

Abstract
KynA is tissue protective in cardiac, cerebral, renal, and retinal ischemia models, but the mechanism is unknown. KynA can bind to multiple receptors, including the aryl hydrocarbon receptor, a7 nicotinic acetylcholine receptor (a7nAChR), multiple ionotropic glutamate receptors, and the orphan G-coupled receptor GPR35. Here, we show that GPR35 activation was necessary and sufficient for ischemic protection by KynA. When bound by KynA, GPR35 activated Gi and G12/13-coupled signaling and trafficked to the outer mitochondria membrane where it bound, apparantly indirectly, to ATP Synthase inhibitory Factor Subunit 1 (ATPIF1). Activated GPR35, in an ATPIF1-dependent and pertussis toxin-sensitive manner, induced ATP synthase dimerization, which prevented ATP loss upon ischemia. These findings provide a rationale for the development of specific GPR35 agonists for the treatment of ischemic diseases.
One Sentence summary:
KynA mediates ischemic protection through orphan GPR35
Kynurenic acid (KynA) protects tissues in preclinical organ ischemia models and mediates cardioprotection in a mouse model of remote ischemic preconditioning, which is a phenomenon whereby an ischemic tissue confers ischemic protection to other tissues at a distance (1–5). Mice, however, are a suboptimal model for cardiac physiology studies because of their small size, extremely rapid heart rates, and short lifespans (6). Therefore, we tested KynA in rabbits. Pretreatment of rabbits with KynA decreased infarct size when their excised, beating, and perfused hearts (Langendorff prep) were subjected to a brief period of ischemia followed by reperfusion to model ischemia-reperfusion (IR) injury (Fig. S1, A through D). KynA also increased cardiac function following IR, as measured by increased fractional shortening, increased left ventricular contractility (dP/dt max) and left ventricular peak developed pressure (LVPDP) during reperfusion (Fig. S1, E through H).
We tested which of the putative KynA receptors (7–13), if any, mediated KynA’s tissue protective effects. Because KynA protects isolated hearts against ischemia, we focused on the Aryl Hydrocarbon Receptor (AhR) and G-protein coupled receptor 35 (GPR35) rather than on ionotropic glutamate receptors, which are linked to neurotransmission. KynA promoted the binding of the AhR to its partner, Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT), and activated AhR-responsive transcription (Fig. S2, A and B). However, cardiac AhR loss in mice did not block KynA-induced cardioprotection in the setting of IR injury in vivo (Fig. S2, C and D). Moreover, stable expression of a constitutively active AHR (AHR-CA) in human-derived induced pluripotent stem cells-cardiomyocytes (hIPS-CMs) did not promote their survival when they were transiently deprived of oxygen and nutrients and then re-exposed to oxygen and nutrients to simulate IR injury ex vivo (Fig. S2, E through G). Therefore, AhR activation is neither necessary nor sufficient for KynA-induced cardioprotection.
GPR35 is broadly expressed, its abundance is increased after hypoxia under the control of the HIFα transcription factor, and its abundance is increased in human heart failure (Fig. S3, A through C) (14, 15). Moreover, GPR35 is linked to tissue protection, although studies disagree as to whether GPR35 agonism or antagonism is protective (16–18). Whether KynA is an authentic endogenous GPR35 ligand is also controversial, partly because its binding affinity for GPR35 varies across species and, in some species, is relatively low (μM) (19).
We confirmed that radiolabeled KynA bound to purified human and mouse GPR35 (Fig. 1A). This was specific, because radiolabeled KynA did not bind to a control GPCR, GPR160, and its binding to GPR35 was competed by excess unlabeled KynA, the known GPR35 activators pamoic acid, zaprinast, and lodoxamide, but not the KynA-derived metabolite quinolinic acid or tryptophan (the parent molecule from which KynA is derived) (Fig. 1B)(20–22). KynA activated Gi and G12/13 in a GPR35-dependent manner, as determined by Gi and G12/13 recruitment, activation of the protein kinase Rho, and suppression of forskolin-induced cAMP abundance and downstream signaling by PKA (Fig. S4, A through E).
Figure 1: KynA binds GPR35 and promotes GPR35 internalization.
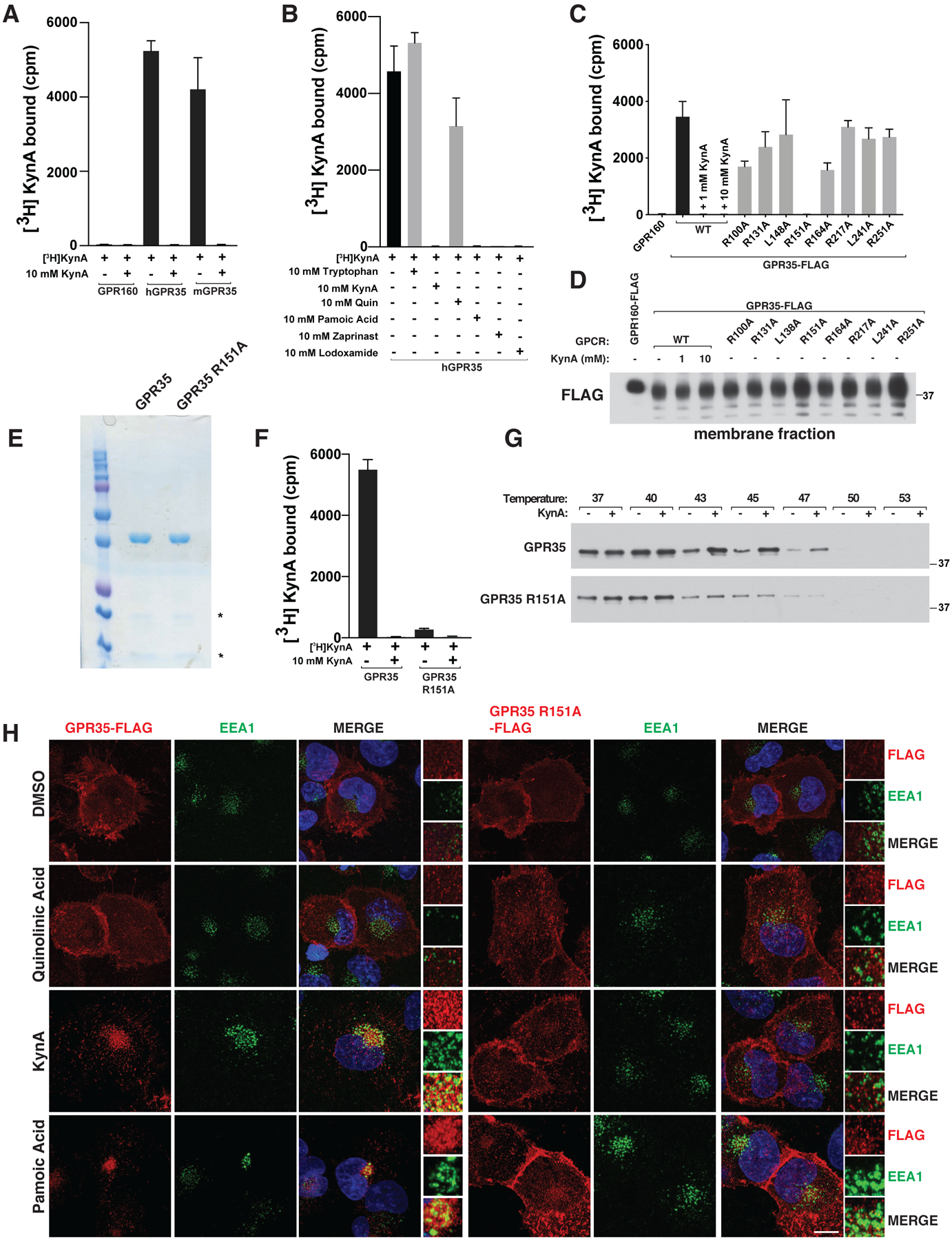
(A, B and F) [3H]-KynA binding assay using tandem affinity purified versions of the indicated HDL-reconstituted HA-FLAG-tagged GPCRs in the presence or absence of the indicated unlabeled competitor compounds (10 mM). (C and D) [3H]-KynA binding (C) and immunoblot (D) assays of isolated membrane fractions from 293T cells expressing FLAG-GPR160 or FLAG-GPR35 (wild-type or mutant). Where indicated, 1 and 10 mM unlabeled KynA was used as a competitor. (E and G) Commassie blue staining (E) and immunoblot (G) assay of the purified GPCRs used in (F). In (G) the proteins were pre-incubated with 1 mM KynA or DMSO for 1 hr at prior to exposure to the indicated temperature. (H) Confocal microscopy of mouse neonatal cardiomyocytes stably expressing wild-type or R151A GPR35-FLAG stimulated with 20 μM quinolinic acid, 20 μM KynA, or 1 μM pamoic Acid for 20 minutes. Scale bar indicates 20 μm. In (A), (B), (C), and (F), values are means −/+ SDs for three technical replicates from one representative experiment.
We sought to create a GPR35 mutant that cannot bind to KynA for use as a specificity control. We made a series of GPR35 missense mutants in which specific residues within GPR35 transmembrane domains III and IV were converted to alanine, motivated by previous homology modeling and ligand docking studies that implicated this region, and particularly specific arginine residues within this region, in ligand binding (23, 24). One such variant, GPR35 R151A, failed to bind to radiolabeled KynA in assays done from GPR35-expressing cell membrane fractions or highly purified GPR35 (Fig. 1, C through F). Similarly, KynA bound to wild-type GPR35, but not GPR35 R151A, in thermal shift assays based on thermostabilization upon ligand binding (Fig. 1G). As these studies used GPR35 that was purified from mammalian cells (293T cells), we cannot yet formally exclude that additional cellular proteins are required for the binding of KynA to GPR35. Many GPCRs, including GPR35, are internalized upon activation (25). We confirmed that wild-type GPR35, but not GPR35 R151A, was internalized in early endosome antigen 1 (EEA1)-containing endosomes in cells treated with KynA or the GPR35 agonists pamoic acid or zaprinast, but not in cells treated with quinolinic acid (Fig. 1H and Fig. S5A) (11, 15). GPR35-containing endosomes also contained Transferrin Receptor (TfR), which is normally present on plasma membranes, consistent with the GPR35 endosomes participating in endosomal trafficking (Fig. S5B). β-Arrestin localization was not regulated by KynA or GPR35 agonist pamoic acid, in the cardiomyocytes we studied, although it is by many other GPCRs that are internalized upon activation (Fig. S5C).
Cardioprotection by KynA administered either 2 or 24 hours prior to injury in vivo or 10 or 30 minutes prior to IR in ex vivo hearts was completely abrogated in GPR35−/− mice we made using CRISPR/Cas9-based gene editing of mouse embryos (Fig. 2, A and B and Fig. S6A) (26). GPR35 loss did not alter, positively or negatively, induction of the hypoxia-inducible transcription factor HIF1α following cardiac IR injury (Fig. S6B). In cell culture experiments performed with murine neonatal cardiomyocytes, KynA, in a GPR35-dependent manner, decreased resting oxygen consumption and, after simulated IR injury, decreased mitochondrial ROS production and preserved mitochondrial membrane potential (Fig. S6, C through E), changes that would be predicted to promote survival after IR.
Figure 2: GPR35 mediates KynA ischemic protection.
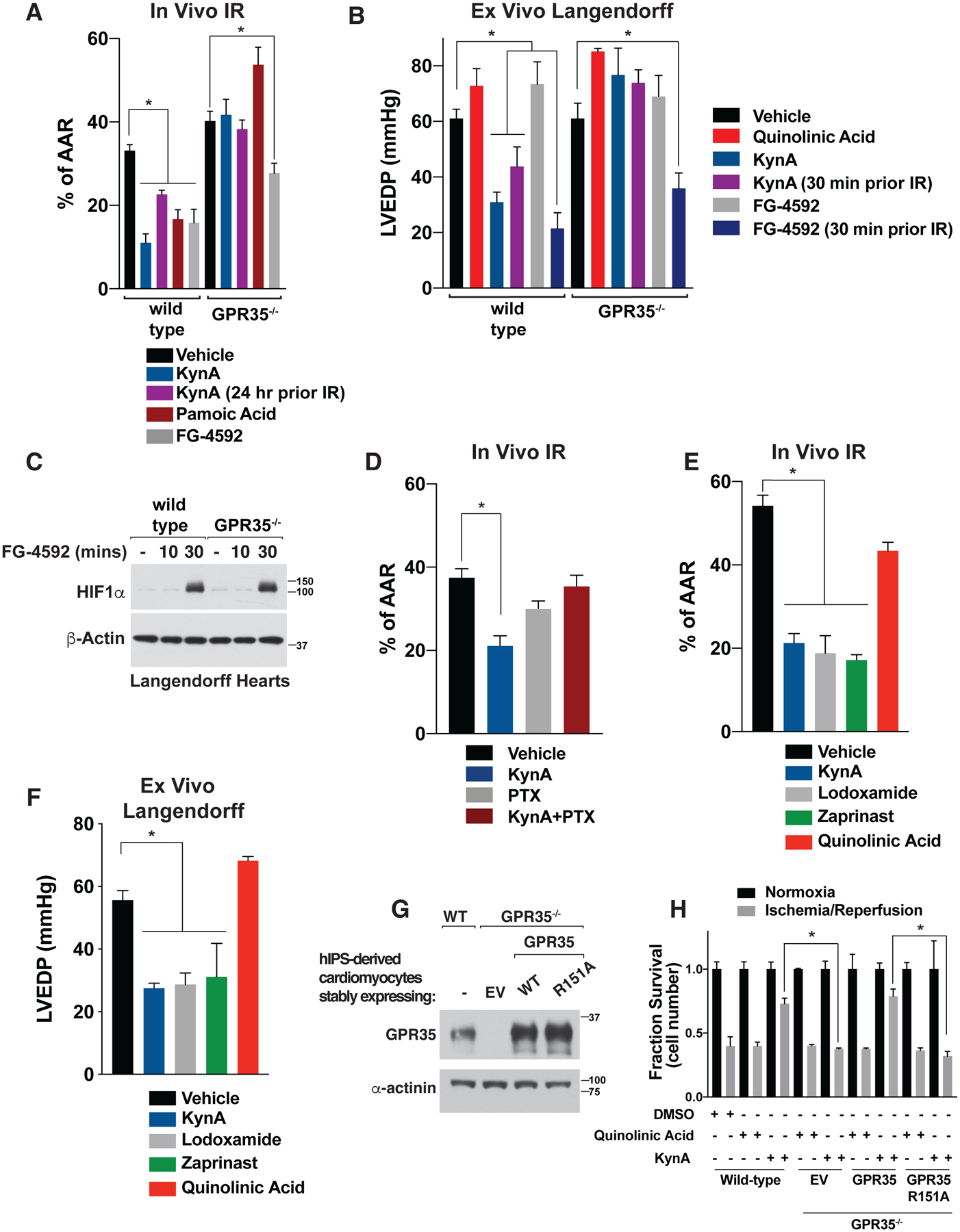
(A) Myocardial infarct size after in vivo cardiac ischemia/reperfusion (I/R) injury in wild-type and GPR35−/− mice. Where indicated mice were given 5 mg/kg KynA, 5 mg/kg pamoic acid, 50 mg/kg FG-4592, 5 mg/kg quinolinic acid, or vehicle by intraperitoneal injection 2 hours (or 24 hours where indicated) prior to ischemia. N=5 per group. (B) LVEDP in Langendorff assays after global I/R injury in wild-type and GPR35−/− mice. Where indicated the hearts were infused with 500 nM KynA, 100 μM FG-4592, or 500 nM quinolinic acid, or vehicle for 10 minutes (or 30 minute where indicated) prior to ischemia. N=4 per group. (C) Immunoblot analysis of lysates from Langendorff hearts perfused with 100 μM FG-4592 for the indicated times. (D) Myocardial infarct size after in vivo cardiac ischemia/reperfusion (I/R) injury in wild-type mice treated with 5 mg/kg KynA, 10 μg/kg pertussis toxin (PTX), both, or vehicle by intraperitoneal injection. Animals were treated with KynA or vehicle 2 hours before the onset of ischemia, while PTX was treated 24 hours prior to ischemia. N=4 per group (E) Myocardial infarct size after in vivo cardiac ischemia/reperfusion (I/R) injury in wild-type mice given GPR35 agonists. Where indicated, mice were given 5 mg/kg KynA, 5 mg/kg lodoxamide, 5 mg/kg zaprinast, 5 mg/kg quinolinic Acid, or vehicle by intraperitoneal injection 2 hours before the onset of ischemia. N=4 per group. (F) LVEDP in Langendorff assays after global I/R injury. Where indicated the hearts were infused with 500 nM KynA, 1 μM lodoxamide, 1 μM zaprinast, 500 nM quinolinic acid or vehicle for 10 minutes prior to ischemia. N=4 per group (G) Immunoblot of wild-type or GPR35−/− human IPS-derived cardiomyocytes stably expressing wild-type or R151A GPR35. (H) Fractional survival after simulated I/R injury ex vivo of human IPS-derived cardiomyocytes modified as in (G). Cell were pretreated with 20 μM KynA, 20 μM quinolinic acid, or DMSO for 1 hr prior to I/R. Shown are mean −/+SD. * indicates P value <0.05.
FG-4592 (Roxadustat), which stabilizes HIF1α by inactivating the EglN prolyl hydroxylases, is cardioprotective in preclinical models (27–29). Cardioprotection is likely an on-target effect of FG-4592 because similar effects have been observed with other structurally unrelated EglN inhibitors and after systemic or cardiac-specific genetic inactivation of EglN1, which is the primary regulator of HIF1α stability (1, 28, 30–32). Although EglN inactivation is thought to cardioprotect by stabilizing HIF1α, systemic inactivation of EglN also increases the plasma concentrations of 2-OG, which is converted to KynA by the liver and released into the circulation (1). Cardiac protection by FG-4592 in vivo was partially abrogated by loss of GPR35 (Fig. 2A). In contrast, FG-4592 was cardioprotective in wild-type and GPR35−/− hearts when administered ex vivo, a setting where KynA would not be produced, prior to IR (Fig. 2, B and C). Notably, protection by FG-4592 was observed when given 30 minutes, but not 10 minutes, prior to IR, which correlated with successful induction of HIF1α 30 minutes after FG-4592 administration (Fig. 2C). In contrast, KynA protected more rapidly (Fig. 2B). These results suggest that cardiac protection in vivo by Roxadustat is due to direct effects of HIFα and indirect effects on GPR35 mediated by endogenous KynA.
Pertussis toxin, a classic Gi and Go protein family inhibitor, abolishes cardiac ischemic preconditioning in rats (33) and likewise inhibited KynA-induced cardiac ischemic protection in vivo, suggesting intact Gi or Go family signaling by GPR35 is required for KynA-induced ischemic protection (Fig. 2D). Multiple structurally unrelated GPR35 agonists, including lodoxamide, zaprinasat, and pamoic acid, were cardioprotective in vivo and ex vivo, whereas quinolinic acid, which fails to activate GPR35, was not (Fig. 2, E and F). Where tested, cardioprotection by these other GPR35 agonists was GPR35-dependent (Fig. 2A).
In order to formally test whether KynA binding to GPR35 is necessary for KynA-induced ischemic protection, we used CRISPR-Cas9 to generate hiPS cells lacking GPR35 and then infected them with a lentivirus encoding wild-type GPR35, GPR35 R151A, or the empty vector (Fig. 2G). These cells were then induced to become cardiomyocytes and subjected to simulated IR ex vivo. KynA pretreatment protected against IR-induced injury and this protection was lost in absence of GPR35. Re-expression of the wild-type GPR35, but not GPR35 R151A, in the GPR35−/− restored protection by KynA (Fig. 2H). Thus, GPR35 mediates KynA-induced ischemic protection in mice and human cells and this protection requires the binding of KynA to GPR35.
GPR35 is annotated to bind to the mitochondrial protein ATP Synthase inhibitory Factor Subunit 1 (ATPIF1) and ATP Synthase in the BioPlex protein-protein interaction database (34). To test whether GPR35 can associate with mitochondria, we reintroduced wild-type or R151A GPR35 into GPR35−/− murine neonatal cardiomyocytes by lentiviral infection. Treatment of the wild-type cells with GPR35 agonists, including KynA, led to colocalization of GPR35 with mitochondrial proteins, such as citrate synthase and the ATP synthase component ATP5H, but not the Golgi apparatus protein, GM130, or the endoplasmic reticulum protein, calreticulin, by confocal microscopy and quantitative image analysis (Fig. 3A, Fig. S7, A through C, and Fig. S8, A through D). This was specific because colocalization with mitochondria was not observed with GPR35 R151A nor with quinolinic acid. KynA and the GPR35 agonist pamoic acid, but not quinolinic acid, also increased GPR35 and TOM20 association in situ based on proximity ligation assays (PLA) (Fig. S7, D through E). To visualize GPR35 trafficking to mitochondria, we transiently over-expressed GPR35-GFP together with mCherry-TOMM20 in GPR35−/− murine cardiomyocytes and performed live-cell imaging. KynA treatment caused GPR35-GFP to become internalized within minutes on cytoplasmic punctae that associated with mCherry-TOMM20 (Fig. S8E). To corroborate these findings, we rapidly isolated mitochondria from cells expressing 3XHA-EGFP-OMP25 (HA-MITO) following KynA treatment and did immunoblot assays (35). We reproducibly detected the association of GPR35 with mitochondria, which was increased in cells treated with KynA or other GPR35 agonists, but not in cells treated with quinolinic acid (Fig. 3B and Fig. S9A). We introduced a biotin ligase (APEX) fused to wild-type or R151A GPR35 into the GPR35−/− murine cardiomyocytes, and captured GPR35-associated proteins using streptavidin agarose after treatment with biotin tyramide and hydrogen peroxide (to activate the APEX enzyme) in the presence or absence of GPR35 agonists (Fig. S9B) (36). Multiple mitochondrial outer membrane proteins, such as VDAC, TOM20, and TOM70 were associated with wild-type, but not R151A, GPR35 in cells treated with KynA (Fig. 3C). Similarly, KynA promoted biotin-labeling of mitochondria, as determined by confocal microscopy using streptavidin-568 as a probe, in cells expressing wild-type GPR35-APEX, but not GPR35 R151A-APEX (Fig. S9C) (37).
Figure 3: KynA promotes GPR35 association with mitochondria.
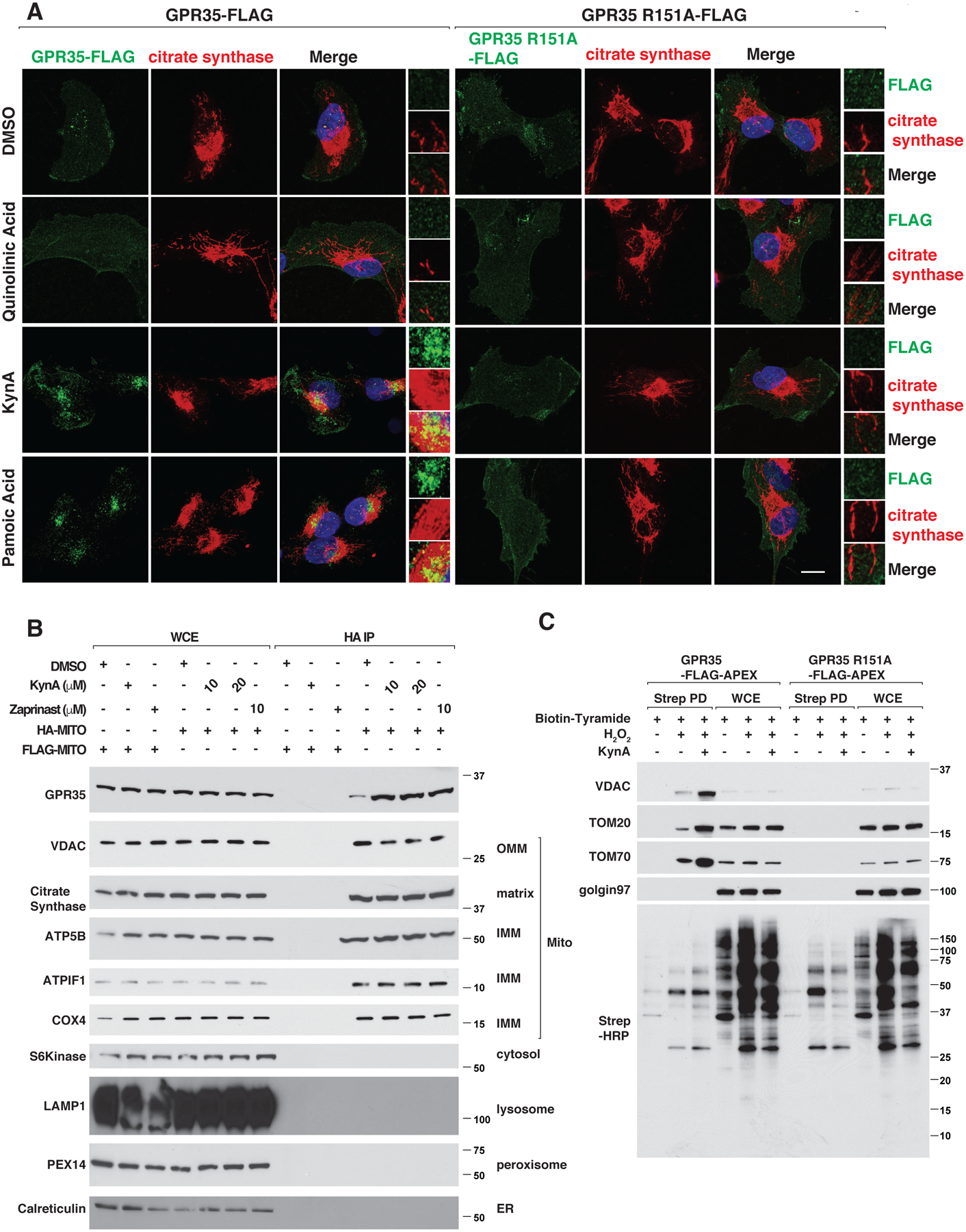
(A) Confocal microscopy of GPR35−/− mouse neonatal cardiomyocytes stably expressing FLAG-tagged wild-type or R151A GPR35 that were treated with 20 μM quinolinic acid, 20 μM KynA, 1 μM pamoic acid or DMSO for 20 mins. Scale bar indicates 20 μm. (B) Immunoblot analysis of whole cell extracts (WCE) or mitochondria that were rapidly immunopurified with an anti-HA antibody (HA-IP) from 293T cells engineered to contain HA-tagged mitochondria (HA-MITO) or, as a control, FLAG-tagged mitochondria (FLAG-MITO). Where indicated the cells were pretreated with 20 μM KynA or 10 μM zaprinast for 20 mins prior to analysis. (C) GPR35−/− mouse cardiomyocytes stably expressing FLAG-APEX-tagged wild-type or R151A GPR35 were treated with Biotin-Tryamide for 30 min and, where indicated, with 20 μM KynA for 20 min and H2O2 for 1 min (to enable biotinylation). Cell lysates (WCE) and biotinylated proteins captured on streptavidin agarose (Strep PD) were resolved by SDS-PAGE, transferred to nitrocellulose, and immunoblotted with antibodies against the indicated proteins or probed with horseradish peroxidase-conjugated streptavidin (Strep-HRP).
We confirmed, using 2 different epitope tags, that epitope tagged human and mouse GPR35 coimmunoprecipitated with endogenous ATPIF1 and ATP Synthase, as determined by immunoblot analysis (Fig. S10, A through D). Consistent with our localization studies, the binding of GPR35 to ATPIF1 was enhanced by KynA and the GPR35 agonists zaprinast and pamoic acid, but not by quinolinic acid (Fig. 4A and Fig. S10A). As additional specificity controls, we did not detect binding to ATPIF1 to cytosolic METAP2, the transmembrane mitochondrial protein TMEM141, GPR160, or to a GPR35 C-terminal truncation mutant (Fig. 4B and Fig. S10B). The electrophoretic mobility of wild-type GPR35 was decreased under these gel conditions compared to the C-terminal truncation mutant because of a slightly higher molecular size and because of glycosylation events that require the GPR35 C-terminus (38).
Figure 4: KynA promotes GPR35-ATPIF1-ATP Synthase interaction.
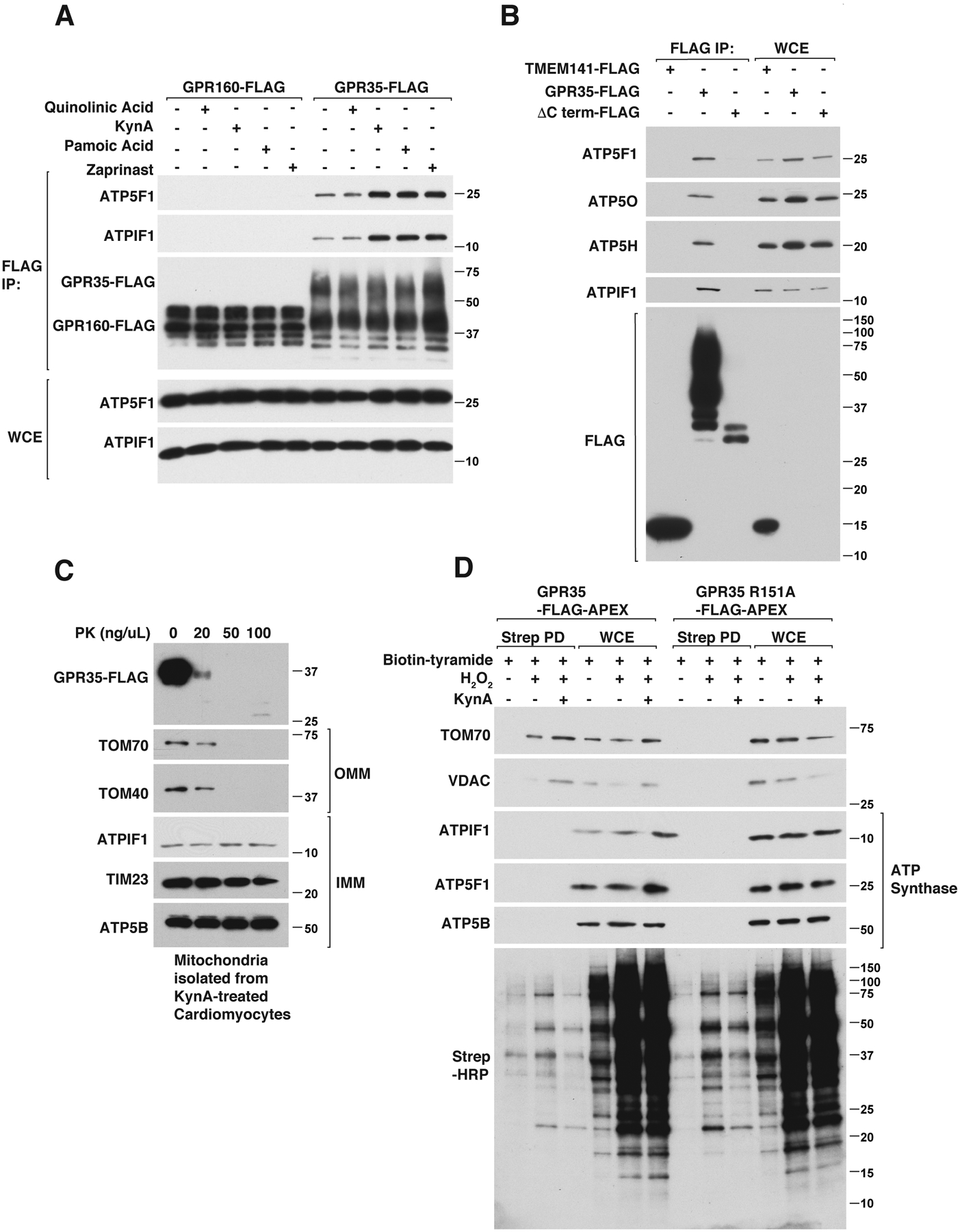
(A) Immunoblot analysis of anti-FLAG immunoprecipitates (FLAG IP) and whole cell extracts (WCE) from mouse neonatal GPR35−/− cardiomyocytes stably expressing GPR160-FLAG or GPR35-FLAG that were treated with 20 μM quinolinic acid, 20 μM KynA, 1 μM pamoic acid, 10 μM zaprinast or DMSO for 20 mins. (B) Immunoblot analysis of anti-FLAG immunoprecipitates from human AC16 cardiomyocytes stably expressing TMEM141-FLAG, GPR35 (wild-type)-FLAG or GPR35 (ΔC-terminus)-FLAG. (C) Immunoblot analysis of mitochondria isolated from mouse neonatal cardiomyocytes stably expressing GPR35-FLAG that were treated with 20 μM KynA for 20 mins. The isolated mitochrondria were incubated with increasing concentrations of proteinase K (PK) for 30 mins. (D) GPR35−/− mouse neonatal cardiomyocytes stably expressing FLAG-APEX-tagged wild-type or R151A GPR35 were treated with Biotin-Tryamide for 30 min and, where indicated, with 20 μM KynA for 20 min and H2O2 for 1 min (to enable biotinylation). Cell lysates (WCE) and biotinylated proteins captured on streptavidin agarose (STREP PD) were resolved by SDS-PAGE, transferred to nitrocellulose, and immunoblotted with antibodies against the indicated proteins or probed with horseradish peroxidase-conjugated streptavidin (Strep-HRP).
ATPIF1 binds to, and regulates, the multimeric ATP synthase complex (39). The ATP synthase subunits ATP5B, ATP5H, ATP5O, and ATP5F1 also coimmunoprecipitated with GPR35 and their abundance, when measured, mirrored that of ATPIF1 (Fig. 4, A and B and Fig. S10, A through C). Given that ATPIF1 and ATP synthase are localized inside mitochondria, we tested whether GPR35 is present inside or outside mitochondria after treatment with KynA. We treated murine cardiomyocytes expressing wild-type GPR35 with KynA and used differential centrifugation to purify mitochondria, which were then exposed to increasing amounts of Proteinase K. Mitochondrially-associated GPR35 was Proteinase K-sensitive, arguing that it is largely associated with the outer mitochondria membrane (Fig. 4C). Consistent with this, KynA promoted GPR35-APEX biotinylation of outer mitochondrial membrane proteins VDAC and TOM70, but did not promote biotinylation of ATPIF1 or ATP Synthase (Fig. 4D). Therefore, KynA promotes the association of GPR35 with ATPIF1 and ATP synthase, but this interaction is likely indirect.
During ischemia ATP synthase shifts from ATP synthesis to ATP hydrolysis, with loss of ATP eventually contributing to cell death (40). Binding of ATPIF1 to ATP synthase promotes the formation of ATP synthase dimers and thereby inhibits ATP hydrolysis, oxidative-phosphorylation, and oxygen consumption (41–44). ATPIF1 mimetic compounds and transgenic overexpression of ATPIF1 both protect tissues in IR models (44, 45). KynA increased the recovery of ATP synthase after immunoprecipitation of ATPIF1 and decreased the recovery of ATPIF1 after the immunoprecipitation of ATP synthase in a pertussis toxin-sensitive manner, consistent with the ATPIF1: ATP synthase stoichiometry shifting from 1:1 to 1:2 (Fig. 5, A and B, and Fig. S11, A through D). We independently confirmed that KynA promotes ATP synthase dimerization by native gel electrophoresis and by immunofluorescence and electron microscopy, which revealed hallmark changes in mitochondrial morphology and mitochondrial cristae, respectively, usually associated with ATP synthase dimerization (Fig. 5, C through E, Fig. S11E, and Fig. S12, A through C) (46).
Figure 5: KynA promotes ATP synthase dimerization and ATPIF1 is required for KynA-induced ischemic protection.
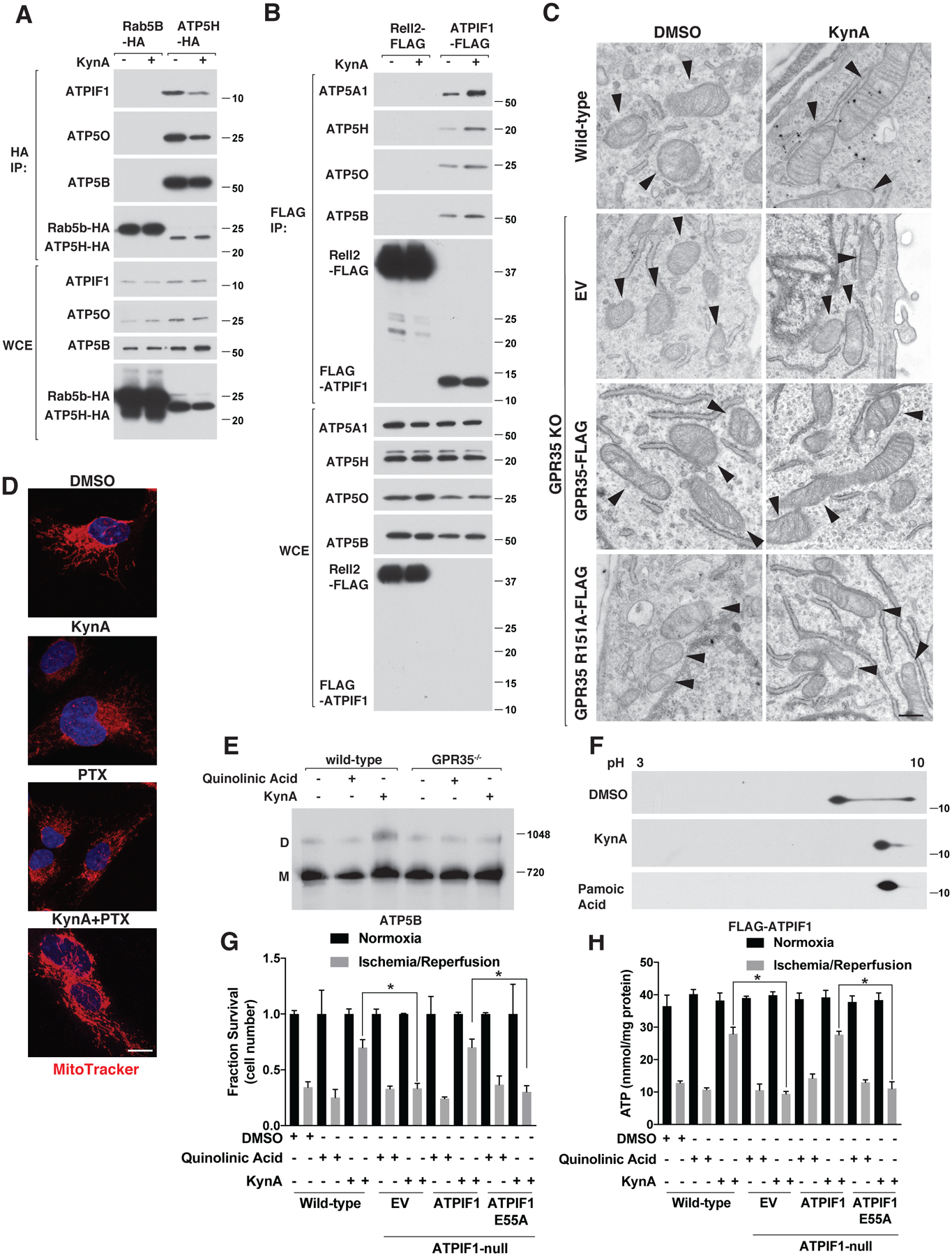
(A) Immunoblot analysis of anti-HA immunoprecipitates (HA-IP) and whole cell extracts (WCE) from mouse neonatal cardiomyocytes stably expressing Rab5B-HA or ATP5H-HA that were or were not treated with 20 μM KynA for 20 mins. (B) Immunoblot analysis of anti-FLAG immunoprecipites (FLAG-IP) and whole cell extracts (WCE) from mouse neonatal cardiomyocytes stably expressing Rell2-FLAG or FLAG-ATPIF1 that were or were not treated with 20 μM KynA for 20 mins. (C) Electron micrographs of wild-type and GPR35 −/− cardiomyocytes stably expressing GPR35-FLAG, R151A GPR35-FLAG, or empty vector (EV) that were treated with 20 μM KynA or DMSO for 20 mins. Arrowheads indicate mitochondria. Scale bar indicates 500 nm. (D) Live cell fluorescence imaging of mouse neonatal cardiomyocytes treated with 20 μM KynA, 200 ng/mL pertussis toxin (PTX), or both prior to staining with 100 nM Mitotracker FM. Scale bar indicates 20 μm. (E) Blue-native page analysis of mitochondria isolated from mouse neonatal cardiomyocytes treated with 20 μM quinolinic acid, 20 μM KynA, or DMSO for 20 mins. (F) Anti-Flag immunoblot analysis after two-dimensional gel-electrophoresis of cell extracts made from mouse neonatal cardiomyocytes stably expressing FLAG-ATPIF1 that were treated with 20 μM KynA, 1 μM pamoic acid, or DMSO for 20 mins. (G and H) Fractional survival (G) and ATP levels (H) of wild-type and ATPIF1−/− human IPS-derived cardiomyocytes stably expressing wild-type or E55A ATPIF1, or the empty vector (EV), that were pretreated with 20 μM KynA, 20 μM quinolinic acid or DMSO for 1 hr prior simulated I/R ex vivo. Shown are mean −/+SD. * indicates P value <0.05.
As phosphorylation of ATPIF1 by PKA prevents it from inhibiting ATP synthase (47) we tested whether GPR35 activation caused dephosphorylation of ATPIF1. Both KynA and the GPR35 agonist pamoic acid blocked ATPIF1 phosphorylation in a GPR35-dependent manner as shown by characteristic electrophoretic mobility changes following 2D-gel electrophoresis (Fig. 5F and Fig. S12D).
We used CRISPR/Cas9 to generate ATPIF1−/− cardiomyocytes from human iPS cells and then infected them with either wild-type ATPIF1, an ATPIF1 point mutant that cannot bind to ATP synthase (ATPIF1 E55A) or an empty vector (EV) (Fig. S13, A and B) (48). Wild-type ATPIF1, but not ATPIF1 E55A, allowed KynA to prevent ATP consumption and preserve cell viability during simulated ischemia ex vivo (Fig. 5, G and H). Collectively, these results show that KynA ischemic protection depends on ATPIF1.
Although GPCRs are typically thought to function at the plasma membrane, there are multiple examples of mitochondrial GPCRs and G-proteins (49, 50). For example, the G-proteins guanine nucleotide binding protein-β subunit 2 (Gβ2) and guanine nucleotide binding protein subunit alpha-12 (Gα12) have roles in mitochondria fission versus fusion (51, 52). Moreover, many GPCRs, including GPR35, are internalized once activated, presumably so that they can signal in a spatially distinct manner (53). The endoplasmic reticulum has important roles in endosomal trafficking and forms multiple contacts with mitochondria and physical interaction between endosomes and mitochondria serve critical functions in cell homeostasis (54–56). The endosomal trafficking of GPR35 to mitochondria is reminiscent of Rab5, a small GTPase that translocates from early endosomes to mitochondria in response to oxidative stress to confer cytoprotection (57).
We found that GPR35 can bind to ATPIF1 and that the latter is necessary for cardioprotection by former. Nonetheless, GPR35 associates with the mitochondrial outer membrane in response to KynA whereas ATPIF1 is a mitochondrial matrix protein (41). Moreover, we did not detect biotinylation of ATPIF1 or ATP Synthase by GPR35-APEX nor was ATPIF1 or ATP Synthase Proteinase K-sensitive. Thus, the interaction of GPR35 with ATPIF1 is not direct, but instead bridged by one or more proteins, perhaps at contact sites that have been demonstrated between outer and inner mitochondrial membranes and endosome-mitochondrial membrane contact sites (55, 58). Pertussis toxin did not block the internalization of GPR35 by KynA but blocked both the KynA-mediated regulated interaction between ATPIF1 and ATP synthase, ATPIF1 dephosphorylation, and cardioprotection, thus the latter two appear to be linked and require Gi/o signaling (Fig. S14A, Fig. S15A, and Fig. 2D). We propose that GPR35, once activated and translocated to mitochondria, inhibits mitochondrial adenylate cyclase and thereby PKA. This, in turn, would allow ATPIF1 to promote ATP synthase dimerization and prevent ATP hydrolysis. Although KynA might have direct effects on mitochondria and may regulate metabolism, the effects we observed clearly depend on GPR35 (59–65).
It is clear that ischemic preconditioning involves both an early and late phase of ischemic protection and we have shown GPR35 is necessary for KynA-mediated cardioprotection whether administered acutely or 24 hours prior to IR injury (Fig. 2A) (66). While our data shows GPR35 is necessary in both settings, it remains unknown whether ATPIF1 is required at longer timescales.
Ischemic diseases such as myocardial infarction and stroke are major causes of death in the developed world. Remote ischemic preconditioning has been demonstrated in animal models, but attempts to harness it for therapeutic purposes in man, such as through repeated hyperinflation of a limb blood pressure cuff prior to elective heart surgery, have not proven successful (67). Without an understanding of the underlying mechanism, however, it was impossible to know whether the responsible tissue protective factor was properly induced by these interventions and whether its effects would be mitigated by other factors. Our findings support further exploration of KynA, and more broadly GPR35 agonists, for the prevention and treatment of ischemic injuries. In this regard, a number of chemically synthetic GPR35 agonists have now been described, including compounds with far greater potency than KynA (20, 21). In addition, our findings provide a unifying explanation for the tissue protective effects of kynurenine hydroxylase and kyurenine monooxygenase inhibitors. Although these inhibitors were originally developed to prevent the production of neuroexcitatory/neurotoxic tryptophan metabolites, they also promote accumulation of KynA (68, 69).
Supplementary Material
Acknowledgements:
We thank the members of the Kaelin laboratory and J. Moslehi (UCSF) for helpful discussions and critical reading of the manuscript. Special thanks to Cardiovascular Physiology Core (Longwood) at BWH/HMS for assistance with cardiac physiology experiments, as well as B. Olenchock, and R. Liao for experimental advice. We thank Thomas Sakmar, M.D. for critical reading of the manuscript. We thank Vamsi Mootha, M.D. for helpful discussions and experimental advice.
Funding:
W.G.K. is supported by an NIH 5R35CA210068 and is an HHMI investigator. G.A.W. is a HHMI Fellow of the Jane Coffin Childs Memorial Fund for Medical Research. I.P.D. is a Fellow of the George and Marie Vergottis Foundation. J. D. M. is supported by Richard A. and Susan F. Smith President’s Innovation Award, the Sidman Family Foundation, the Michael B. Rukin Charitable Foundation, the Kenneth C. Griffin Charitable Research Fund, The George and Marie Vergottis Foundation and the Boston Investment Council.
Footnotes
Competing Interests: W.G.K. has financial interests in Lilly Pharmaceuticals, Fibrogen, Cedilla Therapeutics, IconOvir Bio, Nextech Invest, Tango Therapeutics, LifeMine Therapeutics, Circle Pharma, and Casdin Capital. J. D. M. is a consultant for Cellvie Scientific Inc. The authors declare they have no other competing interests.
Data and materials availability:
All data needed to evaluate and reproduce the conclusions in the paper are present in the paper and/or the Supplementary Materials. All plasmids are available from the authors at request.
References
- 1.Olenchock BA et al. , EGLN1 Inhibition and Rerouting of alpha-Ketoglutarate Suffice for Remote Ischemic Protection. Cell 164, 884–895 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murry CE, Jennings RB, Reimer KA, Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Zheng X et al. , Kynurenine 3-monooxygenase is a critical regulator of renal ischemia-reperfusion injury. Exp Mol Med 51, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gellert L et al. , Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur J Pharmacol 667, 182–187 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Nahomi RB et al. , Kynurenic Acid Protects Against Ischemia/Reperfusion-Induced Retinal Ganglion Cell Death in Mice. Int J Mol Sci 21, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milani-Nejad N, Janssen PM, Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacol Ther 141, 235–249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hilmas C et al. , The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci 21, 7463–7473 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone TW, Kynurenic acid blocks nicotinic synaptic transmission to hippocampal interneurons in young rats. Eur J Neurosci 25, 2656–2665 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Mok MH, Fricker AC, Weil A, Kew JN, Electrophysiological characterisation of the actions of kynurenic acid at ligand-gated ion channels. Neuropharmacology 57, 242–249 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Resta F et al. , Kynurenic acid and zaprinast induce analgesia by modulating HCN channels through GPR35 activation. Neuropharmacology 108, 136–143 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Wang J et al. , Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem 281, 22021–22028 (2006). [DOI] [PubMed] [Google Scholar]
- 12.DiNatale BC et al. , Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci 115, 89–97 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D et al. , Functional metabolomics reveal the role of AHR/GPR35 mediated kynurenic acid gradient sensing in chemotherapy-induced intestinal damage. Acta Pharm Sin B 11, 763–780 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Min KD et al. , Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem Biophys Res Commun 393, 55–60 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Ronkainen VP et al. , Hypoxia-inducible factor 1-induced G protein-coupled receptor 35 expression is an early marker of progressive cardiac remodelling. Cardiovasc Res 101, 69–77 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Chen K et al. , Inhibition of GPR35 Preserves Mitochondrial Function After Myocardial Infarction by Targeting Calpain 1/2. J Cardiovasc Pharmacol 75, 556–563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharmin O et al. , Activation of GPR35 protects against cerebral ischemia by recruiting monocyte-derived macrophages. Sci Rep 10, 9400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olivares-Gonzalez L et al. , cGMP-Phosphodiesterase Inhibition Prevents Hypoxia-Induced Cell Death Activation in Porcine Retinal Explants. PLoS One 11, e0166717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milligan G, Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends Pharmacol Sci 32, 317–325 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Taniguchi Y, Tonai-Kachi H, Shinjo K, Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett 580, 5003–5008 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Zhao P et al. , Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and beta-arrestin2 with antinociceptive activity. Mol Pharmacol 78, 560–568 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKenzie AE et al. , The antiallergic mast cell stabilizers lodoxamide and bufrolin as the first high and equipotent agonists of human and rat GPR35. Mol Pharmacol 85, 91–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jenkins L et al. , Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Galpha(1)(3) and beta-arrestin-2. Br J Pharmacol 162, 733–748 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao P et al. , Crucial positively charged residues for ligand activation of the GPR35 receptor. J Biol Chem 289, 3625–3638 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moo EV, van Senten JR, Brauner-Osborne H, Moller TC, Arrestin-Dependent and -Independent Internalization of G Protein-Coupled Receptors: Methods, Mechanisms, and Implications on Cell Signaling. Mol Pharmacol 99, 242–255 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Wang H, Jaenisch R, Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 9, 1956–1968 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Deguchi H et al. , Roxadustat Markedly Reduces Myocardial Ischemia Reperfusion Injury in Mice. Circ J 84, 1028–1033 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Bao W et al. , Chronic inhibition of hypoxia-inducible factor prolyl 4-hydroxylase improves ventricular performance, remodeling, and vascularity after myocardial infarction in the rat. J Cardiovasc Pharmacol 56, 147–155 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Yeh TL et al. , Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem Sci 8, 7651–7668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nwogu JI et al. , Inhibition of collagen synthesis with prolyl 4-hydroxylase inhibitor improves left ventricular function and alters the pattern of left ventricular dilatation after myocardial infarction. Circulation 104, 2216–2221 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Vogler M et al. , Pre- and post-conditional inhibition of prolyl-4-hydroxylase domain enzymes protects the heart from an ischemic insult. Pflugers Arch 467, 2141–2149 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Holscher M et al. , Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J Biol Chem 286, 11185–11194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thornton JD, Liu GS, Downey JM, Pretreatment with pertussis toxin blocks the protective effects of preconditioning: evidence for a G-protein mechanism. J Mol Cell Cardiol 25, 311–320 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Huttlin EL et al. , Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen WW, Freinkman E, Wang T, Birsoy K, Sabatini DM, Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337 e1311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam SS et al. , Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SY et al. , APEX Fingerprinting Reveals the Subcellular Localization of Proteins of Interest. Cell Rep 15, 1837–1847 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Dong C, Filipeanu CM, Duvernay MT, Wu G, Regulation of G protein-coupled receptor export trafficking. Biochim Biophys Acta 1768, 853–870 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujikawa M, Imamura H, Nakamura J, Yoshida M, Assessing actual contribution of IF1, inhibitor of mitochondrial FoF1, to ATP homeostasis, cell growth, mitochondrial morphology, and cell viability. J Biol Chem 287, 18781–18787 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grover GJ et al. , Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. Am J Physiol Heart Circ Physiol 287, H1747–1755 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Garcia JJ, Morales-Rios E, Cortes-Hernandez P, Rodriguez-Zavala JS, The inhibitor protein (IF1) promotes dimerization of the mitochondrial F1F0-ATP synthase. Biochemistry 45, 12695–12703 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Formentini L, Sanchez-Arago M, Sanchez-Cenizo L, Cuezva JM, The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol Cell 45, 731–742 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Sanchez-Cenizo L et al. , Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem 285, 25308–25313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Formentini L et al. , In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J 33, 762–778 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivanes F et al. , The compound BTB06584 is an IF1 -dependent selective inhibitor of the mitochondrial F1 Fo-ATPase. Br J Pharmacol 171, 4193–4206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Aguilar A, Cuezva JM, A Review of the Inhibition of the Mitochondrial ATP Synthase by IF1 in vivo: Reprogramming Energy Metabolism and Inducing Mitohormesis. Front Physiol 9, 1322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Bermudez J et al. , PKA Phosphorylates the ATPase Inhibitory Factor 1 and Inactivates Its Capacity to Bind and Inhibit the Mitochondrial H(+)-ATP Synthase. Cell Rep 12, 2143–2155 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Ichikawa N et al. , The region from phenylalanine-17 to phenylalanine-28 of a yeast mitochondrial ATPase inhibitor is essential for its ATPase inhibitory activity. J Biochem 130, 687–693 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Valenzuela R et al. , Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis 7, e2427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belous A et al. , Mitochondrial P2Y-Like receptors link cytosolic adenosine nucleotides to mitochondrial calcium uptake. J Cell Biochem 92, 1062–1073 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Zhang J et al. , G-protein beta2 subunit interacts with mitofusin 1 to regulate mitochondrial fusion. Nat Commun 1, 101 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA, G alpha12 is targeted to the mitochondria and affects mitochondrial morphology and motility. FASEB J 22, 2821–2831 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lobingier BT, von Zastrow M, When trafficking and signaling mix: How subcellular location shapes G protein-coupled receptor activation of heterotrimeric G proteins. Traffic 20, 130–136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P, Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 110, 125–132 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Todkar K, Chikhi L, Germain M, Mitochondrial interaction with the endosomal compartment in endocytosis and mitochondrial transfer. Mitochondrion 49, 284–288 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Elbaz-Alon Y et al. , PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat Commun 11, 3645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsu F et al. , Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reichert AS, Neupert W, Contact sites between the outer and inner membrane of mitochondria-role in protein transport. Biochim Biophys Acta 1592, 41–49 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Baran H et al. , Effects of Various Kynurenine Metabolites on Respiratory Parameters of Rat Brain, Liver and Heart Mitochondria. Int J Tryptophan Res 9, 17–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baran H et al. , Kynurenic acid influences the respiratory parameters of rat heart mitochondria. Pharmacology 62, 119–123 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Baran H et al. , Kynurenines and the respiratory parameters on rat heart mitochondria. Life Sci 72, 1103–1115 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Lee DY et al. , Kynurenic acid attenuates MPP(+)-induced dopaminergic neuronal cell death via a Bax-mediated mitochondrial pathway. Eur J Cell Biol 87, 389–397 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Juhasz L et al. , Divergent Effects of the N-Methyl-D-Aspartate Receptor Antagonist Kynurenic Acid and the Synthetic Analog SZR-72 on Microcirculatory and Mitochondrial Dysfunction in Experimental Sepsis. Front Med (Lausanne) 7, 566582 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schneditz G et al. , GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump. Sci Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Agudelo LZ et al. , Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab 27, 378–392 e375 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Bolli R, The late phase of preconditioning. Circ Res 87, 972–983 (2000). [DOI] [PubMed] [Google Scholar]
- 67.Hausenloy DJ et al. , Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. The New England journal of medicine 373, 1408–1417 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Zwilling D et al. , Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 145, 863–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cozzi A, Carpenedo R, Moroni F, Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61–8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab 19, 771–777 (1999). [DOI] [PubMed] [Google Scholar]
- 70.Whorton MR et al. , A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A 104, 7682–7687 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao R, Podesser BK, Lim CC, The continuing evolution of the Langendorff and ejecting murine heart: new advances in cardiac phenotyping. Am J Physiol Heart Circ Physiol 303, H156–167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vasavda C, Zaccor NW, Scherer PC, Sumner CJ, Snyder SH, Measuring G-protein-coupled Receptor Signaling via Radio-labeled GTP Binding. J Vis Exp, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hung V et al. , Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCully JD et al. , Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol 296, H94–H105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate and reproduce the conclusions in the paper are present in the paper and/or the Supplementary Materials. All plasmids are available from the authors at request.