α-Tetrahydrocyclobut[16,17]androstanes. Chem. Pharm. Bull. 1979, 27, 1679–1682. 10.1248/cpb.27.1679. [DOI] [Google Scholar]; c Turro N. J.; Lee T.-J. Intramolecular Photoreduction of Alkyl α-Diketones. J. Am. Chem. Soc. 1969, 91, 5651–5652. 10.1021/ja01048a045. [DOI] [Google Scholar]
- Pb(OAc)4, HIO4, and mCPBA have been used as oxidants:; Maulide N.; Markó I. E. Synthesis and Ring Expansions of Functionalized Spirocyclobutanones. Org. Lett. 2007, 9, 3757–3760. 10.1021/ol7015753. [DOI] [PubMed] [Google Scholar]; See also refs (5a), (6b), and (6c).
- a Herrera A. J.; Rondón M.; Suárez E. Stereocontrolled Photocyclization of 1,2-Diketones: Application of a 1,3-Acetyl Group Transfer Methodology to Carbohydrates. J. Org. Chem. 2008, 73, 3384–3391. 10.1021/jo702663w. [DOI] [PubMed] [Google Scholar]; b Herrera A. J.; Rondón M.; Suárez E. Stereocontrolled Photocyclization of 1,2-Diketones Applied to Carbohydrate Models: A New Entry to C-Ketosides. Synlett 2007, 2007, 1851–1856. 10.1055/s-2007-984523. [DOI] [Google Scholar]
- a Kamijo S.; Hoshikawa T.; Inoue M. Regio- and Stereoselective Acylation of Saturated Carbocycles via Norrish–Yang Photocyclization. Tetrahedron Lett. 2010, 51, 872–874. 10.1016/j.tetlet.2009.12.027. [DOI] [Google Scholar]; b Yoshioka S.; Nagatomo M. M.; Inoue M. Application of Two Direct C(sp3)–H Functionalizations for Total Synthesis of (+)-Lactacystin. Org. Lett. 2015, 17, 90–93. 10.1021/ol503291s. [DOI] [PubMed] [Google Scholar]; c Kawamata T.; Nagatomo M.; Inoue M. Total Synthesis of Zaragozic Acid C: Implementation of Photochemical C(sp3)–H Acylation. J. Am. Chem. Soc. 2017, 139, 1814–1817. 10.1021/jacs.6b13263. [DOI] [PubMed] [Google Scholar]; d Chen S.; Zhang Z.; Jiang C.; Zhao C.; Luo H.; Huang J.; Yang Z. Stereoselective Synthesis of trans-Decalin-Based Spirocarbocycles via Photocyclization of 1,2-Diketones. ACS Omega 2021, 6, 18848–18859. 10.1021/acsomega.1c02054. [DOI] [PMC free article] [PubMed] [Google Scholar]; For other syntheses of natural products using Norrish–Yang reactions of 1,2-diketones, see:; e Zhang Z.; Chen S.; Tang F.; Guo K.; Liang X.-T.; Huang J.; Yang Z. Total Synthesis of (+)-Cyclobutastellettolide B. J. Am. Chem. Soc. 2021, 143, 18287–18293. 10.1021/jacs.1c08880. [DOI] [PubMed] [Google Scholar]; f Wakita F.; Ando Y.; Ohmori K.; Suzuki K. Model Reactions for the Enantioselective Synthesis of γ-Rubromycin: Stereospecific Intramolecular Photoredox Cyclization of an ortho-Quinone Ether to a Spiroacetal. Org. Lett. 2018, 20, 3928–3932. 10.1021/acs.orglett.8b01475. [DOI] [PubMed] [Google Scholar]
- a Álvarez-Dorta D.; León E. I.; Kennedy A. R.; Martín A.; Pérez-Martín I.; Riesco-Fagundo C.; Suárez E. Photochemistry of α-Diketones in Carbohydrates: Anomalous Norrish Type II Photoelimination and Norrish–Yang Photocyclization Promoted by the Internal Carbonyl Group. Chem. Eur. J. 2014, 20, 2663–2671. 10.1002/chem.201303843. [DOI] [PubMed] [Google Scholar]; b Álvarez-Dorta D.; León E. I.; Kennedy A. R.; Martín A.; Pérez-Martín I.; Riesco-Fagundo C.; Suárez E. Sequential Norrish Type II Photoelimination and Intramolecular Aldol Cyclization of α-Diketones: Synthesis of Polyhydroxylated Cyclopentitols by Ring Contraction of Hexopyranose Carbohydrate Derivatives. Chem. Eur. J. 2013, 19, 10312–10333. 10.1002/chem.201301230. [DOI] [PubMed] [Google Scholar]; c Álvarez-Dorta D.; León E. I.; Kennedy A. R.; Riesco-Fagundo C.; Suárez E. Sequential Norrish Type II Photoelimination and Intramolecular Aldol Cyclization of 1,2-Diketones in Carbohydrate Systems: Stereoselective Synthesis of Cyclopentitols. Angew. Chem., Int. Ed. 2008, 47, 8917–8919. 10.1002/anie.200803696. [DOI] [PubMed] [Google Scholar]; d Mohr S. Reactions in organic crystals. IV Norrish type II fragmentation of crystalline spirodiketones and spirodiketoneimines. Tetrahedron Lett. 1980, 21, 593–594. 10.1016/S0040-4039(01)85565-9. [DOI] [Google Scholar]; e Bishop R. Norrish Type II Photoelimination of 1,2-Diketones. J. Chem. Soc., Chem. Commun. 1972, 1288–1289. 10.1039/c39720001288. [DOI] [Google Scholar]
- a Wagner P. J.; Klán P.. Norrish Type II Photoelimination of Ketones: Cleavage of 1,4-Biradicals Formed by γ-Hydrogen Abstraction. In Handbook of Organic Photochemistry and Photobiology; Horspool W. M., Lenci F., Eds.; CRC Press: Boca Raton, 2004; Vol. 1, Chapter 52; pp 1–31. 10.1201/9780203495902. [DOI] [Google Scholar]; b Scheffer J. R.; Scott C.. Crystal Structure–Solid-State Reactivity Relationships: Toward a Greater Understanding of Norrish/Yang Type II Photochemistry. In Handbook of Organic Photochemistry and Photobiology; Horspool W. M., Lenci F., Eds.; CRC Press: Boca Raton, 2004; Vol. 1, Chapter 54; pp 1–25. 10.1201/9780203495902. [DOI] [Google Scholar]; c Hasegawa T.Norrish Type II Processes of Ketones: Influence of Environment. In Handbook of Organic Photochemistry and Photobiology; Horspool W. M., Lenci F., Eds.; CRC Press: Boca Raton, 2004; Vol. 1, Chapter 55; pp 1–14. 10.1201/9780203495902. [DOI] [Google Scholar]; d Wessig P.Regioselective Photochemical Synthesis of Carbo- and Heterocyclic Compounds: The Norrish/Yang Reaction. In Handbook of Organic Photochemistry and Photobiology; Horspool W. M., Lenci F., Eds.; CRC Press: Boca Raton, 2004; Vol. 1, Chapter 57; pp 1–20. 10.1201/9780203495902. [DOI] [Google Scholar]; e Wagner P. J.Yang Photocyclization: Coupling of Biradicals Formed by Intramolecular Hydrogen Abstraction of Ketones. In Handbook of Organic Photochemistry and Photobiology; Horspool W. M., Lenci F., Eds.; CRC Press: Boca Raton, 2004; Vol. 1, Chapter 58; pp 1–70. 10.1201/9780203495902. [DOI] [Google Scholar]; f Griesbeck A. G.; Heckroth H. Stereoselective Synthesis of 2-Aminocyclobutanols via Photocyclization of α-Amido Alkylaryl Ketones: Mechanistic Implications for the Norrish/Yang Reaction. J. Am. Chem. Soc. 2002, 124, 396–403. 10.1021/ja0111941. [DOI] [PubMed] [Google Scholar]
- a Ignatenko V. A.; Tochtrop G. P. Approach for Expanding Triterpenoid Complexity via Divergent Norrish–Yang Photocyclization. J. Org. Chem. 2013, 78, 3821–3831. 10.1021/jo400275p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Renata H.; Zhou Q.; Baran P. S. Strategic Redox Relay Enables a Scalable Synthesis of Ouabagenin, a Bioactive Cardenolide. Science 2013, 339, 59–63. 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Braga D.; Chen S.; Filson H.; Maini L.; Netherton M. R.; Patrick B. O.; Scheffer J. R.; Scott C.; Xia W. 1,4-Hydroxybiradical Behavior Revealed through Crystal Structure–Solid-State Reactivity Correlations. J. Am. Chem. Soc. 2004, 126, 3511–3520. 10.1021/ja039076w. [DOI] [PubMed] [Google Scholar]; b Wagner P. J. Solvent Effects on Type II Photoelimination of Phenyl Ketones. J. Am. Chem. Soc. 1967, 89, 5898–5901. 10.1021/ja00999a028. [DOI] [Google Scholar]
- a Ramamurthy V.; Venkatesan K. Photochemical Reactions of Organic Crystals. Chem. Rev. 1987, 87, 433–481. 10.1021/cr00078a009. [DOI] [Google Scholar]; b Photochemistry in Organized and Constrained Media; Ramamurthy V., Ed.; VCH Publishers: New York, 1991. [Google Scholar]; c Tanaka K.; Toda F. Solvent-Free Organic Synthesis. Chem. Rev. 2000, 100, 1025–1074. 10.1021/cr940089p. [DOI] [PubMed] [Google Scholar]; d Scheffer J. R.; Xia W. Asymmetric Induction in Organic Photochemistry via the Solid-State Ionic Chiral Auxiliary Approach. Top Curr. Chem. 2005, 254, 233–262. 10.1007/b101000. [DOI] [Google Scholar]; e Dalal A.; Khanna R.; Kumar D.; Jindal P.; Chaudhary A.; Kamboj R. C. Photochemical Intramolecular H-Abstractions: A Brief Account on their Synthetic Utility. Curr. Org. Chem. 2015, 19, 2156–2195. 10.2174/1385272819666150730215450. [DOI] [Google Scholar]
- a Hipwell V. M.; Garcia-Garibay M. A. Mechanistic Studies of Adamantylacetophenones with Competing Reaction Pathways in Solution and in the Crystalline Solid State. J. Org. Chem. 2019, 84, 11103–11113. 10.1021/acs.joc.9b01720. [DOI] [PubMed] [Google Scholar]; b Renata H.; Zhou Q.; Dünstl G.; Felding J.; Merchant R. R.; Yeh C.-H.; Baran P. S. Development of a Concise Synthesis of Ouabagenin and Hydroxylated Corticosteroid Analogues. J. Am. Chem. Soc. 2015, 137, 1330–1340. 10.1021/ja512022r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ng D.; Yang Z.; Garcia-Garibay M. A. Total Synthesis of (±)-Herbertenolide by Stereospecific Formation of Vicinal Quaternary Centers in a Crystalline Ketone. Org. Lett. 2004, 6, 645–647. 10.1021/ol0499250. [DOI] [PubMed] [Google Scholar]; d Vishnumurthy K.; Cheung E.; Scheffer J. R.; Scott C. Enhanced Regioselectivity of Yang Photocyclization in the Crystalline State. Org. Lett. 2002, 4, 1071–1074. 10.1021/ol017134h. [DOI] [PubMed] [Google Scholar]; e Gudmundsdottir A. D.; Lewis T. J.; Randall L. H.; Scheffer J. R.; Rettig S. J.; Trotter J.; Wu C.-H. Geometric Requirements for Hydrogen Abstractability and 1,4-Biradical Reactivity in the Norrish/Yang Type II Reaction: Studies Based on the Solid State Photochemistry and X-ray Crystallography of Medium-Sized Ring and Macrocyclic Diketones. J. Am. Chem. Soc. 1996, 118, 6167–6184. 10.1021/ja953420a. [DOI] [Google Scholar]; f Wagner P. J.; Pabon R.; Park B. S.; Zand A. R.; Ward D. L. The Regioselectivity of Internal Hydrogen Abstraction by Triplet o-tert-Amylbenzophenone. J. Am. Chem. Soc. 1994, 116, 589–596. 10.1021/ja00081a020. [DOI] [Google Scholar]
- a Turowska-Tyrk I.; Bakowicz J.; Scheffer J. R. Monitoring Structural Transformations in Crystals. 11. Yang Photocyclizations – One Type of Reaction, but Diversity of Structural Changes. Acta Crystallogr. B Struct. Sci. 2007, B63, 933–940. 10.1107/S0108768107051336. [DOI] [PubMed] [Google Scholar]; b Cheung E.; Rademacher K.; Scheffer J. R.; Trotter J. An Investigation of the Solid-State Photochemistry of α-Mesitylacetophenone Derivatives: Asymmetric Induction Studies and Crystal Structure–Reactivity Relationships. Tetrahedron 2000, 56, 6739–6751. 10.1016/S0040-4020(00)00496-8. [DOI] [Google Scholar]; c Ihmels H.; Scheffer J. R. The Norrish Type II Reaction in the Crystalline State: Toward a Better Understanding of the Geometric Requirements for γ-Hydrogen Atom Abstraction. Tetrahedron 1999, 55, 885–907. 10.1016/S0040-4020(98)01019-9. [DOI] [Google Scholar]
- a Evans S. V.; Trotter J. Crystal Structure and Photochemistry of α-Adamantylacetophenone and Two Polymorphs of α-Adamantyl-p-chloroacetophenone. Acta Crystallogr. 1989, B45, 159–162. 10.1107/S0108768188012352. [DOI] [Google Scholar]; b Lewis T. J.; Rettig S. J.; Scheffer J. R.; Trotter J. The Chemical Consequences of Conformational Polymorphism: The Phase-Transition-Dependent Photochemistry of Crystalline 1,14-Cyclohexacosanedione. J. Am. Chem. Soc. 1991, 113, 8180–8181. 10.1021/ja00021a061. [DOI] [Google Scholar]; c Jones R.; Scheffer J. R.; Trotter J.; Yang J. Crystal Structures, Chiralities and Photochemistry of Two Polymorphs of L-Prolinolium α-Adamantylacetophenone-p-carboxylate. Acta Crystallogr. 1994, B50, 601–607. 10.1107/S0108768194002582. [DOI] [Google Scholar]
- For solid state photochemistry of two cyclic 1,2-diketones, see:; a Olovsson G.; Scheffer J. R.; Trotter J.; Wu C.-H. Novel Differences Between the Solid State and Solution Phase Photochemistry of 1,2-Cyclodecanedione. Tetrahedron Lett. 1997, 38, 6549–6552. 10.1016/S0040-4039(97)01657-2. [DOI] [Google Scholar]; b See also ref (7d). For a diaryl 1,2-diketone, see:; c Frey J.; Faraggi E.; Rappoport Z.; Kaftory M. Crystal Structure of the Solid State Photoreactive 2,2′,4,4′,6,6′-Hexaisopropylbenzil. J. Chem. Soc., Perkin Trans. 1995, 2, 1745–1748. 10.1039/P29950001745. [DOI] [Google Scholar]; For some examples of the solid state photochemistry of related keto amides, see:; d Roberts C. A.; Park B.; Xu L.-P.; Roque J. B.; Yeung C. S.; Musaev D. G.; Sarpong R.; LaLonde R. L. Sequential Norrish–Yang Cyclization and C–C Cleavage/Cross-Coupling of a [4.1.0] Fused Saturated Azacycle. J. Org. Chem. 2021, 86, 12436–12442. 10.1021/acs.joc.1c01466. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Roque J. B.; Kuroda Y.; Jurczyk J.; Xu L.-P.; Ham J. S.; Göttemann L. T.; Roberts C. A.; Adpressa D.; Saurí J.; Joyce L. A.; Musaev D. G.; Yeung C. S.; Sarpong R. C–C Cleavage Approach to C–H Functionalization of Saturated Aza-Cycles. ACS Catal. 2020, 10, 2929–2941. 10.1021/acscatal.9b04551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández R.; León E. I.; Moreno P.; Riesco-Fagundo C.; Suárez E. Synthesis of Highly Functionalized Chiral Nitriles by Radical Fragmentation of α-Hydroxy Azides. Convenient Transformation of Aldononitriles into 1,4- and 1,5-Iminoalditols. J. Org. Chem. 2004, 69, 8437–8444. 10.1021/jo049026p. [DOI] [PubMed] [Google Scholar]
- a Verheijdt P. L.; Cerfontain H. Photochemistry of 1,2-Diketones.4. Photochemistry of alpha,alpha,omega,omega-Tetramethyl-1,2-cycloalkanediones; Influence of the Conformation of the Diketo Moiety on the Photochemical Behavior. Recl. Trav. Chim. Pays-Bas 1983, 102, 173–181. 10.1002/recl.19831020309. [DOI] [Google Scholar]; b Hamer N. K.; Samuel C. Photocyclisation of 1-o-Alkylphenylpropane-1,2-diones: Stereochemistry. J. Chem. Soc., Perkin Trans. 1973, 2, 1316–1321. 10.1039/p29730001316. [DOI] [Google Scholar]; c Maruyama K.; Ono K.; Osugi J. Photochemical Reaction of alpha-Diketones. Bull. Chem. Soc. Jpn. 1972, 45, 847–851. 10.1246/bcsj.45.847. [DOI] [Google Scholar]
- Shkrob I. A.; Wishart J. F.. Free Radical Chemistry in Room-Temperature Ionic Liquids. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu C., Studer A., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2012; Vol. 1, Chapter 17, pp 433–448. 10.1002/9781119953678.rad013. [DOI] [Google Scholar]
- Das D.; Dasgupta A.; Das P. K. Improved Activity of Horseradish Peroxidase (HRP) in ‘Specifically Designed’ Ionic Liquid. Tetrahedron Lett. 2007, 48, 5635–5639. 10.1016/j.tetlet.2007.06.022. [DOI] [Google Scholar]
- For a study on the influence of ionic liquids on the stereoselectivity of the Norrish–Yang cyclization in monoketones, see:; Yamada S.; Iwaoka A.; Fujita Y.; Tsuzuki S. Tetraalkylammonium-Templated Stereoselective Norrish–Yang Cyclization. Org. Lett. 2013, 15, 5994–5997. 10.1021/ol4028732. [DOI] [PubMed] [Google Scholar]
- a Konieczny K.; Bąkowicz J.; Turowska-Tyrk I. Structural Transformations in Crystals Induced by Radiation and Pressure. Part 2. The Path of a Photochemical Intramolecular Reaction in Crystals at Different Pressures. CrystEngComm 2015, 17, 7693–7701. 10.1039/C5CE01238D. [DOI] [Google Scholar]; b Yang C.; Xia W. J.; Scheffer J. R. Asymmetric Synthesis of Tricyclic Tetralin Derivatives via an Intramolecular Photoreaction. Tetrahedron 2007, 63, 6791–6795. 10.1016/j.tet.2007.04.072. [DOI] [Google Scholar]; c Koshima H.; Fukano M.; Uekusa H. Diastereospecific Photocyclization of a Isopropylbenzophenone Derivative in Crystals and the Morphological Changes. J. Org. Chem. 2007, 72, 6786–6791. 10.1021/jo071005i. [DOI] [PubMed] [Google Scholar]; d Koshima H.; Kawanishi H.; Nagano M.; Yu H.; Shiro M.; Hosoya T.; Uekusa H.; Ohashi Y. Absolute Asymmetric Photocyclization of Isopropylbenzophenone Derivatives Using a Cocrystal Approach Involving Single-Crystal-to-Single-Crystal Transformation. J. Org. Chem. 2005, 70, 4490–4497. 10.1021/jo0500784. [DOI] [PubMed] [Google Scholar]; e Koshima H.; Matsushige D.; Miyauchi M. Enantiospecific Single Crystal-to-Single Crystal Photocyclization of 2,5-Diisopropyl-4’-carboxybenzophenone in the Salt Crystals with (S)- and (R)-Phenylethylamine. CrystEngComm 2001, 3, 141–143. 10.1039/B103517G. [DOI] [Google Scholar]
- Molecular mechanics calculations were performed with AMBER* force field as implemented in MacroModel v 9.7 with the GB/SA solvent model for CHCl3, Schrödinger, LLC, New York. The minimum for each conformational isomer was calculated performing a coordinated scan calculation of C18–C13–C17–O dihedral in increments of 1°. The dicarbonyl system deviates appreciably from trans coplanarity. An intercarbonyl dihedral angle of 134.5° as measured in the X-ray structure was used for the study; see:; a Cerfontain H.; Kruk C.; Rexwinkel R.; Stunnenberg F. Determination of the Intercarbonyl Dihedral Angle of 1,2-Diketones by 17O NMR. Can. J. Chem. 1987, 65, 2234–2237. 10.1139/v87-372. [DOI] [Google Scholar]; b Sander R.; Bettermann H. Force Field Calculations for Ethanedial, Butanedione and some Cyclic 1,2-Diketones with Respect to the Influence of the Intercarbonyl Dihedral Angle on their Vibrational Frequencies. J. Mol. Struct. 1991, 263, 123–132. 10.1016/0022-2860(91)80060-H. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.