

Abstract
PLIN4‐myopathy is a recently identified autosomal dominant muscular disorder caused by the coding 99 bp repeat expansion in PLIN4, presenting with distal or proximal weakness. Here, we report one family and one sporadic case of adult‐onset PLIN4‐associated limb‐girdle weakness, whose diagnoses were achieved by a comprehensive genetic analysis workup. We provided additional evidence that the combination of subsarcolemmal/cytoplasmic ubiquitin/p62 positive deposits and rimmed vacuoles could serve as a strong indicator of PLIN4‐myopathy. Moreover, we found novel myopathological features that were ultrastructural subsarcolemmal filamentous materials and membrane‐bound granulofilamentous inclusions formed by the co‐deposition of disrupted lipid droplets and p62 protein aggregates.
Introduction
Autophagic vacuolar myopathies are an emerging group of heterogeneous myopathies sharing histopathological features on muscle pathology, in which genetic diagnosis remains challenging. 1 Recently, two studies reported an autosomal dominant myopathy with rimmed ubiquitin‐positive autophagic vacuolation associated with the coding 99 bp repeat expansion in PLIN4, presenting with either distal myopathy or predominantly proximal muscle weakness. Identical pathological characteristics include ubiquitin‐positive rimmed vacuoles (RVs) and increased perilipin‐4/p62/FK2 positivity in both subsarcolemmal regions and vacuoles. 2 , 3 , 4
Here, we report one more family and the first sporadic patient with PLIN4‐associated adult‐onset limb‐girdle weakness. Additionally, we found subsarcolemmal filamentous materials and membrane‐bound granulofilamentous inclusions between myofibrils formed by protein‐lipid deposits under electron microscopy (EM), which might be related to the pathogenesis of the 99 bp repeat expansion of PLIN4.
Methods and Results
Patients and clinical data
This study was approved by the Ethics Committee at Peking University First Hospital. Seven individuals from four successive generations of one family (Patient F) and one sporadic patient (Patient S) suffered predominantly proximal muscle weakness in their forties to sixties (Fig. 1A). The familial and sporadic cases were referred to Peking University First Hospital in 2009 and 2019, respectively. The initial symptom in all of these cases was proximal muscle weakness of the lower limbs. Muscle weakness developed slowly in affected individuals with disease progression, manifesting as muscle involvement in the proximal upper limb, and the distal muscles are also partially involved. The sporadic case also presented with chest distress. Muscle imaging showed severe fatty replacement, with the thigh muscles more severally affected than the calves. The adductor, vastus intermedius, vatus medialis, soleus, and gastrocnemius were most frequently affected, while the rectus femoris and gracilis were spared (Fig. 1B–E). The pulmonary function test of the sporadic patient showed restrictive ventilation dysfunction. Brain MRI of F‐III3 revealed no special findings. Table 1 presents the detailed clinical characteristics of the patients.
Figure 1.
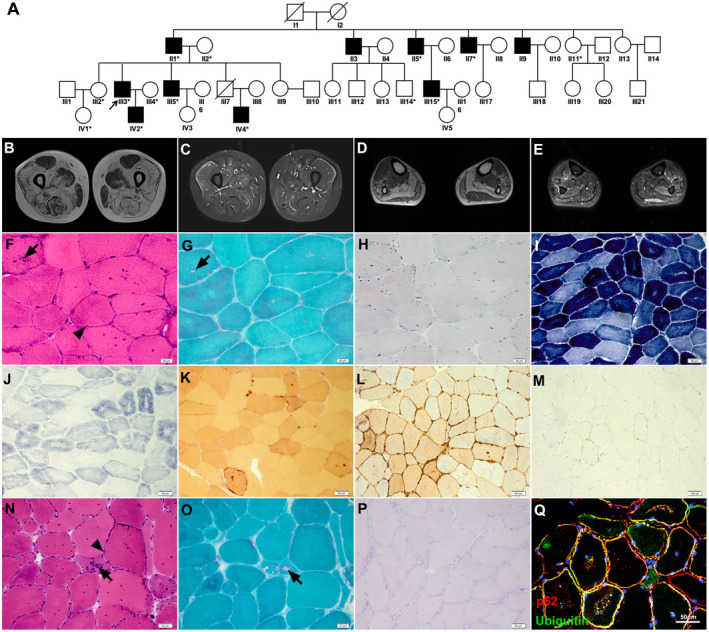
Pedigree, muscle MRI, and pathology. (A) Pedigree chart of the PLIN4‐related family. All the asterisked individuals had available blood DNA. (B–E) Muscle imaging showed severe fatty replacement and mild muscular edema, with the thigh muscles more severally affected than the calves, and rectus femoris and gracilis relative spared. H&E, mGT, and ORO staining of muscles biopsies of Patient S (F–H) and F‐III3 (N–P), NADH‐TR (I), SDH (G), NSE (K), MHC‐I (L), and MAC (M) staining and immunofluorescence of p62/ubiquitin (Q) from Patient S showed amorphous basophilic materials distributed beneath the sarcolemma (arrowhead) and RVs (arrow), with focal areas devoid of NADH‐TR and SDH and increased MHC‐I and MAC expression. ORO staining was normal. Immunohistochemistry showed p62/ubiquitin‐positive protein deposition in both the subsarcolemmal region and in the cytoplasm. Scale bars = 20 μm (F, G, H, and O), 50 μm (I, J, K, N, P, and Q), 100 μm (L and M).
Table 1.
The Clinical features of 4 individuals with PLIN4 repeat expansion mutation.
| Patient | F‐II7 | F‐III3 | F‐IV2 | S |
|---|---|---|---|---|
| Sex | M | M | M | M |
| Age of onset | 49 | 40 | 22 | 49 |
| Disease duration (years) | 15 | 11 | 1 | 11 |
| Initial symptom | Weakness in climbing upstairs | Waddling gait | ‐ | Weakness in climbing upstairs |
| Muscle strength | ||||
| Neck flexion weakness | ‐ | ‐ | ‐ | + |
| Limb muscle weakness | P > D, L > U | P > D, L > U | ‐ | P > D, L > U |
| Deep tendon reflexes | Absent | Absent | Brisk | Absent |
| Skeletal deformities | Winging scapula | ‐ | ‐ | ‐ |
| Maximal mobility | Sit | Walk with support | Run | Sit |
| Other clinical features | ||||
| Ophthalmoplegia | ‐ | ‐ | ‐ | ‐ |
| Facial weakness | ‐ | ‐ | ‐ | ‐ |
| Dysphagia | ‐ | ‐ | ‐ | ‐ |
| Dysarthria | ‐ | ‐ | ‐ | ‐ |
| Respiratory abnormalities | ‐ | ‐ | ‐ | Restrictive ventilation |
| Cardiac abnormalities | ‐ | ‐ | ‐ | ‐ |
| Serum creatine kinase 1 | 822 IU/L | 752 IU/L | 267 IU/L | 256 IU/L |
| EMG pattern | MP+ CRDs | MP+ CRDs | MP + CRDs | MP+ CRDs |
| Muscle magnetic resonance | ||||
| Fatty infiltration |
Thigh: VIM, VM, ADDMAG, ADDBRE, SM Calf: SOL, GAS, TA, EDL, EHL, PL, PB |
Thigh: VIM, VM, VL, SAR, ADDLON, ADDMAG, SM |
Thigh: ‐ Calf: ‐ |
Thigh: VIM, VM, VL, ADDMAG, ADDBRE, SAR, SM, ST, BF Calf: SOL, GAS, PB |
| Edema |
Thigh: VIM, VM, ADDMAG, ADDBRE, SM, BF Calf: GAS, TA, EDG, EHL, PL, PB |
Calf: SOL, GAS, TA, EDL, EHL, PB Thigh: VIM, VM, ADDMAG, ADDBRE, SM, BF Calf: SOL, PL, EHL |
Thigh: ‐ Calf: SOL, GAS |
Thigh: VIM, VM, VL Calf: SOL, GAS, PL |
Abbreviations: ADDBRE, adductor brevis; ADDLON, adductor longus; ADDMAG, adductor magnus; BF, biceps femoris; CRDs, complex repetitive discharges; D, distal muscle; EDL, extensor digitorum longus; EHL, extensor hallucis longus; F, family patient; F, female; GAS, gastrocnemius; L, lower limbs; M, male; MP, myopathic pattern; P, proximal muscle; PB, peroneus brevis; PL, peroneus longus; S, sporadic patient; SAR, sartorius; SM, semimembranosus; SOL, soleus; ST, semitendinosus; TA, tibialis anterior; U, upper limbs; VIM, vastus intermedius; VL, vastus lateralis; VM, vastus medialis.
Normal limits: 70–170 IU/L.
Pathological features
Biopsies of the tibialis anterior muscle from F‐III3, biceps brachii muscles from F‐II7, and Patient S were obtained according to a standard procedure. 5 The pathological features of two family members and Patient S were highly consistent.
For histological examination, hematoxylin and eosin (H&E) (Fig. 1F and N) and modified Gomori trichrome (mGT) staining (Fig. 1G and O) of the three patients revealed myopathic changes characterized by RVs, mild fiber size variation, and internal nucleation. In particular, we found amorphous basophilic materials beneath the sarcolemma and within myofibers, with strong activity for non‐specific esterase (NSE), which highlighted the phagocytic areas (Fig. 1K). Nicotinamide adenine dinucleotide tetrazolium reductase (NADH‐TR) and succinate dehydrogenase (SDH) staining revealed central areas devoid of activity (Fig. 1I and J). Increased MHC‐I expression and membrane attack complex (MAC) deposition were also observed in all patients with minor inflammatory infiltrates (Fig. 1L and M). Oil red O (ORO) staining was normal (Fig. 1H and P).
Immunofluorescence showed p62/ubiquitin‐positive protein was deposited under the sarcolemma and within myofibers (Fig. 1Q).
EM was done as in the previous study. 6 Vacuoles containing numerous membranous or myeloid bodies were found to be located beneath the basal lamina under electron microscopy (EM) (Fig. 2D). Remarkably, EM revealed filamentous materials beneath the basal membrane (Fig. 2H and I) and numerous elongated or round membrane‐bound inclusions containing granulofilamentous materials between myofibrils (Fig. 2E‐G). Some clustered mitochondria were observed around inclusions. Lipid droplets appeared to be normally distributed without aggregation.
Figure 2.
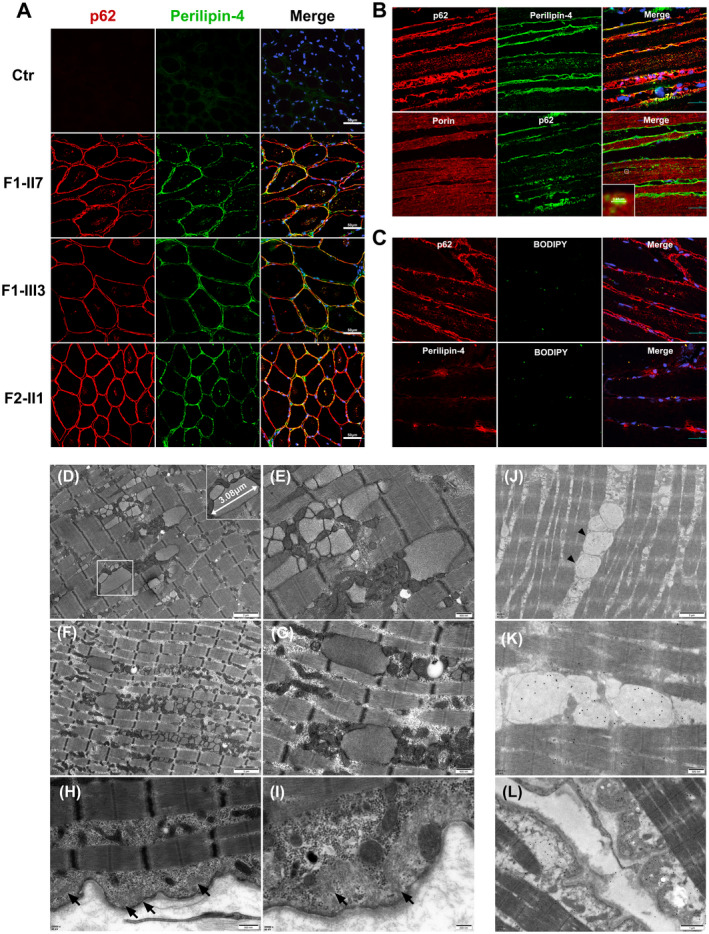
Immunofluorescence, electron microscopy, and immunoelectron microscopy on muscle biopsy samples. (A and B) Co‐localization of perilipin‐4 and p62 in subsarcolemmal regions and within the cytoplasm of myofibers (A) and p62‐positive perilipin‐4 deposition (mean size 3.48 μm) distributed longitudinally within muscular fibers adjacent to porin‐positive mitochondria of F‐III3(B). (C) Colocalization of lipid droplets with p62 and perilipin‐4 in subsarcolemma and cytoplasm of myofibers in Patient S. (D) Vacuoles containing numerous membranous or myeloid bodies and (E–G) numerous membrane‐bound inclusions (mean size 3.08 μm) containing granulofilarmentous materials appeared between myofibrils under electron microscopy of Patient S (E) and F‐III3 (F and G) with clustered mitochondria around the inclusions. (H and I) Filamentous materials beneath basal membrane in Patient S (arrow). (J, K, and L) Immunofluorescence and immunoelectron microscopy (IEM) of Patient S revealed that the membrane‐bound inclusions and subsarcolemmal filamentous materials were p62 positive (arrowhead). Scale bars = 200 nm (I), 500 nm (E, G, H, and K), 1 μm (D and L), 2 μm (F and J), and 50 μm (A, B, and C).
Genetic analysis
Whole‐exome sequencing of F‐II7, F‐III3, and Patient S in 2019 revealed negative results. Noting that the distinctive p62/ubiquitin‐positive staining pattern was the same as that in PLIN4‐associated myopathy published in 2020, we re‐analyzed PLIN4 using next‐generation sequencing (NGS), and its Integrative Genomics Viewer (IGV) visualization revealed an unusually high coverage in exon 3. 3 , 4 Polymerase chain reaction (PCR) amplification of this region showed a wild‐type band in unaffected relatives and a second ~1000 bp higher band in patients. Linkage analysis showed a 3.39 Mb candidate region at 19p13.3 (chr19:2635061–6033965) with a maximum logarithm of odds score of 3.0408. Finally, Oxford Nanopore long‐read sequencing (LRS) was performed on F‐III3. In the candidate region, short tandem repeat (STR) analysis revealed negative findings, while the variable number of tandem repeats (VNTR) analysis showed reads with an abnormal 39 × 99‐neucleotide sequence in exon 3 of PLIN4 in F1‐III3, and the repeat was 29 times that of the control. Consistent with the previous study, 7 our LRS data indicated that the VNTR in PLIN4 was composed of consecutive repetitive DNA with hypervariable motif composition (Fig. S1).
Immunofluorescence and immunoelectron microscopy
Further immunofluorescence and Immunofluorescence and immunoelectron microscopy (IEM) were performed to confirm the correlation between PLIN4 repeat expansion and histopathological changes. Muscle sections were immunostained with primary antibodies against perilipin‐4 (ProteinTech Group, 55404‐1‐AP, anti‐rabbit), p62 (Abcam, ab56416, anti‐mouse), p62 (ProteinTech Group, 18420‐1‐AP, anti‐rabbit), porin (Abcam, ab34726, anti‐mouse), BODIPY (Invitrogen, H34477). All the sections were subsequently incubated with Alexa Fluor488/555 secondary antibodies. Images were acquired with confocal microscopy (Nikon A1MP). IEM was conducted with polyclonal rabbit anti‐p62 (ProteinTech Group, 18420‐1‐AP, anti‐rabbit) or anti‐perilipin‐4 (ProteinTech Group, 55404‐1‐AP, anti‐rabbit), and a 25‐nm gold‐conjugated secondary antibody.
First, immunofluorescence showed perilipin‐4 positive protein deposition in both the subsarcolemmal and intracytoplasmic regions, which co‐localized with p62 and ubiquitin in three PLIN4‐affected individuals (F‐II7, F‐III3, and S) (Fig. 2A). We further performed immunofluorescence of p62 and perilipin‐4 in the longitudinal section of the skeletal muscles. The results revealed rod‐shaped p62‐positive perilipin‐4 deposition distributed longitudinally within myofibers, which were adjacent to the mitochondria, with a mean size similar to that of membrane‐bound inclusions under EM (Fig. 2B). IEM was performed with specific anti‐p62, anti‐perilipin‐4 antibody and secondary anti‐rabbit IgG conjugated to 25 nm gold particles, which revealed that the subsarcolemmal filamentous materials and membrane‐bound inclusions were p62/perilipin‐4 positive (Fig. 2J–L, Fig. S2). Together, the results indicated that the 99 bp repeat expansion of PLIN4 caused protein accumulation. Second, considering that perilipin‐4 is a coat protein of lipid droplets (LDs) related to its stability, 8 immunofluorescence of BODIPY/p62 and BODIPY/perilipin‐4 was conducted, which indicated colocalization of lipid droplets with p62 and perilipin‐4 in the subsarcolemma and cytoplasm of myofibers (Fig. 2C). These findings suggest that subsarcolemmal filamentous materials and membrane‐bound inclusions might originate from abnormal lipid droplet formation, resulting in the co‐deposition of residual lipids and perilipin‐4/p62 protein aggregates.
Discussion
Our results expanded the clinical spectrum of 99 bp repeat expansions in PLIN4. Perilipin‐4 is mostly expressed in adipose cells, the brain, and the skeletal and heart muscles. 9 , 10 We reported the predominantly proximal muscle weakness supported by clinical and muscle imaging consistent with another Chinese study, compared to distal myopathy in the Italian cohort. 3 , 4 Patient S also presented with respiratory dysfunction which has never been reported. The central nervous system was not involved in our patients with normal brain MRI performance. As for skeletal muscle involvement, we found that adductor‐muscle, vastus intermedius‐medialis, soleus, and gastrocnemius were the most affected, accompanied by muscular edema, while rectus femoris and gracilis were little affected, which may partially facilitate diagnosis. EMG revealed a myopathic pattern with myotonic discharge and/or CRD as other vacuolar myopathies such as oculopharyngodistal myopathy (OPDM), myofibrillar myopathy (MFM), but the relationship between them needs to be explored. 11 , 12 The reason for distinct clinical phenotypes in PLIN4‐myopathy may be related to the hypervariable motif composition in VNTR as the important genetic modifiers. 13
Consistent with a previous study, immunohistochemical workup showed perilipin‐4/p62 accumulation in the subsarcolemmal regions and RVs. We proposed that the subsarcolemmal p62/perilipin4 positivity is related to protein physiological distribution because perilipin‐4 is mainly located in the subsarcolemmal region. 14 Previous studies demonstrated that repeat expansion is associated with increased perilipin‐4 aggrephagy. 3 , 4 We observed overexpression of inflammatory markers and clustered mitochondria on muscle biopsy in all PLIN4‐associated patients, suggesting secondary muscle inflammation. 15
Abnormal aggregation and autophagy dysfunction contribute to the pathogenesis of different vacuolar myopathies with RVs; however, membrane‐bound inclusions and subsarcolemmal filamentous materials have not been previously observed in these neuromuscular disorders. 16 We assumed that the special pathogenesis underlying the repeat expansion in PLIN4 was responsible for the distinctive pathological features. Our preliminary study suggested that the co‐deposition of disrupted lipid droplets and protein aggregates might play a role in the pathogenesis of the coding 99 bp repeat expansion in PLIN4. However, further studies on cellular and animal models are required to investigate the underlying mechanisms of perilipin‐4 aggregates in this disease.
Taken together, we report the fourth family and the first sporadic patient with a 99 bp repeat expansion mutation of PLIN4. The distinctive subsarcolemmal and cytoplasmic ubiquitin/p62 positive deposits and RVs significantly facilitated our diagnosis and prompted us to revisit the NGS data of PLIN4 exon3. In addition, we identified subsarcolemmal filamentous materials and membrane‐bound inclusions, providing morphological diagnostic clues for PLIN4‐myopathy. We suggest that the distinctive pathological features combined with NGS analysis may help identify PLIN4 repeat expansion.
Author Contributions
All authors contributed to the study conception and design. QW and MY drafted the manuscript. YY, JD and ZW take full responsibility for the paper. Material preparation, data collection and analysis were performed by WZ, QG, LM, VL, ZJ. ZX, CZ and DW checked all the genetic analyses.
Conflict of Interest
None.
Supporting information
Figure S1 Identification of the 99 bp repeat expansion of PLIN4. (A) Genetic linkage analysis of 14 individuals in the family indicated a 3.39 Mb candidate region at 19p13.3 (chr19:2635061–6033965) with a maximum logarithm of odds score of 3.0408. (B) IGV visualization of NGS data showed a high peak of coverage in PLIN4 exon 3 in patients (arrows), not present in healthy control. (C) PCR amplification of the repeat region in exon 3 showed a wild‐type 3000 bp band and a second ~4000 bp band in affected individuals, and a single band (~ 3000 bp) in unaffected individuals. (D) LRS detection showed multiple reads with abnormal 39 × 99‐neucleotide sequence in exon 3 of PLIN4 in F‐III3, while the repeat was 29 times in control. (E) Repeat expansion of PLIN4 (NM_001080400.1).
Figure S2 Immunoelectron microscopy of perilipin‐4. IEM of Patient S revealed that the membrane‐bound inclusions were perilipin‐4 positive.
Acknowledgments
We would like to thank the patients and their family members for their participation in this study. We thank Mr Lijun Chai and Jin Xu (Laboratory of Electron Microscopy, Peking University First Hospital) for kindly providing technical assistance in electron microscopy. This study was approved by the Local Ethics Committee of Peking University First Hospital.
Funding Information The work was funded by the National Natural Science Foundation of China (No. 81571219, 82071409 and U20A20356 to Z.W. and No. 82171846 to J.D..) and Capital's Funds for Health Improvement and Research (2022‐4‐40716 to J.D.).
Contributor Information
Jianwen Deng, Email: jianwendeng@pkufh.com.
Yun Yuan, Email: yuanyun2002@126.com.
Zhaoxia Wang, Email: drwangzx@163.com.
References
- 1. Mair D, Biskup S, Kress W, et al. Differential diagnosis of vacuolar myopathies in the NGS era. Brain Pathol. 2020;30:877‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Di Blasi C, Moghadaszadeh B, Ciano C, et al. Abnormal lysosomal and ubiquitin‐proteasome pathways in 19p13.3 distal myopathy. Ann Neurol. 2004;56(1):133‐138. [DOI] [PubMed] [Google Scholar]
- 3. Ruggieri A, Naumenko S, Smith MA, et al. Multiomic elucidation of a coding 99‐mer repeat‐expansion skeletal muscle disease. Acta Neuropathol. 2020;140:231‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang K, Zeng YH, Qiu YS, et al. Expanding the phenotype and genotype spectra of PLIN4‐associated myopathy with rimmed ubiquitin‐positive autophagic vacuolation. Acta Neuropathol. 2022;143:733‐735. [DOI] [PubMed] [Google Scholar]
- 5. Deng J, Gu M, Miao Y, et al. Long‐read sequencing identified repeat expansions in the 5′UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. Med Genet. 2019;56:758‐764. [DOI] [PubMed] [Google Scholar]
- 6. Deng J, Wang P, Chen X, et al. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc Natl Acad Sci USA. 2018;115:E9678‐E9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu T‐Y; Human Genome Structural Variation Consortium , Chaisson MJP. Profiling variable‐number tandem repeat variation across populations using repeat‐pangenome graphs. Nat Commun. 2021;12(1):4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gimenez‐Andres M, Emersic T, Antoine‐Bally S, et al. Exceptional stability of a perilipin on lipid droplets depends on its polar residues, suggesting multimeric assembly. Elife. 2021;10:e61401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dalen KT, Schoonjans K, Ulven SM, et al. Adipose tissue expression of the lipid droplet‐associating proteins S3‐12 and perilipin is controlled by peroxisome proliferator‐activated receptor‐gamma. Diabetes. 2004;53(5):1243‐1252. [DOI] [PubMed] [Google Scholar]
- 10. Wolins NE, Skinner JR, Schoenfish MJ, Tzekov A, Bensch KG, Bickel PE. Adipocyte protein S3‐12 coats nascent lipid droplets. Biol Chem. 2003;278(39):37713‐37721. [DOI] [PubMed] [Google Scholar]
- 11. Zhao J, Liu J, Xiao J, et al. Clinical and muscle imaging findings in 14 mainland chinese patients with oculopharyngodistal myopathy. PLoS One. 2015;10(6):e0128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pou Serradell A, Lloreta Trull J, Corominas Torres J, Guicheney P. Familial myopathy with desmin storage seen as a granulo‐filamentar, electron‐dense material without mutation of the alphabeta‐crystallin gene. Neurologia. 2001;16(5):195‐203. [PubMed] [Google Scholar]
- 13. Schule B, McFarland KN, Lee K, et al. Parkinson's disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis. 2017;3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pourteymour S, Lee S, Langleite TM, et al. Perilipin 4 in human skeletal muscle: localization and effect of physical activity. Physiol Rep. 2015;3:e12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naddaf E, Barohn RJ, Dimachkie MM. Inclusion body myositis: update on pathogenesis and treatment. Neurotherapeutics. 2018;15:995‐1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Margeta M. Autophagy defects in skeletal myopathies. Annu Rev Pathol. 2020;15:261‐285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Identification of the 99 bp repeat expansion of PLIN4. (A) Genetic linkage analysis of 14 individuals in the family indicated a 3.39 Mb candidate region at 19p13.3 (chr19:2635061–6033965) with a maximum logarithm of odds score of 3.0408. (B) IGV visualization of NGS data showed a high peak of coverage in PLIN4 exon 3 in patients (arrows), not present in healthy control. (C) PCR amplification of the repeat region in exon 3 showed a wild‐type 3000 bp band and a second ~4000 bp band in affected individuals, and a single band (~ 3000 bp) in unaffected individuals. (D) LRS detection showed multiple reads with abnormal 39 × 99‐neucleotide sequence in exon 3 of PLIN4 in F‐III3, while the repeat was 29 times in control. (E) Repeat expansion of PLIN4 (NM_001080400.1).
Figure S2 Immunoelectron microscopy of perilipin‐4. IEM of Patient S revealed that the membrane‐bound inclusions were perilipin‐4 positive.