

Abstract
Background:
IL-33 is one of the most consistently associated gene candidates for asthma identified by GWAS. Studies in mice and in human cells have confirmed the importance of IL-33 in inducing type-2 cytokine production from both group 2 innate lymphoid cells (ILC2) and Th2 cells. However, there are no pharmacologic agents known to inhibit IL-33 release from airway cells.
Objective:
To determine the effect of glucagon like peptide receptor-1 GLP-1R signaling on aeroallergen-induced airway IL-33 production and release and on innate type-2 airway inflammation.
Methods:
BALB/c mice were challenged intranasally with Alternaria extract for 4 consecutive days. GLP-1R agonist or the vehicle was administered starting either 2 days before the first Alternaria extract-challenge or 1 day after the first Alternaria extract-challenge.
Results:
GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge decreased IL-33 release in the BAL fluid and DUOX1 mRNA expression 1 h after the first Alternaria extract-challenge, and IL-33 expression in lung epithelial cells 24 h after the last Alternaria extract-challenge. Further, GLP-1R agonist significantly decreased the number of ILC2 expressing IL-5 and IL-13, the lung protein expression of type-2 cytokines and chemokines, the number of perivascular eosinophils, mucus production, and airway responsiveness compared with vehicle treatment. GLP-1R agonist treatment starting one day after the first Alternaria extract-challenge also significantly decreased eosinophilia and type-2 cytokine and chemokine expression in the airway after 4 days of Alternaria extract-challenge.
Conclusion:
These results reveal that GLP-1R signaling may be a potential therapy to reduce IL-33 release and inhibit the ILC2 response to protease-containing aeroallergens, such as Alternaria.
Keywords: Glucagon-like peptide-1 receptor (GLP-1R), Liraglutide, Group 2 innate lymphoid cells (ILC2), IL-33, Alternaria
Capsule Summary
GLP-1R signaling inhibited IL-33 release and innate allergic airway inflammation, including eosinophilia, mucus hypersecretion and AR following Alternaria extract-challenge. The GLP-1R agonists may constitute a novel therapeutic approach for allergic asthma mediated by ILC2 activation.
Graphical Abstract
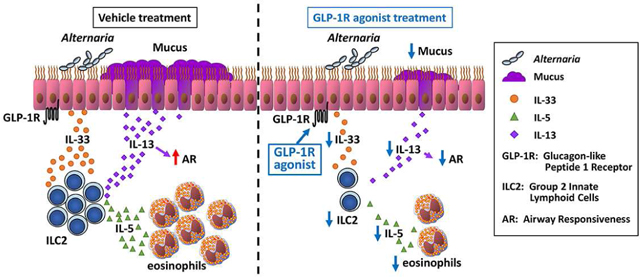
Introduction
IL-33 is one of the most consistently associated gene candidates for asthma identified by genome wide association studies (GWAS) in diverse ethnic groups.1–4 IL-33 is predominantly produced by epithelial cells in response to protease containing aeroallergens and its release is mediated by dual oxidase 1 (DUOX1).5 IL-33 activates group 2 innate lymphoid cells (ILC2) to produce the type-2 cytokines IL-5 and IL-13 that initiate innate immunity-driven allergic responses.6, 7 In addition, IL-33 polarizes naïve CD4 T cells to differentiate into effector T helper 2 (Th2) cells, which produce IL-4, IL-5, and IL-13 that are responsible for adaptive immunity-mediated allergen-induced responses.8 Therefore, IL-33 is a central mediator of both innate and adaptive immunity regulated allergic inflammation in the lung that have a role in the pathogenesis of conditions such as asthma, and IL-33 has been deemed to be an important therapeutic target in inhibiting allergic diseases.9 However, to our knowledge, there have been no reports identifying pharmacologic agents which inhibit lung IL-33 protein release or expression.
Glucagon-like peptide-1 (GLP-1) is a peptide hormone synthesized and released by enteroendocrine L-cells in the ileum and large intestine following oral food intake.10 GLP-1 has a role in glycemic control by inducing glucose-dependent insulin secretion from β-cells and inhibiting glucagon release from α-cells in the pancreas.11, 12 GLP-1 also induces weight loss by promoting satiety.13 GLP-1 receptor (GLP-1R) agonists, such as liraglutide and exenatide are approved by the Food and Drug Administration (FDA) for treatment of type 2 diabetes (T2D).14, 15 Several studies report that GLP-1R agonists had anti-inflammatory effects in multiple disorders including T2D. For instance, GLP-1R agonist administration decreased TNFα and IL-6 production by peripheral blood mononuclear cells (PBMC) of obese patients with T2D16 and diabetic mouse adipose tissue.17 In addition, the GLP-1R agonist, exendin-4, reduced serum inflammatory cytokines during LPS-induced endotoxemia,18 liver inflammation, and aortic atherosclerosis in a rodent model.19 These data indicate that GLP-1R agonists down-regulate innate inflammatory responses to endotoxins or endogenous inflammatory mediators. Further, it has been reported that liraglutide attenuated bleomycin-induced pulmonary fibrosis20 and OVA-induced chronic airway inflammation.21 These results suggest the possibility of novel therapeutic strategies for chronic lung diseases using GLP-1R agonists. However, no studies have reported the effect of GLP-1R agonists on lung IL-33 expression or release, or the effect of GLP-1R agonists on the innate allergic inflammatory response that is mediated by ILC2.
In this study, we performed airway challenge in mice with an extract of Alternaria alternata, an aeroallergen which has protease activity and which is associated with severe asthma exacerbations.22, 23 GLP-1R agonist or the vehicle treatment was started either 2 days before the first Alternaria extract-challenge or 1 day after the first Alternaria extract-challenge. We found that treatment with the GLP-1R agonist, liraglutide, starting 2 days before the first Alternaria extract-challenge inhibited IL-33 expression and release in the lung or into the BAL fluid. Further, GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge inhibited lung DUOX1 expression, providing a potential mechanism by which GLP-1R signaling inhibits lung IL-33 release in response to aeroallergen challenge. In addition, we found that GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge inhibited IL-5 and IL-13 production from lung ILC2, blunted airway mucus and responsiveness, and reduced lung eosinophilia. GLP-1R agonist treatment starting 1 day after the first Alternaria extract-challenge also reduced the eosinophilia and type-2 cytokines and chemokines in the airway. This report is the first to identify an FDA approved pharmacologic agent that inhibits lung IL-33 release, providing an alternative to biologic therapies that target IL-33-mediated diseases.
Methods
Mice
Nine- to twelve-week old female wild type (WT) BALB/c mice were obtained from Jackson Laboratories (Bar Harbor, ME). IL-33Citrine/+ reporter mice were generated by crossbreeding WT BALB/c mice and IL-33Citrine/Citrine (IL-33 KO) mice that are the kind gift of Dr. Andrew N.J. McKenzie.24 Transgenic mice expressing mApple protein under the control of the GLP-1R promoter were generated as previously described.25 Animal experiments were approved by the Institutional Animal Care and Use Committee at Vanderbilt University, and were conducted according to the guidelines for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources, National Research Council.
GLP-1R agonist treatment and Alternaria extract- or house dust mite extract-challenge in a mouse model of allergic inflammation
In the protocol where GLP-1R agonist treatment was started 2 days before the first Alternaria extract-challenge (Fig 2, A), the GLP-1R agonist, liraglutide (Novo Nordisk Inc., Bagsvaerd, Denmark) or 50 μl 0.1 % BSA/PBS as vehicle was administered subcutaneously 4 h before and 4 h after Alternaria extract-, house dust mite (HDM) extract- (Dermatophagoides pteronyssinus, GREER, Lenoir, NC), or PBS-challenge from day −2 to day 3 (Fig 2, A, Fig 3, A, Fig E5, A and C). In the protocol where GLP-1R agonist was started 1 day after the first Alternaria extract-challenge, the vehicle was administered from day −2 to day 0, and then either GLP-1R agonist or the vehicle was administered from day 1 to day 3 (Fig 7, A). From day 0 to day 3, either 5 μg (protein amount) of Alternaria extract in 100 μl PBS, 25 μg (protein amount) of HDM extract in 100 μl PBS, or 100 μl of PBS as vehicle was used as an intranasal challenge to mice anesthetized with ketamine/xylazine. The mice were sacrificed and bronchoalveolar lavage (BAL) fluid and whole lungs were harvested 1 h after the first challenge of Alternaria extract or the vehicle (for IL-33 release), or 24h (for cytokine measurement) or 48 h (for histopathology and airways responsiveness measurement) after the last challenge of Alternaria extract, HDM extract, or PBS.
FIG 2.

A, Mice were challenged with Alternaria extract intranasally for 4 consecutive days. GLP-1R agonist or its vehicle was administered subcutaneously on day −2 and −1, and then every 4 h before and after Alternaria extract-challenge on day 0–3. The dose of GLP-1R agonist was 0.05 mg/kg on day −2, 0.1 mg/kg on day −1, and 0.2 mg/kg on day 0 −3. B, Representative histograms of citrine (IL-33) expression on CD45−EpCAM+ cells in the lungs from WT mice (filled gray area) or IL-33Citrine/+ reporter mice challenged with vehicle-PBS (black line), GLP-1R agonist-PBS (orange line), vehicle-Alternaria extract (blue line), and GLP-1R agonist-Alternaria extract (green line). C-E, The MFI, the percentage, and the number of Citrine+CD45−EpCAM+ cells in the lung. The results are representative of 2 independent experiments, and shown as mean ± S.E.M. of 3 mice in PBS-challenged groups and 5 mice in Alternaria extract-challenged groups. Veh=vehicle. PBS=phosphate buffered saline. EC=epithelial cells * P< 0.05
FIG 3.

A, GLP-1R agonist or its vehicle was administered subcutaneously on day −2 and −1, and then 4 h before Alternaria extract-challenge on day 0. The dose of GLP-1R agonist was 0.05 mg/kg on day −2, 0.1 mg/kg on day −1, and 0.2 mg/kg on day 0. The BAL fluid was harvested 1 h after the Alternaria extract-challenge on day 0. B, The protein level of IL-33 in the BAL fluid was measured by ELISA. n=4 for PBS-challenged groups, and n=8 for Alternaria extract-challenged groups. C, Quantity of DUOX1 mRNA expression in the lung was measured by real-time PCR. n=5 for each groups. The all results are combined with 2 independent experiments, and shown as mean ± S.E.M. Veh=vehicle. PBS=phosphate buffered saline. * P< 0.05
FIG 7.


A, Mice were challenged with Alternaria extract intranasally for 4 consecutive days (from day 0 to day 3). The mice of vehicle treatment group and GLP-1R agonist treatment starting 2 days before the first Alternaria challenge group were treated with vehicle or GLP-1R agonist respectively from day −2 to day 3. The mice of GLP-1R agonist treatment starting 1 day after the first Alternaria challenge group were treated with vehicle from day-2 to day0, and then treated with GLP-1R agonist from day 1 to day 3. B, Cell differentials in BAL fluid harvested 24 h after the last Alternaria extract-challenge. C, BAL fluid and D, lungs were harvested 24 h after the last Alternaria extract-challenge to measure the protein expression of IL-4, IL-5, IL-9, IL-13, CCL11, CCL17, CCL22, and CCL24 by ELISA. The results are combined with 2 independent experiments, and shown as mean ± S.E.M. of 10 mice in each groups. * P< 0.05
Flow cytometry
The harvested lungs were minced and digested with collagenase IV (1 mg/ml, SIGMA-Aldrich, St. Louis, MO) and DNase I (50 IU/ml, SIGMA-Aldrich) in RPMI1640 with 5% FBS for 45 minutes at 37 °C and a single cell suspension was obtained by grinding and passing the digested lung through a 70 μm cell strainer. The cells were stimulated with PMA (10 ng/ml), ionomycin (1 μM) and BD GolgiStop™ (BD Biosciences, San Jose, CA) at the manufacturer’s recommended concentration for 5 hours. Then both cell surface and intracellular cytokine staining were performed. Antibodies used for flow cytometry are shown in Table E1. All samples were measured on a BD LSR II Flow Cytometer and analyzed using FlowJo Software.
ELISA and LDH activity assay
DuoSet ELISA kits (CCL11, CCL17, CCL22, CCL24, IL-4, IL-9, and IL-33) from R&D Inc. (Minneapolis, MN) and Ready-Set-GO!® ELISA kits (IL-5 and IL-13) from Affymetrix eBioscience (San Diego, CA) were used to measure the concentration of cytokines and chemokines in BAL fluid and lung homogenates according to manufacturer’s instructions. Cysteinyl Leukotriene ELISA kit and Prostaglandin D2 ELISA Kit from Cayman chemical (Ann Arbor, MI) were used to measure cysteinyl leukotrienes (CysLTs), which include LTC4, LTD4, and LTE4, and Prostaglandin D2 (PGD2) in BAL fluid. LDH cytotoxity assay kit (Thermo scientific, Waltham, MA) was used to evaluate Alternaria extract-induced cell damage associated with IL-33 release. Values of ELISA below the detection limit were assigned a value at half of the lower limit of detection to allow for statistical analysis.
Statistical analysis
All data were analyzed with GraphPad Prism 5 (GraphPad Software, La Jolla, CA). The p values were calculated by one-way analysis of variance (ANOVA) with Bonferroni-multiple pairs comparisons test. Values of p < 0.05 were considered significant between two groups.
Additional detail on the methods is available in an Online Repository
Results
GLP-1R expression on lung epithelial cells
We sought to examine if GLP-1R expression was present on lung epithelial cells to determine whether GLP-1 could directly modulate epithelial cell function. GLP-1R mRNA expression was detected in normal human lung as well as mouse lung (Fig 1, A and B). Further, GLP-1R protein expression was also detected on mouse lung epithelial cells and endothelial cells (Fig 1, C and D) using GLP-1R/mApple reporter mice. These results provide a rationale for investigating the effect of GLP-1R signaling in response to airway Alternaria extract-challenge.
FIG 1.

A and B, RT-PCR of GLP-1R in human lung and mouse lung. Results are representative of 3 different samples. RT= reverse transcriptase. C, The gating strategy of epithelial cells and endothelial cells. D, Representative histograms of mApple (GLP-1R) expression on CD45− CD146+EpCAM− endothelial cells and CD45−CD146+EpCAM+ epithelial cells in the lungs from WT mice (filled gray area) or GLP-1R/mApple reporter mice (black line). Results are representative of 3 different mice.
GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge suppresses Alternaria extract-induced IL-33 expression in lung epithelial cells
IL-33 is a key activator of the innate allergic airway response, and in particular IL-33 stimulates ILC2.6 Therefore, we tested if GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge decreases IL-33 protein expression following airway Alternaria extract-challenge for 4 consecutive days (Fig 2, A). To confirm the localization of IL-33 expression and the effect of GLP-1R agonist on a per cell IL-33 expression level, we used IL-33Citrine/+ reporter mice. We found that Alternaria extract-challenge significantly increased mean fluorescence intensity (MFI) of citrine fluorescence as a marker of IL-33 expression by lung epithelial cells (CD45− EpCAM+), and the percentage and the number of the citrine+ epithelial cells. GLP-1R agonist treatment significantly decreased the Alternaria extract-induced MFI of citrine in the lung epithelial cells, the percentage and the number of citrine+ epithelial cells compared with the vehicle treatment (Fig 2, B-E). These results indicate that GLP-1R agonist treatment reduced the number of lung epithelial cells expressing IL-33 and the level of IL-33 expression by individual cells following Alternaria extract-challenge.
GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge suppresses Alternaria extract-induced IL-33 release, but not LDH activity in the BAL fluid
Airborne allergens, such as Alternaria have proteolytic activity which induces IL-33 release in BAL fluid 1 h after airway administration through PAR2 signaling.26 We therefore measured IL-33 protein level in BAL fluid 1 h after first Alternaria extract-challenge following GLP-1R agonist treatment or vehicle treatment starting 2 days before the first Alternaria extract-challenge (Fig 3, A). Alternaria extract-challenge significantly increased the protein level of IL-33 in BAL fluid compared with PBS-challenged groups. The Alternaria extract-induced IL-33 in BAL fluid was significantly decreased by GLP-1R agonist treatment compared with the vehicle treatment (Fig 3, B). Next, we measured lactate dehydrogenase (LDH) activity in BAL fluid to assess the effect of GLP-1R agonist treatment for lung cell damage during IL-33 release. Alternaria extract-challenge significantly increased LDH activity in BAL fluid compared with PBS-challenged groups (Fig E1, B). Although the Alternaria extract-induced LDH activity was not decreased in GLP-1R agonist treatment group compared with vehicle treatment group (Fig E1, B), Alternaria extract-induced IL-33 protein level was significantly decreased in BAL fluid from the GLP-1R agonist treatment group compared with the vehicle treatment group (Fig E1, A). These results indicate that the effect of GLP-1R signaling on IL-33 release pathway is independent of Alternaria extract-induced lung cell damage.
A previous study reported that Alternaria extract directly and quickly induced mast cell degradation and activation deriving CysLTs and PGD2.27 Therefore, we measured CysLTs and PGD2 in BAL fluid to assess an effect of GLP-1R agonist treatment for Alternaria extract-induced mast cell or the other CysLTs and PGD2 productive cell activation. Alternaria extract-challenge significantly increased the CysLTs and PGD2 in BAL fluid compared with PBS-challenged groups; however, GLP-1R agonist treatment did not decrease the Alternaria extract-induced CysLTs and PGD2 compared with vehicle treatment (Fig E1, C and D). In addition, we measured GLP-1R protein expression on bone marrow-derived cultured mast cells (BMCMC) using GLP-1R/mApple reporter mice. The BMCMC gating strategy was shown in Fig E1, E. GLP-1R/mApple expression was not detected on the BMCMC (Fig E1, F), supporting that GLP-1R agonist does not have a direct effect on mast cell biology. Further, the induction of LDH activity, CysLTs and PGD2 by Alternaria extract-challenge was observed in IL-33 KO mice as well as WT mice (Fig E1, B-D). Therefore, the Alternaria extract-induced cell damage and production of the lipid mediators are independent of biological effects of endogenous IL-33. Taken together, these results reveal that GLP-1R agonist treatment suppresses acute IL-33 release after Alternaria extract-challenge, but does not suppress lung cell damage and production of CysLTs and PGD2 from lung resident cells such as mast cells.
GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge suppresses DUOX1 mRNA expression in the Alternaria extract-challenged lung
DUOX1 is a critical mediator of IL-33 release from lung epithelial cells following aeroallergen challenge.5 We therefore measured mRNA expression level of DUOX1in the lung 1 h after Alternaria extract-challenge, the time of peak IL-33 release, following GLP-1R agonist or vehicle treatment. Lung DUOX1 mRNA expression was significantly decreased by GLP-1R agonist treatment compared with vehicle treatment (Fig 3, C). This result suggests a possible mechanism by which GLP-1R agonist suppressed IL-33 release.
GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge suppresses the number of mouse lung ILC2 expressing IL-5 and IL-13 in response to airway Alternaria extract-challenge.
Since IL-33 is an important stimulus for the innate allergic immune response, we sought to determine if GLP-1R signaling inhibited ILC2 function. First, we measured GLP-1R expression on lung ILC2 using GLP-1R/mApple reporter mice to reveal whether the GLP-1R agonist directly regulates the ILC2. The gating strategy for ILC2 was shown in Fig E1. ILC2 were identified as lineage (lin)−CD3−CD45+CD25+CD127+ cells by cell surface staining. In contrast to epithelial cells and endothelial cells, GLP-1R/mApple expression was not detected on the lung ILC2 (Fig 4, A). Next, we enumerated the number of total, IL-5+ and IL-13+ lung ILC2 following Alternaria extract-challenge for 4 consecutive days to determine the effect of IL-33 reduction by GLP-1R agonist treatment. The gating strategy for ILC2 and representative flow cytometry dot plots of IL-5 and IL-13 intracellular staining are shown in Fig E2. ILC2 were identified as lineage (lin)−CD3−CD45+CD25+ and ICOS+. Alternaria extract-challenge significantly increased the number of total, IL-5 + and IL-13+ lung ILC2 compared with PBS-challenged mice (Fig 4, B-D). GLP-1R agonist treatment significantly suppressed the number of Alternaria extract-induced total, IL-5+, and IL-13+ lung ILC2 (Fig 4, B-D) compared with vehicle treatment.
FIG 4.

A, Representative histograms of mApple (GLP-1R) expression on lung ILC2 from WT mice (filled gray area) or GLP-1R/mApple reporter mice (black line). Result is a representative of 3 different mice. B-D, Flow cytometric analysis of the number of total, IL-5+, and IL-13+ ILC2 in the lung. The results are combined with 2 independent experiments, and shown as mean ± S.E.M. of 4 mice in PBS-challenged groups and 8 mice in Alternaria extract-challenged groups. Veh=vehicle. PBS=phosphate buffered saline. * P< 0.05
To compare the contribution of CD4 T cells to IL-5 and IL-13 expression, intracellular cytokine staining was performed by flow cytometry. The gating strategy and dot plots of CD4 T cell expressing IL-5 and IL-13 are shown in Fig E3. The number of IL-5+ and IL-13+ CD4 T cell were significantly increased in Alternaria extract-challenged groups compared to PBS-challenged groups. GLP-1R agonist treatment significantly suppressed the number of Alternaria extract-induced CD4 T cells expressing IL-13, but not IL-5. In the Alternaria extract-challenged groups, there was a statistically significant 10-fold increase in the number of IL-5+ ILC2 (Fig 4, C) compared to IL-5+ CD4 T cells (Fig E3, C), and a 2.4-fold increase in the number of IL-13+ ILC2 (Fig 4, D) compared to IL-13+ CD4 T cells (Fig E3, D). The fold change between the number of ILC2 and CD4 T cells expressing IL-5 and IL-13 are similar to our previous report.28 Therefore, these results indicate that ILC2, but not CD4 T cell, are the major source of IL-5 and IL-13 in response to this 4 consecutive day airway Alternaria extract-challenge protocol, and that GLP-1R signaling likely inhibits this early innate allergic immune response by the reduction of IL-33 release in the lung.
GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge suppresses Alternaria extract-induced cytokine and chemokine expression in the lung and airway.
Next, we measured the protein level of type-2 cytokines and chemokines in BAL fluid and lung homogenate. Alternaria extract-challenge significantly increased the protein expression of IL-4, IL-5, IL-13, CCL11 (eotaxin), CCL17 (TARC), CCL22 (MDC), and CCL24 (eotaxin-2) in both the BAL fluid and lung homogenates compared with PBS-challenged groups. IL-9 was significantly increased in the lung homogenate, but not BAL fluid following Alternaria extract-challenge. We found a statistically significant decrease in Alternaria extract-induced protein expression of IL-5, IL-13, CCL17, CCL 22 and CCL24 in both the BAL fluid and lung homogenates from GLP-1R agonist treated mice compared with vehicle treated mice (Fig 5). In addition, Alternaria extract-induced IL-4, IL-9, and CCL11 protein expression was also decreased in the lung homogenates from GLP-1R agonist treated mice compared with vehicle treated mice.
FIG 5.

BAL fluid and lungs were harvested 24 h after the last Alternaria extract-challenge to measure the protein expression of IL-4, IL-5, IL-9, IL-13, CCL11, CCL17, CCL22, and CCL24 by ELISA. The results are combined with 2 independent experiments, and shown as mean ± S.E.M. of 6 mice in PBS-challenged groups and 11 mice in Alternaria extract-challenged groups. Veh=vehicle. PBS=phosphate buffered saline. * P< 0.05
GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge suppresses eosinophilia, mucus production, and airway responsiveness in response to airway Alternaria extract-challenge.
Based on our finding that GLP-1R agonist treatment significantly decreased protein expression of Alternaria extract-induced IL-5, CCL11, and CCL24 that are associated with eosinophil recruitment, we hypothesized that GLP-1R agonist treatment decreases eosinophil accumulation in the lung following Alternaria extract-challenge. To test this hypothesis, we measured BAL cell counts and differentials, and assessed perivascular eosinophils by immunohistopathology. Alternaria extract-challenge significantly increased the number of total BAL cells, macrophages, eosinophils, lymphocytes and neutrophils compared with PBS-challenged groups. GLP-1R agonist treatment significantly decreased the number of Alternaria extract-induced total BAL cells, macrophages, eosinophils, and lymphocytes compared with vehicle treatment (Fig 6, A). Further, we found that GLP-1R agonist treatment resulted in a significant decrease of Alternaria extract-induced perivascular eosinophils in the lung compared with vehicle treatment by immunohistopathology (Fig 6, B and C).
FIG 6.


A, Cell differentials in BAL fluid harvested 24 h after the last Alternaria extract-challenge. n=6 for PBS-challenged groups, and n=11 for Alternaria extract-challenged groups. B, Representative sections and C, eosinophil score as determined by anti-MBP antibody staining. n=5 for each group. D, Representative sections and E, mucus score as determined by PAS staining. n=4 for PBS-challenged groups, and n=10 for Alternaria extract-challenged groups. The lungs for histological analysis were harvested 48 h after the last Alternaria extract-challenge. F, Airway resistance to increasing dose of methacholine challenge. n=7 for PBS-challenged groups, and n=16 for Alternaria extract-challenged groups (* P< 0.05 compared with vehicle-PBS. ** P< 0.05 compared with vehicle-Alternaria). The all results are combined with 2 independent experiments, and shown as mean ± S.E.M. Veh=vehicle. PBS=phosphate buffered saline. Mch= methacholine. * P< 0.05
IL-13 is a critical inducer of airway mucus production.29 We found that GLP-1R agonist treatment decreased protein level of IL-13 in the lung. Therefore, we tested the hypothesis that GLP-1R agonist treatment suppresses mucus production in the airway following Alternaria extract-challenge. There was no mucus or mucous producing cells in the lungs of mice challenged with PBS. Alternaria extract-challenge significantly increased mucus production on large airway epithelial cells. GLP-1R agonist treatment significantly decreased the Alternaria extract-induced mucus production compared with vehicle treatment (Fig 6, D and E). These results indicate that GLP-1R agonist treatment suppresses acute eosinophilic lung inflammation and airway mucus production in innate allergic immune responses to Alternaria extract-challenge.
We found that GLP-1R agonist treatment significantly decreased asthma-like airway inflammation following Alternaria extract-challenge for 4 consecutive days compared with vehicle treatment. We next tested the hypothesis that GLP-1R agonist treatment decreases airway responsiveness (AR) in Alternaria extract-challenged mice. Alternaria extract-challenge significantly increased AR compared with PBS challenge. GLP-1R agonist treatment significantly decreased the Alternaria extract-induced AR compared with vehicle treatment (Fig 6, F).
GLP-1R agonist treatment starting 2 days before the first HDM extract-challenge suppresses HDM extract-induced IL-5, IL-13 and IL-33 in the lung.
To assess the effects of GLP-1R agonist treatment in a different antigen-induced innate allergic inflammation model, we used HDM extract because HDM is one of common antigens associated with allergic asthma and atopic dermatitis.30 First, we measured IL-33 protein level and LDH activity in BAL fluid 1 h after first HDM extract-challenge following GLP-1R agonist or vehicle treatment (Fig E5, A). However, IL-33 protein was not detected in either the BAL fluid from HDM extract-challenged groups or the PBS-challenged groups (Fig E5, B); and there was no difference in BAL fluid LDH activity between HDM extract- and PBS-challenged groups (Fig E5, B). These results indicate that HDM extract has no cytolytic activity to induce IL-33 release. Meanwhile, 4 consecutive days of HDM extract-challenge increased protein level of IL-5, IL-13 and IL-33 in the lung homogenates 24 h after the last HDM extract-challenge compared to the PBS-challenge (Fig E5, D). The HDM extract-induced IL-5, IL-13 and IL-33 were significantly decreased in GLP-1R agonist treatment group compared with vehicle treatment group (Fig E5, D). This result revealed that GLP-1R agonist treatment decreased type-2 cytokine and IL-33 expression caused by HDM extract-induced innate inflammation.
GLP-1R agonist treatment starting 1 day after the first Alternaria extract-challenge as well as treatment starting 2 days before the first Alternaria extract-challenge suppresses Alternaria extract-induced eosinophilia, and type-2 cytokine and chemokine expression in the airway.
To determine the effect of GLP-1R agonist treatment as a potential therapeutic use, we compared the effect of GLP-1R agonist treatment starting 1 day after the first Alternaria extract-challenge against treatment starting 2 days before the first Alternaria extract-challenge in Alternaria extract-induced innate allergic inflammation (Fig 7, A). GLP-1R agonist treatment starting 1 day after the first Alternaria extract-challenge significantly decreased the number of Alternaria extract-induced total BAL cells, eosinophils, and lymphocytes compared with vehicle treatment similar to GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge (Fig 7, B). Alternaria extract-induced IL-5, CCL11, CCL17, CCL22, and CCL24 in both the BAL fluid and lung homogenates were significantly decreased GLP-1R agonist treatment in both protocols compared with vehicle treatment (Fig 7, C and D). Alternaria extract-induced IL-13 in the BAL fluid and IL-9 in the lung homogenates were also significantly decreased in GLP-1R agonist treatment group starting 1 day after the first Alternaria extract-challenge compared with vehicle treatment (Fig 7, C and D). These results suggest that GLP-1R agonist treatment after initial antigen challenge also suppresses Alternaria extract-induced airway eosinophilia and type-2 cytokine and chemokine expression in the airway and lung.
Discussion
Our results show that a GLP-1R agonist inhibited lung epithelial expression and airway release of IL-33 in response to the clinically relevant, protease-containing, ubiquitous aeroallergen, Alternaria alternata. This is the first report of an FDA approved pharmacologic agent inhibiting allergen-induced lung IL-33 expression and release, and this finding has significant implications as it may provide an alternative to biologic therapies such as monoclonal antibodies or receptor antagonists that target IL-33-mediated diseases. In addition, DUOX1 mRNA was also decreased by GLP-1R agonist treatment during IL-33 release following Alternaria extract-challenge. Further, we found that GLP-1R agonist treatment 2 days before the first Alternaria extract-challenge significantly decreased the number of lung ILC2 expressing IL-5 and IL-13 following 4 consecutive days of Alternaria extract-challenge. GLP-1R agonist treatment 2 days before the first Alternaria extract-challenge significantly decreased IL-4, IL-5, IL-9, IL-13, CCL11, CCL17, CCL22 and CCL24 protein expression in the lung homogenates, the number of eosinophils in the BAL fluid and perivascular eosinophil accumulation, mucus production, and AR induced by the Alternaria extract-challenge. Likewise, GLP-1R agonist starting 1 day after the first Alternaria extract-challenge significantly decreased eosinophilia and IL-5, IL-13, CCL11, CCL17, CCL22 and CCL24 in the airway. These results suggest that GLP-1R signaling not only has an effect on the initial allergen-induced IL-33 release in the airway, but also inhibits established airway inflammation.
GLP-1R is abundantly expressed in human and mouse lung, heart, brain and kidney as well as pancreas,31, 32 and many studies indicate the therapeutic effect of GLP-1R signaling for diseases in multiple tissues. For instance, a GLP-1R agonist, exendin-4, improved atherosclerosis by reducing macrophage adhesion to endothelial cells as well as by reducing serum glucose level.32 GLP-1 peptide treatment decreased irradiation- or LPS-induced proinflammatory cytokine expression in the brain and astrocytes in a mouse model.33, 34 In addition, GLP-1R agonist decreased chronic lung inflammation in a mouse model of OVA sensitization combined with 66 days of OVA challenge,21 and reduced mortality and airway resistance in a mouse model of obstructive lung disease induced by OVA and LPS challenge.35
Binding of GLP-1 to its G protein coupled receptor causes adenylate cyclase activation resulting in the formation of cyclic adenosine monophosphate (cAMP). Subsequent activation of protein kinase A (PKA) and the cAMP-regulated guanine nucleotide exchange factor II (cAMP-GEFII) leads to an elevation of intracellular calcium concentrations, and enhanced exocytosis of insulin-containing granules in pancreatic β-cells.36 Further, β2 adrenergic receptor signaling also activate the cAMP/PKA signaling pathway, and then relaxed airway smooth muscle and reduced airway mucus secretion.37 Our group reported that prostaglandin I2 (PGI2) receptor signaling down-regulated DC, T cell, and ILC2 activation mediated by cAMP induction.38–40 In addition, GLP-1R agonist reduced inflammatory responses in mouse peritoneal macrophages stimulated with LPS through cAMP signaling pathway.41 These results suggest that cAMP induction by GLP-1R agonist as well as β2 adrenergic agonist or PGI2 may have suppressive effects on the activation of immune cells that express GLP-1R.
In this study, we found that a GLP-1R agonist treatment down-regulated Alternaria extract-induced IL-33 release and expression. IL-33 plays important roles in type-2 innate immunity following helminth infection or after exposure to protease containing aeroallergens.26, 42 Biologically active full length IL-33 is released into the extracellular space after cell damage such as necrosis, and then IL-33 activates many types of immune cells, including CD4 T cells, mast cells, basophils, macrophages, DCs, and epithelial cells as well as ILC2.43, 44 IL-33 is likely to be a first alarm signal due to its constitutive expression in normal epithelial and endothelial cells,44 and is ready to be released following infection or mechanical cell damage. We found that GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge reduced IL-33 release and DUOX1 mRNA expression, but not LDH activity after the first Alternaria extract-challenge. These results suggest that GLP-1R signaling down-regulates the IL-33 release pathway in lung epithelial cells expressing IL-33 without suppression of the cell damage. Hristova and colleagues reported that Alternaria induced IL-33 release in cultured primary human bronchial epithelial cells (HBE) depended critically on DUOX1-mediated activation.5 Shiraki and colleagues reported that GLP-1R signaling inhibited NADPH oxidase activation in primary human endothelial cells.45 Taken together, our findings suggest that GLP-1R signaling decreases DUOX1 expression in the lung epithelial cells, thus inhibiting Alternaria-induced IL-33 secretion.
The reduction in IL-33 protein level to airway Alternaria extract-challenge mediated by the GLP-1R agonist is one possible mechanism by which GLP-1R signaling reduced the number of IL-5 and IL-13 expressing ILC2 and protein levels of CCL11, CCL24, TARC, and MDC in the airway and lung following 4 consecutive days Alternaria extract-challenge. These findings are consistent with previous reports that IL-33 induced IL-5 and IL-13 expression by ILC2 in dose-dependent fashion,46 CCL11 expression by fibroblast,47 and TARC expression by DCs.48 Although GLP-1R signaling reduced the number of IL-5 and IL-13 expressing ILC2 following 4 consecutive days Alternaria extract-challenge, the percentage of IL-5 and IL-13 expressing ILC2 was not decreased. Intracellular cytokine staining is appropriate to determine the cells that have ability to express IL-5 and IL-13; however, cell activation by such a strong stimulus as PMA and ionomycin might prevent the evaluation of the precise quantitative cytokine expression level in individual cells.
Meanwhile, we found that GLP-1R agonist treatment starting 2 days before the first Alternaria extract-challenge significantly decreased IL-5, IL-13, and IL-33 expression in the lung after 4 consecutive days of HDM extract-challenge. This HDM extract-induced innate inflammation was induced without IL-33 release. Further, GLP-1R agonist treatment starting 1 day after the first Alternaria extract-challenge also significantly decreased eosinophilia, and type-2 cytokine and chemokine expression in the airway. These in vivo results suggest a mechanism for downstream of IL-33 release by which GLP-1R signaling down-regulates innate allergic inflammation. However, we could not detect GLP-1R protein expression on ILC2 in the lung and BMCMC from naïve GLP-1R/mApple reporter mice, raising a concern about the specificity of the anti-GLP-1R antibody that has been used in immunohistochemistry and flow cytometry. Previous studies reported that GLP-1R was expressed in mouse peritoneal macrophages, the human monocyte cell line THP-1,41 and the human invariant natural killer T (iNKT) cell line.49 Since GLP-1R protein expression level on immune cells is not fully known, further experiments are necessary to determine the mechanisms of the anti-inflammatory effect of GLP-1R signaling.
In our 4 consecutive days of Alternaria extract-challenge model, the number of IL-5 and IL-13 expressing CD4 T cells was statistically significantly less than the number of IL-5 and IL-13 expressing ILC2, and our previous report showed that total IgE and antigen specific IgG1 in serum were no different between naïve mice and mice challenged with Alternaria extract for 4 consecutive days.40 This supports the concept that the innate immune system, and not adaptive immunity, initiate and promotes type-2 allergic airway inflammation in this mouse model of allergen challenge. Taken together, our findings reveal that GLP-1R signaling suppresses aeroallergen-driven IL-33 release and inhibits ILC2-dependent innate type-2 airway inflammation. Currently, biological agents are in development to antagonize IL-33 for the treatment of atopic dermatitis, food allergy, and asthma.9 Our results suggest that a currently FDA approved GLP-1R antagonist inhibits allergen-induced IL-33 release and expression.
In conclusion, we found that GLP-1R agonist treatment down-regulated IL-33 release and expression, and innate allergic airway inflammation, including lung eosinophilia, mucus hypersecretion and AR following 4 consecutive days of Alternaria extract-challenge. Our results suggest that GLP-1R agonists may constitute a novel therapeutic approach for allergic asthma induced by protease-containing aeroallergens.
Supplementary Material
Key message.
GLP-1R agonist inhibits IL-33 release and expression in response to aeroallergen challenge
GLP-1R signaling inhibited aeroallergen-induced ILC2 cytokine production, eosinophilic inflammation, mucus metaplasia, and airway responsiveness.
A current FDA approved pharmacologic agent, GLP-1R agonist may be a novel therapeutic approach for allergic airway diseases through a blocking IL-33 release into the airway and an inhibition of type-2 inflammation.
Acknowledgement
The authors are grateful to Dr. Andrew N. J. McKenzie, MRC Laboratory of Molecular Biology, Cambridge University, Cambridge, UK for providing IL-33Citrine/Citrine mice.
Supported by Department of Veterans Affairs 2I01BX000624, NIH AI 095227-02, AI 111820, AI124456, and HL 090664-04.
Abbreviations
- ILC2
group 2 innate lymphoid cells
- GLP-1
glucagon-like peptide-1
- GLP-1R
glucagon-like peptide-1receptor
- Th2
T helper 2
- FDA
Food and Drug Administration
- T2D
type 2 diabetes
- WT
wild type
- HDM
house dust mite
- BAL
bronchoalveolar lavage
- LDH
lactate dehydrogenase
- BMCMC
bone marrow-derived cultured mast cells
- DUOX1
dual oxidase 1
- MFI
mean fluorescence intensity
- AR
airway responsiveness
- cAMP
cyclic adenosine monophosphate
- PKA
protein kinase A
- HBE
human bronchial epithelial cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li X, Ampleford EJ, Howard TD, Moore WC, Torgerson DG, Li H, et al. Genome-wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J Allergy Clin Immunol 2012; 130: 861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nieuwenhuis MA, Siedlinski M, van den Berge M, Granell R, Li X, Niens M, et al. Combining genomewide association study and lung eQTL analysis provides evidence for novel genes associated with asthma. Allergy 2016; 71: 1712–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ober C, Yao T-C. The genetics of asthma and allergic disease: a 21st century perspective. Immunological Reviews 2011; 242:10–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savenije OE, Mahachie John JM, Granell R, Kerkhof M, Dijk FN, de Jongste JC, et al. Association of IL33–IL-1 receptor–like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol 2014; 134: 170–177. [DOI] [PubMed] [Google Scholar]
- 5.Hristova M, Habibovic A, Veith C, Janssen-Heininger YM, Dixon AE, Geiszt M, et al. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol 2015; 137:1545–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halim Timotheus YF, Krauß Ramona H, Sun Ann C, Takei F. Lung Natural Helper Cells Area a Critical Source of Th2 Cell-Type Cytokines in Protease Allergen-Induced Airway Inflammation. Immunity 2012; 36: 451–463. [DOI] [PubMed] [Google Scholar]
- 7.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage− CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol 2012; 188: 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol 2008; 181: 4780–90. [DOI] [PubMed] [Google Scholar]
- 9.Borish L. The immunobiology of asthma: Asthma phenotypes and theor implications for personalized treatment. Ann Allergy Asthma Immunol 2016; 117:108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim GE, Brubaker PL. Glucagon-Like Peptide 1 Secretion by the L-Cell: The View From Within. Diabetes 2006; 55: S70–S77. [Google Scholar]
- 11.Vilsboll T, Toft-Nielsen MB, Krarup T, Madsbad S, Dinesen B, Holst JJ. Evaluation of beta-cell secretory capacity using glucagon-like peptide 1. Diabetes Care 2000; 23: 807–812. [DOI] [PubMed] [Google Scholar]
- 12.De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, et al. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 2010; 11: 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spreckley E, Murphy KG. The L-Cell in Nutritional Sensing and the Regulation of Appetite. Front Nutr 2015; 2: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan AG, Blind E, Dunder K, de Graeff PA, Hummer BT, Bourcier T, et al. Pancreatic safety of incretin-based drugs-FDA and EMA assessment. N Engl J Med 2014; 370: 794–797. [DOI] [PubMed] [Google Scholar]
- 15.Divino V, DeKoven M, Hallinan S, Varol N, Wirta SB, Lee WC, et al. Glucagon-like Peptide-1 receptor agonist treatment patterns among type 2 diabetes patients in six European countries. Diabetes Ther 2014; 5: 499–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogan AE, Gaoatswe G, Lynch L, Corrigan MA, Woods C, O’Connell J, et al. Glucagon-like peptide 1 analogue therapy directly modulates innate immune-mediated inflammation in individuals with type 2 diabetes mellitus. Diabetologia 2014; 57: 781–784. [DOI] [PubMed] [Google Scholar]
- 17.Lee YS, Park MS, Choung JS, Kim SS, Oh HH, Choi CS, et al. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia 2012; 55: 2456–2468. [DOI] [PubMed] [Google Scholar]
- 18.Yanay O, Bailey AL, Kernan K, Zimmerman JJ, Osborne WR. Effects of exendin-4, a glucagon like peptide-1 receptor agonist, on neutrophil count and inflammatory cytokines in a rat model of endotoxemia. J Inflamm Res 2015; 8: 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Parlevliet ET, Geerling JJ, van der Tuin SJ, Zhang H, Bieghs V, et al. Exendin-4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. Br J Pharmacol 2014; 171: 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gou S, Zhu T, Wang W, Xiao M, Wang XC, Chen ZH. Glucagon like peptide-1 attenuates bleomycin-induced pulmonary fibrosis, involving the inactivation of NF-kappaB in mice. Int Immunopharmacol 2014; 22: 498–504. [DOI] [PubMed] [Google Scholar]
- 21.Zhu T, Wu XL, Zhang W, Xiao M. Glucagon Like Peptide-1 (GLP-1) Modulates OVA-Induced Airway Inflammation and Mucus Secretion Involving a Protein Kinase A (PKA)-Dependent Nuclear Factor-kappaB (NF-kappaB) Signaling Pathway in Mice. Int J Mol Sci 2015; 16: 20195–20211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol 2011; 186: 4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Hollaren MT, Yunginger JW, Offord KP, Somers MJ, O’Connell EJ, Ballard DJ, et al. Exposure to an aeroallergen as a possible precipitating factor in respiratory arrest in young patients with asthma. N Engl J Med 1991; 324: 359–63. [DOI] [PubMed] [Google Scholar]
- 24.Hardman CS, Panova V, McKenzie ANJ. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. European Journal of Immunology 2013; 43: 488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy IA, Pino JA, Weikop P, Osses N, Sorensen G, Bering T, et al. Glucagon-like peptide 1 receptor activation regulates cocaine actions and dopamine homeostasis in the lateral septum by decreasing arachidonic acid levels. Transl Psychiatry 2016; 6: e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snelgrove RJ, Gregory LG, Peiró T, Akthar S, Campbell GA, Walker SA, et al. Alternaria-derived serine protease activity drives IL-33–mediated asthma exacerbations. Journal of Allergy and Clinical Immunology 2014; 134: 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bankova LG, Lai J, Yoshimoto E, Boyce JA, Austen KF, Kanaoka Y, et al. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc Natl Acad Sci U S A 2016; 113:6242–6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toki S, Goleniewska K, Reiss S, Zhou W, Newcomb DC, Bloodworth MH, et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax 2016; 71: 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression ofinterleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest 1999; 103: 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrant M, Loucoubar C, Boufkhed S, Bassene H, Sarr FD, Baril L, et al. Risk factors associated with asthma, atopic dermatitis and rhinoconjunctivitis in a rural Senegalese cohort. Allergy Asthma Clin Immunol 2015; 11: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pyke C, Heller RS, Kirk RK, Ørskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 Receptor Localization in Monkey and Human Tissue: Novel Distribution Revealed With Extensively Validated Monoclonal Antibody. Endocrinology 2014; 155: 1280–1290. [DOI] [PubMed] [Google Scholar]
- 32.Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of Monocyte Adhesion to Endothelial Cells and Attenuation of Atherosclerotic Lesion by a Glucagon-like Peptide-1 Receptor Agonist, Exendin-4. Diabetes 2010; 59: 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parthsarathy V, Hölscher C. The type 2 diabetes drug liraglutide reduces chronic inflammation induced by irradiation in the mouse brain. European Journal of Pharmacology 2013; 700: 42–50. [DOI] [PubMed] [Google Scholar]
- 34.Iwai T, Ito S, Tanimitsu K, Udagawa S, Oka J-I. Glucagon-like peptide-1 inhibits LPS-induced IL-1β production in cultured rat astrocytes. Neuroscience Research 2006; 55: 352–360. [DOI] [PubMed] [Google Scholar]
- 35.Viby N-E, Isidor MS, Buggeskov KB, Poulsen SS, Hansen JB, Kissow H. Glucagon-Like Peptide-1 (GLP-1) Reduces Mortality and Improves Lung Function in a Model of Experimental Obstructive Lung Disease in Female Mice. Endocrinology 2013; 154: 4503–4511. [DOI] [PubMed] [Google Scholar]
- 36.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007; 87: 1409–1439. [DOI] [PubMed] [Google Scholar]
- 37.Billington CK, Ojo OO, Penn RB, Ito S. cAMP regulation of airway smooth muscle function. Pulmonary Pharmacology & Therapeutics 2013; 26: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou W, Blackwell TS, Goleniewska K, O’Neal JF, Fitzgerald GA, Lucitt M, et al. Prostaglandin I2 analogs inhibit Th1 and Th2 effector cytokine production by CD4 T cells. J Leukoc Biol 2007; 81: 809–817. [DOI] [PubMed] [Google Scholar]
- 39.Zhou W, Hashimoto K, Goleniewska K, O’Neal JF, Ji S, Blackwell TS, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol 2007; 178: 702–710. [DOI] [PubMed] [Google Scholar]
- 40.Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, et al. Prostaglandin I2 Signaling and Inhibition of Group 2 Innate Lymphoid Cell Responses. Am J Respir Crit Care Med 2016; 193: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of Monocyte Adhesion to Endothelial Cells and Attenuation of Atherosclerotic Lesion by a Glucagon-like Peptide-1 Receptor Agonist, Exendin-4. Diabetes 2010; 59: 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hung LY, Lewkowich IP, Dawson LA, Downey J, Yang Y, Smith DE, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci U S A 2013; 110: 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurowska-Stolarska M, Hueber A, Stolarski B, McInnes IB. Interleukin-33: a novel mediator with a role in distinct disease pathologies. Journal of Internal Medicine 2011; 269: 29–35. [DOI] [PubMed] [Google Scholar]
- 44.Cayrol C, Girard J-P. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Current Opinion in Immunology 2014; 31: 31–37. [DOI] [PubMed] [Google Scholar]
- 45.Shiraki A, Oyama J, Komoda H, Asaka M, Komatsu A, Sakuma M, Kodama K et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-alpha-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis 2012; 221: 375–382. [DOI] [PubMed] [Google Scholar]
- 46.Mohapatra A, Dyken SJV, Schneider C, Nussbaum JC, Liang H-E, Locksley RM. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol 2016; 1: 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurokawa M, Matsukura S, Kawaguchi M, Ieki K, Suzuki S, Odaka M, et al. Expression and Effects of IL-33 and ST2 in Allergic Bronchial Asthma: IL-33 Induces Eotaxin Production in Lung Fibroblasts. International Archives of Allergy and Immunology 2011; 155: 12–20. [DOI] [PubMed] [Google Scholar]
- 48.Besnard A-G, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. European Journal of Immunology 2011; 41: 1675–1686. [DOI] [PubMed] [Google Scholar]
- 49.Hogan AE, Tobin AM, Ahern T, Corrigan MA, Gaoatswe G, Jackson R. et al. Glucagon-like peptide-1 (GLP-1) and the regulation of human invariant natural killer T cells: lessons from obesity, diabetes and psoriasis. Diabetologia 2011; 54: 2745–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.