

Abstract
Objective
Ornithine transcarbamylase deficiency (OTC‐D) is an X‐linked metabolic disease and the most common urea cycle disorder. Due to high phenotypic heterogeneity, ranging from lethal neonatal hyperammonemic events to moderate symptoms and even asymptomatic individuals, the prediction of the disease course at an early disease stage is very important to individually adjust therapies such as medical treatment or liver transplantation. In this translational study, we developed a severity‐adjusted classification system based on in vitro residual enzymatic OTC activity.
Methods
Applying a cell‐based expression system, residual enzymatic OTC activities of 71 pathogenic OTC variants were spectrophotometrically determined and subsequently correlated with clinical and biochemical outcome parameters of 119 male individuals with OTC‐D (mOTC‐D) as reported in the UCDC and E‐IMD registries.
Results
Integration of multiple data sources enabled the establishment of a robust disease prediction model for mOTC‐D. Residual enzymatic OTC activity not only correlates with age at first symptoms, initial peak plasma ammonium concentration and frequency of metabolic decompensations but also predicts mortality. The critical threshold of 4.3% residual enzymatic activity distinguishes a severe from an attenuated phenotype.
Interpretation
Residual enzymatic OTC activity reliably predicts the disease severity in mOTC‐D and could thus serve as a tool for severity‐adjusted evaluation of therapeutic strategies and counselling patients and parents.
Introduction
Ornithine transcarbamylase deficiency (OTC‐D) is an X‐linked urea cycle disorder (UCD) caused by deficiency of the mitochondrial enzyme ornithine transcarbamylase (OTC; OMIM# 311250) due to pathogenic variants in the ornithine transcarbamylase gene (OTC) located on chromosome Xp21.1. OTC‐D is the most common UCD with an estimated incidence of 1: 56,500–63,000 newborns. 1 , 2 , 3 Since the urea cycle is the only metabolic pathway of irreversible elimination of excess nitrogen through ureagenesis in humans, disruption of this pathway leads to accumulation of ammonia and glutamine in plasma and thus, affected individuals often present with severe life‐threatening hyperglutaminemia and hyperammonemic events (HAE) during the neonatal period (early onset, EO, defined as first 28 days of life) facing mortality rates as high as 25%–50%. Moreover, survivors often suffer from significant cognitive deficits, and the (cognitive) outcome has not improved within the last decades in neonatal‐onset disease. 4 , 5 , 6 , 7 , 8 Some individuals with OTC‐D and other UCDs, however, may be less severely affected since the clinical phenotype is highly variable, ranging from severe HAE to mild‐to‐moderate symptoms with disease onset any time after the neonatal period (late onset, LO). Symptoms of LO disease characteristics comprise headache, recurrent vomiting, liver dysfunction, psychiatric symptoms or cognitive impairment, even in the absence of recurrent HAE. 9 , 10 , 11
Clinical approaches to predict the disease course and cognitive outcome in individuals with OTC‐D identified disease onset as well as the severity of initial HAE [peak plasma ammonium concentration during initial HAE (NH4 + max)] as determinants. However, a considerable variability persists throughout these clinical correlations. 2 , 4 , 5 , 9 , 12 , 13 , 14 , 15 Importantly, on the genomic level, OTC variants leading to complete loss of enzymatic OTC function are thought to cause higher disease burden as reflected by EO of symptoms, NH4 + max, frequency of HAE, cognitive impairment and mortality as opposed to partly preserved enzymatic activity. 4 , 12 , 16 Nevertheless, methods to systematically investigate enzymatic OTC function are still lacking since in silico modelling of pathogenic variants to predict protein function and stability is still not reliable and kinetic characterization based on liver biopsies or purified recombinant human OTC expressed in bacteria has not yet been adapted for systematic measurements. 16
Due to the heterogeneity in various clinical prediction models and existing methodological shortcomings regarding the impact of the genotypic background on the clinical outcome, we developed a novel in vitro expression system for male individuals with OTC‐D (mOTC‐D) that enables to predict major clinical outcome parameters and fosters a precision‐based medical approach for future evaluation of diagnostic and therapeutic interventions which are supposed to alter the clinical disease course.
Materials and Methods
Research strategy
Recently, we demonstrated that the clinical disease course and neurocognitive outcome of individuals with citrullinemia type 1 (CTLN1) and argininosuccinic aciduria (ASA), the most prevalent cytosolic UCDs, can be reliably predicted by the enzymatic activity of argininosuccinate synthetase 1 (ASS1) and argininosuccinate lyase (ASL), respectively, as determined by a newly established biallelic mammalian expression system. 17 , 18 As we wanted to know if enzymatic OTC activity can also predict clinical outcome in individuals with OTC‐D, the most prevalent mitochondrial UCD and the only X‐linked UCD, we systematically investigated all unequivocal pathogenic exonic OTC variants and correlated the spectrophotometrically determined enzymatic OTC activity with individuals' clinical, biochemical and neurological follow‐up data. Data were retrieved from the North American Urea Cycle Disorders Consortium (UCDC; https://www.rarediseasesnetwork.org/cms/ucdc) and the European registry and network for Intoxication‐type Metabolic Diseases (E‐IMD; https://www.eimd‐registry.org/), the two largest observational natural history studies on UCDs worldwide.
Eligibility criteria
Only hemizygous male individuals carrying known pathogenic exonic variations in the OTC gene, who were confirmed by molecular genetic testing and were enrolled in the observational longitudinal studies of UCDC or E‐IMD, were included in this study. Due to individual variation of X‐inactivation, the methodological approach used in this study cannot reliably determine enzymatic OTC activity in female individuals with heterozygous variations in the OTC gene. The data model of both registries, information on written informed consent as well as the follow‐up protocols used have been previously described in detail. 14 , 19 All procedures were in accordance with the ethical standards of the Helsinki Declaration of 1975, as revised in 2013. Written informed consent was given by patients or their legal guardians before enrollment in this study. Data were retrieved from the UCDC and E‐IMD electronic databases with the cut‐off date for data retrieval being 10 October 2018. The UCDC database is registered at the US National Library of Medicine (https://clinicaltrials.gov, NCT00237315), whereas the E‐IMD registry is recorded on the German Clinical Trials Register (https://www.drks.de, DRKS00013085).
Plasmids
To generate tagged wild‐type OTC expression vectors, the OTC coding sequence has been modified with an MYC‐tag introduced in between the N‐terminal signalling peptide and the core protein, which was cloned into HindIII‐ and NotI‐restriction sites in the open‐reading frame of the eukaryotic expression vector pcDNA5/FRT/TO (Thermo Fisher Scientific). The inserted OTC coding sequence corresponds to NCBI reference sequence NM_000531.6 (https://www.ncbi.nlm.nih.gov/nucleotide/NM_000531.6). Using the QuickChange II site‐directed mutagenesis kit (Agilent), pathogenic OTC gene variants reported in mOTC‐D individuals from the E‐IMD and UCDC registries were introduced into the tagged OTC expression vector according to the manufacturer's protocol. The correct nucleotide sequence of every inserted variant was confirmed by Sanger‐sequencing. All OTC variants reported in this study and their provided nomenclature have been checked by applying the Mutalyzer 2.0.34 software (https://mutalyzer.nl/). pSV‐β‐Galactosidase control vector was kindly provided by N. Himmelreich (Heidelberg University, Germany).
Cell culture and transfections
COS‐7 cells were maintained as adherent cell culture in 100 mm culture dishes using a DMEM medium (Thermo Fisher Scientific) supplemented with 10% heat‐inactivated fetal calf serum in a humified incubator at 37 °C and 5% CO2. Transfections were performed using Lipofectamine 2000 reagent (Thermo Fisher Scientific) with 5 μg of the wild‐type or mutated MYC‐tagged OTC expression vector, 2.5 μg of each ASS1 and ASL expression vector and 1 μg of β‐galactosidase control vector. Forty‐eight hours after transfection, cells were lysed and lysates were used to perform Western blot analysis or spectrophotometric determination of enzymatic OTC activity.
Western blot
Forty‐eight hours after transfection, COS‐7‐cells were washed with ice‐cold phosphate‐buffered saline (PBS) and lysed in RIPA buffer (Sigma‐Aldrich) followed by sonification. After centrifugation at 13,000g max and 4°C for 10 min, supernatants were used for Western blotting according to standard laboratory protocols. For protein visualization, PVDF membranes were incubated with the following primary antibodies: anti‐MYC (1:2000; Cell Signaling) or anti‐β‐actin (1:2000; Sigma‐Aldrich).
Spectrophotometric analysis of OTC enzyme activity
Purified citrate synthase, malate dehydrogenase and fumarase from the porcine heart were purchased from Sigma‐Aldrich. Transfected COS‐7 cells were washed in ice‐cold PBS and lysed in a buffer containing 10 mmol/L potassium phosphate, 10 mmol/L TRIS–HCl at pH 7.4 and by additional sonification. OTC enzymatic activity was determined as NAD+ reduction at λ = 340–400 nm measured in 40 μl of lysates (triplicates) diluted in 100 μl of the above‐mentioned buffer supplemented with 2 mmol/L ornithine, 2 mmol/L carbamyl phosphate, 2 mmol/L aspartic acid, 330 μmol/L NAD+, 100 μmol/L acetyl‐CoA and 2 mmol/L ATP, 660 mU/ml fumarase, 1800 mU/ml malate dehydrogenase and 560 mU/ml citrate synthase, adjusted to pH 7.4. Lysates of COS‐7 transfected cells with only β‐galactosidase control plasmid served as a negative control. Negative control values were subtracted from OTC activities to adjust for unspecific background activity. OTC activity values were normalized to β‐galactosidase activity in each sample as determined by the β‐galactosidase enzyme assay system (Promega, Germany) following the manufacturer's protocol to control for transfection efficacy. Adjusted OTC activities were then normalized to the protein content of each sample. Enzymatic activities are depicted as a percentage of the total (%) by dividing the normalized OTC activity of the respective variant by the normalized wild‐type OTC activity.
Clinical variables used for data analyses
Data on the following numerical clinical variables were collected: HAE (defined as plasma ammonium concentration (NH4 +) > 100 μmol/L), age at first symptoms, age at death, initial NH4 + max (defined as peak plasma ammonium concentration during first HAE), number of HAE per year of observation (defined as the time between the date of birth and the last regular visit), initial L‐citrulline and L‐glutamine concentrations in plasma and initial orotic acid concentration in urine. Moreover, the cognitive standard deviation score (SDS) at the most recent visit was calculated using the normative data from the standardization sample of each cognitive test. The most recent visit was chosen to include the latest developmental data in the analysis. For each Wechsler Adult Intelligence Scale (n = 3), Wechsler Abbreviated Scale of Intelligence (n = 11), Wechsler Intelligence Scale for Children (n = 6) and Wechsler test, unknown version (n = 1) full scale IQ were selected. For Bayley Scales of Infant Development (n = 15), mental developmental index and cognitive scale were used. For the Adaptive Behavior Assessment System (n = 4), the general adaptive composite was used.
As for categorical clinical variables, data of the following items were collected: mortality, disease onset (EO, LO, asymptomatic), presence or absence of movement disorders (dystonia and/or chorea and/or ataxia), tone change (muscular hypotonia and/or muscular hypertonia and/or spasticity), hepatocellular injury (alanine aminotransferase ≥250 U/L or aspartate aminotransferase ≥250 U/L), liver transplantation (LTx) and kidney dysfunction (full age spectrum glomerular filtration rate (FAS‐GFR) < 90 mL/min/1.73 m2). For symptomatic individuals with reported HAE during the initial presentation, the biochemical data of initial NH4 + max, initial plasma L‐glutamine and urinary orotic acid concentrations represent the highest values, respectively, and the lowest value for initial plasma L‐citrulline concentration prior to initiation of treatment. For symptomatic individuals without reported HAE during initial presentation, initial NH4 + max was defined as the upper limit of the normal range (50 μmol/L). For untreated asymptomatic individuals, NH4 + max values represent the highest reported follow‐up values during the observation period; plasma L‐citrulline, plasma L‐glutamine and urinary orotic acid concentrations represent the arithmetic mean of all reported follow‐up values. Asymptomatic individuals with mOTC‐D receiving therapeutic agents (scavenger, for L‐citrulline concentration analysis additionally L‐citrulline and/or L‐arginine substitution) were not considered for biochemical sub‐analysis in order to prevent a confounding bias with iatrogenic interventions.
We investigated the impact of the residual enzyme activity of OTC in vitro as determined by the established expression system on clinical outcome parameters outlined above.
Data availability statement
The data sets generated and analysed during the current study are not publicly available due to existing data protection laws. Furthermore, data ownership is retained by the members of the UCDC and E‐IMD consortia making data only available for specific research purposes. Data availability is subject to the consent of both consortia upon request.
Statistical analyses
All analyses were performed using R (http://www.r‐project.org). To evaluate the association between a continuous dependent variable and the residual enzyme in vitro activity of OTC as predictor variable, a linear regression or generalized additive regression model (GAM) with automated smoothing selections was used. In GAM, the linear relationship between the response variable and predictors is replaced by non‐linear smooth functions to evaluate an apparently non‐linear relationship between dependent and predictor variables. The R package ‘mgcv’ was used to fit GAM regressions. To compare a numeric dependent variable between two groups, a t‐test with Welch correction was applied. We used unbiased recursive partitioning to determine cut‐off values for the impact of the enzymatic OTC activity on outcome variables. 20 Cox‐proportional hazard regression and Kaplan–Meier estimates were used to analyse mortality. p values reported were two‐sided. p ≤ 0.05 was considered statistically significant.
Results
Description of the study population and OTC protein functions
In 119 male individuals with OTC‐D and a total of 71 different OTC gene variants, we systematically determined OTC protein expression levels by Western Blot and enzymatic activities using the newly established OTC enzymatic assay. Results of Western blot analysis are shown in Figure S1. Residual enzymatic OTC activities per variant are illustrated in Figure S2. Detailed descriptive characteristics of the study population for each subsequent analysis are specified in Table S1.
Enzymatic activity is inversely correlated to the severity of the initial presentation
First, we studied whether age at first symptoms and biochemical parameters such as initial NH4 + max, L‐glutamine, L‐citrulline in plasma and orotic acid level in urine correlate with in vitro enzymatic activity. Age at first symptoms correlated with residual enzymatic OTC activity (n = 100, p < 0.0001, R 2 = 0.17, linear regression), demonstrating that individuals with high residual enzymatic OTC activity present with first symptoms later in life than those with low residual enzymatic OTC activity (Fig. 1). This association is particularly strong for variants leading to residual enzymatic activities below 10%. In analogy, the disease onset of mOTC‐D is strongly associated with residual enzymatic OTC activity as determined by the enzymatic assay. Individuals with early onset (n = 50) showed significantly lower enzymatic activities than those with LO (n = 56, p < 0.001, Tukey multiple comparison of means) and asymptomatic individuals (n = 10, p < 0.01, Tukey multiple comparison of means) (Fig. S3).
Figure 1.
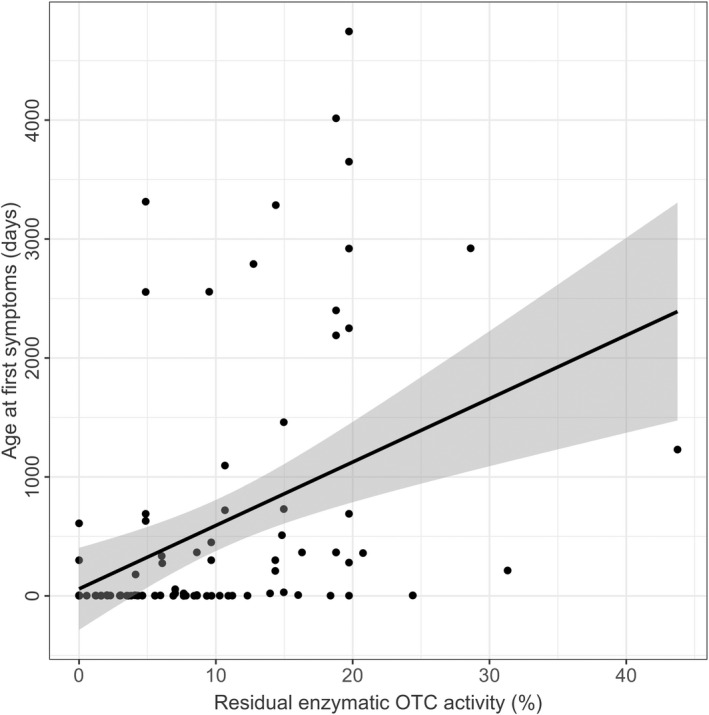
Residual enzymatic OTC activity predicts age at first symptoms. Age at first symptoms (days) subject to residual enzymatic OTC activity (%). Each point represents a single patient (n = 100). The grey line displays the estimated regression curve. Linear regression, p < 0.0001, R 2 = 0.16.
Initial NH4 + max is negatively correlated with residual OTC activity (n = 98, p < 0.0001, R 2 = 0.10, GAM analysis). Moreover, recursive partitioning identified a threshold separating mOTC‐D individuals with a severe initial decompensation from those with an attenuated form. Residual enzymatic OTC activity of below or equal to 4.6% was associated with a more severe decompensation (n = 98, p < 0.001), showing mean initial NH4 + max of 1,703 μmol/L in comparison to 652 μmol/L (mean) for residual activities >4.6% (Fig. 2). Unlike age at first symptoms and initial NH4 + max, there was no significant correlation between residual enzymatic OTC activity and initial plasma L‐glutamine (n = 32, p = 0.06, Pearson's product–moment correlation), L‐citrulline (n = 25, p = 0.20, r = 0.26, Pearson's product–moment correlation) or urinary orotic acid concentrations (n = 34, p = 0.91, r = 0.02, Pearson's product–moment correlation).
Figure 2.
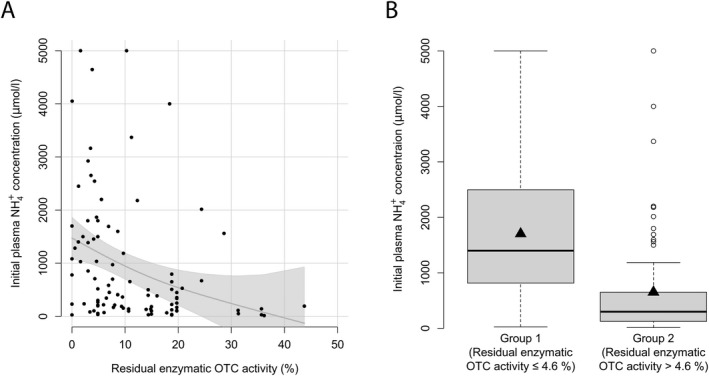
Residual enzymatic OTC activity correlates with initial plasma NH4 + max. (A) Initial plasma NH4 + max (μmol/L) subject to residual enzymatic OTC activity (%) as determined in the expression system. Each point represents a single patient (n = 98). The grey line displays the estimated regression curve. GAM analysis, p = 0.004, R 2 = 0.102. (B) Box plot illustrating initial NH4 + max (μmol/L) with residual enzymatic OTC activity below or equal to 4.6% (n = 27) and above 4.6% (n = 71). Data are shown as median (black thick line) and mean (triangle), length of the box corresponds to interquartile range (IQR) and upper and lower whiskers correspond to the max. of 1.5 × IQR, each point represents an outlier. Recursive partitioning, p = 0.001.
Residual OTC activity correlates with the frequency of hyperammonemic events
In the next step, we assessed whether residual enzymatic OTC activity could also predict the severity of disease course and found a correlation with the number of HAE per year of observation (n = 52, p < 0.05, R 2 = 0.06, GAM analysis), showing a higher number of HAE below the threshold of 16.0% (n = 52, p < 0.05, recursive partitioning) (Fig. 3). In mean, individuals with residual enzymatic activities below or equal to 16.0% (n = 35) present with 3.2 HAE per year of observation, whereas individuals with residual OTC activities above 16.0% (n = 17) present with 0.3 HAE per year of observation.
Figure 3.
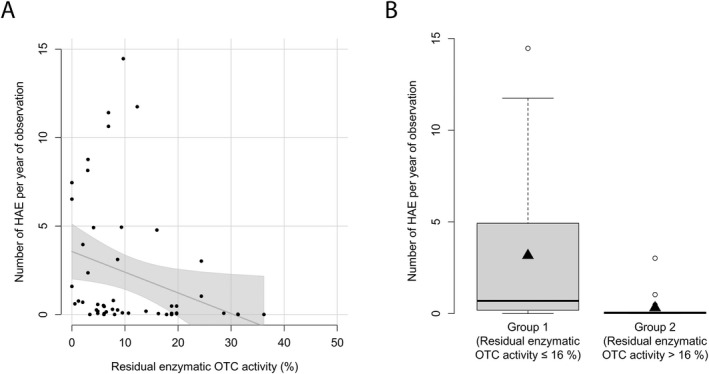
Residual enzymatic OTC activity predicts number of HAE per year. (A) Number of HAE (NH4 + max ≥ 100 μmol/L) per year of observation subject to residual enzymatic OTC activity (%). Each point represents a single patient (n = 52). The grey line displays the estimated regression curve. GAM‐analysis, p = 0.04, R 2 = 0.06. (B) Boxplot illustrating number of HAE (NH4 + max ≥ 100 μmol/L) per year of observation with residual enzymatic OTC activity below or equal to 16.0% (n = 35) or above 16.0% (n = 17). Data are shown as median (black thick line) and mean (triangle), length of the box corresponds to IQR and upper and lower whiskers correspond to the max. of 1.5 × IQR, each point represents an outlier. Recursive partitioning, p = 0.04.
Low residual OTC activity increases the risk of mortality
Mortality negatively correlates with residual enzymatic OTC activity (n = 16/119, p = 0.005, odds ratio = 0.87, Cox Proportional Hazard). Male individuals with residual OTC activity below or equal to 4.3% had a higher risk of mortality than those with higher residual activities (n ≤4.3% = 9/28, n >4.3% = 7/91, p = 0.003, recursive partitioning) (Fig. 4). Corresponding mortality rates are 32.1% for residual OTC activities below or equal to 4.3% and 7.7% for residual activities above 4.3%.
Figure 4.
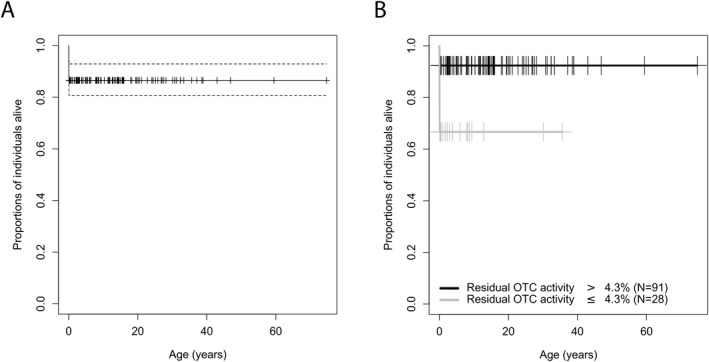
Residual enzymatic OTC activity correlates with mortality. Kaplan–Meier curve illustrating estimated overall survival (A) in total and (B) with residual enzymatic OTC activity below or equal to 4.3% (n = 28, grey) or above 4.3% (n = 91, black). Censored individuals are marked with a “+”. Of the two remaining (non‐representative) individuals above the age of 55 years, one individual has been censored who died at the age of 59 years due to an epileptic state. Dotted lines are indicating the confidence interval of 95%. Recursive partitioning, p = 0.003.
A correlation between residual OTC activity and cognitive SDS at last follow‐up test (n = 40, p = 0.12, R2 = 0.04, linear regression) was not found. However, sub‐analyses showed that cognitive SDS was associated with initial NH4 + max (n = 29, p = 0.002, R2 = 0.40, multiple linear regression) but not with age at first symptoms (n = 29, p = 0.56, R2 = 0.40, multiple linear regression).
Residual enzymatic activity was not associated with tone change (N = 93, p = 0.06, Welch two‐sample t test), movement disorder (n = 90, p < 0.93, Welch two‐sample t‐test), liver transplantation (n = 119, p = 0.09, Welch two‐sample t‐test), kidney dysfunction (n = 105, p = 0.44, Welch two‐sample t‐test) or hepatocellular injury (n = 83, p = 0.44, Welch two‐sample t‐test).
Discussion
The major aim of this study was to evaluate whether residual enzymatic activity of mutated OTC protein could precisely predict disease severity. To this end, we compared in vitro enzymatic activity of unequivocal exonic OTC variants with clinical outcome data reported in the UCDC and E‐IMD databases. Residual enzymatic OTC activity reliably predicts phenotypic severity as reflected by the initial presentation, metabolic disease course and mortality for male individuals. We found for male individuals with OTC‐D that (I) residual enzymatic OTC activity correlates with age at first symptoms, (II) enzymatic OTC activity below or equal to 4.3% is associated with higher initial plasma NH4 + max and higher mortality rates and (III) residual enzymatic OTC activity below or equal to 16.0% is associated with a higher number of HAE per year of observation.
Residual enzymatic OTC activity qualifies as a predictive biomarker for disease severity
As in vitro residual enzymatic OTC activity reflects disease severity and reliably predicts major clinical and biochemical outcome parameters, it qualifies as a prognostic and predictive biomarker as defined by Katz 21 and the US Food and Drug Administration. 22 As there are currently no approved pharmacotherapies aiming at increasing enzymatic OTC activity, the required reflection of net treatment effects on the outcome to truly serve as a validated surrogate marker cannot be addressed at present. 23 However, the demonstrated correlations could further encourage the development of new therapies targeting the increase of enzymatic OTC activity. In particular, the established disease prediction model enables a precision‐based medical approach as it indicates to which level residual enzymatic OTC activity needs to be increased to convert the phenotype from severe to attenuated.
Using the identified robust cut‐off values of residual enzymatic OTC activities enables risk stratification of mOTC‐D phenotypes and their underlying OTC variants. According to the above‐mentioned thresholds, we suggest a new classification comprising two clinical phenotypes: Severe phenotype for OTC variants leading to residual enzymatic activity below or equal to 4.3% (high initial NH4 + max and mortality rate) and attenuated phenotype for OTC variants leading to residual enzymatic activity above 4.3% (lower initial NH4 + max and mortality rate).
Showing that the onset type is associated with residual enzymatic OTC activity allowed us to validate the current classification system based on EO, LO and asymptomatic individuals. A major shortcoming of the existing clinical classification system is the lack of predictive options since patients will be attributed retrospectively to one class after the onset of first symptoms. The here presented complementary stratification system is able to further specify and predict the evolving phenotype before first symptoms might have manifested and is based on the genotypic background of affected individuals.
This new system of risk stratification allows counselling of patients and their families with regard to relevant clinical and biochemical long‐term outcome parameters as early as genetic testing is completed. However, with regard to counselling patients and families, it is important to emphasize that this disease prediction model cannot predict the exact individual disease course but is of importance to anticipate the expected outcome in an evidence‐based manner. Moreover, it serves as a useful tool for the severity‐adjusted evaluation of (future) therapies and care concepts and allows the evaluation of existing diagnostic concepts (e.g. newborn screening) in a standardized and severity‐adjusted manner.
Modelling evidence‐based thresholds for clinical health outcomes in male OTC‐D
It has recently been shown that residual enzymatic activity of the cytosolic urea cycle enzymes ASS1 and ASL reliably predict disease severity of individuals with CTLN1 and ASA, respectively. 17 , 18 As we wanted to know if these associations also hold true for OTC‐D, the most prevalent urea cycle disorder, we investigated residual activities of the mitochondrial enzyme OTC in vitro. Comparable to CTLN1 and ASA, residual OTC activity correlates with initial NH4 + max and the number of annual HAE in mOTC‐D. Intriguingly, it predicts further relevant clinical endpoints, which are mortality and age at first symptoms. Although individuals with mOTC‐D present a higher metabolic disease burden with more severe and more frequent HAE and higher mortality rates, the neurocognitive outcome in survivors seems to be better than in individuals with distal UCDs, suggesting additional underlying pathomechanisms other than NH4 +‐dependent neurotoxic alterations alone. 14 , 18
In the context of determining the accuracy of our expression system, the cut‐off value for mortality of 4.3% can be seen as similar to the one for initial peak plasma NH4 + max (4.6%), reinforcing the clinical relevance of this threshold. This goes in line with the clinical observation that the most severely affected individuals with mOTC‐D still frequently die during their first metabolic decompensation in the neonatal period despite improved intensive care as well as efficacious intravenous and extracorporeal detoxification strategies. Thus, it is tempting to speculate that therapeutic interventions that are able to increase the residual enzymatic activity at an asymptomatic state early in life might be a very promising option to reduce mortality.
This is reinforced by the interesting finding that the threshold for a lethal disease course in mOTC‐D is similar for different mammalian species. Knocking out the OTC gene in mice results in high and early mortality, while different currently used mouse models (spf, spf‐j and spf ash ) with residual OTC activities of 5%–20% present milder phenotypes with low mortality rates and onslight biochemical alterations. 24 , 25 , 26 , 27
Our results support the notion of higher mortality rates among individuals carrying an OTC variant leading to complete loss of OTC function. 2 , 4 The study cohort shows one‐third of mortality rates for individuals with residual enzymatic OTC activities below or equal to 4.3% as opposed to mortality rates of less than 10% for individuals with higher residual enzymatic OTC activities. Moreover, only two of the observed deaths occurred after the neonatal period. This fact strengthens the need for early diagnosis and stratification of fatal mutations in order to reduce mortality.
Low plasma L‐citrulline, high plasma L‐glutamine and the detection of orotic acid in urine are highly indicative of OTC‐D in clinical practice. 9 , 10 However, these biochemical parameters do not seem to qualify as reliable predictive biomarkers for the severity of the initial decompensation or the subsequent disease course. Similar limitations account for neurological alterations, such as tone change and movement disorders.
Intriguingly, residual enzymatic OTC activity showed no correlation with the cognitive outcome as measured by IQ testing. However, in line with recent works, a sub‐analysis of our cohort revealed an association between cognitive SDS and initial NH4 + max. 12 , 13 , 14 These findings indicate that the height of the initial NH4 + max appears not only to be determined by the underlying residual enzymatic activity but also by other non‐standardized factors, such as but not limited to (1) timepoint and intensity of initial medical management and (2) effect size of the cause(s) of the initial decompensation (e.g. catabolic state, protein load, etc.), which are very heterogeneous between various individuals having similar or the same residual enzymatic activities.
Although the UCDC and E‐IMD registries have been systematically collecting follow‐up data of UCD individuals since 2003 and 2011, respectively, we are still missing intra‐individual long‐term data on cognitive functioning and cerebral brain imaging to improve our understanding of the natural cognitive disease course and to reveal morphological changes as well as their underlying pathomechanisms of neurological impairment.
Directions for future research
The exploratory data on alterations of protein expression of OTC variants captured in this study can be used as a basis for further characterization of underlying molecular pathomechanisms of the respective pathogenic variants.
For more than two decades, researchers have aimed at developing gene therapies for OTC‐D. 28 , 29 , 30 , 31 , 32 , 33 Recently, a phase 1/2 study of adeno‐associated virus 8 gene therapy in adults has been conducted (https://clinicaltrials.gov/, Identifier: NCT02991144). Using the newly established expression system could be of help to assess the treatment effects of new therapeutic approaches (e.g. by application of gene therapies, chaperones or antisense nucleotides). Chaperones improving the stability of translated protein and antisense nucleotides targeting altered splicing of the precursor mRNA to increase the concentration of correctly translated protein have shown beneficial effects in other inborn diseases. 34 , 35 Achieving an increase of residual enzymatic OTC activity above the threshold of 5% by therapeutic means might not only reflect a reduction of mortality but also the phenotypic conversion from a severe to an attenuated disease course.
Limitations
The here presented stratification system is not able to predict exact maximal heights of NH4 +, number of HAE or age at onset. It appears likely that other factors like diagnostic and therapeutic delay or severity of catabolic state influenced by protein intake, infections, drugs, awareness of preceding symptoms or compliance to life‐long therapy modify the clinical severity of an individual affected by OTC‐D. It has been reported that even the same mutations can cause clinically variable appearances in different individuals. 1 , 5 , 16 , 36 The study results need to be evaluated prospectively in the future.
Moreover, our study has some inherent limitations. First, due to the applied cloning technique utilizing the OTC coding sequence, we were not able to design intronic mutations or those variants associated with defective splicing. Second, variants that affect substrate binding affinity (K m ‐variants) could not be investigated as our assay uses supraphysiological substrate concentrations leading to artificially increased enzymatic activities in those variants. Third, we could not include female individuals with OTC‐D because of variable X‐inactivation. Our study does not claim to systematically analyse the underlying molecular events in every OTC variant including impaired protein folding or decreased protein stability. Rather, the results of protein expression levels should be considered somewhat exploratory and encourage further investigation in future studies.
Furthermore, the structure of the UCDC and E‐IMD registries did not enable the inclusion of data from both registries for every subanalysis. Despite systematic requirements for data collection, the given data density and quality reduced test power for some analyses. Further intraindividual long‐term data are crucial to substantiate our findings, and longer observation periods and the inclusion of more individuals might help to reveal further and strengthen given genotype–phenotype correlations, in particular, with regard to classify cognitive alterations or to identify asymptomatic individuals. It seems likely that the group of male individuals with an attenuated phenotype comprises different subphenotypes, such as life‐long asymptomatic individuals, which might be revealed by evaluating long‐term data in the future.
Conclusion
Given the genotype–phenotype correlations revealed in this study, we suggest a new severity‐adjusted classification system and prediction model for male individuals with OTC‐D dividing affected individuals into two phenotypic classes: severe (residual enzymatic OTC activity ≤4.3%) and attenuated phenotypes (residual enzymatic OTC activity >4.3%). This classification system can be considered complementary to the current clinical classification system for OTC‐D based on the disease onset (EO, LO and asymptomatic). Given the impact of enzymatic OTC, ASS1 and ASL activities as predictive and prognostic biomarkers for the clinical disease course and outcome of individuals with OTC‐D, CTLN1 and ASA, respectively, we are confident that this approach can be adapted and applied to other monogenetic inborn errors of metabolism. This would not only enable early identification of severely affected individuals preferably before irreversible organ dysfunctions might have manifested but could also facilitate the development and implementation of individual therapeutic care concepts adjusted to the anticipated disease severity.
Author Contributions
The STROBE statement was used when preparing this manuscript. SS, RP and MZ contributed to the conception and design of the study. SS, RP, SFG, FG, MJS, ACD, JGO, ALG, SCSN, GFH, SK, MZ and all individual contributors from the UCDC and E‐IMD consortia study group (Table S2) contributed to the acquisition and analysis of data. SS, RP, SFG, SK and MZ contributed to drafting the text and preparing the figures.
Conflict of Interest
SK received EU funding for the European registry and network for Intoxication type Metabolic Diseases (E‐IMD; CHAFEA agreement no. 2010 12 01). SK receives funding from Immedica Pharma AB for the European Post‐Authorization Registry for Ravicti® (glycerol phenylbutyrate) oral liquid in partnership with the E‐IMD (RRPE) (EU PAS Register no. EUPAS17267; http://www.encepp.eu/). SK and GFH receive funding from the Dietmar Hopp Foundation (St. Leon‐Rot, Germany) for a pilot study on extended newborn screening evaluating the technical feasibility, diagnostic process quality and health benefits for 28 inherited metabolic diseases including UCDs (NBS 2025, project no. 1DH1911376, 1DH2011117). GFH received lecture fees from Swedish Orphan Biovitrum GmbH. RP receives consultancy fees from Immedica Pharma AB. The sponsors have in no way influenced the design, conductance, analysis and report of the present study. All other authors declare that they have no conflict of interest.
Supporting information
Figure S1. Overview of protein expression levels per variant.
Figure S2. Overview of residual enzymatic OTC activity per variant.
Figure S3. Correlation between disease onset and residual enzymatic OTC activity.
Table S1. Descriptive characteristics of study cohort and subanalyses.
Table S2. Additional members and affiliations of the UCDC and E‐IMD consortia study group.
Acknowledgements
All UCDC and E‐IMD sites contributed to the data sets of the longitudinal studies used in this publication. Principal investigators and personnel with key contributions are listed as UCDC and E‐IMD consortia study group members. Furthermore, we gratefully acknowledge subsequent study coordinators—Jennifer Seminara, Saima Ali, Sondra Bloxam, Kia Bryan, Sara Elsbecker, Joan Hart, Melanie Horn, Elijah Kravets, Audrey Lynn, Mary Mullins, Maya Muldowney, Kendall Parks, Ulrike Mütze, Thu Quan, Kara Simpson, Julia Smith, Suzanne Hollander and Hayden Vreugdenhil—and study neuropsychologists—Fabienne Dietrich Alber, Talin Babikian, Heidi Bender, Christopher Boys, David Breiger, Mina Nguyen‐Driver, Benjamin Goodlett, Elizabeth Kerr, Magdalena E. Kowoll, Casey Krueger, Eva Mamak, Jacqueline H. Sanz, David Schwartz, Arianna K. Stefanatos, Rachel Tangen and Greta N. Wilkening. We would also like to acknowledge the contributions of (former) longitudinal study principal investigators: Mark L. Batshaw, Stephen Cederbaum, Annette Feigenbaum, Douglas S. Kerr, Brendan Lee, Uta Lichter‐Konecki, Margretta R. Seashore, Marshall L. Summar, Peter Burgard, Curtis R. Coughlin II, Gregory Enns, Renata C. Gallagher, Cynthia Le Mons, Shawn E. McCandless, Tamar Stricker, Mendel Tuchman, Susan Waisbren and James D. Weisfeld‐Adams. In particular, we are indebted to all our UCD individuals and their families for their trust, patience and participation in both longitudinal registry studies for many years.
The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through a collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The Urea Cycle Disorders Consortium is also supported by the O'Malley Foundation, the Rotenberg Family Fund, the Dietmar Hopp Foundation, the Kettering Fund and the National Urea Cycle Disorders Foundation. In addition, support for neuropsychological testing is provided by an NIH grant for Intellectual and Developmental Disability Research Centers (U54HD090257). This work was also supported in part by the Clinical Translational Core at Baylor College of Medicine which is supported by the IDDRC (grant number U54HD083092) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The E‐IMD patient registry has received funding from the European Union (E‐IMD; EAHC no 2010 12 01; coordinator: SK), in the framework of the Health Program. After the end of the EU funding period, the E‐IMD patient registry has been sustained by funding from the Kindness‐for‐Kids Foundation (Munich, Germany), the Kettering Fund, and the Dietmar Hopp Foundation. No external funding was secured for this study. MZ (Heidelberg, Germany) was supported by the Physician Scientist Program at the University of Heidelberg and by a Career Development Fellowship provided by the Heidelberg Research Center for Molecular Medicine (HRCMM) in the framework of Excellence Initiative II of the German Research Foundation. Open Access funding enabled and organized by Projekt DEAL. WOA Institution: UNIVERSITATSKLINIKUM HEIDELBERG Consortia Name : Projekt DEAL
Funding Information
The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through a collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The Urea Cycle Disorders Consortium is also supported by the O'Malley Foundation, the Rotenberg Family Fund, the Dietmar Hopp Foundation, the Kettering Fund and the National Urea Cycle Disorders Foundation. In addition, support for neuropsychological testing is provided by an NIH grant for Intellectual and Developmental Disability Research Centers (U54HD090257). This work was also supported in part by the Clinical Translational Core at Baylor College of Medicine which is supported by the IDDRC (grant number U54HD083092) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The E‐IMD patient registry has received funding from the European Union (E‐IMD; EAHC no 2010 12 01; coordinator: SK), in the framework of the Health Program. After the end of the EU funding period, the E‐IMD patient registry has been sustained by funding from the Kindness‐for‐Kids Foundation (Munich, Germany), the Kettering Fund, and the Dietmar Hopp Foundation. No external funding was secured for this study. MZ (Heidelberg, Germany) was supported by the Physician Scientist Program at the University of Heidelberg and by a Career Development Fellowship provided by the Heidelberg Research Center for Molecular Medicine (HRCMM) in the framework of Excellence Initiative II of the German Research Foundation.
Funding Statement
This work was funded by Dietmar Hopp Foundation; European Union Health Program grant EAHC no 2010 12 01; Intellectual and Developmental Disability Research Centers grant U54HD090257; Kettering Fund ; Kindness‐for‐Kids Foundation (Munich, Germany); National Institutes of Health (NIH) Rare Diseases Clinical Research Network grant U54HD061221; National Urea Cycle Disorders Foundation; O'Malley Foundation; Rotenberg Family Fund; Research Foundation ; University of Heidelberg ; IDDRC ; Baylor College of Medicine ; the Kettering Fund ; Eunice Kennedy Shriver National Institute of Child Health and Human Development .
Contributor Information
Matthias Zielonka, Email: matthias.zielonka@med.uni-heidelberg.de.
for the Urea Cycle Disorders Consortium (UCDC) and the European registry and network for Intoxication type Metabolic Diseases (E‐IMD) Consortia Study Group:
Nicholas Ah Mew, Matthias R. Baumgartner, Gerard T. Berry, Susan A. Berry, Lindsay Burrage, George A. Diaz, Can Ficicioglu, Genya Kisin, Laura Konczal, Christina Lam, Shawn E. McCandless, J. Lawrence Merritt, Andreas Schulze, Magdalena E. Walter, Ashley Wilson, Derek Wong, Florence Arnaudo, Persephone Augoustides‐Savvopoulou, Ivo Barić, Annet M. Bosch, Aline Cano, Yin‐Hsiu Chien, Carlo Dionisi‐Vici, Dries Dobbelaere, Francois Eyskens, Peter Freisinger, Angeles Garcia‐Cazorla, Tomas Honzik, Daniela Karall, Allan M. Lund, Elaine Murphy, René Santer, Manuel Schiff, Anastasia Skouma, Jolanta Sykut‐Cegielska, Frits A. Wijburg, and Jiri Zeman
References
- 1. Batshaw ML, Tuchman M, Summar M, Seminara J, Members of the urea cycle disorders C . A longitudinal study of urea cycle disorders. Mol Genet Metab. 2014;113(1‐2):127‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nettesheim S, Kolker S, Karall D, et al. Incidence, disease onset and short‐term outcome in urea cycle disorders ‐cross‐border surveillance in Germany, Austria and Switzerland. Orphanet J Rare Dis. 2017;12(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110(1‐2):179‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ah Mew N, Krivitzky L, McCarter R, Batshaw M, Tuchman M. Urea cycle disorders consortium of the rare diseases clinical research N. clinical outcomes of neonatal onset proximal versus distal urea cycle disorders do not differ. J Pediatr. 2013. Feb;162(2):324‐9 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003. Jun;162(6):410‐416. [DOI] [PubMed] [Google Scholar]
- 6. Burgard P, Kolker S, Haege G, Lindner M, Hoffmann GF. Neonatal mortality and outcome at the end of the first year of life in early onset urea cycle disorders—review and meta‐analysis of observational studies published over more than 35 years. J Inherit Metab Dis. 2016;39(2):219‐229. [DOI] [PubMed] [Google Scholar]
- 7. Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival after treatment with phenylacetate and benzoate for urea‐cycle disorders. N Engl J Med. 2007;356(22):2282‐2292. [DOI] [PubMed] [Google Scholar]
- 8. Waisbren SE, Stefanatos AK, Kok TMY, Ozturk‐Hismi B. Neuropsychological attributes of urea cycle disorders: a systematic review of the literature. J Inherit Metab Dis. 2019;42(6):1176‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42(6):1192‐1230. [DOI] [PubMed] [Google Scholar]
- 10. Kolker S, Garcia‐Cazorla A, Valayannopoulos V, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015;38(6):1041‐1057. [DOI] [PubMed] [Google Scholar]
- 11. Kolker S, Valayannopoulos V, Burlina AB, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J Inherit Metab Dis. 2015;38(6):1059‐1074. [DOI] [PubMed] [Google Scholar]
- 12. Buerger C, Garbade SF, Dietrich Alber F, et al. Impairment of cognitive function in ornithine transcarbamylase deficiency is global rather than domain‐specific and is associated with disease onset, sex, maximum ammonium, and number of hyperammonemic events. J Inherit Metab Dis. 2019;42(2):243‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Posset R, Garcia‐Cazorla A, Valayannopoulos V, et al. Age at disease onset and peak ammonium level rather than interventional variables predict the neurological outcome in urea cycle disorders. J Inherit Metab Dis. 2016;39(5):661‐672. [DOI] [PubMed] [Google Scholar]
- 14. Posset R, Gropman AL, Nagamani SCS, et al. Impact of diagnosis and therapy on cognitive function in urea cycle disorders. Ann Neurol. 2019;86(1):116‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21‐year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97(10):1420‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype‐phenotype correlations in ornithine Transcarbamylase deficiency: a mutation update. J Genet Genomics. 2015;42(5):181‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zielonka M, Kolker S, Gleich F, et al. Early prediction of phenotypic severity in citrullinemia type 1. Ann Clin Transl Neurol. 2019;6(9):1858‐1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zielonka M, Garbade SF, Gleich F, et al. From genotype to phenotype: early prediction of disease severity in argininosuccinic aciduria. Hum Mutat. 2020;41:946‐960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Posset R, Garbade SF, Boy N, et al. Transatlantic combined and comparative data analysis of 1095 patients with urea cycle disorders‐a successful strategy for clinical research of rare diseases. J Inherit Metab Dis. 2019;42(1):93‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hothorn T, Hornik K, Zeileis A. Unbiased recursive partitioning: a conditional inference framework. J Comput Graph Stat. 2006;15(3):651‐674. [Google Scholar]
- 21. Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx. 2004;1(2):189‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. FDA UFaDACfDEaR . Guidance for Industry and Staff; Qualification Process for Drug Development Tools. 2014. Available from: https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/qualification‐process‐drug‐development‐tools‐guidance‐industry‐and‐fda‐staff [Google Scholar]
- 23. Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8(4):431‐440. [DOI] [PubMed] [Google Scholar]
- 24. Allegri G, Deplazes S, Rimann N, et al. Comprehensive characterization of ureagenesis in the spf(ash) mouse, a model of human ornithine transcarbamylase deficiency, reveals age‐dependency of ammonia detoxification. J Inherit Metab Dis. 2019;42(6):1064‐1076. [DOI] [PubMed] [Google Scholar]
- 25. Batshaw ML, Yudkoff M, McLaughlin BA, et al. The sparse fur mouse as a model for gene therapy in ornithine carbamoyltransferase deficiency. Gene Ther. 1995;2(10):743‐749. [PubMed] [Google Scholar]
- 26. Tarasenko TN, Rosas OR, Singh LN, Kristaponis K, Vernon H, McGuire PJ. A new mouse model of mild ornithine transcarbamylase deficiency (spf‐j) displays cerebral amino acid perturbations at baseline and upon systemic immune activation. PLoS One. 2015;10(2):e0116594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang L, Bell P, Morizono H, et al. AAV gene therapy corrects OTC deficiency and prevents liver fibrosis in aged OTC‐knock out heterozygous mice. Mol Genet Metab. 2017;120(4):299‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunetti‐Pierri N, Clarke C, Mane V, et al. Phenotypic correction of ornithine transcarbamylase deficiency using low dose helper‐dependent adenoviral vectors. J Gene Med. 2008;10(8):890‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Sabbata G, Boisgerault F, Guarnaccia C, et al. Long‐term correction of ornithine transcarbamylase deficiency in Spf‐ash mice with a translationally optimized AAV vector. Mol Ther Methods Clin Dev. 2021;12(20):169‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ginn SL, Amaya AK, Liao SHY, et al. Efficient in vivo editing of OTC‐deficient patient‐derived primary human hepatocytes. JHEP Rep. 2020;2(1):100065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kiwaki K, Kanegae Y, Saito I, et al. Correction of ornithine transcarbamylase deficiency in adult spf(ash) mice and in OTC‐deficient human hepatocytes with recombinant adenoviruses bearing the CAG promoter. Hum Gene Ther. 1996;7(7):821‐830. [DOI] [PubMed] [Google Scholar]
- 32. Wang L, Morizono H, Lin J, et al. Preclinical evaluation of a clinical candidate AAV8 vector for ornithine transcarbamylase (OTC) deficiency reveals functional enzyme from each persisting vector genome. Mol Genet Metab. 2012;105(2):203‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L, Yang Y, Breton C, et al. A mutation‐independent CRISPR‐Cas9‐mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci Adv. 2020;6(7):eaax5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chiriboga CA, Darras BT, Iannaccone ST, et al. Results from a phase 1 study ofnusinersen (ISIS‐SMNRx) in children with spinal muscular atrophy. Neurology. 2016;86:890‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Germain DP, Nicholls K, Giugliani R, et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat‐amenable variants: data from the phase 3 randomized, multicenter, double‐blind clinical trial and extension study. Genet Med. 2019;21(9):1987‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Silvera‐Ruiz SM, Arranz JA, Haberle J, et al. Urea cycle disorders in argentine patients: clinical presentation, biochemical and genetic findings. Orphanet J Rare Dis. 2019;14(1):203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overview of protein expression levels per variant.
Figure S2. Overview of residual enzymatic OTC activity per variant.
Figure S3. Correlation between disease onset and residual enzymatic OTC activity.
Table S1. Descriptive characteristics of study cohort and subanalyses.
Table S2. Additional members and affiliations of the UCDC and E‐IMD consortia study group.
Data Availability Statement
The data sets generated and analysed during the current study are not publicly available due to existing data protection laws. Furthermore, data ownership is retained by the members of the UCDC and E‐IMD consortia making data only available for specific research purposes. Data availability is subject to the consent of both consortia upon request.