

Abstract
HTT full‐penetrance pathogenic repeat expansions, the genetic cause of Huntington's disease (HD), have been recently reported in a minority of frontotemporal dementia/amyotrophic lateral sclerosis (ALS) patients (0.13%). We analyzed HTT CAG repeats in an Italian cohort of ALS patients (n = 467) by repeat‐primed polymerase chain reaction. One patient harbored two expanded alleles in the HTT gene (42 and 37 CAG repeats). The absence of HD typical symptoms and the clinical picture consistent with ALS, corroborated by the diagnostic assessment, apparently excluded a misdiagnosis of HD.
Introduction
Dewan and colleagues have recently reported HTT full‐penetrance pathogenic repeat expansions in three probands (0.12%) out of 2442 frontotemporal dementia (FTD)/amyotrophic lateral sclerosis (ALS), patients. 1 After expanding the analysis to an independent cohort of 3674 FTD/ALS patients, five additional carriers of HTT pathogenic expansions were identified (0.14%). Comparing these data to the prevalence of pathogenic HTT repeat expansions in the general population (0.03%), 2 , 3 the authors concluded that the carrier rate was significantly higher in FTD/ALS patients.
Thomas and colleagues have recently challenged this finding, highlighting several points which argue against the role of HTT pathogenic expansions in FTD/ALS. 4 Among them, the authors cited a previously published work which reported a 0.18% carrier rate of HTT repeat expansions in the general population. Accordingly, they suggested that the occurrence of HTT pathogenic expansions in FTD/ALS might merely reflect their prevalence among the general population. 5 Furthermore, Thomas and colleagues questioned the lack of clinical description of the cases. Indeed, they could have been misdiagnosed due to the clinical heterogeneity of HD, especially in juvenile forms, and to the age‐dependent penetrance of HTT pathogenic expansions. 6 , 7 Regarding neuropathology, Thion and coauthors stated that the absence of neostriatal atrophy was coherent with the small repeat expansions of the investigated patients, and that the co‐existence of huntingtin and TAR DNA‐binding protein 43 (TDP‐43) aggregates and motor neuron loss had been previously reported in HD brains. 8 , 9
In this scenario, we performed a screening analysis of CAG repeats in HTT in our cohort of ALS patients, to further assess huntingtin role in motor neuron disorders (MNDs).
Methods
A screening analysis of HTT CAG repeats was conducted by repeat‐primed polymerase chain reaction (RP‐PCR) in a cohort of Italian patients, who were diagnosed with ALS according to the El Escorial criteria, 10 as previously described. 11 A group of 298 Italian healthy subjects (aged >60 years) was used as control group. The length of expanded alleles was estimated by RP‐PCR and confirmed by fluorescent PCR. Sanger sequencing was used to exclude repeat interruptions and modifications in the polyproline sequence following CAG repeats. Carriers of expanded HTT alleles underwent a targeted next‐generation sequencing (NGS) panel to analyze genes implicated in MNDs (Table S1). The library was generated by using a 150‐bp amplicon‐based approach (Haloplex, Agilent) and sequenced on MiSeq instrument (Illumina). Reads were aligned to the human genome (assembly hg19), and the identified variants were annotated (ANNOVAR) and filtered, focusing on rare variants (≤0.5% in public databases), causing changes potentially damaging for the protein function.
Results
Within our cohort (n = 467), ALS patients' mean age at onset was 60.6 years (SD 13.7), and 60.9% were males. Familial cases were 6.6%. The distribution of CAG repeats in the ALS cohort was evaluated and compared to a control group of Italian healthy subjects (n = 298, age at sampling >60 years) (Fig. 1). The difference observed between the two groups failed to reach statistical significance (Mann–Whitney U‐test, p = 0.056). The repeat frequency of normal (CAG < 27) and unstable intermediate (27 < CAG ≤ 35) alleles was not different in the two groups (Fisher's exact test, p > 0.05). The proportion of intermediate alleles was identical in the control (14/298 = 4.7%) and in the ALS (22/267 = 4.7%) groups and comparable to that observed in an independent cohort of Italian control subjects (5.3%). 12
Figure 1.
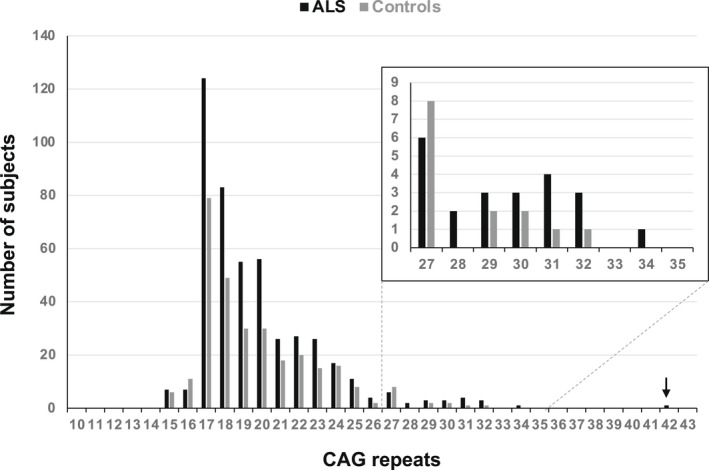
HTT CAG repeat length (largest allele) in 466 ALS patients and 292 Italian control subjects (age at sampling >60 years). The insert shows a zoomed view of HTT intermediate alleles (27–35 CAG repeats). The arrow indicates the ALS Patient harboring a full‐penetrant pathogenic HTT allele described in the text.
Interestingly, we identified one ALS patient who harbored two expanded alleles in the HTT gene with 42 and 37 CAG repeats (Fig. 2A and Fig. S1). By performing targeted NGS panel analysis, no additional disease‐causing mutations were detected in genes implicated in MNDs in this patient (Subject II‐2 in Fig. 2B). The hexanucleotide pathogenic expansion in chromosome 9 open reading frame 72 (C9ORF72) was also excluded.
Figure 2.
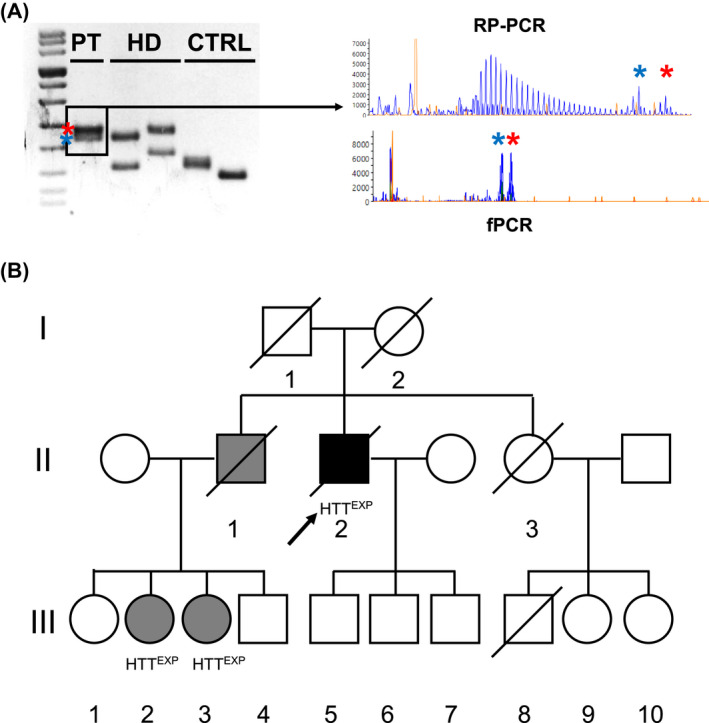
(A) (left) PCR amplicons encompassing HTT CAG repeat regions electrophoresed through agarose gel amplified starting from DNA obtained of the patient described in manuscript (PT), two HD subjects (size of the largest alleles: 38 and 42, respectively) and two control subjects. (right) Electropherograms obtained from patient's DNA after repeat‐primed (RP‐PCR) and fluorescent (fPCR) PCR amplification, displaying the saw‐tooth pattern and the expanded HTT alleles (37 CAG in blue, 42 CAG in red). (B) Pedigree of the family described in the text. Roman and Arab numbers are used to indicate the generation and the subject within each generation. The arrow indicates our proband. Black and gray symbols indicate disease status (clinical diagnosis of ALS and HD, respectively). HTTexp indicates a positive molecular testing for CAG repeats expanded HTT alleles.
At the age of 61, the patient started complaining of muscle weakness in the upper limbs, mainly on the right. Neurological evaluation showed signs of muscle wasting and asymmetric brisk deep tendon reflexes at the upper limbs. Electromyography detected chronic neurogenic changes and acute denervation signs in the distal muscles of both upper limbs. Brain and spinal cord magnetic resonance imaging (MRI) ruled out alternative diseases, and a diagnosis of possible ALS was formulated. Eighteen months later, he developed progressive dysphagia and severe respiratory impairment. Therefore, percutaneous endoscopic gastrostomy (PEG) and tracheostomy were placed. No cognitive impairment nor psychiatric disturbances were reported. He died of respiratory insufficiency at 64. The proband was born to Italian, nonconsanguineous parents. The father died at 35 years during the Second World War. The mother was asymptomatic for neurological disorders and died at 80 years of age. The patient had two siblings. His sister (II‐3) died at 60 years of lung carcinoma, with no signs or symptoms of nervous system dysfunction. She had one son (III‐8), who died in a car accident at 30 years, and two currently healthy daughters, of 62 and 60 (III‐9, III‐10). The proband's brother (II‐1) died at 70 years of age. At 55, he had been diagnosed with HD. One daughter (III‐1) and the only son (III‐4) of, respectively, 67 and 50 years have not been tested. She is asymptomatic for neurological or psychiatric disorders, while he recently developed psychiatric symptoms. The remaining two daughters, currently of 61 and 59 years of age, carry full‐penetrance pathogenic repeat expansions in HTT. The older daughter (III‐2) developed psychiatric symptoms at 35 years of age, choreic movements, and extrapyramidal motor signs at 50, and she later developed cognitive impairment. Genetic testing revealed 42 and 18 CAG repeats in HTT alleles. The younger daughter (III‐3) developed depression at age 45, followed by choreic movements. Our proband has three sons, respectively, 48, 42, and 36 years old (III‐5, III‐6, III‐7). They have not shown signs of neurological involvement so far, and they refused to pursue the predictive genetic testing.
Discussion
In our case, the positive family history for HD and the absence of MND in the relatives who harbor a pathogenic HTT repeat expansion might suggest a “double‐trouble” in the proband. Our patient could be affected, at least apparently, by sporadic, rapidly progressive ALS, which led to death before the appearance of HD signs or symptoms. Although we ruled out molecular defects in a large panel of genes involved in MNDs (including all the ALS causative genes), an undisclosed genetic or environmental predisposing factor might have increased the susceptibility to develop a MND phenotype. The absence of HD in our proband might be explained by the wide age at onset variation observed among HD patients with CAG repeats between 40 and 42. Indeed, the age at onset of Italian HD patients with 42 CAG repeats is 39–69 years. 13
However, the remarkable difference of age at onset within the family (about 20 years) and the absence of psychiatric or motor symptoms consistent with HD in our proband, who reached the seventh decade, deserve further reflections.
Our proband harbored two expanded HTT alleles. Subjects harboring two HTT expanded alleles are rare with estimated prevalence ranging from 0.1% 14 to 0.4% 15 within HD cohorts. The largest HTT allele, usually considered the main determinant for disease onset and progression, is likely shared by the patient's brother and nieces. The minor allele (37 repeats) confers a low‐penetrance risk for HD and could potentially be involved in modifying the clinical presentation. Indeed, HTT alleles with 37 repeats are considered pathogenic with reduced penetrance (37 < CAG < 40). Nevertheless, biallelic HD patients do not display significant differences in disease onset or progression compared to those harboring a single mutated allele, 16 supporting the “complete dominance” of HD.
Several reports have described the occurrence of ALS in patients with a family history of HD and a positive genetic testing for high‐penetrance pathogenic CAG repeat expansion in HTT. 17 , 18 , 19 , 20 Interestingly, in two cases the available post‐mortem tissues displayed concomitant ALS and HD pathology, 21 , 22 a finding recently paralleled in larger independent HD brain cohorts. 18
These findings suggest that HTT expansion might predispose a subset of individuals to develop clinical and pathological features consistent with ALS. This hypothesis cannot be definitively excluded in our patient. Considering the clinical picture consistent with MND, corroborated by the diagnostic assessment, and the absence of HD typical symptoms, we exclude a misdiagnosis of HD at the time of clinical evaluation of our patient. The presence of a positive family history for HD differentiates our case from those described by Dewan and colleagues. 1
Our study indicates that CAG abnormal repeats in HTT might be extremely rare in Italian ALS patients. The absence of pathological HTT repeat expansions in the remaining cohort might have been influenced by its demographic features, especially by the low rate of ALS familial cases (6.6%). Indeed, out of eight carriers of abnormal HTT CAG repeats reported by Dewan and colleagues, four had a known family history of FTD/ALS. This finding suggests that HTT pathogenic expansions might be more common in familial rather than sporadic FTD/ALS. 1 We did not observe a higher rate of intermediate (27–35) HTT repeat expansions in ALS patients compared to control subjects, although the difference in the distribution of CAG repeats between these two groups was nearly significant.
The association of expanded HTT alleles with heterogeneous clinical presentations might acknowledge the variable expressivity of this molecular defect. Otherwise, it might indicate that clinically distinct neurodegenerative disorders such as ALS and HD could share a common genetic basis. A similar hypothesis has been postulated for other repeat expansion disorders. Hexanucleotide repeat expansions in C9ORF72, the main genetic cause of ALS/FTD, have been linked to different neurodegenerative disorders, including dementia and parkinsonism, 23 and represent the most common genetic cause of HD phenocopies. 24 Notably, pathological CAG expansions in HTT can also induce TDP‐43 and huntingtin co‐aggregation in vitro, likely through a direct interaction between poly‐Q residues and TDP‐43 C‐terminal domain. 8 Therefore, HTT expansion could represent an additional example of the pathogenic link connecting poly‐Q expansions and TDP‐43 proteinopathies, including ALS/FTD.
In summary, we believe that the direct involvement of HTT in ALS pathogenesis, emerged by novel sequencing technologies, should be interpreted with great prudence. Although it is possible that HD and ALS are part of the same neurodegenerative disease landscape and that HTT expansion might produce ALS features as the primary manifestation in a small minority of HD individuals, available data so far are limited and fragmented. A replication of these studies in further cohorts is required to confidently confirm the pathogenic role of HTT repeat expansions in ALS before moving to common clinical practice.
Author Contributions
AM: conceptualization; data curation; formal analysis; investigation; writing–original draft preparation; writing–review and editing. DG: conceptualization; data curation; writing–original draft preparation; writing–review and editing. MM: writing–review and editing. SA: investigation. RDB: writing–review and editing. CS: writing–review and editing. GPC: writing–review and editing. SC: writing–review and editing. DR: conceptualization; data curation; formal analysis; investigation; writing–review and editing.
Conflict of Interest
RDB, GPC, and SC are members of the FALS Sequencing Consortium. The other authors declare no existing conflict of interests.
Supporting information
Figure S1. Sanger sequencing chromatograms of the HTT CAG repeat expansion. HTT CAG repeat length in the patient described was assessed by cloning followed by Sanger sequencing. Electropherograms showing the two alleles containing 37 and 42 CAG repeats, respectively.
Table S1. List of genes included in our NGS panel.
Acknowledgements
This work was partially supported by Italian Ministry of Health (Ministero della Salute), Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico Grant Ricerca Corrente 2020 to GPC. We would like to thank “Associazione Centro Dino Ferrari” for its support.
Funding Information
This work was partially supported by Italian Ministry of Health (Ministero della Salute), Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico Grant Ricerca Corrente 2020 to GPC.
Funding Statement
This work was funded by Italian Ministry of Health (Ministero della Salute) ; Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico Grant Ricerca Corrente.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Dewan R, Chia R, Ding J, et al. Pathogenic huntingtin repeat expansions in patients with frontotemporal dementia and amyotrophic lateral sclerosis. Neuron. 2021;109(3):448‐460.e4. https://linkinghub.elsevier.com/retrieve/pii/S0896627320308837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gardiner SL, Boogaard MW, Trompet S, et al. Prevalence of carriers of intermediate and pathological polyglutamine disease–associated alleles among large population‐based cohorts. JAMA Neurol. 2019;76(6):650. doi: 10.1001/jamaneurol.2019.0423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peplow M. The 100,000 genomes project. BMJ. 2016;i1757. doi: 10.1136/bmj.i1757 [DOI] [PubMed] [Google Scholar]
- 4. Thomas Q, Coarelli G, Heinzmann A, Le Ber I, Amador MM, Durr A. Questioning the causality of HTT CAG‐repeat expansions in FTD/ALS. Neuron. 2021;109(12):1945‐1946. https://linkinghub.elsevier.com/retrieve/pii/S0896627321002609 [DOI] [PubMed] [Google Scholar]
- 5. Thion MS, Tézenas du Montcel S, Golmard J‐L, et al. CAG repeat size in Huntingtin alleles is associated with cancer prognosis. Eur J Hum Genet. 2016;24(9):1310‐1315. http://www.nature.com/articles/ejhg201613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee J‐M, Ramos EM, Lee J‐H, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78(10):690‐695. doi: 10.1212/WNL.0b013e318249f683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paulsen JS, Miller AC, Hayes T, Shaw E. Cognitive and behavioral changes in Huntington disease before diagnosis. Handb Clin Neurol. 2017;144:69‐91. https://linkinghub.elsevier.com/retrieve/pii/B9780128018934000067 [DOI] [PubMed] [Google Scholar]
- 8. Coudert L, Nonaka T, Bernard E, Hasegawa M, Schaeffer L, Leblanc P. Phosphorylated and aggregated TDP‐43 with seeding properties are induced upon mutant Huntingtin (mHtt) polyglutamine expression in human cellular models. Cell Mol Life Sci. 2019;76(13):2615‐2632. doi: 10.1007/s00018-019-03059-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phillips O, Squitieri F, Sanchez‐Castaneda C, et al. The corticospinal tract in Huntington's disease. Cereb Cortex. 2015;25(9):2670‐2682. doi: 10.1093/cercor/bhu065 [DOI] [PubMed] [Google Scholar]
- 10. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord. 2000;1(5):293‐299. doi: 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- 11. Jama M, Millson A, Miller CE, Lyon E. Triplet repeat primed PCR simplifies testing for Huntington disease. J Mol Diagn. 2013;15(2):255‐262. https://linkinghub.elsevier.com/retrieve/pii/S152515781200308X [DOI] [PubMed] [Google Scholar]
- 12. Mongelli A, Magri S, Salvatore E, et al. Frequency and distribution of polyQ disease intermediate‐length repeat alleles in healthy Italian population. Neurol Sci. 2020;41(6):1475‐1482. doi: 10.1007/s10072-019-04233-3 [DOI] [PubMed] [Google Scholar]
- 13. Capiluppi E, Romano L, Rebora P, et al. Late‐onset Huntington's disease with 40–42 CAG expansion. Neurol Sci. 2020;41(4):869‐876. doi: 10.1007/s10072-019-04177-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kremer B, Goldberg P, Andrew SE, et al. A worldwide study of the Huntington's disease mutation: the sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994;330(20):1401‐1406. doi: 10.1056/NEJM199405193302001 [DOI] [PubMed] [Google Scholar]
- 15. Alonso M, Yescas P, Rasmussen A, et al. Homozygosity in Huntington's disease: new ethical dilemma caused by molecular diagnosis. Clin Genet. 2002;61(6):437‐442. doi: 10.1034/j.1399-0004.2002.610607.x [DOI] [PubMed] [Google Scholar]
- 16. Cubo E, Martinez‐Horta S‐I, Santalo FS, et al. Clinical manifestations of homozygote allele carriers in Huntington disease. Neurology. 2019;92. doi: 10.1212/WNL.0000000000007147 [DOI] [PubMed] [Google Scholar]
- 17. Oskarsson B, Wheelock V, Benatar M, et al. A case of familial ALS due to multi‐system proteinopathy 1 and Huntington disease. Amyotroph Lateral Scler Front Degener. 2015;16(1–2):124‐126. doi: 10.3109/21678421.2014.952238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hickman RA, Dewan R, Cortes E, et al. Amyotrophic lateral sclerosis is over‐represented in two Huntington's disease brain bank cohorts: further evidence to support genetic pleiotropy of pathogenic HTT gene expansion. Acta Neuropathol. 2022;143(1):105‐108. doi: 10.1007/s00401-021-02385-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bernard E, Mouzat K, Leblanc P, et al. Amyotrophic lateral sclerosis in Huntington disease gene carrier. Rev Neurol. 2017;173(10):670‐671. https://linkinghub.elsevier.com/retrieve/pii/S0035378716302016 [DOI] [PubMed] [Google Scholar]
- 20. Smith AL, Teener JW, Callaghan BC, Harrington J, Uhlmann WR. Amyotrophic lateral sclerosis in a patient with a family history of huntington disease: genetic counseling challenges. J Genet Couns. 2014;23(5):725‐733. doi: 10.1007/s10897-014-9715-6 [DOI] [PubMed] [Google Scholar]
- 21. Rubio A, Steinberg K, Figlewicz DA, et al. Coexistence of Huntington's disease and familial amyotrophic lateral sclerosis: case presentation. Acta Neuropathol. 1996;92(4):421‐427. doi: 10.1007/s004010050539 [DOI] [PubMed] [Google Scholar]
- 22. Tada M, Coon EA, Osmand AP, et al. Coexistence of Huntington's disease and amyotrophic lateral sclerosis: a clinicopathologic study. Acta Neuropathol. 2012;124(5):749‐760. doi: 10.1007/s00401-012-1005-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cooper‐Knock J, Shaw PJ, Kirby J. The widening spectrum of C9ORF72‐related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol. 2014;127(3):333‐345. doi: 10.1007/s00401-014-1251-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hensman Moss DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. 2014;82(4):292‐299. doi: 10.1212/WNL.0000000000000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sanger sequencing chromatograms of the HTT CAG repeat expansion. HTT CAG repeat length in the patient described was assessed by cloning followed by Sanger sequencing. Electropherograms showing the two alleles containing 37 and 42 CAG repeats, respectively.
Table S1. List of genes included in our NGS panel.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.