

Summary
CCL8 (MCP-2) is a chemoattractive cytokine associated with various immune-related pathologies. Recent studies show that CCL8 is significantly stimulated during acute respiratory distress syndrome in severely ill patients with COVID-19, making the inhibition of CCL8 activity a promising treatment. Lipopolysaccharide (LPS)-induced lung injury was evaluated in mice using a neutralizing antibody (1G3E5) against human CCL8. Pharmacokinetic studies indicated that following IP administration, 1G3E5 was sustained at higher levels and for a longer period compared to IV administration. CCL8 expression in the lungs was not enhanced by LPS, but CCR2 and CCR5 receptors were significantly stimulated. 1G3E5-mediated inhibition of CCL8 was associated with the reduction of pulmonary inflammation and suppression of various pro-inflammatory cytokines. These results point to a previously unrecognized, permissive role for CCL8 in mediating cytokine induction and ultimately sustaining inflammation. Disruption of CCL8 activity may provide a strategy for mitigating pulmonary inflammation during lung injury when related to abnormal cytokine induction.
Subject areas: Biological sciences, Immunology, Immunology specialty
Graphical abstract
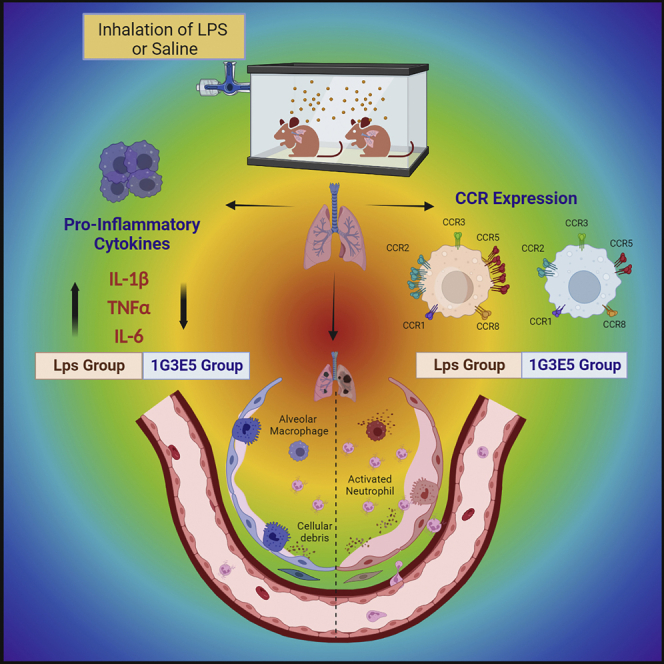
Highlights
-
•
CCL8 is involved in immune-related pathologies, including COVID-19
-
•
A neutralizing antibody was developed for CCL8 and tested in LPS-treated mice
-
•
Inhibition of CCL8 alleviated inflammation and suppressed cytokine response
Biological sciences; Immunology; Immunology specialty
Introduction
Acute respiratory distress syndrome (ARDS) is a life-threatening complication associated with many conditions, including sepsis/septic shock, pneumonia, inhalation of toxic agents, traumas, burns, while rarely is idiopathic and recently is the major complication of SARS-CoV-2 infection that causes the ongoing COVID-19 pandemic.1,2,3,4,5,6 It is associated with severe lung injury and may develop lung fibrosis.1,7,8 The uncontrolled pro-inflammatory response is considered essential for the pathogenesis of ARDS and fibrosis, and indeed suppression of inflammation is used as the strategy of choice for the management of the pathological consequences of lung injury.9,10 In the context of the ongoing COVID-19 pandemic, the eventual surge in fibrosis cases is anticipated with the development of respiratory insufficiency presenting major concerns for public health even when SARS-CoV2 infection rates eventually get under control.11,12 Despite the progress in our understanding of disease pathogenesis, the therapeutic options available for the management of severe ARDS remain limited. Thus, the discovery of novel disease targets and the development of agents modulating their activity remains a priority.
CCL8 is a small chemokine located in chromosome 17q12 in humans and is involved in monocyte migration and inflammation. CCL8 recently attracted attention due to the recognition of its role in carcinogenesis, primarily in the breast and other tissues.13,14,15,16 Nevertheless, a role for CCL8 in pathologies other than cancer has long been suggested and included graft versus host disease17,18,19 microbial infections,20,21 and other conditions relevant to the execution of an aberrant pro-inflammatory response. In pulmonary fibrosis, CCL8 has been identified as a candidate gene target for the differential diagnosis and prediction of patient survival.22,23 More recently, a role of CCL8 in the ARDS that develops in severely ill patients with COVID-19 has also been proposed.24,25,26 Although this evidence strongly supports the beneficial activity of CCL8 inhibition in conditions related to lung injury, no studies exist evaluating the therapeutic potential of CCL8 suppression. In addition, no tools are available that can provide the basis for anti-CCL8 therapeutics.
A potential limitation in extending the results of preclinical studies to the clinic in relation to CCL8 is probably related to the relatively low similarity between human and mouse CCL8. It is possible that this limitation does not permit direct assessment in mice of the value of therapeutics that were generated to target human CCL8 activity. In the present study, we evaluated the consequences of acute inhibition of CCL8 in lipopolysaccharide (LPS)-induced pulmonary inflammation using a novel anti-CCL8 antibody we have developed. For our studies, we focused on outbred deer mice (Peromyscus maniculatus); since CCL8 in these animals is more similar to human than mouse CCL8, it can be targeted more effectively by anti-CCL8 therapeutics that have been developed against human CCL8, allowing the assessment of CCL8 inhibition in the context of a genetically diverse population at which groups of siblings could be interrogated.27,28,29 Our results suggest an important role for CCL8 during lung injury consistent with a permissive function for this chemokine during acute lung injury.
Results
Genetic deficiency of CCL8 mitigates pulmonary inflammation in mice
To test if CCL8 is implicated in inflammation at lung injury, we initially performed a pilot study in which wild type (wt) and mice deficient for CCL8 (CCL8KO)13,14 received LPS via nebulizer to evaluate the extent of lung inflammation.30,31,32 The histological assessment indicated moderate inhibition of inflammation in the lungs of CCL8-deficient mice that remained marginally insignificant (p = 0.0790) compared to the wt controls (Figures 1A and 1B). Evaluation of IL6, IL1β, and TNFα, which are linked to the cytokine cascade at lung injury, showed that TNFα was significantly suppressed in the lungs of CCL8KO mice following the administration of LPS (Figure 1C).
Figure 1.

Effect of CCL8 genetic ablation in LPS-induced pulmonary inflammation in mice
Animals received 2.5 mg LPS via a nebulizer and were sacrificed 6 hrs later.
(A) Representative microphotographs of H&E-stained sections of lungs of wt and CCL8KO mice that received LPS showing the development of hyaline membranes (yellow arrows), the presence of neutrophils (red arrows), and the formation of proteinaceous debris in the alveolar spaces (blue arrows) (scale bar: 25 uM).
(B) Scoring of inflammation was evaluated histologically in wt (n = 5) and CCL8KO (n = 8) mice. Side by side dot plot is shown. p value is indicated (Mann-Whitney U test).
(C) Expression of IL1β, IL6, and TNFα in the lungs of wt (n = 5) and CCL8KO mice (n = 7) following LPS administration as described. Results were normalized versus GAPDH expression, expressed in arbitrary units (AU), and analyzed by unpaired t-test. Data are represented as mean ± SEM. P values are indicated.
Anti-CCL8 antibody 1G3E5 inhibits CCL8 activity
We hypothesized that the moderate impact of CCL8 ablation recorded in CCL8KO mice might be related to the fact that CCL8 deficiency is constitutive, implying in turn that the acute inhibition of CCL8 may have more pronounced effects during acute lung injury. To that end, we developed a neutralizing antibody against human CCL8 that could potentially be used to inhibit CCL8 activity in human patients. Sequencing of the hybridomas revealed the presence of a single H and two L chains suggesting that 3 antibody species, corresponding to the 2 L chains and their hybrid, may be present and active (Figure S1). In view of the low homology between human and mouse CCL8 (71%, Figure 2A), especially at the antigenic peptide used for the development of 1G3E5 (6 out of 14 amino acid identities, Figure 2A), we explored if other experimental rodent models encode for a CCL8 ortholog that possesses higher similarity to hCCL8 than mCCL8. We identified Peromyscus maniculatus (North American deer mouse) with 80% similarity with human CCL8 retained in the antigenic peptide (10 out of 14 amino acid identities, Figure 2A). To explore the potency of 1G3E5 in inhibiting pCCL8, we compared its neutralizing activity against hCCL8-and pCCL8-induced migration of RAW macrophages. As shown in Figure 2B, clone 1G3E5 significantly inhibited RAW macrophage migration that was stimulated by both human and P. maniculatus CCL8 (hCCL8 and pCCL8, respectively). Thus, we proceeded with in vivo studies using outbred deer mice as a rodent model.
Figure 2.

Neutralizing activity of 1G3E5 against CCL8-induced migration
(A) Alignment between human CCL8 and either mouse (Mus) or deer mouse (Peromyscus maniculatus) CCL8. Homologies are shown. The antigenic peptide is indicated by the dashed blue square.
(B) Anti-CCL8 antibody 1G3E5 inhibits RAW macrophage migration that is induced by either human or Peromyscus CCL8. RAW cells migrated in the presence of hCCL8 or pCCL8 at 10 ng/mL alone or combined with 1G3E5 at 1.5 ug/mL. Results are expressed as mean ± SEM (n = 4) and analyzed by unpaired t-test. ∗, p < 0.05; ∗∗∗, p < 0.001.
1G3E5 potently inhibits lung inflammation in deer mice treated with lipopolysaccharides
To evaluate if acute suppression of CCL8 by neutralizing antibody 1G3E5 interferes with lung inflammation, we administered LPS alone or combined with 1G3E5 to genetically diverse deer mice, and after 6 hrs, we evaluated lung histology. For this study, available sibling pairs were distributed evenly in the LPS and the 1G3E5+LPS groups to reduce effect bias due to genetic differences of the experimental animals. Administration of 1G3E5 was well tolerated, and no evidence of toxicity was recorded. As shown in Figure 3, LPS significantly induced lung inflammation with the presence of several neutrophils, the formation of hyaline membranes, the presence of proteinaceous debris in the alveolar spaces, and the thickening of the alveolar septa, and this effect was mitigated by 1G3E5 (p < 0.05).
Figure 3.

Effect of 1G3E5 in LPS-induced pulmonary inflammation in deer mice (P. maniculatus)
Animals received 2.5 mg LPS via a nebulizer and were sacrificed 6 hrs later.
(A) Representative microphotographs of H&E-stained sections of lungs of animals that received LPS showing the development of hyaline membranes (yellow arrows), the presence of neutrophils (red arrows), and the formation of proteinaceous debris in the alveolar spaces (blue arrows). These histological findings were abolished in the group that received LPS+1G3E5 (scale bar: 25 uM).
(B) Scoring of inflammation evaluated histologically in animals that received saline (n = 3), 1G3E5 at 2.5 mg/kg (n = 3), LPS (n = 5), or 1G3E5 at 2.5 mg/kg plus LPS (n = 5). Results were analyzed by the Kruskal-Wallis test. P value is indicated.
Then, we evaluated the effects of LPS alone or in combination with 1G3E5 by measuring the expression of CCL8, IL1β, IL6, and TNFα in the lungs. As shown in Figure 4, LPS stimulated at variable levels the expression of IL1β, IL6, and TNFα, with more pronounced stimulation recorded for IL1β. This is likely due to the genetic diversity of the animals. 1G3E5 caused an insignificant inhibition of these cytokines that was significant when administered in combination with LPS. Interestingly, while in line with earlier findings,18 CCL8 expression was not stimulated by LPS, and 1G3E5 inhibited its expression in both the control and the LPS-treated animals. The fact that sibling pairs were included in the LPS and the LPS+1G3E5 groups facilitated paired analysis in cytokine expression to absorb the variation in expression levels due to the specimens’ genetic heterogeneity. This analysis indicated significant inhibition of all cytokines, including CCL8, by 1G3E5.
Figure 4.

Effect of 1G3E5 in cytokine expression in deer mice
Expression of IL1β, IL6, TNFα, and CCL8 in the lungs of deer mice that received saline (n = 7), 1G3E5 at 2.5 mg/kg (n = 7), LPS (n = 8), and 1G3E5 (n = 8) at 2.5 mg/kg plus LPS. Expression was normalized versus GAPDH expression and expressed in arbitrary units (AU). Comparisons between all groups were performed by ANOVA, and the results are expressed as mean ± SEM. Significance is indicated as follows: #, p < 0.05; ###, p < 0.001. Comparisons between siblings in the groups that received LPS alone or combined with 1G3E5 were performed by Wilcoxon non-parametric test. Significance was indicated as follows: ∗, p < 0.05; ∗∗, p < 0.01. Inlets in dashed black ovals show the individual expression levels in the siblings that received LPS or LPS plus 1G3E5. The mating number of parents is indicated. Results were analyzed by paired t-test, and p values are indicated.
The expression of CCR1, CCR2, CCR3, CCR5, and CCR8 receptors that mediate the effects of CCL833,34,35,36,37 was also evaluated by semiquantitative qPCR in the lungs following LPS administration. Significant stimulation of CCR2 and CCR5 expression was detected after the administration of LPS. Some reduction in receptors’ levels was detected in the 1G3E5 group but remained insignificant (Figure 5A).
Figure 5.

Effect of LPS and 1G3E5 in CCL8 receptors’ expression in deer mice
(A) Expression of CCR1, 2, 3, 5, and 8 in the lungs of deer mice that received saline (n = 7), 1G3E5 at 2.5 mg/kg (n = 7), LPS (n = 8), and 1G3E5 (n = 8) at 2.5 mg/kg plus LPS. Expression was normalized versus GAPDH expression and expressed in arbitrary units (AU). Comparisons between all groups were performed by ANOVA, and the results are expressed as mean ± SEM. Significance vs ctr is indicated as follows: ∗, p < 0.05; ∗∗, p < 0.01, ∗∗∗∗, p < 0.00001.
(B and C) The heatmap correlation matrix depicts the degree of co-expression between chemokine receptors, CCL8, and pro-inflammatory cytokines in sibling pairs of deer mice that received LPS alone (B) or combined with 1G3E5 (C). Correlation coefficients are indicated.
The expression of CCL8 and chemokine receptors is correlated
The functional association between different regulators of inflammation can be unveiled by assessing their degree of co-expression in pairwise comparisons. As shown in Figure 5B, in the LPS group, the expression of chemokine receptors was highly coregulated, which is consistent with the execution of an orchestrated response to LPS and the coregulation of the corresponding chemokine receptors. Similarly, IL1β and IL6, but not TNFα, exhibited coregulation. It is noteworthy that CCL8 was consistently coregulated with the CCRs but not with IL1β, IL6, and TNFα, suggesting that its regulation during inflammation proceeds independently of these pro-inflammatory cytokines. During 1G3E5 treatment, these overall patterns were retained, but the degree of coregulation decreased, implying that the inhibition of CCL8 activity disrupts the execution of the pro-inflammatory response (Figure 5C).
1G3E5 suppresses the long-term effects of lipopolysaccharides in the lungs
The fact that lung inflammation is associated with the induction of pulmonary fibrosis prompted us to explore if the inhibitory activity of 1G3E5 in cytokine expression and inflammation also influenced the subsequent induction of fibrosis following LPS administration. To that end, animals were treated for 3 consecutive days with LPS alone, in combination with 1G3E5, and non-specific IgG. Histology was evaluated four weeks later. Induction of inflammation was detected in the lungs of the animals that received LPS alone and in combination with non-specific IgG, but not in the animals treated with 1G3E5 (Figures 6 and S2). Fibrosis of the alveolar sacs and septa was also occasionally recorded (positive trichrome staining) in the LPS and LPS + IgG groups, while staining was negative in the group treated with 1G3E5 (Figure 7). Similar were the results when LPS was administered every other day, 5 times, for 10 days (not shown).
Figure 6.

Long-term effects of 1G3E5 in LPS-induced lung inflammation in deer mice (P. maniculatus)
Animals received LPS (2.5 mg/5 mL saline) via nebulizer for three consecutive days and were sacrificed 4 weeks later. 1G3E5 was administered following the administration of LPS. Microphotographs show representative H&E-stained sections of the lungs at which prominent mononuclear inflammatory infiltration and thickening of the alveolar septa are seen in the animals that received LPS alone or combined with non-specific IgG (scale bar: 25 μM). In the animals that received 1G3E5, the histopathology caused by LPS was abolished. Results are shown in the lower right panel and analyzed by Kruskal Wallis test. p value is indicated. Bar diagram shows the density of inflammation scores for individual animals that represent sibling pairs and received LPS in combination with either non-specific IgG or 1G3E5. The results were analyzed by Wilcoxon signed rank test, and P-value is shown.
Figure 7.

Trichrome staining (blue) for collagen
Representative images of trichrome-stained sections of lungs from the LPS + IgG and LPS+1G3E5 groups (scale bar: 25 uM) highlight the presence of fibrosis in the LPS + IgG as opposed to the LPS+1G3E5 group. Sections from 2 littermates are shown corresponding to mating #2094 (from Figure 6). Positivity consistent with fibrosis is indicated by yellow arrows.
Beneficial effects of 1G3E5 after the induction of inflammation
The results presented above suggest that 1G3E5 protects against LPS-induced inflammation but do not indicate whether it is beneficial when administered after the inflammation has been established. To explore this deer mice (n = 6 per group) were injected with 1G3E5 24 hours after receiving LPS by inhalation. A histological assessment performed 24 h later indicated that inflammation had been resolved (Figure 8).
Figure 8.

Effect of 1G3E5 administered after the induction of LPS-induced pulmonary inflammation in deer mice (P. maniculatus)
Animals received 2.5 mg LPS via a nebulizer and 24 h later received either IgG or 1G3E5. Animals were sacrificed 24 h later and lung histology was evaluated.
(A) Representative microphotographs of H&E-stained sections of lungs of animals that received LPS + IgG or LPS+1G3E5 (scale bar: 25 uM).
(B) Scoring of inflammation was evaluated histologically (n = 6 per group). Results were analyzed by the Mann Whitney U test.
Pharmacokinetics of 1G3E5
The mean plasma concentration-time profiles observed after the intravenous and intraperitoneal administration of 1G3E5 (2.5 mg /kg) are shown in Figure 9, and the corresponding pharmacokinetic parameters are summarized in Table 1. In brief, peak concentrations were achieved at approximately 0.08 and 0.5 hours after i.v. and i.p. administration. 1G3E5 concentrations declined after Tmax with a mean terminal elimination half-life of 6 and 15.5 hours following i.v. and i.p. administration. MRT, the average time the drug stays in the body after i.p. administration, was significantly longer than i.v. The high bioavailability of 1G3E5 (80%) and long Tmax (4 hrs) after i.p. injection indicated that 1G3E5 was slowly absorbed. The area under the serum concentration versus time curve (AUC) was calculated using the trapezoidal method. The AUMC0–∞ was markedly higher after i.p. administration than i.v., indicating 1G3E5 has long retention after i.p. injection.
Figure 9.

Mean serum concentration-time profiles of 1G3E5 following a single (2.5 mg/kg) intravenous (i.v.) and intraperitoneal (i.p.) injection, n = 6 per data point
Table 1.
Pharmacokinetic parameters of 1G3E5 after intraperitoneal and intravenous administration
| Pharmacokinetic Parameters | i.v. (2.5 mg/kg) | i.p. (2.5 mg/kg) |
|---|---|---|
| AUC0-inf (μg.h/mL) | 85.73 | 178.1 |
| AUMC0-inf (μg.hr2/mL) | 269.74 | 2139.69 |
| MRT (hr) | 3.14 | 12 |
| Tmax (hr) | 0.08 | 4 |
| Cmax (μg/mL) | 14.15 | 9.69 |
| t1/2 (hr) | 7.1 | 15.5 |
| F % | – | 80 |
| Vd (mL) | 5.96 | 6.29 |
| Total Cl (mL/hr) | 0.58 | 0.28 |
AUC, area under the curve; AUMC, area under the moment curve; Cmax, maximum concentration; Tmax, time to reach Cmax; t1/2, elimination half-life; MRT, mean residence time; Vd, volume of distribution; Cl, total clearance; F, bioavailability. (F, %) was calculated at Cmax for intraperitoneal administration using AUC0-4 of intraperitoneal/AUC0–4 of intravenous × 100. Cmax value for intravenous administration was observed at the first point of blood sampling, 0.08 hr
Discussion
The pro-inflammatory role of CCL8 has been established in the context of several pathologies that range from autoimmune disorders, cancer, and more recently COVID-19-associated complications. Nevertheless, despite the abundance of experimental findings pointing to the beneficial effects of CCL8 inhibition, no therapeutic modalities have been developed yet to block CCL8 activity for disease management. In the present study, we report the development and preclinical assessment of a novel neutralizing mouse monoclonal antibody, 1G3E5, that inhibits the chemoattractive activity of hCCL8.
The beneficial activity of 1G3E5 has been evaluated in a rodent model of LPS-induced acute lung injury. Instead of using conventional laboratory mice (genus Mus), we used outbred deer mice P. maniculatus that offer some unique advantages: First, they express CCL8 that is more similar to human CCL8 than that of Mus; therefore, they facilitate the assessment of a pharmaceutical agent that has been developed to block human CCL8 in an animal model that resembles people more accurately in that regard. Second, by being outbred, they mimic natural human populations that exhibit high genetic diversity. This becomes particularly relevant in the context of chemokine-based therapeutics, especially for assessing immune system responses that are highly diverse in people.38 Thus, they provide a system that permits the capture and appreciation of the potential therapeutic value of anti-CCL8 therapeutics in a context that resembles human populations. Finally, by being animals bred in captivity, they allow experimentation in sibling pairs, which in turn permits adequate pairwise comparison of the experimental findings in a context where the power of statistical analysis is not compromised while genetic diversity and response heterogeneity are maintained.
In our studies, 1G3E5 was well tolerated and its serum levels remained detectable 1day after IP administration in deer mice, while following IV administration its levels in the serum decreased considerably. In vivo, 1G3E5 was highly effective in mitigating the LPS-induced cytokine storm and the subsequent development of pulmonary inflammation. These beneficial effects were recorded when 1G3E5 was administered both simultaneously with LPS and when administered 24 h later after lung injury had been established. Similar, albeit less pronounced, findings were also recorded in mice that were deficient for CCL8. It is plausible that constitutive suppression of CCL8 activity in CCL8KO mice activates alternative pro-inflammatory pathways that bypass the requirement for CCL8 in the induction of inflammation.
The fact that CCL8 was not elevated after LPS administration despite the strong effects of 1G3E5 in inhibiting pulmonary inflammation suggests that CCL8 is only permissive for inducing the inflammatory response. In addition, the enhanced expression after the LPS administration of at least CCR2 and CCR5 that mediate the effects of CCL8 may be responsible for the beneficial effects of anti-CCL8 therapy. To that end, it is plausible that under these conditions of experimental lung injury, CCL8 activity does not change considerably, but the abundance of cells expressing CCL8 receptors does, and therefore, the inhibition of CCL8 activity becomes beneficial. Inhibition of CCL8 activity either by a neutralizing antibody in deer mice or by genetic ablation of CCL8 in mice inhibited the expression of TNFα, causing the disruption of a CCL8-TNFα network. This disruption was sufficient to inhibit TNFα expression and pulmonary inflammation.39,40
The significance of CCL8 in inducing and sustaining inflammation is also illustrated by the reduced coregulation between the different chemokine receptors in the 1G3E5-treated mice. While CCRs and CCL8 were co-expressed following LPS treatment, co-expression was abolished in the presence of 1G3E5, suggesting that the execution of a pro-inflammatory response had been disrupted.
The observation that the anti-CCL8 1G3E5 treatment was not associated with elevated levels of endogenous CCL8 in the lungs of deer mice is of particular significance in view of earlier findings that attributed the unsuccessful outcome of MCP-1 (CCL2) neutralizing strategies to the elevation in the expression of the endogenous ligand.41,42,43,44 In this study, treatment with 1G3E5 resulted in the reduction of CCL8 in the lungs of deer mice. This raises the possibility that besides the blockade of CCL8 activity, anti-CCL8 treatment may cause either depletion of CCL8-positive cells or downregulation of CCL8 expression by the CCL8-expressing cells.
These results collectively identify a significant role of CCL8 in acute lung injury and associated pro-inflammatory response. They suggest that the inhibition of CCL8 activity may represent a strategy of choice to manage pathologies related to pulmonary inflammation. Our study also illustrates the power of outbred deer mice in testing therapeutics in the context of genetically diverse populations and for targets at which conventional laboratory mice may be inadequate.
Limitations of the study
We recognize that to appreciate the therapeutic potential of anti-CCL8 treatment for lung injury, follow-up studies are needed to evaluate lung histology and function after longer time periods. In addition, the study would greatly benefit from bronchoalveolar lavage cellular analysis and differential, and by flow cytometric analyses to quantify various inflammatory cell populations. Finally, cloning of the antibody and its production as recombinant proteins to identify the most active clone would also be required.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse IgG kappa binding protein (m-IgGκ BP)/HRP | Santa Cruz Biotechnology, Inc. | Cat#sc-516102; RRID:AB_2687626 |
| 1G3E5 | GeneScript Biotech | N/A |
| Biological samples | ||
| Mice blood samples | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| CCL8 antigen | GeneScript Biotech | N/A |
| Human CCL8 | Cell Guidance Systems | Cat#GFH130-100 |
| Peromyscus CCL8 | GeneScript Biotech | N/A |
| Lipopolysaccharides from Escherichia coli O111:B4 | Sigma-Aldrich | Cat#L2630-100MG |
| RPMI 1640 media, with L-glutamine | Cytiva Life Sciences | Cat#SH30027.01 |
| FBS | Cytiva Life Sciences | Cat#SH30070.03 |
| Trypsin-EDTA | Cytiva Life Sciences | Cat#SH30236.01 |
| PBS | Cytiva Life Sciences | Cat#SH30013.03 |
| 10% Formaldehyde | VWR | Cat#89370-094 |
| Critical commercial assays | ||
| iScript cDNA Synthesis Kit | Bio-Rad | Cat#1708891 |
| RNeasy Mini kit | Qiagen | Cat#74106 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | Cat#1725122 |
| Experimental models: Cell lines | ||
| RAW 264.7 Murine Macrophage cells | ATCC | N/A |
| Experimental models: Organisms/strains | ||
| Peromyscus maniculatus mice | Peromyscus Genetic Stock Center | N/A |
| CCL8-deficient mice | University of South Carolina | N/A |
| C57BL/6 mice | University of South Carolina | N/A |
| Oligonucleotides | ||
| qPCR Primers (See Table S1) | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 | Graph Pad | N/A |
| BioRender | BioRender | https://biorender.com |
Resource availability
Lead contact
Further information and reasonable requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hippokratis Kiaris (hk@sc.edu).
Materials availability
This study developed a novel anti-CCL8 antibody.
Experimental model and subject details
Animals
Male 3-4 months old wild type and CCL8KO mice in C57BL6 background were described before and maintained in our colony.13,14 Male P. maniculatus aged 18-24 weeks were obtained from the Peromyscus Genetic Stock Center (Columbia, SC) (RRID:SCR_002769). The animals were housed in a temperature and humidity-controlled room with a 12-h day/night cycle and free access to food and water. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of South Carolina (approval no 2489-101521-051920 and 2474-101463-102819).
Cell lines
RAW 264.7 Murine Macrophage cells were cultured in DMEM with 10% Fetal Bovine Serum (Corning). Cell lines were frequently tested for mycoplasma contamination using a commercially available Mycoplasma detection kit (Myco Alert kit, Lonza, Walkersville, MD, USA).
Method details
Antibody development
Antibody development was performed by Genscript (Piscataway, NJ). Briefly, the peptide CINRKIPIQRLESYT-KLH was used to immunize BALB/c mice. Hybridomas were developed by using SP2/0 myeloma cells. Different clones were evaluated, and 1G3E5 (IgG1, k) was selected for subsequent studies. For all experiments, low endotoxin (<3 EU/mg) protein A purified antibody was used. The concentration of the Ab was determined indirectly by ELISA using a coating antigen CINRKIPIQRLESYT. Peroxidase-AffiniPure Goat Anti-Mouse IgG, Fcγ Fragment Specific (min X Hu, Bov, Hrs Sr Prot) was used as a secondary antibody. Sequencing of H and L chains of hybridomas was performed by Genscript (Piscataway, NJ).
Migration assay
About 1.0 × 106 RAW 264.7 Murine Macrophage cells were seeded on the top chamber of transwells (6-well format, with 8-μm pore size insert, Costar, Waltham, MA, USA) in serum-free media and inserted in the 6-well plate. Bottom chamber of wells contained 10 ng/mL of human CCL8 (Cell Guidance Systems St Louis, MO, USA), 10 ng/mL of Peromyscus CCL8 (GeneScript Biotech Piscataway, NJ, USA), 1.5 μg/mL 1G3E5 antibody (GeneScript Biotech Piscataway, NJ, USA), a mixture of 10 ng/mL human CCL8 with 1.5 μg/mL 1G3E5 antibody and a mixture of 10 ng/mL Peromyscus CCL8 with1.5 μg/mL 1G3E5 antibody for 24 h. All conditions were added in DMEM medium supplemented with 10% FBS, including control. Experiments were performed in duplicates. After 24 h, the transwells were removed, and cells in the bottom chamber were collected and centrifuged for 5 min at 800 g. Supernatant was removed, and cells were resuspended by adding 100 μL DMEM. Cells were counted by placing in Counting Slides, Dual Chamber (Bio-Rad Laboratories, Hercules, CA), then into TC20 Automated Cell Counter (Bio-Rad Laboratories, Hercules, CA). CCL8 peptides were purchased from Cell Guidance Systems LLC, St. Louis MO (hCCL8), or synthesized by Genscript (Piscataway, NJ) (pCCL8).
Inhalation of LPS
Animals were exposed to nebulized saline or LPS. The animals were placed in a polysulfone cage with a filter on the top, connected to a nontoxic PVC tube at one end, and attached to the nebulizer on the other. A total 5 mL volume LPS (Escherichia coli (serotype O111: B4; Sigma-Aldrich) at 500 μg/mL in 0.9% saline was aerosolized for 30 min. Aerosols were generated by a compressed-air nebulizer at an airflow of 7 L/min, yielding a particle size of 0.5 μm. For the control group, 5 mL of saline solution, 0.9% NaCl was nebulized. We used two identical nebulization systems to prevent contamination of the control group.
In vivo studies
Antibody 1G3E5 (anti-CCL8 neutralizing antibody) was administered intraperitoneally at a dose of 2.5 mg/kg immediately after the animal received LPS through inhalation. Experimental groups (n = 7–8/group) were designed as follows for the acute response study: Control (saline solution-nebulized and received 100 μL saline i.p.), LPS (LPS-nebulized (dose: 2.5 mg/5 mL saline) for 30 min and received 100 μL saline i.p.), SAL + 1G3E5 (saline solution-nebulized and 1G3E5 was administered i.p. at a dose of 2.5 mg/kg), LPS + 1G3E5 (LPS-nebulized (dose: 2.5 mg/5 mL saline) for 30 min and 1G3E5 was administered i.p. at a dose of 2.5 mg/kg). All the animals were sacrificed after 6 hours, and the lung tissue was collected for histology and qPCR assay. To assess fibrosis in the lungs, three experimental groups (n = 5/group) were designed as follows: All animals were exposed for 3 consecutive days to nebulized LPS (dose: 2.5 mg/5 mL saline) for 30 min, then the LPS group received 100 μL saline, i.p.; The LPS + IgG group received IgG 2.5 mg/kg i.p., and the LPS + 1G3E5 group received 2.5 mg/kg 1G3E5 i.p. After four weeks, the animals were sacrificed for histological examination. To evaluate the therapeutic potential of 1G3E5 animals (n = 6 per group) received LPS as described by inhalation and then 1G3E5 or IgG i.p. 24 h later. Animals were sacrificed for histological analyses 24 h later.
Measurement of cytokine expression
The expression level of IL1β, IL6, TNFα, and CCL-8 in the lungs were evaluated using real-time PCR, as previously reported.13 The expression of GAPDH was used as an internal control. primer sequences used for P. maniculatus and Mus musculus mice can be found in Table S1.
Tissue collection
The animals in each group were euthanized at the end of the study via isoflurane inhalation overdose for tissue collection. Lungs were removed and fixed for 48 h in 10% formalin and subsequently embedded in paraffin blocks via a standard protocol.
Histopathology
Formalin-fixed tissues were embedded in paraffin blocks and sectioned at 5-7 μm. The tissue sections were stained with hematoxylin and eosin (H&E) for histological evaluation. The histopathological analyses of the lungs were performed blindly by a certified pathologist. Lung injury was assessed based on the scoring system suggested by Matute-Bello et al.45 The scale used was from 0 to 2 for each of the following criteria: A) neutrophils in the alveolar space, B) neutrophils in the interstitial space, C) hyaline membranes, D) proteinaceous debris filling the airspaces, and E) the extent of the septal thickening. The final score (0-2) was the result of the following calculation based on the Score = [(20xA)+(14xB)+(7xC)+(7xD)+(2xE)]/number of fieldsx100).45 For the experiment at which the animals were left for almost a month after the administration with LPS, LPS + IgG, or LPS+1G3E5, histological evaluation was based on the number of the mononuclear cell infiltration and the thickening of the septa. Trichrome staining (Abcam, Connective Tissue Stain, ab150686) was also performed to evaluate lung fibrosis. Representative images were taken at 40x using a Leica ICC50W camera on a Leica DM 1000 microscope.
Pharmacokinetics study
Male P. maniculatus aged 20-24 weeks (20-24 g) were used for pharmacokinetics analysis. The mice were divided into two groups for i.p. and i.v. injection; n = 6. In both groups, mice were treated with a single dose of 1G3E5 at 2.5 mg/kg in 100 μL saline. Blood samples (20 μL) were collected 0.08, 0.5, 2, 4, 6, 9, 12, 24, and 48 h after i.v. and 0.5, 2, 4, 6, 9, 12, 24, and 48 h after i.p. administration. Samples were centrifuged at 5,000 rpm for 10 min at 4°C and sera stored at −80∘ C until analyzed by ELISA.
ELISA for 1G3E5
Invitrogen (Nunc MaxiSorp® flat-bottom) 96-well plates were coated with 1 μg/mL of CCL8 antigen diluted in coating buffer (0.05M carbonate buffer, pH = 9.6), and incubated at 4°C overnight. Afterward, the plates were washed three times with 200 μL/well of washing buffer and then blocked with 200 μL/well of 1% non-fat dry milk in PBS; PH = 7.4 for 2 h at room temperature (RT) to prevent an unspecific reaction. Then, the plates were washed four times using 200 μL/well of washing buffer, and the standards and diluted (100 times with blocking buffer) serum samples (100 μL/well) were added and incubated for 75 min at RT with gentle continual shaking. For labeling, the plates were washed four times and treated with mice IgG HRP-conjugated antibody diluted in blocking buffer (1:5.000) for 30 min at RT. The plates were washed and revealed with 100 μL/well of TMB for 30 min. This reaction was stopped by adding 100 μL/well of stop solution, and the absorbance was read at 450 nm using Bio-Rad iMarkTM Microplate Reader. Using the standard curves, total counts of absorbance units were converted to micrograms per milliliters (μg/mL) of 1G3E5.
Quantification and statistical analysis
Statistical analysis was performed by ANOVA or Kruskal Wallis test and unpaired t-tests or Mann Whitney U test as indicated, and results were considered significant if p < 0.05. For pairwise comparisons between siblings, Wilcoxon signed rank test or paired t-test was performed, as shown in the results and figure legends.
Acknowledgments
This study was supported by NSF (Award Number: OIA1736150) and a grant from the South Carolina Research Authority (SCRA).
Author contributions
Conceptualization, A.N., E.F., I.C., and H.K.; methodology, A.N., E.F., I.C., and H.K.; investigation, A.N., E.F., B.C., E.S., N.D., and I.C.; resources, V.K., and Y.Z.; formal analysis, C.C.; writing – original draft, A.N. and H.K.; writing – review & editing, A.N., E.F., I.C., B.C., E.S., N.D., V.K., Y.Z., C.C., and H.K.
Declaration of interests
The University of South Carolina has filed patent applications for the 1G3E5 antibody. AN, EF, VK, IC, and HK are designated as inventors in these applications.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. We support inclusive, diverse, and equitable conduct of research.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105520.
Supplemental information
Data and code availability
Data reported in this paper and 1G3E5 antibody will be shared by the lead contact upon request. Peromyscus animals are distributed by the PGSC.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Fernandez I.E., Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380:680–688. doi: 10.1016/S0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- 2.Schwaiblmair M., Behr W., Haeckel T., Märkl B., Foerg W., Berghaus T. Drug induced interstitial lung disease. Open Respir. Med. J. 2012;6:63–74. doi: 10.2174/1874306401206010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenkins G.I.P.F. American Thoracic Society; 2017. Moving from Idiopathic to Infectious Pulmonary Fibrosis? pp. 125–127. [DOI] [PubMed] [Google Scholar]
- 4.Azadeh N., Limper A.H., Carmona E.M., Ryu J.H. The role of infection in interstitial lung diseases: a review. Chest. 2017;152:842–852. doi: 10.1016/j.chest.2017.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallelli L., Zhang L., Wang T., Fu F. Severe acute lung injury related to COVID-19 infection: a review and the possible role for escin. J. Clin. Pharmacol. 2020;60:815–825. doi: 10.1002/jcph.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan E., Beitler J.R., Brochard L., Calfee C.S., Ferguson N.D., Slutsky A.S., Brodie D. COVID-19-associated acute respiratory distress syndrome: is a different approach to management warranted? Lancet Respir. Med. 2020;8:816–821. doi: 10.1016/S2213-2600(20)30304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King T.E., Jr., Pardo A., Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 8.Gilbert J.A. Advancing towards precision medicine in ARDS. Lancet Respir. Med. 2018;6:494–495. doi: 10.1016/S2213-2600(18)30156-5. [DOI] [PubMed] [Google Scholar]
- 9.Baudouin S. BMJ Publishing Group Ltd; 2006. Manipulation of Inflammation in ARDS: Achievable Goal or Distant Target? pp. 464–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mason C., Dooley N., Griffiths M. Acute respiratory distress syndrome. Clin. Med. 2016;16:s66–s70. doi: 10.7861/clinmedicine.16-6s-s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tale S., Ghosh S., Meitei S.P., Kolli M., Garbhapu A.K., Pudi S. Post-COVID-19 pneumonia pulmonary fibrosis. QJM: Int. J. Med. 2020;113:837–838. doi: 10.1093/qjmed/hcaa255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraser E. British Medical Journal Publishing Group; 2020. Long Term Respiratory Complications of Covid-19. [DOI] [PubMed] [Google Scholar]
- 13.Farmaki E., Chatzistamou I., Kaza V., Kiaris H. A CCL8 gradient drives breast cancer cell dissemination. Oncogene. 2016;35:6309–6318. doi: 10.1038/onc.2016.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farmaki E., Kaza V., Chatzistamou I., Kiaris H. CCL8 promotes postpartum breast cancer by recruiting M2 macrophages. iScience. 2020;23:101217. doi: 10.1016/j.isci.2020.101217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cassetta L., Fragkogianni S., Sims A.H., Swierczak A., Forrester L.M., Zhang H., Soong D.Y.H., Cotechini T., Anur P., Lin E.Y., et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas J.K., Mir H., Kapur N., Bae S., Singh S. CC chemokines are differentially expressed in Breast Cancer and are associated with disparity in overall survival. Sci. Rep. 2019;9:4014–4112. doi: 10.1038/s41598-019-40514-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Igarashi K., Takei N., Hori T., Yamamoto M., Kokai Y. CCL8 deficiency in host strongly inhibits early mortality and morbidity of graft-versus-host disease in mice. Blood. 2014;124:1096. [Google Scholar]
- 18.Hori T., Naishiro Y., Sohma H., Suzuki N., Hatakeyama N., Yamamoto M., Sonoda T., Mizue Y., Imai K., Tsutsumi H., Kokai Y. CCL8 is a potential molecular candidate for the diagnosis of graft-versus-host disease. Blood. 2008;111:4403–4412. doi: 10.1182/blood-2007-06-097287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igarashi K., Hori T., Yamamoto M., Sohma H., Suzuki N., Tsutsumi H., Kawasaki Y., Kokai Y. CCL8 deficiency in the host abrogates early mortality of acute graft-versus-host disease in mice with dysregulated IL-6 expression. Exp. Hematol. 2022;106:47–57. doi: 10.1016/j.exphem.2021.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Liu H., Liu Z., Chen J., Chen L., He X., Zheng R., Yang H., Song P., Weng D., Hu H., et al. Induction of CCL8/MCP-2 by mycobacteria through the activation of TLR2/PI3K/Akt signaling pathway. PLoS One. 2013;8:e56815. doi: 10.1371/journal.pone.0056815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Severa M., Islam S.A., Waggoner S.N., Jiang Z., Kim N.D., Ryan G., Kurt-Jones E., Charo I., Caffrey D.R., Boyartchuk V.L., et al. The transcriptional repressor BLIMP1 curbs host defenses by suppressing expression of the chemokine CCL8. J. Immunol. 2014;192:2291–2304. doi: 10.4049/jimmunol.1301799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J.-U., Cheong H.S., Shim E.-Y., Bae D.-J., Chang H.S., Uh S.-T., Kim Y.H., Park J.-S., Lee B., Shin H.D., Park C.S. Gene profile of fibroblasts identify relation of CCL8 with idiopathic pulmonary fibrosis. Respir. Res. 2017;18:3–12. doi: 10.1186/s12931-016-0493-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luzina I.G., Salcedo M.V., Rojas-Peña M.L., Wyman A.E., Galvin J.R., Sachdeva A., Clerman A., Kim J., Franks T.J., Britt E.J., et al. Transcriptomic evidence of immune activation in macroscopically normal-appearing and scarred lung tissues in idiopathic pulmonary fibrosis. Cell. Immunol. 2018;325:1–13. doi: 10.1016/j.cellimm.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thoutam A., Breitzig M., Lockey R., Kolliputi N. Coronavirus: a shift in focus away from IFN response and towards other inflammatory targets. J. Cell Commun. Signal. 2020;14:469–470. doi: 10.1007/s12079-020-00574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suhre K., Sarwath H., Engelke R., Sohail M.U., Cho S.J., Whalen W., Alvarez-Mulett S., Krumsiek J., Choi A.M.K., Schmidt F. Identification of robust protein associations with COVID-19 disease based on five clinical studies. Front. Immunol. 2022;12:781100. doi: 10.3389/fimmu.2021.781100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Havighorst A., Zhang Y., Farmaki E., Kaza V., Chatzistamou I., Kiaris H. Differential regulation of the unfolded protein response in outbred deer mice and susceptibility to metabolic disease. Dis. Model. Mech. 2019;12:dmm037242. doi: 10.1242/dmm.037242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Havighorst A., Crossland J., Kiaris H. Seminars in Cell & Developmental Biology. Elsevier; 2017. Peromyscus as a model of human disease; pp. 150–155. [DOI] [PubMed] [Google Scholar]
- 29.Chatzistamou I., Farmaki E., Kaza V., Kiaris H. The value of outbred rodent models in Cancer research. Trends Cancer. 2018;4:468–471. doi: 10.1016/j.trecan.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 30.de Souza Xavier Costa N., Ribeiro Júnior G., dos Santos Alemany A.A., Belotti L., Zati D.H., Frota Cavalcante M., Matera Veras M., Ribeiro S., Kallás E.G., Nascimento Saldiva P.H., et al. Early and late pulmonary effects of nebulized LPS in mice: an acute lung injury model. PLoS One. 2017;12:e0185474. doi: 10.1371/journal.pone.0185474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domscheit H., Hegeman M.A., Carvalho N., Spieth P.M. Molecular dynamics of lipopolysaccharide-induced lung injury in rodents. Front. Physiol. 2020;11:36. doi: 10.3389/fphys.2020.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafikov R., Dimitropoulou C., Aggarwal S., Kangath A., Gross C., Pardo D., Sharma S., Jezierska-Drutel A., Patel V., Snead C., et al. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J. Biol. Chem. 2014;289:4710–4722. doi: 10.1074/jbc.M114.547596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao Q., Vadgama J.V., Wang P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020;18:82. doi: 10.1186/s12964-020-00589-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ge B., Li J., Wei Z., Sun T., Song Y., Khan N.U. Functional expression of CCL8 and its interaction with chemokine receptor CCR3. BMC Immunol. 2017;18:54–58. doi: 10.1186/s12865-017-0237-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Islam S.A., Chang D.S., Colvin R.A., Byrne M.H., McCully M.L., Moser B., Lira S.A., Charo I.F., Luster A.D. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ TH 2 cells. Nat. Immunol. 2011;12:167–177. doi: 10.1038/ni.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halvorsen E.C., Hamilton M.J., Young A., Wadsworth B.J., LePard N.E., Lee H.N., Firmino N., Collier J.L., Bennewith K.L. Maraviroc decreases CCL8-mediated migration of CCR5+ regulatory T cells and reduces metastatic tumor growth in the lungs. OncoImmunology. 2016;5:e1150398. doi: 10.1080/2162402X.2016.1150398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Struyf S., Proost P., Vandercappellen J., Dempe S., Noyens B., Nelissen S., Gouwy M., Locati M., Opdenakker G., Dinsart C., Van Damme J. Synergistic up-regulation of MCP-2/CCL8 activity is counteracted by chemokine cleavage, limiting its inflammatory and anti-tumoral effects. Eur. J. Immunol. 2009;39:843–857. doi: 10.1002/eji.200838660. [DOI] [PubMed] [Google Scholar]
- 38.Satija R., Shalek A.K. Heterogeneity in immune responses: from populations to single cells. Trends Immunol. 2014;35:219–229. doi: 10.1016/j.it.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parameswaran N., Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradley J.R. TNF-mediated inflammatory disease. J. Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 41.Haringman J.J., Gerlag D.M., Smeets T.J.M., Baeten D., Van den Bosch F., Bresnihan B., Breedveld F.C., Dinant H.J., Legay F., Gram H., et al. A randomized controlled trial with an anti-CCL2 (anti–monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:2387–2392. doi: 10.1002/art.21975. [DOI] [PubMed] [Google Scholar]
- 42.Raghu G., Martinez F.J., Brown K.K., Costabel U., Cottin V., Wells A.U., Lancaster L., Gibson K.F., Haddad T., Agarwal P., et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur. Respir. J. 2015;46:1740–1750. doi: 10.1183/13993003.01558-2014. [DOI] [PubMed] [Google Scholar]
- 43.Loberg R.D., Ying C., Craig M., Yan L., Snyder L.A., Pienta K.J. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia. 2007;9:556–562. doi: 10.1593/neo.07307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J., Patel L., Pienta K.J. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010;21:41–48. doi: 10.1016/j.cytogfr.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matute-Bello G., Downey G., Moore B.B., Groshong S.D., Matthay M.A., Slutsky A.S., Kuebler W.M., Acute Lung Injury in Animals Study Group An officialAmerican Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper and 1G3E5 antibody will be shared by the lead contact upon request. Peromyscus animals are distributed by the PGSC.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.