

Keywords: aging, airway remodeling, asthma, fibrosis, senescence
Abstract
Senescent cells can drive age-related tissue dysfunction partially via a senescence-associated secretory phenotype (SASP) involving proinflammatory and profibrotic factors. Cellular senescence has been associated with a structural and functional decline during normal lung aging and age-related diseases such as chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Asthma in the elderly (AIE) represents a major healthcare burden. AIE is associated with bronchial airway hyperresponsiveness and remodeling, which involves increased cell proliferation and higher rates of fibrosis, and resistant to standard therapy. Airway smooth muscle (ASM) cells play a major role in asthma such as remodeling via modulation of inflammation and the extracellular matrix (ECM) environment. Whether senescent ASM cells accumulate in AIE and contribute to airway structural or functional changes is unknown. Lung tissues from elderly persons with asthma showed greater airway fibrosis compared with age-matched elderly persons with nonasthma and young age controls. Lung tissue or isolated ASM cells from elderly persons with asthma showed increased expression of multiple senescent markers including phospho-p53, p21, telomere-associated foci (TAF), as well as multiple SASP components. Senescence and SASP components were also increased with aging per se. These data highlight the presence of cellular senescence in AIE that may contribute to airway remodeling.
INTRODUCTION
Lung function gradually declines with aging and often plays an unappreciated role in healthy aging, as a decreased pulmonary reserve is a risk factor for morbidity and mortality (1). Aging is a risk factor for the development of chronic lung diseases such as obstructive pulmonary disease (COPD) (2, 3), and idiopathic pulmonary fibrosis (IPF) (4). There is now increasing recognition that asthma in the elderly (AIE) is also a major health issue with ∼7% prevalence, higher rates of airway hyperresponsiveness, severe presentation, and resistant to standard therapy. Thus, the mechanisms that contribute to AIE become relevant but are not well understood.
Cellular senescence is a well-recognized feature of organismal aging. It is characterized by permanent cell cycle arrest associated with high expression of proinflammatory cytokines, chemokines, and extracellular matrix (ECM)-degrading proteins, collectively known as senescence-associated secretory phenotype (SASP). Although senescence serves important roles in regulating wound repair and suppressing oncogenesis (5), it is now established that senescent cells can also have detrimental influences on the pathogenesis of several age-related diseases. Senescent cells can influence naïve neighboring cells or even have remote effects by secreting SASP (6).
Senescent cells are thought to play an important role in aging and age-related disease by impairing tissue regeneration and inducing chronic inflammation via SASP. Senescent cells have been shown to accumulate during aging in the lung and are associated with diseases such as COPD (2, 3), IPF (4), and bronchiectasis (7). Conversely, clearance of senescent cells has been shown to improve lung compliance, structure, and elasticity in middle-aged mice (4) as well as to improve phenotypes in fibrotic lung disease (4), suggesting that targeting senescent cells may be a strategy to counteract age-related lung pathophysiology (8). Whether senescence plays a role in asthma per se or AIE is not clear.
Beyond senile emphysema, imaging studies show aging bronchial airways are also thicker and fibrotic (9–13) highlighting the idea that bronchial remodeling may be important in both normal aging and AIE, a distinct entity with its own diagnostic and therapeutic challenges (14). Accordingly, a key question becomes what are the cell types in airways that promote remodeling with aging and contribute to AIE? Although multiple cell types within bronchial airway play significant roles in airway hyperreactivity and remodeling, airway smooth muscle (ASM) cells play an integral role in this regard. Particularly, besides changes in the epithelial layer, remodeling can involve ASM hypertrophy (15, 16) and hyperplasia (15–17), and a thickened, fibrotic subepithelial basement membrane (14, 18). Patients with asthma show greater remodeling with changes in collagen subtype deposition, adhesion proteins such as fibronectin and tenascin, and other related ECM proteins (18–28). Importantly, increased ASM proliferation and inflammatory cytokine production are also characteristic of asthma (15, 17). These make ASM potentially significant in aging airways and in AIE. Whether senescent ASM cells accumulate in AIE or contribute to structural and functional changes in this condition is unknown. We tested this hypothesis by investigating the extent of cellular senescence within the ASM layer of bronchial airways and isolated ASM cells from young persons with nonasthma (<45 yr old), elderly persons with nonasthma (>65 yr old), and elderly patients with asthma (>65 yr old).
MATERIALS AND METHODS
Human Lung Tissue Specimens
Lung and airway specimens were obtained from patients undergoing thoracic surgery at Mayo Clinic under an IRB-approved protocol and with patient written informed consent obtained during presurgery evaluation. Patients undergoing surgery for pneumonectomies, lobectomies, or noninfectious diseases were included, whereas disseminated cancers or infectious causes were excluded. Normal areas distant from (typically) tumors were identified by the surgical pathologist. Patient medical data were collected, but patient identifiers were not stored (unique numbers assigned to samples). Table 1 lists patient details on samples included in this study.
Table 1.
All patient data
| Young |
Old |
Old Asthma |
||||
|---|---|---|---|---|---|---|
| Lung Tissue | ASM Cells | Lung Tissue | ASM Cells | Lung Tissue | ASM Cells | |
| Age, yr | 33.7 ± 2.0 | 31.0 ± 2.3 | 72.4 ± 0.9 | 71.7 ± 1.2 | 71.3 ± 1.2 | 70.6 ± 1.9 |
| Females, n | 7 | 8 | 8 | 5 | 10 | 5 |
| Males, n | 5 | 4 | 4 | 10 | 2 | 5 |
Values are means ± SE or n. ASM, airway smooth muscle.
ASM Cell Isolation
Techniques for isolating human ASM cells have been described previously (29–32). Briefly, the epithelial layer was separated from bronchial airways, the ASM layer dissected, and ASM cells enzymatically dissociated. Cell pellets were resuspended in DMEM/F12 medium containing 10% FBS (R&D Systems, Minneapolis, MN), 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, and 250 ng·mL−1 amphotericin B (Gibco), and cultured under standard conditions. Experiments were limited to cells from two subcultures.
IHC Staining for Collagen and Fibronectin Deposition
Formalin-fixed paraffin-embedded (FFPE) human lung tissues containing airways of different sizes were cut at 6 µm, mounted on glass slides, and processed for immunohistochemistry (IHC) staining [hematoxylin-eosin (H&E) staining for initial evaluation] following a standard deparaffinization, rehydration, and antigen retrieval procedure.
Masson’s trichome (MT) staining was performed following standard protocol. Briefly, tissue sections were fixed using Bouin’s solution at 56°C for an hour, followed by washing in running tap water for 1 min. Next, tissue sections were stained in Weigert’s iron hematoxylin working solution for 10 min followed by washing in running tap water for 5 min and then Biebrich scarlet-acid fuchsin solution for 2 min. Tissue sections were washed using distilled water and incubated for 10 min in a phosphomolybdic-phosphotungstic acid solution. Subsequently, tissue sections were transferred into aniline blue for 20 min, rinsed with distilled water, and placed in 1% acetic acid solution for 4 min. Next, tissue sections were washed and dehydrated in 95% and 100% ethanol (2 times each) and two times xylene for 5 min each. Finally, tissue sections were mounted using Cytoseal 60 mounting media (Thermo Fisher Scientific; Waltham, MA) and covered.
For IHC, primary antibodies used were mouse monoclonal anti-CDKN2A/p16INK4a (ab54210; Abcam, Waltham, MA; 1:300) (29); rabbit polyclonal anti-fibronectin (ab2413; Abcam; 1:150), and mouse monoclonal anti-smooth muscle actin (M0851; Agilent Dako, Santa Clare, CA; 1:500). Isotype control antibodies were anti-mouse IgG2b (401202, Biolegend, San Diego, CA) for p16 and anti-rabbit IgG (7074; Cell Signaling, Danvers, MA) for fibronectin. Standard overnight primary antibody incubation was done at 4°C. An ImmPRESS Duet Double Staining Polymer Kit was used (Vector Laboratories, Burlingame, CA). Horseradish peroxidase and anti-mouse IgG-brown and alkaline phosphatase enzyme polymers conjugated to anti-rabbit IgG-magenta were used.
Stained tissue sections were digitally scanned. Borders of bronchial airways were identified (29) and quantified using Orbit Image Analysis software (Idorsia Pharmaceuticals Ltd.; Allschwil, Switzerland) for collagen and fibronectin analysis following protocols established by the software manufacturer (33, 34). Briefly, tissue sections were differentiated using a pixel-based classification model to differentiate tissue from the background (exclusion) and quantify the amount of collagen or fibronectin staining (inclusion). Subsequently, the model was trained and used to categorize stained tissue. The region of interest [ROI; bronchial airways (29–32)] was measured, and the result of the ratio was multiplied by 100. All airways within a section were quantified regardless of shape or size to minimize bias.
ASM mass area was measured (35–38) using Aperio ImageScope (12.4.3; Leica Biosystems, Deer Park, IL) following protocols available within the software. Briefly, ASM mass area (µm2) of each airway was measured and divided by the total area of the airway (µm2); the result of the ratio was then multiple by 100. All airways within the tissue section for each patient were measured regardless of shape or size to minimize bias.
Quantitative Pathology and Bioimage Analysis software (QuPath; Center for Cancer Research and Cell Biology, Queen’s University Belfast) was used for p16INK4a quantification according to software specifications; see Supplemental Material for script codes (1a and 1b; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.21215183) (39). ASM versus epithelial cells were identified by morphology and relative location within the airway as previously described (29–32). All bronchial airways were quantified regardless of shape or size to minimize bias.
Telomere-Associated DDR Foci Staining
Telomere-associated DNA damage response (DDR) foci (TAF) staining techniques were previously developed by our group to identify and quantify senescent cells in aging and senescence, for use in vitro and in vivo (40). Briefly, TAF staining involves immunostaining for γH2A.X (marker of DNA damage) followed by in situ hybridization of telomere-specific markers that provide a more specific indication of cellular senescence specific to DNA damage pathways. Lung tissue sections were stained and quantified as previously described (7, 41). Briefly, FFPE tissue sections were deparaffinized, rehydrated, and followed by antigen retrieval. Tissue sections were blocked using normal goat serum (1:60 prepared in BSA/PBS) for 30 min and incubated overnight at 4°C with rabbit anti-γH2A.X (mAb 9718, Cell Signaling; 1:200). A biotinylated secondary antibody (BA-1000-1.5, Vector Laboratories; 1:200) was then incubated for 45 min and followed by a tertiary Cy5 streptavidin for 20 min (SA-1500-1, Vector Laboratories; 1:500 in PBS). Sections were washed and cross-linked using 4% paraformaldehyde in PBS and dehydrated in a series of cold ethanol solutions. Hybridization solution (10 µL) containing 70% deionized formamide (Sigma), 25 mM MgCl2, 1 M Tris pH 7.2, 5% blocking reagent (Roche), 25 µg/mL Cy-3-labeled telomere-specific (C-rich probe repeats CCCTAA; F1002 PNAprobe; Panagene) was added and placed in the oven at 80°C for 10 min. Tissue sections were washed in a series of 70% formamide in 2× saline-sodium citrate (SSC) for 10 min, 2× SSC for 10 min, and PBS for 10 min. Tissue sections were mounted using Prolong mounting media with DAPI to counterstain cell nuclei (Thermo Fisher Scientific). Slides were allowed for 2 days to dry and then imaged using Nikon confocal microscope with optical sectioning and Z stacking (minimum of 30 optical slices using a ×100 lens). FIJI-ImageJ (42, 43) was used for TAF quantifications within ASM layer of bronchial airways; see Supplemental Materials (1c).
Western Blot Analysis
Lung tissues were homogenized using 1× cell lysis buffer (Cell Signaling) supplemented with a pierce protease inhibitor tablet (Thermo Fisher). Protein concentrations were measured using a DC protein assay kit (Bio-Rad), loaded at 25 µg per sample into Criterion TGX Precast Gels. Gels were transferred onto 0.2-µm nitrocellulose membrane (Trans-Blot TurboTM). Intercept Blocking Buffer (LI-COR Biosciences, Lincoln, NE) was used. Primary antibodies were: rabbit polyclonal anti-phospho-p53 (Ser15) (53 kDa, 9284; Cell Signaling), rabbit monoclonal anti-p21 (21 kDa, ab109520; Abcam), and rabbit monoclonal anti-GAPDH (37 kDa, 2118; Cell Signaling). Primary antibodies were diluted according to company recommendations. Secondary antibody IRDaye 800CW goat anti-rabbit (926–32211; LI-COR; 1:10,000) was used, and membranes were imaged and bands quantified using a LI-COR Odyssey XL system. All biological replicates were run on the same gel.
Quantitative Real-Time PCR
Quantitative real-time PCR (qRT-PCR) was performed using standard methods (29). Briefly, RNA was extracted using a Qiagen kit, and concentrations were measured using a Thermo Scientific NanoDrop. qRT-PCR was completed in duplicate for each sample with normalization to S16 (LightCycler 96, Roche Diagnostics Corporation, Indianapolis, IN). All primers were obtained from IDT (Newark, NJ) or Qiagen. Fold changes were calculated using ΔΔCT method (2−ΔΔCT). For homogenized lung tissue, young, old, and old persons with asthma were normalized to young age groups. For ASM cell culture, young, old, and old persons with asthma on day 7 were normalized to the young age groups on day 0.
Senescence-Associated β-Galactosidase Activity
ASM cells from different groups were cultured in 24-well plates at a final seeding density of 2 × 104 mL−1. On day 3, cells were washed with 1× Dulbecco’s phosphate-buffered saline (DPBS; Gibco), followed by senescence-associated β-galactosidase activity (SA-βGal) staining according to the manufacturer’s protocol (Cell Signaling), and counterstained with DAPI (Thermo Fisher Scientific). A Cytation 5 imaging system (BioTek Instruments, Winooski, VT) was used to count the number of SA-βGal positive cells, normalized to total cell count.
ELISA and Gene Expression
On day 7 of ASM cell culture, the supernatant was collected to assess SASP profile (IL-6 and IL-8) using ELISA kits (R&D Systems); and ASM cells were then processed for qRT-PCR.
Statistical Analysis
Three groups including both females and males in each group were used: young (<45 yr old), old (>65 yr old), and old persons with asthma (>65 yr old); see Table 1. Experiments were performed using a set of 12 patient samples, although not every patient sample was used for all protocols. Details regarding the patient samples used for specific protocols are provided in the Supplemental Tables S1–S5. Data analysis was performed using GraphPad Prism (8.4.3) with one-way ANOVA with Holm–Sidak correction for multiple comparisons. For a nonparametric test, Kruskal–Wallis analysis for multiple comparisons was performed. Robust regression and outlier removal method (ROUT) was used to identify and remove outliers (44). Values are expressed as means ± SE; P values <0.05 were considered significantly different.
RESULTS
Airway Remodeling Is Increased in Aging Lungs and Exacerbated in AIE
Bronchial collagen deposition was quantified in MT-stained lung sections using Orbit Image Analysis. Greater percentages of bronchial collagen were detected in elderly persons with nonasthma and elderly persons with asthma compared with young age group; P < 0.05 and P < 0.01, respectively (Fig. 1A).
Figure 1.

Airway remodeling in aging and asthma in the elderly (AIE). A: representative images of Masson’s trichrome staining of bronchial airway sections from young, elderly persons with nonasthma, and elderly patients with asthma (see methods and Table 1 for details). More abundant collagen (yellow arrow) was seen within bronchial airways particularly in the airway smooth muscle (ASM) layer (black arrow) of elderly persons with nonasthma and further increased in elderly patients with asthma compared with young (n = 5 patients/group). B: representative images of chromogenic double staining for fibronectin (magenta) and smooth muscle actin (dark brown) with cell nuclei counterstained with hematoxylin (blue). Higher fibronectin (yellow arrow) was seen within the ASM layer (black arrow) of elderly patients with asthma (n = 4–6 patients/group). C: greater ASM mass (brown) was seen in airways of elderly person with nonasthma and asthma (n = 4–6 patients/group). Scale bar = 60 µm. *P < 0.05; **P < 0.01. Means ± SE.
Compared with young, a higher percentage of bronchial fibronectin was detected in elderly persons with asthma (P < 0.05), although there was no statistical difference between elderly persons with nonasthma versus young (Fig. 1B). ASM mass area within bronchial airways was also increased in elderly persons with asthma and nonasthma compared with the young age group (Fig. 1C).
Senescent Cell Markers Are Increased in Aging Lungs and Exacerbated in AIE
Senescence markers were selected on their broad expression in senescent cells in different organs or having been identified in other lung diseases such as COPD and IPF (45). Although there is no gold standard marker for cellular senescence, two tumor suppressors are often used since they represent interacting but independent signaling pathways: p53-p21CIP1 and p16Ink4a-Rb (45). Downstream, these pathways promote permanent cell cycle arrest at the G1/S transitional phase (45).
In whole lung tissue, Western blot analysis of tumor suppressor protein phospho-p53 and cyclin-dependent kinase inhibitor p21CIP1 showed higher p53 phosphorylation in lungs of elderly persons with asthma compared with elderly persons with nonasthma or young age group (P < 0.01 and P < 0.01, respectively; Fig. 2A). p21CIP1 was also higher in elderly persons with asthma compared with old persons with nonasthma and young age groups (P < 0.05 and P < 0.05, respectively; Fig. 2A). qRT-PCR analyses of mRNA for senescence-associated markers revealed higher expression in lungs of elderly persons with asthma for p16 and p21, compared with elderly persons with nonasthma or younger age group (P < 0.05 and P < 0.05 respectively; Fig. 2B). Interestingly, no substantial change in p53 mRNA expression was observed in elderly persons with asthma compared with elderly persons with nonasthma or young (Fig. 2B).
Figure 2.

Senescent cell burden in bronchial airways with normal aging and asthma in the elderly (AIE). A: Western blot analysis of lungs of elderly patients with asthma show higher phospho-p53 (53 kDa) and p21CIP1 (21 kDa) protein expression compared with persons with nonasthma age-matched and young age groups (n = 9–12 patients/group), each patient was normalized to its loading control, GAPDG (37 kDa). All biological replicates (per group, young, old, and old asthma) were run on the same gel. B: qRT-PCR analysis showed significantly higher expression of senescent markers p21 and p16 in lungs of elderly person with asthma compared with young age groups. C: similarly, higher levels of SASP-related gene expression (PAI-1, TNFα, MMP1, and CCL2) were observed in lungs of elderly persons with asthma compared with nonasthma age-matched and young (n = 4–10 patients/group). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Means ± SE.
The senescence-associated secretory proteins (SASP) profile is cell- and context-specific, but there are common elements (45) including inflammatory mediators such as TNFα, IL-6 and IL-8, ECM proteins, and modulators such as plasminogen activator inhibitor-1 (PAI-1) and matrix metalloproteinases (MMPs) (45). Analysis of SASP-related genes in homogenized lung tissues shows substantial changes in PAI-1, MMP1, and CCL2 expression in elderly persons with asthma compared with elderly persons with nonasthma and young age group while TNF-α was significantly higher in elderly persons with nonasthma and asthma compared with young (Fig. 2C).
ASM Cells Show Increased Senescence with Aging, Exacerbated in AIE
Based on the data that senescence-associated markers increase in the whole lung with aging, especially in elderly persons with asthma, we sought to investigate if ASM cells are a contributing cell type, given their role in airway hyperreactivity and remodeling. IHC chromogenic staining of senescence-associated marker p16INK4A showed a higher number of p16INK4A positive ASM nuclei in elderly persons with nonasthma (P < 0.05) and patients with asthma (P < 0.001) compared with young (Fig. 3A).
Figure 3.
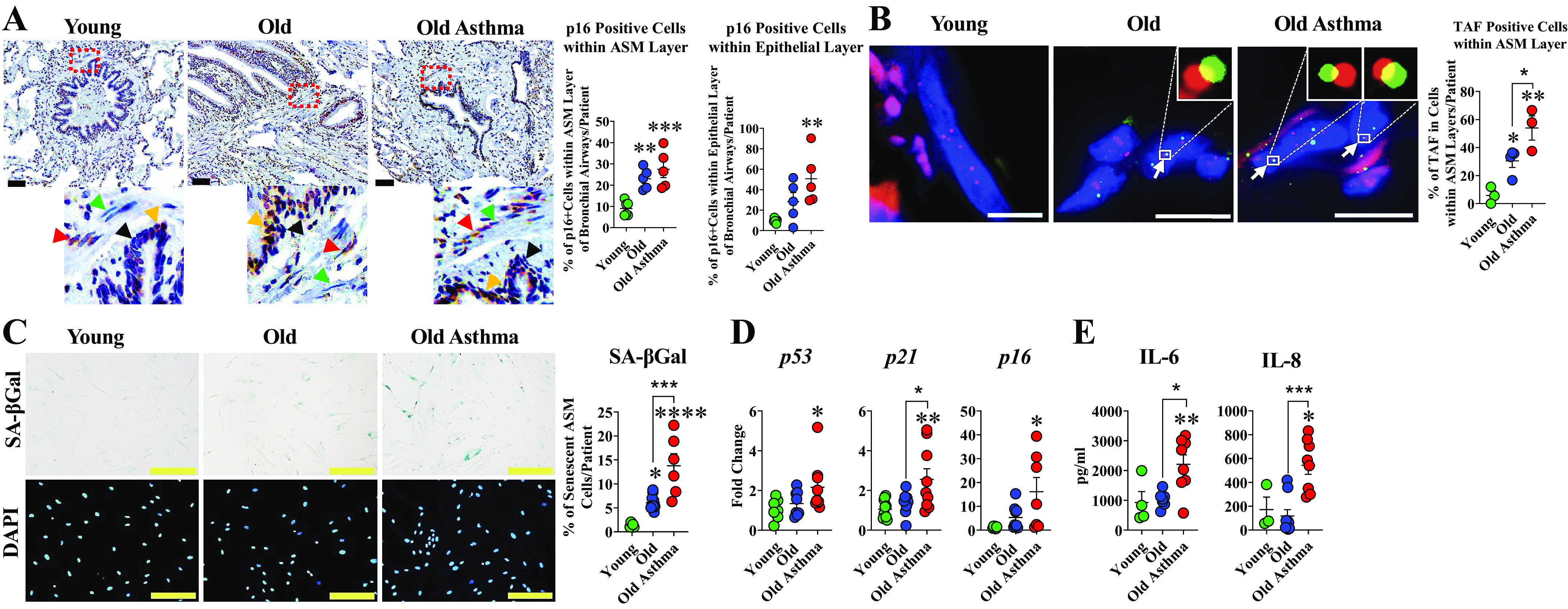
Senescence in airway smooth muscle (ASM) cells in aging and asthma in the elderly (AIE). A: representative images of chromogenic staining for p16INK4A (dark brown) with cell nuclei counterstained with hematoxylin (blue). Red and orange arrows show p16INK4A positive cells while green and black arrows show p16INK4A negative cells within ASM and epithelial layers, respectively (n = 5 or 6 patients/group). Scale bar = 60 µm. B: representative images of quantitative fluorescence in situ hybridization (FISH) combined with immunofluorescence staining for γH2AX (red) and telomeres (green); cell nuclei counterstained with DAPI (blue). Accumulation of cell nuclei containing telomere-associated foci (TAF) within ASM layer was significantly higher in elderly asthmatic bronchial airway compared with elderly patients with nonasthma and young. White arrows point to TAF of colocalization at a single plane of Z-stack at higher magnification (top right, elderly and elderly asthma) (n = 3 or 4 patients/group). C: SA–βGal staining was increased in ASM cells from aging airways of elderly persons with asthma and elderly persons with nonasthma compared with young (n = 5–10 patients/group); Scale bar = 200 µm. D: qRT-PCR analysis showed significant changes in senescence-associated genes p53, p21, and p16 in ASM cells of elderly persons with asthma compared with young (n = 6–10 patients/group). E: ELISA of supernatants from cultured ASM cells of elderly persons with asthma showed increased IL-6 and IL-8 (n = 3–9). (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Means ± SE.
Telomere shortening and DNA damage response (DDR) are hallmarks of cellular senescence (40). Fluorescence in situ hybridization (FISH) of telomerase combined with immunofluorescence staining of γ-H2AX allows for localization of telomere-associated foci (TAF), and has been used to identify senescent cells (3, 45): a technique developed by the Passos group (40). Critically short telomeres induce senescence via the activation of a DNA damage response (DDR), and telomere/DNA damage is irreparable and leads to a persistent DDR during cellular senescence (40). We, therefore, investigated telomere dysfunction in ASM cells using Immuno-FISH to quantify colocalization between DDR proteins γH2A.X and telomeres, i.e., TAF. Increased ASM nuclei containing TAF were observed in elderly persons with asthma compared with elderly persons with nonasthma and young (P < 0.05 and P < 0.01, respectively; Fig. 3B).
ASM cells isolated from elderly patients with asthma also showed elevated levels of senescence markers. Senescence-associated β-galactosidase (SA-βgal) is widely utilized as a marker to distinguish senescent cells in vitro and in vivo (45, 46). The blue chromagenic assay relies on lysosomal enzyme activity at a pH of 6.0 present in senescent cells (46). We observed a greater percentage of SA-βGal positive ASM cells in elderly persons with asthma compared with elderly persons with nonasthma or young (P < 0.001 and P < 0.0001, respectively; Fig. 3C). qRT-PCR analysis of senescence-associated genes revealed higher expression for p53 and p21 in ASM cells of elderly persons with asthma compared with elderly persons with nonasthma cells or young (Fig. 3D). Higher expression of p16 was also observed in ASM of elderly persons with asthma (P < 0.001) compared with nonasthma or young (Fig. 3D).
ELISA analysis of the supernatants from ASM cell culture showed significantly higher levels of IL-6 in elderly persons with asthma compared with nonasthma or young (Fig. 3E). A similar pattern for IL-8 was observed in elderly persons with asthma compared with elderly persons with nonasthma or young (P < 0.01; Fig. 3E).
A Pearson correlation was performed to explore the strength and direction of association between senescence markers or ECM deposition. Interestingly, although we found no correlation between p16INK4A positive cells within ASM layer and collagen deposition, we found a significant correlation between the percentage of fibronectin and p16INK4A positive cells within the ASM layer of bronchial airways, suggesting that senescent cells may be drivers of airway fibrosis (Fig. 4).
Figure 4.

Correlation between senescence and extracellular matrix (ECM). The percentage of fibronectin staining within bronchial airways and p16 positive airway smooth muscle (ASM) cells was higher in elderly patients with asthma. Pearson’s correlation coefficient (r) and P value for <0.05 significance are shown.
DISCUSSION
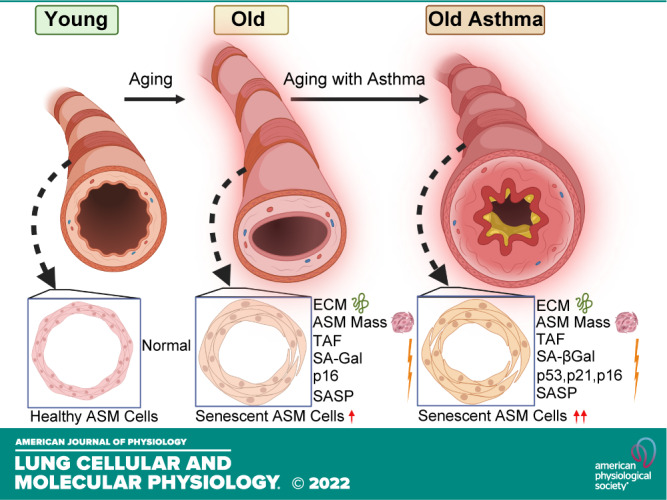
With an increasing elderly population (47–49), understanding aging-associated structural and functional changes in airways and lung parenchyma becomes important toward improving health (50–52) as well as understanding diseases such as asthma (53–56), COPD, and IPF (57–61) that disproportionately affect the elderly. Although aging is usually associated with “senile emphysema” and thus the alveolar compartment, imaging studies show that aging bronchial airways are also thicker and more fibrotic (9–13), highlighting the idea that bronchial remodeling may be important in both normal aging and AIE. Accordingly, key questions in the field become what are the cell types in airways that promote remodeling with aging and contribute to AIE, and what are the mechanisms at play? In this study, we report novel data on cellular senescence and remodeling changes that occur with AIE by comparing three groups, young (<45 yr of age), elderly persons with nonasthma, and elderly persons with asthma (>65 yr of age), toward demonstrating clinical significance of our studies. Our results indicate that aging results in increased expression of senescence-associated markers and SASP components in ASM, with greater ECM that could contribute to the thicker and stiffer airways found in the aging lung. Importantly, we find that AIE involves exacerbated senescence, SASP, and ECM, suggesting that cellular senescence may play a role in the enhanced fibrosis of the aging asthmatic airway (Fig. 5).
Figure 5.

Schematic summarizing increased senescent-associated markers and extracellular matrix (ECM) in aging airways, exacerbated in asthma in the elderly (AIE). ASM, airway smooth muscle; SA-Gal, senescence-associated β-galactosidase; SASP, senescence-associated secretory phenotype; TAF, telomere-associated foci. [Image created with BioRender.com and published with permission.]
Although multiple cell types usually contribute to structural and functional changes in the bronchial airway that occur in asthma, ASM cells are recognized to be important given their role in contractility as well as in remodeling via cell proliferation and fibrosis as occurs in diseases such as asthma (62–64), COPD (65), and even IPF (66). However, there is very little known about ASM cells and ECM deposition with aging or in AIE. Most data investigating aging and ECM deposition demonstrate a role for lung fibroblasts during aging (4, 67–70). We previously showed that aging increases ASM deposition of collagen III and fibronectin, whereas the ECM modifying proteins MMP2 and MMP9 are decreased, suggesting reduced ECM turnover (71). In the present study, we also find aging-associated increases in collagen and fibronectin in the bronchial airways as well within the ASM layer, along with exacerbated increases in AIE, underlining the potential role of ASM cells in the pathogenesis of fibrosis in elderly persons with asthma. Indeed, airway remodeling in elderly patients with asthma involves a thickened, fibrotic subepithelial basement membrane (14, 18) with increased ASM mass. The latter can occur via ASM hypertrophy (15, 16) and hyperplasia (15–17). We previously showed that aging is associated with increased ASM proliferation (71). Whether senescent cells within the aging airway contribute to such proliferation, exacerbated in AIE, is unknown.
Cellular senescence contributes to aging-associated structural and functional changes within organs (1, 72). Senescent cells release SASP that can include proinflammatory and profibrotic elements, including IL-6 and IL-8 that we explored, leading to altered cell proliferation, upregulation of endoplasmic reticulum stress, disruption of the unfolded protein response, mitochondrial dysfunction (73–75), fibrosis (4, 8, 76, 77), and inflammation (4, 8, 29, 77). The presence of senescent cells in the lung or in lung diseases is recognized (78–82). In pulmonary fibrosis, epithelial cells express p21CIP1 and p53 (83) and fibrotic foci show p16INK4A (84). Fibroblasts in pulmonary fibrosis produce SASP that enhances ECM production by naïve fibroblasts (4). In mice, removal of senescent cells reverses bleomycin-induced injury, p21CIP1, and SASP (85). Senescent fibroblasts and epithelial cells (86–88) are also increased in COPD (1, 3, 89). Compared with these findings, senescence in adult asthma has not been well studied, although senescence is suggested by increased p16INK4A, p21CIP1, and SA-βgal-staining in the asthmatic epithelium (90, 91). The contribution of ASM cells in this regard is entirely unknown. Accordingly, our data showing increased senescence within airways and particularly in ASM with aging become significant. ASM cells show an upregulation of proteins associated with activation of senescent pathways such as p21CIP1, p16INK4A, and SA-βgal. This increase in senescence-associated markers expression is accompanied by changes in SASP components such as IL-6 and IL-8. Overall, these novel data highlight the presence of ASM senescence in aging airways and in AIE.
Our data show that senescent ASM cells accumulate in elderly patients with as well as without asthma, as demonstrated within stained lung tissues and in isolated ASM cells. Mechanistically, senescence can involve multiple pathways, and there are cell- and context-specific differences in the contribution of these pathways. We found that the tumor suppressor protein p16INK4A, which regulates the cell cycle and is often used as a senescence-associated marker (92) to be increased in ASM nuclei of aging airways and AIE compared with young age. Quantitative analysis of TAF staining revealed a significant increase in ASM cells from aging and in elderly patients with asthma highlighting the accumulation of senescent cells containing dysfunctional telomeres. This novel finding is consistent with the evidence of increased p53 and p21CIP1 elements in lung and ASM since this pathway plays a substantial role in stress-associated DNA damage response (93–96). The transcriptional factor p16 is also significantly upregulated in elderly patients with asthma, as are several SASP-related genes (TNFα, PAI-1, CCL2, and MMP1). These results suggest that classic senescence pathways including telomere shortening to DNA damage, p53 phosphorylation, p21CIP1 (97, 98), and p16INK4A (98) activation following downstream inhibition of cell cycle progression are involved in AIE. These pathways are also elevated in ASM in this group suggesting a role for this cell type: a functional aspect that remains to be explored. Our results are also consistent with previous reports suggesting increased p21CIP1 and p16INK4A expression in the asthmatic epithelium of adult patients (90, 91).
Our studies demonstrate that cellular senescence occurs in bronchial airways with aging, exacerbated in AIE, and that senescence of ASM cells follows this pattern by age and disease. Furthermore, we show that increased airway fibrosis occurs with aging and AIE. However, our studies do not functionally or mechanistically link increased ASM senescence to the increased airway fibrosis. Nonetheless, previous data and current understanding of senescent cell biology suggest this is likely the case. Senescent cells are now known to exert their detrimental effects by releasing proinflammatory and profibrotic factors, including TNFa, IL-6, and IL-8. Such factors have been previously implicated in ASM remodeling and contractility per se (29, 99). Thus, an increase in these SASP modulators, produced by senescent ASM cells, for example, could lead to increased fibrosis in aging or AIE. We have previously demonstrated that aging is associated with increased ASM proliferation, and thus it is possible that senescent cells are one driver for these changes in the aging airway, with increased effects in AIE. Exploring these causal links will necessitate approaches to removing senescent cells via novel drugs (senolytics) (45) and transgenic mouse models where INK4A positive senescent cells can be selectively eliminated (45): topics for future studies.
Our current study focused on senescence in ASM cells. However, airway remodeling certainly involves other cell types including epithelium and fibroblasts. And in this regard, senescence in these cell types can also be expected. Indeed, epithelial cell senescence has been shown in pulmonary fibrosis (83, 84), as has been fibroblast senescence and their SASP effects (4). Senescent fibroblasts and epithelial cells in COPD have also been shown (1, 3, 89). We also find p16-positive cells within the airway epithelial layer with aging and asthma (Fig. 3A). Thus, it is possible that in aging bronchial airways, paracrine effects of multiple senescent cell types on multiple cell types occur and overall lead to the observed remodeling. Thus, our study is the first to introduce the idea of ASM as one cell type that could play a role in remodeling in the context of aging and asthma. Future studies, potentially involving cell-type-specific transgenic mouse models of senescence can help address the relative roles of different cell types in aging and asthma.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental script codes (1a, 1b, and 1c) and Supplemental Tables S1–S5: https://doi.org/10.6084/m9.figshare.21215183.
GRANTS
This work was supported by NIH Grants T32 HL105355 (A. Aghali), HL088029 and HL158532 (Y. S. Prakash), HL142061 (C. M. Pabelick), 1R01AG068048-01 (to J. F. Passos), and 1UG3CA268103-01 (to J. F. Passos).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.A., J.F.P., and Y.S.P. conceived and designed research; A.A., L.K., A.B.L., and L.Y.D. performed experiments; A.A., L.K., J.J.T., and Y.S.P. analyzed data; A.A., J.F.P., and Y.S.P. interpreted results of experiments; A.A., A.B.L., J.F.P., and Y.S.P. prepared figures; A.A. drafted manuscript; A.A., J.F.P., and Y.S.P. edited and revised manuscript; A.A., C.M.P., J.F.P., and Y.S.P. approved final version of manuscript.
ACKNOWLEDGMENTS
Graphical abstract image created with BioRender.com and published with permission.
REFERENCES
- 1. Parikh P, Wicher S, Khandalavala K, Pabelick CM, Britt RD Jr, Prakash YS. Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol 316: L826–Ll842, 2019. doi: 10.1152/ajplung.00424.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Antony VB, Thannickal VJ. Cellular senescence in chronic obstructive pulmonary disease: multifaceted and multifunctional. Am J Respir Cell Mol Biol 59: 135–136, 2018. doi: 10.1165/rcmb.2018-0061ED. [DOI] [PubMed] [Google Scholar]
- 3. Birch J, Anderson RK, Correia-Melo C, Jurk D, Hewitt G, Marques FM, Green NJ, Moisey E, Birrell MA, Belvisi MG, Black F, Taylor JJ, Fisher AJ, De Soyza A, Passos JF. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 309: L1124–L1137, 2015. doi: 10.1152/ajplung.00293.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hemann MT, Narita M. Oncogenes and senescence: breaking down in the fast lane. Genes Dev 21: 1–5, 2007. doi: 10.1101/gad.1514207. [DOI] [PubMed] [Google Scholar]
- 6. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118, 2010. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Birch J, Victorelli S, Rahmatika D, Anderson RK, Jiwa K, Moisey E, Ward C, Fisher AJ, De Soyza A, Passos JF. Telomere dysfunction and senescence-associated pathways in bronchiectasis. Am J Respir Crit Care Med 193: 929–932, 2016. doi: 10.1164/rccm.201510-2035LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, Kirkland JL. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40: 554–563, 2019. doi: 10.1016/j.ebiom.2018.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niewoehner DE, Kleinerman J. Morphologic basis of pulmonary resistance in the human lung and effects of aging. J Appl Physiol 36: 412–418, 1974. doi: 10.1152/jappl.1974.36.4.412. [DOI] [PubMed] [Google Scholar]
- 10. Janssens JP, Pache JC, Lp N. Physiological changes in respiratory function associated with ageing. Eur Respir J 13: 197–205, 1999. doi: 10.1034/j.1399-3003.1999.13a36.x. [DOI] [PubMed] [Google Scholar]
- 11. Hochhegger B, Souza G, Meirelles P, Irion K, Zanetti G, Garcia E, Moreira J, Marchiori E. The chest and aging: radiological findings. J Bras Pneumol 38: 656–665, 2012. doi: 10.1590/s1806-37132012000500016. [DOI] [PubMed] [Google Scholar]
- 12. Boulet LP, Robitaille C, Deschesnes F, Villeneuve H, Boulay ME. Comparative clinical, physiological, and inflammatory characteristics of elderly subjects with or without asthma and young subjects with asthma. Chest 152: 1203–1213, 2017. doi: 10.1016/j.chest.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 13. Copley SJ. Morphology of the aging lung on computed tomography. J Thorac Imaging 31: 140–150, 2016. doi: 10.1097/RTI.0000000000000211. [DOI] [PubMed] [Google Scholar]
- 14. Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J 17: e85–e93, 2010. doi: 10.1155/2010/318029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson PR. Role of human airway smooth muscle in altered extracellular matrix production in asthma. Clin Exp Pharmacol Physiol 28: 233–236, 2001. doi: 10.1046/j.1440-1681.2001.03426.x. [DOI] [PubMed] [Google Scholar]
- 16. James AL, Elliot JG, Jones RL, Carroll ML, Mauad T, Bai TR, Abramson MJ, McKay KO, Green FH. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med 185: 1058–1064, 2012. doi: 10.1164/rccm.201110-1849OC. [DOI] [PubMed] [Google Scholar]
- 17. Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, Burgess JK, Black JL. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med 164: 474–477, 2001. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- 18. Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet 1: 520–524, 1989. doi: 10.1016/S0140-6736(89)90067-6. [DOI] [PubMed] [Google Scholar]
- 19. Ito JT, Lourenço JD, Righetti RF, Tibério I, Prado CM, Lopes F. Extracellular matrix component remodeling in respiratory diseases: what has been found in clinical and experimental studies? Cells 8: 342–25, 2019. doi: 10.3390/cells8040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mostaço-Guidolin LB, Osei ET, Ullah J, Hajimohammadi S, Fouadi M, Li X, Li V, Shaheen F, Yang CX, Chu F, Cole DJ, Brandsma CA, Heijink IH, Maksym GN, Walker D, Hackett TL. Defective fibrillar collagen organization by fibroblasts contributes to airway remodeling in asthma. Am J Respir Crit Care Med 200: 431–443, 2019. doi: 10.1164/rccm.201810-1855OC. [DOI] [PubMed] [Google Scholar]
- 21. Reeves SR, Kolstad T, Lien TY, Elliott M, Ziegler SF, Wight TN, Debley JS. Asthmatic airway epithelial cells differentially regulate fibroblast expression of extracellular matrix components. J Allergy Clin Immunol 134: 663–670.e1, 2014. doi: 10.1016/j.jaci.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fang CL, Yin LJ, Sharma S, Kierstein S, Wu HF, Eid G, Haczku A, Corrigan CJ, Ying S. Resistin-like molecule-β (RELM-β) targets airways fibroblasts to effect remodelling in asthma: from mouse to man. Clin Exp Allergy 45: 940–952, 2015. doi: 10.1111/cea.12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheng W, Yan K, Xie LY, Chen F, Yu HC, Huang YX, Dang CX. MiR-143-3p controls TGF-β1-induced cell proliferation and extracellular matrix production in airway smooth muscle via negative regulation of the nuclear factor of activated T cells 1. Mol Immunol 78: 133–139, 2016. doi: 10.1016/j.molimm.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 24. Harkness LM, Weckmann M, Kopp M, Becker T, Ashton AW, Burgess JK. Tumstatin regulates the angiogenic and inflammatory potential of airway smooth muscle extracellular matrix. J Cell Mol Med 21: 3288–3297, 2017. doi: 10.1111/jcmm.13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koopmans T, Crutzen S, Menzen MH, Halayko AJ, Hackett TL, Knight DA, Gosens R. Selective targeting of CREB-binding protein/β-catenin inhibits growth of and extracellular matrix remodelling by airway smooth muscle. Br J Pharmacol 173: 3327–3341, 2016. doi: 10.1111/bph.13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kistemaker LEM, Prakash YS. Airway innervation and plasticity in asthma. Physiology (Bethesda) 34: 283–298, 2019. doi: 10.1152/physiol.00050.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Skloot GS, Busse PJ, Braman SS, Kovacs EJ, Dixon AE, Vaz Fragoso CA, Scichilone N, Prakash YS, Pabelick CM, Mathur SK, Hanania NA, Moore WC, Gibson PG, Zieman S, Ragless BB, ATS ad hoc Committee on Asthma in the Elderly. An Official American Thoracic Society Workshop Report: evaluation and management of asthma in the elderly. Ann Am Thorac Soc 13: 2064–2077, 2016. doi: 10.1513/AnnalsATS.201608-658ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chiarella SE, Cardet JC, Prakash YS. Sex, cells, and asthma. Mayo Clin Proc 96: 1955–1969, 2021. doi: 10.1016/j.mayocp.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parikh P, Britt RD Jr, Manlove LJ, Wicher SA, Roesler A, Ravix J, Teske J, Thompson MA, Sieck GC, Kirkland JL, LeBrasseur N, Tschumperlin DJ, Pabelick CM, Prakash YS. Hyperoxia-induced cellular senescence in fetal airway smooth muscle cells. Am J Respir Cell Mol Biol 61: 51–60, 2019. doi: 10.1165/rcmb.2018-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prakash YS, Thompson MA, Pabelick CM. Brain-derived neurotrophic factor in TNF-alpha modulation of Ca2+ in human airway smooth muscle. Am J Respir Cell Mol Biol 41: 603–611, 2009. doi: 10.1165/rcmb.2008-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sathish V, Abcejo AJ, Thompson MA, Sieck GC, Prakash YS, Pabelick CM. Caveolin-1 regulation of store-operated Ca(2+) influx in human airway smooth muscle. Eur Respir J 40: 470–478, 2012. doi: 10.1183/09031936.00090511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vohra PK, Thompson MA, Sathish V, Kiel A, Jerde C, Pabelick CM, Singh BB, Prakash YS. TRPC3 regulates release of brain-derived neurotrophic factor from human airway smooth muscle. Biochim Biophys Acta 1833: 2953–2960, 2013. doi: 10.1016/j.bbamcr.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stritt M, Stalder AK, Vezzali E. Orbit image analysis: an open-source whole slide image analysis tool. PLoS Comput Biol 16: e1007313, 2020. doi: 10.1371/journal.pcbi.1007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fitzgerald S, Wang S, Dai D, Murphree DH Jr, Pandit A, Douglas A, Rizvi A, Kadirvel R, Gilvarry M, McCarthy R, Stritt M, Gounis MJ, Brinjikji W, Kallmes DF, Doyle KM. Orbit image analysis machine learning software can be used for the histological quantification of acute ischemic stroke blood clots. PLoS One 14: e0225841, 2019. doi: 10.1371/journal.pone.0225841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saunders R, Kaul H, Berair R, Gonem S, Singapuri A, Sutcliffe AJ, Chachi L, Biddle MS, Kaur D, Bourne M, Pavord ID, Wardlaw AJ, Siddiqui SH, Kay RA, Brook BS, Smallwood RH, Brightling CE. DP(2) antagonism reduces airway smooth muscle mass in asthma by decreasing eosinophilia and myofibroblast recruitment. Sci Transl Med 11: 1–11, 2019. doi: 10.1126/scitranslmed.aao6451. [DOI] [PubMed] [Google Scholar]
- 36. Khalfaoui L, Symon FA, Couillard S, Hargadon B, Chaudhuri R, Bicknell S, Mansur AH, Shrimanker R, Hinks TSC, Pavord ID, Fowler SJ, Brown V, McGarvey LP, Heaney LG, Austin CD, Howarth PH, Arron JR, Choy DF, Bradding P. Airway remodelling rather than cellular infiltration characterizes both type2 cytokine biomarker-high and -low severe asthma. Allergy 77: 2974–2986, 2022. doi: 10.1111/all.15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Russell RJ, Chachi L, FitzGerald JM, Backer V, Olivenstein R, Titlestad IL, Ulrik CS, Harrison T, Singh D, Chaudhuri R, Leaker B, McGarvey L, Siddiqui S, Wang M, Braddock M, Nordenmark LH, Cohen D, Parikh H, Colice G, Brightling CE; MESOS study investigators. Effect of tralokinumab, an interleukin-13 neutralising monoclonal antibody, on eosinophilic airway inflammation in uncontrolled moderate-to-severe asthma (MESOS): a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Respir Med 6: 499–510, 2018. doi: 10.1016/S2213-2600(18)30201-7. [DOI] [PubMed] [Google Scholar]
- 38. Diver S, Khalfaoui L, Emson C, Wenzel SE, Menzies-Gow A, Wechsler ME, Johnston J, Molfino N, Parnes JR, Megally A, Colice G, Brightling CE. Effect of tezepelumab on airway inflammatory cells, remodelling, and hyperresponsiveness in patients with moderate-to-severe uncontrolled asthma (CASCADE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med 9: 1299–1312, 2021. doi: 10.1016/S2213-2600(21)00226-5. [DOI] [PubMed] [Google Scholar]
- 39. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW. QuPath: open source software for digital pathology image analysis. Sci Rep 7: 16878, 2017. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3: 708–709, 2012. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lagnado A, Leslie J, Ruchaud-Sparagano M-H, Victorelli S, Hirsova P, Ogrodnik M, et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. Embo j 40: 1–19, 2021. doi: 10.15252/embj.2020106048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682, 2012. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Motulsky HJ, Brown RE. Detecting outliers when fitting data with nonlinear regression—a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7: 123–20, 2006. doi: 10.1186/1471-2105-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aghali A, Koloko Ngassie ML, Pabelick CM, Prakash YS. Cellular senescence in aging lungs and diseases. Cells 11: 1781–16, 2022. doi: 10.3390/cells11111781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 4: 1798–1806, 2009. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 47. Vincent G, Velkoff V. The Next Four Decades: The Older Population in the United States: 2010 to 2050, edited by U.S. Dept. of Commerce EaSA. Washington, DC: U.S. Census Bureau, 2010. [Google Scholar]
- 48. Gordon EH, Peel NM, Samanta M, Theou O, Howlett SE, Hubbard RE. Sex differences in frailty: a systematic review and meta-analysis. Exp Gerontol 89: 30–40, 2017. doi: 10.1016/j.exger.2016.12.021. [DOI] [PubMed] [Google Scholar]
- 49. Hadley EC, Kuchel GA, Newman AB; Workshop Speakers and Participants. Report: NIA workshop on measures of physiologic resiliencies in human aging. J Gerontol A Biol Sci Med Sci 72: 980–990, 2017. [Erratum in J Gerontol A Biol Sci Med Sci 73: 995, 2018]. doi: 10.1093/gerona/glx015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Skloot GS. The effects of aging on lung structure and function. Clin Geriatr Med 33: 447–457, 2017. doi: 10.1016/j.cger.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 51. Zeleznik J. Normative aging of the respiratory system. Clin Geriatr Med 19: 1–18, 2003. doi: 10.1016/S0749-0690(02)00063-0. [DOI] [PubMed] [Google Scholar]
- 52. Meiners S, Eickelberg O, Konigshoff M. Hallmarks of the ageing lung. Eur Respir J 45: 807–827, 2015. doi: 10.1183/09031936.00186914. [DOI] [PubMed] [Google Scholar]
- 53. Ventura MT, Scichilone N, Paganelli R, Minciullo PL, Patella V, Bonini M, Passalacqua G, Lombardi C, Simioni L, Ridolo E, Del Giacco SR, Gangemi S, Canonica GW. Allergic diseases in the elderly: biological characteristics and main immunological and non-immunological mechanisms. Clin Mol Allergy 15: 2, 2017. doi: 10.1186/s12948-017-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scichilone N. Comorbidities of lung disease in the elderly. Clin Geriatr Med 33: 597–603, 2017. doi: 10.1016/j.cger.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 55. Braman SS. Asthma in the elderly. Clin Geriatr Med 33: 523–537, 2017. doi: 10.1016/j.cger.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 56. Scichilone N, Pedone C, Battaglia S, Sorino C, Bellia V. Diagnosis and management of asthma in the elderly. Eur J Intern Med 25: 336–342, 2014. doi: 10.1016/j.ejim.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 57. Rangarajan S, Bernard K, Thannickal VJ. Mitochondrial dysfunction in pulmonary fibrosis. Ann Am Thorac Soc 14: S383–S388, 2017. doi: 10.1513/AnnalsATS.201705-370AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Patterson KC, Shah RJ, Porteous MK, Christie JD, D'Errico CA, Chadwick M, Triano MJ, Deshpande C, Rossman MD, Litzky LA, Kreider M, Miller WT Jr.. Interstitial lung disease in the elderly. Chest 151: 838–844, 2017. doi: 10.1016/j.chest.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 59. Selman M, Buendia-Roldan I, Pardo A. Aging and pulmonary fibrosis. Rev Invest Clin 68: 75–83, 2016. [PubMed] [Google Scholar]
- 60. Jo HE, Randhawa S, Corte TJ, Moodley Y. Idiopathic pulmonary fibrosis and the elderly: diagnosis and management considerations. Drugs Aging 33: 321–334, 2016. doi: 10.1007/s40266-016-0366-1. [DOI] [PubMed] [Google Scholar]
- 61. Leung J, Cho Y, Lockey RF, Kolliputi N. The role of aging in idiopathic pulmonary fibrosis. Lung 193: 605–610, 2015. doi: 10.1007/s00408-015-9729-3. [DOI] [PubMed] [Google Scholar]
- 62. Johnson PR, Burgess JK, Ge Q, Poniris M, Boustany S, Twigg SM, Black JL. Connective tissue growth factor induces extracellular matrix in asthmatic airway smooth muscle. Am J Respir Crit Care Med 173: 32–41, 2006. doi: 10.1164/rccm.200406-703OC. [DOI] [PubMed] [Google Scholar]
- 63. Ambhore NS, Kalidhindi RSR, Pabelick CM, Hawse JR, Prakash YS, Sathish V. Differential estrogen-receptor activation regulates extracellular matrix deposition in human airway smooth muscle remodeling via NF-kappaB pathway. FASEB J 33: 13935–13950, 2019. doi: 10.1096/fj.201901340R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Freeman MR, Sathish V, Manlove L, Wang S, Britt RD Jr, Thompson MA, Pabelick CM, Prakash YS. Brain-derived neurotrophic factor and airway fibrosis in asthma. Am J Physiol Lung Cell Mol Physiol 313: L360–L370, 2017. doi: 10.1152/ajplung.00580.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ichimaru Y, Krimmer DI, Burgess JK, Black JL, Oliver BG. TGF-β enhances deposition of perlecan from COPD airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 302: L325–L333, 2012. doi: 10.1152/ajplung.00453.2010. [DOI] [PubMed] [Google Scholar]
- 66. Burgess JK, Mauad T, Tjin G, Karlsson JC, Westergren-Thorsson G. The extracellular matrix—the under-recognized element in lung disease? J Pathol 240: 397–409, 2016. doi: 10.1002/path.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Caporarello N, Meridew JA, Jones DL, Tan Q, Haak AJ, Choi KM, Manlove LJ, Prakash YS, Tschumperlin DJ, Ligresti G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 74: 749–760, 2019. doi: 10.1136/thoraxjnl-2019-213064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eickelberg O, Kohler E, Reichenberger F, Bertschin S, Woodtli T, Erne P, Perruchoud AP, Roth M. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-β3. Am J Physiol Lung Cell Mol Physiol 276: L814–L824, 1999. doi: 10.1152/ajplung.1999.276.5.L814. [DOI] [PubMed] [Google Scholar]
- 69. Sueblinvong V, Neveu WA, Neujahr DC, Mills ST, Rojas M, Roman J, Guidot DM. Aging promotes pro-fibrotic matrix production and increases fibrocyte recruitment during acute lung injury. Adv Biosci Biotechnol 5: 19–30, 2014. doi: 10.4236/abb.2014.51004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alvarez D, Cardenes N, Sellares J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D'Cunha H, Sembrat J, Nouraie M, Shanker S, Caufield C, Shiva S, Armanios M, Mora AL, Rojas M. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol 313: L1164–L1173, 2017. doi: 10.1152/ajplung.00220.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wicher SA, Roos BB, Teske JJ, Fang YH, Pabelick C, Prakash YS. Aging increases senescence, calcium signaling, and extracellular matrix deposition in human airway smooth muscle. PLoS One 16: e0254710–19, 2021. doi: 10.1371/journal.pone.0254710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine 21: 21–28, 2017. doi: 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chapman J, Fielder E, Passos JF. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett 593: 1566–1579, 2019. doi: 10.1002/1873-3468.13498. [DOI] [PubMed] [Google Scholar]
- 74. Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, Birch-Machin MA, Kirkwood TB, von Zglinicki T. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 5: e110, 2007. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly YM, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, von Zglinicki T, Korolchuk VI, Passos JF. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35: 724–742, 2016. doi: 10.15252/embj.201592862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gulati S, Thannickal VJ. The aging lung and idiopathic pulmonary fibrosis. Am J Med Sci 357: 384–389, 2019. doi: 10.1016/j.amjms.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 77. You K, Parikh P, Khandalavala K, Wicher SA, Manlove L, Yang B, Roesler A, Roos BB, Teske JJ, Britt RD Jr, Pabelick CM, Prakash YS. Moderate hyperoxia induces senescence in developing human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 317: L525–L536, 2019. doi: 10.1152/ajplung.00067.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Baker JR, Donnelly LE, Barnes PJ. Senotherapy: a new horizon for COPD therapy. Chest 158: 562–570, 2020. doi: 10.1016/j.chest.2020.01.027. [DOI] [PubMed] [Google Scholar]
- 79. Cho SJ, Stout-Delgado HW. Aging and lung disease. Annu Rev Physiol 82: 433–459, 2020. doi: 10.1146/annurev-physiol-021119-034610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu RM, Liu G. Cell senescence and fibrotic lung diseases. Exp Gerontol 132: 110836, 2020. doi: 10.1016/j.exger.2020.110836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang ZN, Su RN, Yang BY, Yang KX, Yang LF, Yan Y, Chen ZG. Potential role of cellular senescence in asthma. Front Cell Dev Biol 8: 59, 2020. doi: 10.3389/fcell.2020.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Campisi J. Cellular senescence and lung function during aging. Yin and Yang. Ann Am Thorac Soc 13: S402–S406, 2016. doi: 10.1513/AnnalsATS.201609-703AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, Hara N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 154: 477–483, 1996. doi: 10.1164/ajrccm.154.2.8756825. [DOI] [PubMed] [Google Scholar]
- 84. Lomas NJ, Watts KL, Akram KM, Forsyth NR, Spiteri MA. Idiopathic pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int J Clin Exp Pathol 5: 58–71, 2012. [PMC free article] [PubMed] [Google Scholar]
- 85. Hohmann MS, Habiel DM, Coelho AL, Verri WA Jr, Hogaboam CM. Quercetin enhances ligand-induced apoptosis in senescent IPF fibroblasts and reduces lung fibrosis in vivo. Am J Respir Cell Mol Biol 60: 28–40, 2019. doi: 10.1165/rcmb.2017-0289OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Muller KC, Welker L, Paasch K, Feindt B, Erpenbeck VJ, Hohlfeld JM, Krug N, Nakashima M, Branscheid D, Magnussen H, Jorres RA, Holz O. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir Res 7: 32–10, 2006. doi: 10.1186/1465-9921-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhou F, Onizawa S, Nagai A, Aoshiba K. Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir Res 12: 78–18, 2011. doi: 10.1186/1465-9921-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence exacerbates pulmonary inflammation in patients with chronic obstructive pulmonary disease. Respiration 80: 59–70, 2010. doi: 10.1159/000268287. [DOI] [PubMed] [Google Scholar]
- 89. Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: implications for lung ageing and disease. Pharmacol Ther 183: 34–49, 2018. doi: 10.1016/j.pharmthera.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 90. Wu J, Dong F, Wang RA, Wang J, Zhao J, Yang M, Gong W, Cui R, Dong L. Central role of cellular senescence in TSLP-induced airway remodeling in asthma. PLoS One 8: e77795, 2013. doi: 10.1371/journal.pone.0077795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Puddicombe SM, Torres-Lozano C, Richter A, Bucchieri F, Lordan JL, Howarth PH, Vrugt B, Albers R, Djukanovic R, Holgate ST, Wilson SJ, Davies DE. Increased expression of p21(waf) cyclin-dependent kinase inhibitor in asthmatic bronchial epithelium. Am J Respir Cell Mol Biol 28: 61–68, 2003. doi: 10.1165/rcmb.4715. [DOI] [PubMed] [Google Scholar]
- 92. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114: 1299–1307, 2004. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene 18: 7644–7655, 1999. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 94. Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677, 1998. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 95. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334, 1997. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 96. Xu Y, Li N, Xiang R, Sun P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem Sci 39: 268–276, 2014. doi: 10.1016/j.tibs.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 25: 585–621, 1961. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 98. Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 14: 501–513, 2004. doi: 10.1016/S1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 99. Damera G, Panettieri RA Jr.. Does airway smooth muscle express an inflammatory phenotype in asthma? Br J Pharmacol 163: 68–80, 2011. doi: 10.1111/j.1476-5381.2010.01165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental script codes (1a, 1b, and 1c) and Supplemental Tables S1–S5: https://doi.org/10.6084/m9.figshare.21215183.
Data Availability Statement
Data will be made available upon reasonable request.