

Abstract
Background:
miRNA therapeutics have gained attention during the past decade. These oligonucleotide treatments can modulate the expression of miRNAs in vivo and could be used to correct the imbalance of gene expression found in human diseases such as obesity, metabolic syndrome and atherosclerosis. The in vivo efficacy of current anti-miRNAs technologies hindered by physiological and cellular barriers to delivery into targeted cells and the nature of miRNAs that allows one to target an entire pathway that may lead to deleterious off-target effects. For these reasons, novel targeted delivery systems to inhibit miRNAs in specific tissues will be important for developing effective therapeutic strategies for numerous diseases including atherosclerosis.
Methods:
We used pH Low-Insertion Peptides (pHLIP) constructs as vehicles to deliver miR-33 antisense oligonucleotides to atherosclerotic plaques. Immunohistochemistry and histology analysis was performed to assess the efficacy of miR-33 silencing in atherosclerotic lesions. WE also assessed how miR-33 inhibition affects gene expression in monocyte/macrophages by single cell RNA transcriptomics.
Results:
The anti-miR-33 conjugated pHLIP constructs are preferentially delivered to atherosclerotic plaques macrophages. The inhibition of miR-33 using pHLIP-directed macrophage targeting improves atherosclerosis regression by increasing collagen content and decreased lipid accumulation within vascular lesions. Single cell RNA sequencing analysis revealed higher expression of fibrotic genes (Col2a1, Col3a1, Col1a2, Fn1, etc) and tissue inhibitor of metalloproteinase 3 (Timp3), and downregulation of matrix metallopeptidase 12 (Mmp12) in macrophages from atherosclerotic lesions targeted by pHLIP- anti-miR-33.
Conclusions:
This study provides proof of principle for the application of pHLIP for treating advanced atherosclerosis via pharmacological inhibition of miR-33 in macrophages that avoids the deleterious effects in other metabolic tissues. This may open new therapeutic opportunities for atherosclerosis-associated cardiovascular diseases via selective delivery of other protective miRNAs.
Keywords: Basic Science Research, Cell Therapy, Lipids and Cholesterol, Vascular Biology
Graphical abstract
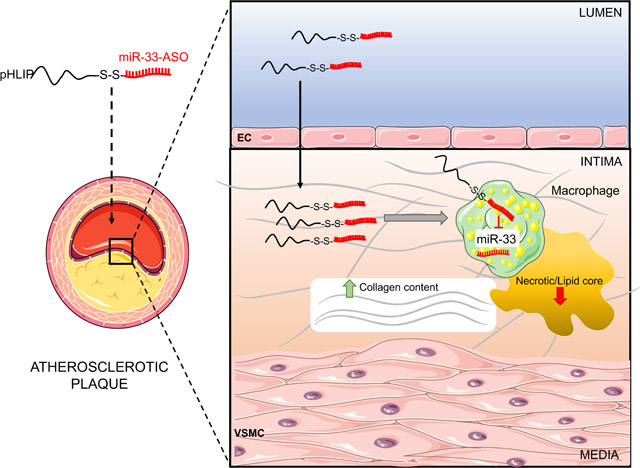
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease characterize by the retention of atheroprone lipoproteins and accumulation of monocyte-derived macrophages triggering maladaptive immune response within the vessel wall of large arteries1. The oxygen supply of vascular cells in atherosclerosis relies on the luminal blood or the adventitial vasa vasorum, and the distance to the deep layer of the intima exceeds the oxygen diffusion threshold resulting in local hypoxia2–5. The uptake of modified lipoproteins by macrophages differentiated from recruited monocytes results in the accumulation of macrophage foam cells in the atherosclerotic lesions. Specifically, hypoxia strongly correlates with the macrophage foam cell clusters surrounding the plaque core6. The higher oxygen demand of activated immune cells and insufficient oxygen supply lead to severe hypoxia and tissue acidification in the macrophage-rich regions of atherosclerotic lesions7, 8. Notably, the acidic extracellular environment in macrophage foam cells impairs the expression of ATP-binding cassette transporter ABCA1, resulting in decreased cholesterol efflux and accelerated lipid accumulation9.
Our group and others initially identified microRNA-33–5p (miR-33) as crucial regulator of cellular lipid homeostasis and lipoprotein metabolism, controlling downstream target genes including ABCA1 and ABCG110–16. The benefits of miR-33 deficiency on atherosclerosis development are attributed to its protective effects in macrophages12. Therapeutic inhibition of miR-33 in the mice and non-human primates raises plasma high density lipoprotein (HDL) levels and inhibits the progression of atherosclerosis by increasing HDL levels/functionality or enhancing cholesterol efflux through induction of ABCA1 and ABCG1 in macrophages14–17. Notably, specific disruption of Abca1 targeting by miR-33 is sufficient to mimic the effects of miR-33 deficiency on cholesterol efflux and atherogenesis18. However, long-term silencing of miR-33 increases circulating triglycerides levels and lipid accumulation in the liver through upregulation of genes involved in fatty acid synthesis when mice are fed a high-fat diet, indicating deleterious effects of moderate hepatic steatosis and hypertriglyceridemia19. Moreover, genetic models of miR-33 deficiency showed a strong predisposition to obesity and metabolic dysfunction12, 13, 19, 20. Thus, specific targeting of miR-33 in the macrophages of atherosclerotic lesions could provide an effective therapeutic strategy for atherosclerosis that avoids the deleterious effects in other metabolic tissues. In this study, we show that pH-Low Insertion Peptides (pHLIP) are an excellent vehicle to deliver anti-sense oligonucleotides to atherosclerotic lesions. Notably, we demonstrate that pHLIP conjugated with anti-miR-33 oligonucleotides significantly improves the capacity and specificity of miR-33 inhibitors to promote the regression of atherosclerosis.
METHODS
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
Wide-type (WT) and low-density lipoprotein receptor knockout (Ldlr−/−) were obtained from The Jackson Laboratory. Ldlr−/− mice were fed a high-fat and high-cholesterol Western-type diet (WD, 40% fat and 1.25% cholesterol, #D12108, Research Diets, Inc., New Brunswick, NJ) for 1 or 3 months to induce atherosclerosis. Mice used in all experiments were sex and age matched, kept in individually ventilated cages in pathogen-free facility. Male mice were used in atherosclerosis experiments and both sexes were used for macrophage isolation. All the experiments were approved by the Institutional Animal Care Use Committee of Yale University School of Medicine. To power our atherosclerotic studies, sample size was predefined as n=7 or greater per group for morphometric analyses n=6 or greater per group for immunostaining analysis based on a priori analysis of the inherent variability in our analyses and the ability to detect a 25% difference (a=0.05, power=80%).
Synthesis of PNA
The synthesis of anti-miR-33 and scramble PNA was performed as reported previously21, 22. Boc-PNA regular monomers used for the synthesis were purchased from ASM Chemicals and Research (Germany). Solid phase synthesis of PNA was carried out on 4-methylbenzylhydramine (MBHA) resin and cysteine was conjugated on N-terminus of PNAs. After completion of synthesis, PNA was cleaved from the resin using cleavage cocktail (Trifluoromethanesulfonic acid: trifluoroacetic acid: dimethyl sulfide: m-cresol, 2:6:1:1) and precipitated using diethyl ether. Further purification of PNA was performed using reverse phase high performance liquid chromatography (RP-HPLC). Mass spectroscopy (matrix assisted laser desorption/ ionization time of flight, MALDI) was used to confirm the molecular weight and concentration of PNAs was determined using UV-spectroscopy.
Synthesis and fluorescent labeling of pHLIP-PNA
pHLIP-PNA conjugations were performed as previously described23, 24. Briefly, Aldrithiol-2 (Millipore Sigma) was conjugated with pHLIP-Cys (ADDQNPWRAYLDLLFPTDTLLLDLLWCG, CS Bio) in dimethylformamide (DMF) at have a molar ratio of 1:1. The reaction mix was then mixed with 100 mM sodium phosphate and 150 mM NaCl buffer, pH 7.2 (saturated with argon), incubated at RT for 1 hour, purified by reverse-phase HPLC (Zorbax SB-C18 columns, 9.4 × 250 mm, 5 μm; Agilent Technologies, the gradient consisting of a binary solvent system using water and acetonitrile with 0.05% trifluoroacetic acid [TFA] for 20%–70% over 40 minutes), lyophilized, and characterized by surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry. Subsequently, PNAs were added to pHLIP-Cys(Pys) in DMSO (molar ratio 1:3) in the presence of 0.1 M ammonium bicarbonate, pH 8.8 (saturated with argon), incubated overnight at 30°C, purified by reverse-phase HPLC, lyophilized, and characterized by SELDI-TOF mass spectrometry. pHLIP (ACDDQNPWRAYLDLLFPTDTLLLDLLWA) and K-pHLIP (ACKKQNPWRAYLKLLFPTKTLLL KLLW) were conjugated at the N-terminus with Alexa Fluor 546-, 633- and Alexa Fluor 750-maleimide (Life Technologies) in DMF at a ratio of 1:1 at room temperature for about 2 hours. Constructs were purified by reverse-phase HPLC, lyophilized, and characterized by SELDI-TOF mass spectrometry. The concentrations of the constructs were determined by their absorbance using the following molar extinction coefficients: ε554 = 93,000 M/cm (for Alexa Fluor 546-pHLIPs), ε633 = 159,000 M/cm (for Alexa Fluor 633-pHLIPs), and ε753 = 290,000 M/cm (for Alexa Fluor 750-pHLIPs).
Assessment of pHLIP delivery
To determine the specificity of pHLIP delivery, animals were injected intravenously with pHLIP-A750-Var3 or the non-inserting control peptide pHLIP-A750–5K-Var3 (4 nmol). Animals were sacrificed after 4, 12 and 24 hours, and uptake in different tissues was determined by near-infrared fluorescence imaging, performed on an IVIS Spectrum system (Caliper Life Science) with appropriate excitation (Ex) and emission (Em) filter sets (Ex/Em = 745/800 nm). For flow cytometry analysis, entire aortas (from the root to the iliac bifurcation) were harvested 4 hours after injection with 4 nmol A546-Var3–5K or A546-Var3. The aortas were cut into small pieces and subjected to enzymatic digestion with 400 U/ml collagenase I, 125 U/ml collagenase XI, 60 U/ml DNAse I and 60 U/ml hyaluronidase (Sigma-Aldrich) for 1 h at 37ºC while shacking. Macrophages and monocytes were identified with the following antibodies (all from Biolegend): Lineage-PE (CD3, CD90.2 clone 53–2.1, CD19, Ly6G clone 1A8, NK1.1 clone PK126, Ter119 clone Ter119, CD11c clone HL3), CD11b-PacificBlue clone M1/70, F4/80-APC (BM8) and Ly6-C-FITC (clone AL-21). Flow cytometry was performed using a BD LSRII (BD Biosciences), and data were analyzed using FlowJo software v8.7 (Tree Star, Inc.).
Anti-miR-33pHLIP treatment
Ldlr−/− mice were fed a WD for 3 months to induce atherosclerosis and then transferred to chow diet (CD) for 1 month. anti-miR-33pHLIP and ScrpHLIP constructs were administered by intravenous injection at a dose of 1 mg/kg body weight in PBS with 5% DMSO. Injections were performed once every week (totally 5 times) during CD feeding before harvest. The mice fed with a WD for 3 months were harvested as Baseline for atherosclerosis analysis. Liver, spleen, lung and kidney were harvested for the analysis of miR-33 and its target genes, ABCA1 and ABCG1, expression. For in vitro pHLIP treatment, macrophages were incubated with 2 μM ScrpHLIP and anti-miR-33pHLIP for 2 hrs and changed to complete medium. The cell viability was analyzed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) analysis 24 hrs after pHLIP loading.
Plasma lipids, lipoprotein profile and leukocytes analysis
After 3-month WD and 1-month CD feeding, mice were fasted for 12–14 h before blood samples were collected by retro-orbital venous plexus puncture. Plasma was separated by centrifugation and stored at −80°C until analysis. Plasma total cholesterol (TC), high density lipoprotein-cholesterol (HDL-C) and triglyceride (TG) concentrations were determined by standard enzymatic methods (Wako Chemicals, USA). Plasma cholesterol fractions (VLDL, IDL/LDL and HDL) were detected by fast-performance liquid chromatography (FPLC) gel filtration on Superose 6 HR 10/30 size-exclusion column (Pharmacia) as described previously.25 White blood cells (WBC) counting in circulation was determined from EDTA-anticoagulated blood using a hemocytometer (Hemavet Counter HV950FS). Circulating aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were analyzed using commercial enzymatic assays.
Histology and morphometric analysis
Following anesthesia (100 mg/kg ketamine; 10 mg/kg xylazine), thoracic cavity was exposed immediately and in situ perfusion fixation through the left cardiac ventricle was performed by thorough perfusion with PBS and 4% paraformaldehyde (PFA). Subsequently, hearts and aortas were harvested and fixed in 10% formaldehyde solution overnight. Hearts were embedded in OCT after dehydration with 30% sucrose and serial sections were cut at 6 μm thickness using a cryostat. Every third slide from the serial sections was stained with haematoxylin and eosin (H&E) and each consecutive slide was stained with oil red O (ORO) for quantification of lesion area. Aortic lesion size of each animal was obtained by averaging the lesion areas in four sections from the same mouse. Collagen content was assessed by Picro Sirus Red staining of consecutive slides from serial sections and quantified as a percentage of the total plaque area.
Immunofluorescence staining
The atherosclerotic sections were fixed with 4% PFA and incubated overnight with primary antibodies for CD68 (Serotec; #MCA1957) and Actin, α-smooth muscle-Cy3 (Sigma, #C6198) after blocking with blocker buffer (5% Donkey Serum, 0.5% BSA, 0.3% Triton X-100 in PBS) for 1 hour at RT, followed by incubation with Alexa Fluor secondary antibody (Invitrogen, Carlsbad, CA) for 1 hour at RT. Rat IgG2a negative control antibody (Bio-Rad, MCA1212) and FITC-conjugated Goat anti-Rat IgG secondary antibody (ThermoFisher, #31629) were used to validate antibody specificity and distinguish genuine target staining from background. The stained sections were captured using a Carl Zeiss scanning microscope Axiovert 200M imaging system and images were digitized under constant exposure time, gain, and offset. Results are expressed as the percent of the total plaque area stained measured with the Image J software (ImageJ version 1.51, Yale software library, Yale University).
Aortic cells isolation from atherosclerotic lesions
To obtain the whole cells in the atherosclerotic lesions, the aorta was digested (from the root to the diaphragm) with 1 mg/ml Collagenase A (Roche, Cat 11088785103) for 7 min at 37 °C to remove the adventitia under microscope. Aortic tissue was cut into small pieces and subjected to enzymatic digestion with 1.5 mg/ml Collagenase A and 0.5 mg/ml Elastase (Worthington, Cat LS006365) for 40 min at 37 °C while shaking. The digested aortas were passed through a 70 μm Cell Strainer to obtain single cell suspensions followed by incubation of 10 minutes at 4°C with 10 μg/ml of purified rat anti-mouse FcgRII/III (Biolegend) to block non-specific binding of antibodies to Fc Receptors. Total cell viability was obtained using live/dead viability dye eFluor 450 (Thermo Fisher Scientific). Viable cells were sorted by FACS Aria III (BD Biosciences) and immediately processed for single-cell RNA-seq (sc-RNA-seq).
Droplet-based sc-RNA-seq library construction and sequencing
The sorted viable cells were encapsulated into droplets and processed following manufacturer’s specifications using 10X Genomics GemCode Technology. Equal numbers of cells per sample were loaded on a 10x Genomics Chromium controller instrument to generate single-cell Gel Beads in emulsion (GEMs) at Yale Center for Genome Analysis. Lysis and barcoded reverse transcription of polyadenylated mRNA from single cells were performed inside each GEM followed by cDNA generation using the Single Cell 3’ Reagent Kits v3 (10X Genomics). Libraries were sequenced on an Illumina NovaSeq 6000 as 2 × 100 paired-end reads.
sc-RNA-seq data analysis
Sample demultiplexing, aligning reads to the mouse genome (mouse UCSC mm10 reference genome) with STAR and unique molecular identifier (UMI) processing were processed using CellRanger software (version 4.0.0) as previously described.26 Low quality cells, doublets and potentially dead cells were filtered based on the percentage of mitochondrial genes and number of genes and UMIs expressed in each cell: nFeature_RNA > 500, nCount_RNA < 25,000 and percent.mt < 5. After filtering we identified 7,771 ScrpHLIP cells (a mean of 32,589 reads per cell, and a median 2,072 genes per cell) and 13,424 anti-miR-33pHLIP cells (a mean of 18,886 reads per cell, and a median 1,722 genes per cell) for downstream analysis. Data clustering was performed using Seurat R package (Version 3.0) with filtered genes by barcode expression matrices as inputs.27 Highly variable genes (HVGs) were calculated using Seurat function FindVariableFeatures and used for downstream clustering analysis. Principal component analysis (PCA) was performed with RunPCA function (Seurat) using HVGs for dimensionality reduction and the number of significant principlal components was calculated using JackStraw function. We applied the RunUMAP function to significant principal components (PCs) identified by JackStraw analysis and presented data in two-dimensional coordinates through uniform manifold approximation and projection (UMAP) generated by R package ggplot2. Clustering was done through FindClusters function using 30 significant PCs with a resolution of 0.3. Significantly differentially expressed genes in a cluster were analyzed using Seurat function FindAllMarkers, which were expressed in more than 25% of cells with at least 0.25-fold difference and reach statistical significance of an adjusted p<0.05 as determined by the Wilcox test. Ingenuity Pathway Analysis (Ingenuity Systems QIAGEN, Content version: 47547484, 2019, Redwood City, CA, USA) was used to carry out analyses for pathway with differentially-expressed genes across samples.
In vivo foam cell formation
In vivo foam cell formation was performed as previously described28. Briefly, Ldlr−/− mice were fed a WD for 3 months to establish atherosclerotic plaques, then switched to a chow diet (CD) and injected with ScrpHLIP and anti-miR-33pHLIP (1 mg/Kg) weekly for one month. Peritoneal cells were collected from the peritoneal cavity 4 days after i.p. administration of 3% thioglycollate medium and allowed to adhere for 30 min. After removing non-adherent cells, the adherent macrophages were fixed by 4% PFA in PBS for 1h and stained for 30 min with 0.3% Oil-Red O solution in 60% isopropanol. The mean area of Oil Red O-stained region per cell were quantified with 200 representative cells using Image J software from the NIH.
Western blot analysis
In vivo foam cells were lysed in ice-cold buffer containing 50 mM Tris-HCl, pH 7.4, 0.1 mM EDTA, 0.1 mM EGTA, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 100 mM NaCl, 10 mM NaF, 1 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM Pefabloc, and 2 mg/ml protease inhibitor cocktail (Roche Diagnostics Corp). Protein concentrations were determined using the DC Protein assay kit (Bio-Rad Laboratories). Cell lysates containing 50 μg of protein were analyzed by SDS-PAGE and immunoblotting. Primary antibodies used include the following: anti-ABCA1 (Abcam, ab18180), anti-ABCG1 (Novus Biologicals, NB400–132) and anti-HSP90 (BD Biosciences, #610419). Secondary antibodies were fluorescence-labeled antibodies and bands were visualized using the Odyssey Infrared Imaging System (LI-COR Biotechnology).
qPCR analysis
Total RNA from cells was isolated using TRIzol reagent. One microgram of total RNA was reverse-transcribed using the iScript RT Supermix (Bio-Rad, Hercules, CA, USA), following the manufacturer’s protocol. Quantitative real-time PCR was performed in triplicate using iQ SYBR green Supermix (Bio-Rad) on a Real-Time Detection System (BioRad). The mRNA level was normalized to GAPDH as a housekeeping gene. Real-time PCR was conducted with gene expression levels with oligonucleotides specific for each of the genes.
Flow cytometry
Blood was collected by retro-orbital puncture in heparinized micro-hematocrit capillary tubes. Erythrocytes were lysed with ACK lysis buffer (155 mM Ammonium Chloride, 10 mM Potassium Bicarbonate, 0.01 mM EDTA, pH 7.4). WBC were resuspended in 3% FBS in PBS, blocked with 2 μg/ml of FcgRII/III, then stained with a cocktail of antibodies. Monocytes were identified as CD115hi and subsets as Ly6-Chi and Ly6-Clo; neutrophils were identified as CD115loLy6-ChiLy6-Ghi. The following antibodies were used (all from BioLegend): FITC-Ly6-C (AL-21), PE-CD115 (AFS98), APC-Ly6-G (1A8). Flow cytometry was performed using a BD LSRII (BD Biosciences), and data were analyzed using FlowJo software v8.7 (Tree Star, Inc.).
Ex vivo foam cell formation.
Bone marrow-derived macrophages (BMMs) from WT mice plated in 12-well plates with cover glass were exposure to acetylated LDL [ac-LDL (100 μg/ml] for 48 hours in the presence of 10% lipoprotein deficient serum (LPDS, Biomedical Technologies, Inc) and detected by Oil-Red O staining as described previously29. The mean integrated optical density (IOD) of Oil Red O-stained region per cell was quantified with 200 representative cells using the Image J software.
THP-1-derived macrophages
THP-1 cells obtained from ATCC were cultured according to manufacturer’s instructions and differentiated into macrophages with 160 nM Phorbol 12-myristate 13-acetate (PMA, Sigma) for 12 hours in complete media. 2 μM A633-pHLIP was incubated with THP-1-derived macrophages for 2 hours and the uptake of pHLIP by macrophages was analyzed by flow cytometry. For macrophage foam cell formation, cells were loaded with 2 μM ScrpHLIP and anti-miR-33pHLIP, exposed to 120 μg/ml ac-LDL for 24 hours and analyzed by Oil-red O staining as described above. The expression of ABCA1 and ABCG1 in THP-1-derived macrophages was analyzed by western blot 24 and 48 hours after pHLIP loading.
DiI-Ac-LDL uptake
Macrophage uptake of ac-LDL was performed using ac-LDL labeled with fluorescent probe, 1,1’-dioctadecyl-1 to 3,3,3’,3’- tetramethylindocarbocyanine perchlorate (Dil, Biomedical Technologies, inc) as described previously 30. Macrophages loaded with 2 μM ScrpHLIP and anti-miR-33pHLIP were incubated with 10 μg/ml DiI-ac-LDL for 8 hours at 37℃ in the presence of 10% LPDS to investigate the extent of internalization. Cells were washed with acid wash buffer (pH 2.7; 25 mM Glycine, 3% BSA in PBS) for four times and collected for flow cytometry analysis using BD LSRII (BD Biosciences). Autofluorescence of unlabeled cells was determined and subtracted from total fluorescence to calculate the mean fluorescence intensity (MFI).
Cholesterol efflux experiment
The cellular cholesterol efflux assay was modified from the methods described31. Briefly, macrophages loaded with 2 μM ScrpHLIP and anti-miR-33pHLIP were cholesterol-loaded and labeled for 24 hours in DMEM supplemented with 10% FBS and 2 μCi/ml 3H-cholesterol (GE Healthcare) that had been preincubated for 30 min at 37°C with 75 μg/ml ac-LDL. After cholesterol loading, the cells were washed twice and incubated in DMEM containing 2 mg/ml fatty acid-free BSA for 24 hours to allow equilibration of the label. The cells were then washed three times with DMEM, and cholesterol efflux was determined in the presence of human recombinant apolipoprotein A-I (ApoA1, 50 μg/mL) or fetal bovine serum (FBS, 10%) in serum-free media for 4 hours. The amount of 3H-cholesterol in the media and cell lysates was then measured by liquid scintillation counting. Efflux was expressed as a percentage of 3H-cholesterol in medium/(3H-cholesterol in medium + 3H-cholesterol in cells) × 100%. Efflux to the medium without ApoA1 or FBS was determined and subtracted from the wells containing ApoA1 or FBS.
scRNA-Seq data
Data were deposited in the NCBI’s Gene Expression Omnibus database (GEO) number: GSE191220.
Statistical analysis
GraphPad Prims 7.0 (GraphPad Software, CA) was used for statistical analysis. Data are shown as mean±S.E.M with individual data points. Normality data was first analyzed by Shapiro-Wilk normality test. The statistical differences of normally distributed data were calculated with either unpaired two-sided Student’s t-test, one-way or two-way analysis of variance (ANOVA) followed by the Bonferroni post-test as indicated in figure legends. A nonparametric test (Mann–Whitney) was used when data did not pass the normality test. A value of P≤0.05 was considered statistically significant.
RESULTS
pH-Low Insertion Peptides (pHLIP) target atherosclerotic plaque macrophages
pHLIP are a novel class of water-soluble membrane molecules that target areas of high acidity at the surface of cells, which have been employed to deliver miRNA inhibitors to the acidic environment of tumors and the kidney24, 32. Given the hypoxia in macrophage foam cells and the acidic environment of the lipid core in atherosclerosis, we explore the utility of anti-miR-33 peptide nucleic acid (PNA) delivery vectors (anti-miR-33pHLIP) for specific targeting of the macrophages in vascular lesions (Figure 1A). Notably, near-infrared fluorescence imaging showed highly specific uptake of pHLIP variant 3 conjugated with fluorescent Alexa 750 (A750-Var3) in the aortic arch, an atherosclerotic prone area, of hypercholesterolemic mice. Other tissues characterized by the acidic microenvironment such as the kidney also accumulate the peptide. The targeting of VAR3 to atherosclerotic plaques was specific since a similar peptide with an altered amino acid sequence that prevents insertion across the membranes in acidic conditions (A546–5K-Var3) was unable to target vascular lesions (Figure 1A). The targeting of A546-Var3 into the aortic arch occurred early (4 hours) and was sustained for 24 hours (Figure S1A and S1B). Imaging of whole organs indicated that uptake in the liver was diminished with the A546-Var3 compared to the A546–5K-Var3 mutant (Figure S1A and S1B), while similar fluorescent density was observed in histologic sections (Figure S1C). Higher uptake of A546-Var3 was observed in primary renal tubular cells (Figure S1C). The affinity of A750-Var3 to atherosclerotic aortic arch was confirmed by analyzing aortas in low-density lipoprotein receptor knockout (Ldlr−/−) and WT mice injected with A750-Var3, A750–5K-Var3 or PBS (Figure 1B and C). We further demonstrated the internalization of A546-Var3 in isolated macrophages from atherosclerotic aortas by flow cytometry. The results showed a significant uptake of A546-Var3 in macrophages (CD45+CD11bhighF4/80+Ly-6Clow) and monocytes (CD45+CD11bhighLy-6Chigh), but no difference in endothelial cells (CD45-CD31+) or other CD45- cells from atherosclerotic plaques (Figure 1D and S2). These findings correlate with our previous studies showing a specific uptake of pHLIP in tumor associated macrophages33. The uptake of A546-Var3 in monocyte enrichment tissues such as spleen and lung were significantly lower compared to kidney and aortic lesions (Figure S3A). Like our previous findings in normolipemic mice, splenic monocytes were unable to internalize pHLIP, indicating that the acidic microenvironment is required for the uptake of the peptide in vivo33 (Figure S3B).
Figure 1. pH low insertion peptides (pHLIP) target macrophages in atherosclerotic plaques.

A, Generation of anti-miR-33 PNA-conjugated pH low insertion peptides (pHLIP) variant 3 capable of delivering miR-33 inhibitors to acidic microenvironments. B, Representative images of tissues from low density lipoprotein receptor knockout (Ldlr−/−) mice injected with fluorescently labeled targeted (A750-Var3) or non-inserting control (A750–5K-Var3) constructs. Ldlr−/− mice were fed a western diet (WD) for 3 months to induce atherosclerosis and tissues were harvested 4 hours post injection of constructs. Quantification of the percentage of total intensity distribution across indicated tissues is shown in right panel. Quantification represents the mean ± SEM (n=3). C, Representative images of aortas from WT or Ldlr−/− mice injected with PBS, A750–5K-Var3 or A750-Var3 constructs. Tissues were harvested 4 hours post injection. D, Representative flow cytometry histograms showing fluorescence intensities in macrophages (Mac), monocytes (Mono) and endothelial cells (EC) isolated from atherosclerotic lesions of mice injected with PBS, A546–5K-Var3 or A546-Var3 constructs. Quantification of the median fluorescence intensity [MFI; arbitrary units (a.u.] in macrophages, monocytes and ECs is shown in right panels. Quantification represents the mean ± SEM (n=4 in A546–5K-Var3 and n=5 in A546-Var3 group). A two-way ANOVA with a Bonferroni test for multiple comparisons was used to determine significance.
Anti-miR-33pHLIP increases ABCA1 expression and attenuates lipid accumulation in macrophages
We next assessed the efficacy in delivering fluorescently labeled constructs (A633-Var3) to foamy macrophage in vivo. Notably, we found a marked uptake by in vivo foam cells isolated from Ldlr−/− mice fed a WD for 3 months using both fluorescent microscopy and flow cytometry (Figure 2A). To assess the efficacy of anti-miR-33pHLIP in suppressing miR-33 expression and preventing foam cell formation in vivo, we injected Ldlr−/− mice fed a Western diet (WD) with anti-miR-33pHLIP, and non-targeting antisense oligonucleotide conjugated with pHLIP (ScrpHLIP) and isolated peritoneal macrophages. anti-miR-33pHLIP resulted in significant reduction of miR-33 levels (Figure 2B) and neutral lipid accumulation (Figure 2C, quantified in right panel) compared to macrophages treated ScrpHLIP. The marked reduction in foamy macrophages correlated with an increase of ABCA1 expression in anti-miR-33pHLIP treated macrophages (Figure 2D, quantified in right panel).
Figure 2. Selective targeting of miR-33 using pHLIP attenuates in vivo foam cell formation through induction of ABCA1.

A, Representative images and flow cytometry histograms of in vivo foam cells from mice injected with PBS and fluorescently labeled pHLIP variant 3 constructs (A633-Var3). B, qRT-PCR analysis of miR-33 expression in peritoneal macrophages isolated from mice treated with anti-miR-33 peptide nucleic acid delivery vector (anti-miR-33pHLIP) or scrambled control (ScrpHLIP). Quantification represents the mean ± SEM (n=3). C, Representative Oil-red O (ORO) staining of in vivo foam cells from mice injected with anti-miR-33pHLIP or ScrpHLIP. Quantification of lipid accumulation are shown in the right panel and represents the mean ± SEM (n=5). D, Representative Western blot analysis of ABCA1 and ABCG1 in in vivo foam cells isolated from mice injected with anti-miR-33pHLIP or ScrpHLIP. Quantification is shown in the right panel and represents the mean ± SEM (n=5). Results were analyzed with a nonparametric Mann-Whitney test. Scale bar: 100 μm.
To further explore the effect of anti-miR-33pHLIP in regulating macrophage cholesterol metabolism, we treated mouse primary macrophages anti-miR-33pHLIP and ScrpHLIP, and assess cellular viability, lipoprotein uptake and cholesterol efflux. Incubation of macrophages with both conjugated peptides did not affect cellular viability (Figure S4A) and lipoprotein uptake (Figure S4B) under neutral and acidic conditions. anti-miR-33pHLIP treatment was able to enhance cholesterol efflux to ApoA1 in macrophages cultured at different pH (Figure S4C). Our results in vivo demonstrated a high efficiency in delivering anti-miR-33pHLIP to foamy macrophages. Thus, we wondered whether lipid accumulation might facilitate the uptake of the anti-miR-33pHLIP. To test this possibility, we loaded mouse primary macrophages with acetylated LDL (Ac-LDL) for 24 hours and assessed the uptake of fluorescence pHLIP by flow cytometry. Interestingly, we found a significant increase in pHLIP uptake in foamy macrophages compared to non-foamy cells in vitro (Figure S4D), which is consistent with higher pHLIP uptake in peritoneal macrophages isolated from WD fed mice than that from chow diet (CD) fed mice (Figure S4E). We further analyzed whether pHLIP can be used to deliver antisense oligonucleotides to human monocytes. Notably, we found that pHLIP can be efficiently delivered to THP-1 cells and anti-miR-33pHLIP attenuated neutral lipid accumulation (Figure S5A and S5B). These results correlate with the significant upregulation of ABCA1 and ABCG1 observed in macrophages treated with anti-miR-33pHLIP compared to ScrpHLIP treated macrophages (Figure S5C). Together, these results indicate that pHLIP is an effective and highly specific vehicle to target the expression of miRNAs in foamy macrophages accumulated in atherosclerotic plaques.
Anti-miR-33pHLIP promotes the regression of atherosclerosis
Considering patients are generally not treated in the early stage of atherosclerosis, we next assessed the efficacy of inhibiting miR-33 expression in lesional macrophages during the regression of atherosclerosis. To this end, Ldlr−/− mice were fed a Western diet (WD) for 3 months to establish atherosclerotic plaques, then switched to a chow diet (CD) and injected with ScrpHLIP and anti-miR-33pHLIP (1 mg/Kg) weekly for one month (Figure 3A). In this model, the switch from WD to chow diet results in reduced circulating lipid levels and increased plaque stability, characterized by decreased overall macrophage content and lipid accumulation, increased fibrotic material, reduced apoptosis and a smaller necrotic core in the atherosclerotic lesions, without influencing the plaque size34, 35. Consistent with previous studies, switching Ldlr−/− mice to CD resulted in a significant decrease of circulating total cholesterol (TC) and triglycerides (TG) level and increase in plasma HDL-C (Figure S6A). There was no difference between mice treated with anti-miR-33pHLIP and ScrpHLIP, suggesting that the hepatic delivery of anti-miR-33 using pHLIP was not sufficient to impact regulation of HDL biogenesis by miR-33 in the liver (Figure S6A). Similar lipoprotein profiles (Figure S6B), body weight (Figure S6C) and circulating leukocytes (Figure S7A) were observed in mice treated with anti-miR-33pHLIP and ScrpHLIP. Moreover, we did not observed differences in serum hepatotoxicity markers, aspartate aminotransferase (AST) and alanine aminotransferase (ALT), in mice treated with anti-miR-33pHLIP compared with ScrpHLIP-treated mice (Figure S7B). Together, these results and our previous observations showing that anti-miR-33pHLIP treatment did not affect circulating BUN and creatinine levels24, indicate that pHLIP delivery of antisense oligonucleotides is an effective vehicle for targeting miRNAs in vivo without causing hepatic and renal toxicity.
Figure 3. Targeted silencing of miR-33 improves regression of atherosclerosis.

A, Experimental protocol for atherosclerosis regression. Low density lipoprotein receptor knockout (Ldlr−/−) mice were placed on a western diet (WD) for 3 months, and then switched to a chow diet and received anti-miR-33 peptide nucleic acid delivery vector (anti-miR-33pHLIP) or scrambled control (ScrpHLIP) intravenously at a dose of 1 mg/kg body weight every week for a total of 5 injections. B, Representative histological analysis of cross sections of the aortic root stained with hematoxylin and eosin (H&E) of Ldlr−/− mice injected with anti-miR-33pHLIP or ScrpHLIP vectors. Quantification of plaque size is shown in the right panel and represents the mean ± SEM (n=8 in Baseline and n=13 in anti-miR-33pHLIP or ScrpHLIP group). C, Representative Oil-red O (ORO) staining of cross sections of the aortic root from Ldlr−/− mice injected with anti-miR-33pHLIP or ScrpHLIP vectors. Quantification of lipid accumulation is shown in the right panel and represents the mean ± SEM (n=6 in Baseline and n=5 in anti-miR-33pHLIP or ScrpHLIP group). D, qRT-PCR analysis of Abca1 mRNA expression in atherosclerotic aortas from Ldlr−/− mice injected with anti-miR-33pHLIP or ScrpHLIP vectors. Data represents the mean ± SEM (n=4 in Baseline, n=6 in ScrpHLIP and n=3 in anti-miR-33pHLIP group). E, Representative images of Picrosirus red staining of collagen in cross sections of the aortic root from Ldlr−/− mice injected with anti-miR-33pHLIP or ScrpHLIP vectors. Quantification of collagen content in atherosclerotic lesions is shown in the right panel and represents the mean ± SEM (n=6 in Baseline, n=7 in ScrpHLIP and n=8 in anti-miR-33pHLIP group). Data were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. Scale bar: 100 μm. NS, no significance.
We next sought to determine whether delivery of anti-miR-33 by pHLIP peptides could contribute to the regression of established atherosclerotic plaques. Consistent with our previous studies 35, miR-33 antisense oligonucleotide treatment did not influence plaque size as compared with mice treated with ScrpHLIP (Figure 3B, quantified in right panel). However, we found that anti-miR-33pHLIP treatment promoted a significant decrease of lipid accumulation in atherosclerotic lesions, which correlated with higher expression of Abca1 mRNA in the aorta (Figure 3C and D). No differences in miR-33, ABCA1 and ABCG1 expression levels were found in the liver, spleen, lung and kidney of mice injected with anti-miR-33pHLIP and ScrpHLIP, suggesting that pHLIP direct the specific silencing of miR-33 into the arterial wall (Figure S8A and S8B). We further characterized the plaques by analyzing markers of inflammation and lesion stability. No statistical difference was observed in the quantification of macrophage content in the lesions by CD68+ staining in anti-miR-33pHLIP treated mice compared to controls (Figure 4). Notably, there was a significant increase in lesional collagen (Figure 3E, quantification right panel) without statistical difference on smooth muscle cell content (Figure 4) in mice treated with anti-miR-33pHLIP. Together, these results indicate that targeted inhibition of miR-33 in vascular lesions promotes macrophage cholesterol efflux and atherosclerotic plaque remodeling resulting in a more stable phenotype.
Figure 4. Targeted silencing of miR-33 does not influence macrophage and smooth muscle cell content in atherosclerotic lesions.

Low density lipoprotein receptor knockout (Ldlr−/−) mice were placed on a western diet (WD) for 3 months, and then switched to a chow diet and received anti-miR-33 peptide nucleic acid delivery vector (anti-miR33pHLIP) or scrambled control (ScrpHLIP) at a dose of 1 mg/kg body weight every week for a total of 5 injections. Representative immunofluorescence staining of macrophage (CD68 positive) and smooth muscle cell (α-smooth muscle actin) in cross sections of the aortic root from Ldlr−/− mice injected with anti-miR33pHLIP or ScrpHLIP vectors. Quantification of macrophage and smooth muscle cell content are shown in the right panel and represent the mean ± SEM (n=8 in Baseline, n=12 in ScrpHLIP and n=13 in anti-miR-33pHLIP group). Data were analyzed by one-way ANOVA with Bonferroni correction for multiple comparisons. Scale bar: 100 μm.
Atherosclerotic plaque macrophages from Anti-miR-33pHLIP treated mice show a pro-fibrotic phenotype that favors plaque stabilization
To assess the potential mechanisms by which anti-miR-33pHLIP regulates macrophage function and regression of atherosclerosis, we isolated live whole cells from enzyme-digested aortas of anti-miR-33pHLIP- and ScrpHLIP-treated mice and performed single cell transcriptomics (Figure 5A and Figure S9). A total of 7,771 ScrpHLIP cells and 13,424 anti-miR-33pHLIP cells were obtained from atherosclerotic plaques in each group and run on the 10x Genomics platform. Unsupervised Seurat-based clustering identified 12 distinct cell clusters based on gene expression of established canonical markers including monocytes/macrophages (Mono/Mac, Cluster 2, 10 and 11), endothelial cells (EC, Cluster 3), dendritic cells (DC, Cluster 8), vascular smooth muscle cells (VSMC, Cluster 0, 1 and 4), T cells (Cluster 5 and 7) and fibroblasts (Cluster 6 and 9) (Figure 5A and Figure S9). Given the cellular delivery of anti-miR-33 by pHLIP into macrophages and monocytes in atherosclerotic lesions (Figure 1D), we further analyzed the phenotype of these Mono/Mac populations during the regression of atherosclerosis (Figure S10). Specific gene expression profiles differentiated 5 aortic monocyte and macrophage populations from the Mono/Mac clusters (Figure 5B–C and Figure S9A). These included Trem2high Mac (Cluster 0: Trem2, Cd9, Spp1, Lgals3), F10+ Mono (Cluster 2: F10, Ccr2, low H2-Eb1), Inflammatory Mac (Cluster 3: Il1b, Cxcl2, Nfkbiz, S100a9, S100a8) and Stem-like Mac (Cluster 4: Top2a, Ube2c, Cenpf, Stmn1) (Figure 5C). Interestingly, besides these well-established cell populations found in atherosclerotic lesions (Trem2high Mac, F10+ Mono, Inflammatory Mac and Stem-like Mac),36, 37 we observed one specific macrophage cluster (Cluster 1, herein referred to as “ECMhigh Mac”) that expressed monocyte and macrophage genes (Cd14, Cd68, Adgre1 and Csf1r) (Figure S10A) and was highly enriched for extracellular matrix (ECM)-associated genes, including Col1a2, Col3a2, Col1a1, Fn1, Eln, Lum, Bgn and Dcn (Figure 5C and Figure S10C). The percentage of ECMhigh Mac were increased in the mice treated with anti-miR-33pHLIP as compared to ScrpHLIP (Figure 5D). This intriguing finding indicates that suppression of miR-33 in macrophages during the regression of atherosclerosis promotes a pro-fibrotic phenotype that favour plaque stabilization. Notably, the population of inflammatory macrophages (Cluster 3) characterized by the high expression of pro-inflammatory cytokines and chemokines was decreased, and the “stem-like macrophages” in cluster 4, enriched for cell cycle genes and highly proliferative, were increased in the mice treated with anti-miR-33pHLIP as compared to ScrpHLIP (Figure 5D). We next performed pathway enrichment analysis associated with changes in gene expression in Mono/Mac cells (Table S1). Among all the pathways that were significantly altered in response to anti-miR-33pHLIP treatment, we found upregulated fibrosis, M2 polarization (IL-4 Signaling) and antigen presentation pathways (Figure 5E and Table S2). In addition to an increased signature with extracellular matrix and tissue inhibitors of metalloproteinases (Timp3) (Figure 5F and Table S1), we also observed a decrease of matrix metalloproteinase gene (Mmp12) and an increase of antigen presentation genes (Cd74, H2-Eb1, H2-Aa, H2-Ab1 and H2-K1) in the mice treated with anti-miR-33pHLIP (Fig. 5G). Consistent with our current findings, previous reports from our lab and others observed reduced expression of matrix metalloproteinase 9 and increased expression of COLA1–1 and COL3A-1 16, 38, suggesting the important role of miR-33 in extracellular matrix remodeling. Considering the role of miR-33 in inflammation and the cross talk between inflammation and extracellular matrix remodeling12, 39, the indirect effect of anti-miR-33 delivered by pHLIP in macrophage extracellular matrix components might be attributed to its regulation in inflammation and generation of factors that promote remodeling of the extracellular matrix including transforming growth factor-β, fibronectin and matrix metalloproteinases 40–42. Together, these results indicate that anti-miR-33 treatment by pHLIP peptides induces macrophages towards a more stable phenotype in the atherosclerotic lesions.
Figure 5. Single cell RNA-Seq analysis reveals increased expression of extracellular matrix (ECM) genes in macrophages isolated from anti-miR-33pHLIP treated mice.

A, Uniform manifold approximation and projection (UMAP) representation of aligned gene expression data in single cells extracted from atherosclerotic plaques of anti-miR-33pHLIP and ScrpHLIP-treated mice (n=3 per group). UMAP dimensionality reduction analysis identified 12 major clusters. B, UMAP representation of aligned gene expression data in monocytes and macrophages (Mono/Mac) cluster extracted from atherosclerotic plaques. C, Heatmap of the 20 most upregulated genes in each cluster defined in B and selected enriched genes used for biological identification of each cluster. D, Proportions of defined Mono/Mac populations extracted from the atherosclerotic aortas of anti-miR-33pHLIP and ScrpHLIP-treated mice. E, Pathway enrichment of differentially expressed genes expressed as the log[–P] analyzed by Ingenuity Pathway Analysis. F, Violin plots of the top differentially expressed genes showing statistically significant upregulation of ECM in Mono/Mac population from anti-miR-33pHLIP-treated mice. G, Violin plots of the top differentially expressed genes showing less inflammatory and higher antigen presentation genes in Mono/Mac population from anti-miR-33pHLIP-treated mice.
DISCUSSION
miRNAs represent an elegant mechanism of posttranscriptional regulation of gene expression that serves to fine-tune biological processes. One of the most unique aspects of miRNAs is their ability to target many different mRNAs, which allows them to exert both very nuanced and extremely pronounced effects in different situations. However, this promiscuity has also raised important concerns for both research and clinical applications, especially since the target preferences and impact of miRNAs can vary dramatically in different tissues and cell types. During the last few years, a number of groups have developed different strategic approaches to overcome this challenge. These include the delivery of miRNAs using 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) nanoliposomes, poly(lactic-co-glycolic acid (PLGA), chitosan nanoparticles and reconstituted HDL (rHDL) nanoparticles43–45. While these nanoparticles protect against the degradation of miRNAs by circulating nucleases and facilitates the passive delivery to tissues, methods enabling tissue and cell-selective delivery of miRNA mimics and inhibitors are highly desirable to enhance the efficacy of these pharmacological therapies and avoid deleterious side effects. Previous work from our group and others have demonstrated the efficacy of pHLIP as a vehicle to deliver miRNA mimics and inhibitors to acidic microenvironments such as tumors and the kidney 24, 32, 33. Notably, anti-miR-21 oligonucleotides conjugated with pHLIP directly target tumor associated macrophages and attenuate tumor progression33. In this study, we demonstrate the remarkable selectivity of pHLIP peptides to target macrophages in atherosclerotic lesions based on their affinity for acidic microenvironments. In contrast to other anti-miR-33 therapeutic approaches using chemically modified antisense oligonucleotides that significantly inhibits miR-33 activity in several organs including the liver14, 46, anti-miR-33pHLIP direct the silencing of miR-33 to atherosclerotic lesions, thus avoiding potential off target effects that the anti-miRNA therapy may cause in other tissues. The specific delivery of anti-miR-33 to atherosclerotic plaques using pHLIP peptides promotes a more stable phenotype by induction of ABCA1-mediated cholesterol efflux and extracellular matrix deposition. These findings highlight the therapeutic potential of anti-miR-33pHLIP constructs for the regression of atherosclerosis, while avoiding the potential deleterious effects in other organs. The pHLIP technology can also be applied to the selective delivery of other protective miRNAs to the macrophages from atherosclerotic plaques for the therapy of atherosclerosis-associated cardiovascular diseases. Considering the potential for disparate and possibly adverse effects in different organs, targeted delivery systems such as that described in this work may prove incredibly valuable for the development of safe and reliable miRNA-based therapies for treating atherosclerosis.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Inhibition of miR-33 has been shown to reduce the progression of atherosclerosis by increasing circulating high-density lipoprotein (HDL) and enhancing cholesterol efflux in atherosclerotic macrophages.
Long-term silencing or global genetic deficiency of miR-33 resulted in deleterious effects including obesity, moderate hepatic steatosis and hypertriglyceridemia.
pH-Low Insertion Peptides (pHLIP) has been demonstrated as a specific and effective vehicle to deliver miRNA mimics and inhibitors targeting tumor and kidney.
What New Information Does This Article Contribute?
Anti-miR-conjugated pHLIP constructs deliver anti-sense oligonucleotides to macrophages in atherosclerotic lesions.
Specific targeting of miR-33 using pHLIP improves the regression of atherosclerosis by increasing collagen content and decreasing lipid accumulation.
Silencing of miR-33 using pHLIP peptides does not alter body weight or cause off target effects in other tissues.
Inhibition of miR-33 has been shown to attenuate the progression and accelerate the regression of atherosclerosis, but silencing or genetic deficiency of miR-33 results in deleterious effects including obesity and metabolic dysfunction. In this study, to deliver anti-sense oligonucleotides to atherosclerotic lesions, we generated anti-miR-33 conjugated pHLIP constructs, which have been demonstrated to be effective vehicles to deliver miRNA mimics and inhibitors for the therapy of tumor and renal diseases. Specific targeting of miR-33 using pHLIP in the macrophages of atherosclerotic lesions improves atherosclerosis regression by increasing collagen content and reducing lipid accumulation but does not influence body weight and off target effects in other tissues. Our findings indicate that pHLIP is an effective delivery vehicle of nucleotides to the macrophages accumulated in atherosclerotic lesions that can be used as therapeutic tool to treat atherosclerosis-associated cardiovascular diseases.
Sources of Funding
This work was supported by grants from the National Institutes of Health (R35HL135820 to CF-H; and R35HL155988 to YS and 1K01DK120794 to NP), the American Heart Association (20TPA35490416 to CF-H and 17SDG33110002 to NR).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- ABCA1
ATP-binding cassette transporter A1
- ABCG1
ATP-binding cassette transporter G1
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CD
chow diet
- DC
dendritic cells
- EC
endothelial cells
- ECM
extracellular matrix
- HDL-C
high density lipoprotein-cholesterol
- Ldlr
low-density lipoprotein receptor
- Mac
macrophages
- miR-33
microRNA-33–5p
- Mmp
matrix metalloproteinase
- Mono
monocytes
- pHLIP
pH-Low Insertion Peptides
- PNA
peptide nucleic acid
- Timp
tissue inhibitors of metalloproteinases
- TC
total cholesterol
- TG
triglyceride
- VSMC
vascular smooth muscle cells
- WD
Western-type diet
- WT
Wide-type
Footnotes
Disclosures
O.A.A. and Y.K.R. are founders of pHLIP, Inc. They have shares in the company, but the company did not fund any part of the work reported in this paper, which was carried out in their academic laboratories.
REFERENCES
- 1.Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levin M, Leppanen O, Evaldsson M, Wiklund O, Bondjers G, Bjornheden T. Mapping of atp, glucose, glycogen, and lactate concentrations within the arterial wall. Arterioscler Thromb Vasc Biol. 2003;23:1801–1807 [DOI] [PubMed] [Google Scholar]
- 3.Parathath S, Mick SL, Feig JE, Joaquin V, Grauer L, Habiel DM, Gassmann M, Gardner LB, Fisher EA. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res. 2011;109:1141–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silvola JM, Saraste A, Forsback S, Laine VJ, Saukko P, Heinonen SE, Yla-Herttuala S, Roivainen A, Knuuti J. Detection of hypoxia by [18f]ef5 in atherosclerotic plaques in mice. Arterioscler Thromb Vasc Biol. 2011;31:1011–1015 [DOI] [PubMed] [Google Scholar]
- 5.van der Valk FM, Sluimer JC, Voo SA, Verberne HJ, Nederveen AJ, Windhorst AD, Stroes ES, Lambin P, Daemen MJ. In vivo imaging of hypoxia in atherosclerotic plaques in humans. JACC Cardiovasc Imaging. 2015;8:1340–1341 [DOI] [PubMed] [Google Scholar]
- 6.Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, Cleutjens JP, van den Akker LH, Corvol P, Wouters BG, Daemen MJ, Bijnens AP. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–1265 [DOI] [PubMed] [Google Scholar]
- 7.Khan T, Soller B, Naghavi M, Casscells W. Tissue ph determination for the detection of metabolically active, inflamed vulnerable plaques using near-infrared spectroscopy: An in-vitro feasibility study. Cardiology. 2005;103:10–16 [DOI] [PubMed] [Google Scholar]
- 8.Naghavi M, John R, Naguib S, Siadaty MS, Grasu R, Kurian KC, van Winkle WB, Soller B, Litovsky S, Madjid M, Willerson JT, Casscells W. Ph heterogeneity of human and rabbit atherosclerotic plaques; a new insight into detection of vulnerable plaque. Atherosclerosis. 2002;164:27–35 [DOI] [PubMed] [Google Scholar]
- 9.Lee-Rueckert M, Lappalainen J, Leinonen H, Pihlajamaa T, Jauhiainen M, Kovanen PT. Acidic extracellular environments strongly impair abca1-mediated cholesterol efflux from human macrophage foam cells. Arterioscler Thromb Vasc Biol. 2010;30:1766–1772 [DOI] [PubMed] [Google Scholar]
- 10.Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T. Microrna-33 encoded by an intron of sterol regulatory element-binding protein 2 (srebp2) regulates hdl in vivo. Proc Natl Acad Sci U S A. 2010;107:17321–17326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marquart TJ, Allen RM, Ory DS, Baldan A. Mir-33 links srebp-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A. 2010;107:12228–12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price NL, Rotllan N, Canfran-Duque A, Zhang X, Pati P, Arias N, Moen J, Mayr M, Ford DA, Baldan A, Suarez Y, Fernandez-Hernando C. Genetic dissection of the impact of mir-33a and mir-33b during the progression of atherosclerosis. Cell Rep. 2017;21:1317–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price NL, Singh AK, Rotllan N, Goedeke L, Wing A, Canfran-Duque A, Diaz-Ruiz A, Araldi E, Baldan A, Camporez JP, Suarez Y, Rodeheffer MS, Shulman GI, de Cabo R, Fernandez-Hernando C. Genetic ablation of mir-33 increases food intake, enhances adipose tissue expansion, and promotes obesity and insulin resistance. Cell Rep. 2018;22:2133–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of mir-33a/b in non-human primates raises plasma hdl and lowers vldl triglycerides. Nature. 2011;478:404–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. Mir-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rotllan N, Ramirez CM, Aryal B, Esau CC, Fernandez-Hernando C. Therapeutic silencing of microrna-33 inhibits the progression of atherosclerosis in ldlr−/− mice--brief report. Arterioscler Thromb Vasc Biol. 2013;33:1973–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horie T, Baba O, Kuwabara Y, Chujo Y, Watanabe S, Kinoshita M, Horiguchi M, Nakamura T, Chonabayashi K, Hishizawa M, Hasegawa K, Kume N, Yokode M, Kita T, Kimura T, Ono K. Microrna-33 deficiency reduces the progression of atherosclerotic plaque in apoe−/− mice. J Am Heart Assoc. 2012;1:e003376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price NL, Rotllan N, Zhang X, Canfran-Duque A, Nottoli T, Suarez Y, Fernandez-Hernando C. Specific disruption of abca1 targeting largely mimics the effects of mir-33 knockout on macrophage cholesterol efflux and atherosclerotic plaque development. Circ Res. 2019;124:874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goedeke L, Salerno A, Ramirez CM, Guo L, Allen RM, Yin X, Langley SR, Esau C, Wanschel A, Fisher EA, Suarez Y, Baldan A, Mayr M, Fernandez-Hernando C. Long-term therapeutic silencing of mir-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol Med. 2014;6:1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, Usami S, Izuhara M, Sowa N, Yahagi N, Shimano H, Matsumura S, Inoue K, Marusawa H, Nakamura T, Hasegawa K, Kume N, Yokode M, Kita T, Kimura T, Ono K. Microrna-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun. 2013;4:2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen L, Fitzpatrick R, Gildea B, Petersen KH, Hansen HF, Koch T, Egholm M, Buchardt O, Nielsen PE, Coull J, Berg RH. Solid-phase synthesis of peptide nucleic acids. J Pept Sci. 1995;1:175–183 [DOI] [PubMed] [Google Scholar]
- 22.Malik S, Lim J, Slack FJ, Braddock DT, Bahal R. Next generation mirna inhibition using short anti-seed pnas encapsulated in plga nanoparticles. J Control Release. 2020;327:406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andreev OA, Dupuy AD, Segala M, Sandugu S, Serra DA, Chichester CO, Engelman DM, Reshetnyak YK. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proc Natl Acad Sci U S A. 2007;104:7893–7898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price NL, Miguel V, Ding W, Singh AK, Malik S, Rotllan N, Moshnikova A, Toczek J, Zeiss C, Sadeghi MM, Arias N, Baldan A, Andreev OA, Rodriguez-Puyol D, Bahal R, Reshetnyak YK, Suarez Y, Fernandez-Hernando C, Lamas S. Genetic deficiency or pharmacological inhibition of mir-33 protects from kidney fibrosis. JCI Insight. 2019;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez CM, Zhang X, Bandyopadhyay C, Rotllan N, Sugiyama MG, Aryal B, Liu X, He S, Kraehling JR, Ulrich V, Lin CS, Velazquez H, Lasuncion MA, Li G, Suarez Y, Tellides G, Swirski FK, Lee WL, Schwartz MA, Sessa WC, Fernandez-Hernando C. Caveolin-1 regulates atherogenesis by attenuating low-density lipoprotein transcytosis and vascular inflammation independently of endothelial nitric oxide synthase activation. Circulation. 2019;140:225–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das R, Ganapathy S, Mahabeleshwar GH, Drumm C, Febbraio M, Jain MK, Plow EF. Macrophage gene expression and foam cell formation are regulated by plasminogen. Circulation. 2013;127:1209–1218, e1201–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, Zhu X, Chen B. Inhibition of collar-induced carotid atherosclerosis by recombinant apoa-i cysteine mutants in apoe-deficient mice. J Lipid Res. 2010;51:3434–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y, Ishigami M, Kuriyama H, Kishida K, Nishizawa H, Hotta K, Muraguchi M, Ohmoto Y, Yamashita S, Funahashi T, Matsuzawa Y. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class a scavenger receptor expression in human monocyte-derived macrophages. Circulation. 2001;103:1057–1063 [DOI] [PubMed] [Google Scholar]
- 31.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, Svoronos A, Braddock DT, Glazer PM, Engelman DM, Saltzman WM, Slack FJ. Microrna silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahraei M, Chaube B, Liu Y, Sun J, Kaplan A, Price NL, Ding W, Oyaghire S, Garcia-Milian R, Mehta S, Reshetnyak YK, Bahal R, Fiorina P, Glazer PM, Rimm DL, Fernandez-Hernando C, Suarez Y. Suppressing mir-21 activity in tumor-associated macrophages promotes an antitumor immune response. J Clin Invest. 2019;129:5518–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burke AC, Huff MW. Regression of atherosclerosis: Lessons learned from genetically modified mouse models. Curr Opin Lipidol. 2018;29:87–94 [DOI] [PubMed] [Google Scholar]
- 35.Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ, Fisher EA. Mir33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res. 2014;115:759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin JD, Nishi H, Poles J, Niu X, McCauley C, Rahman K, Brown EJ, Yeung ST, Vozhilla N, Weinstock A, Ramsey SA, Fisher EA, Loke P. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. 2019;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. Single-cell rna-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. 2018;122:1661–1674 [DOI] [PubMed] [Google Scholar]
- 38.Nakao T, Horie T, Baba O, Nishiga M, Nishino T, Izuhara M, Kuwabara Y, Nishi H, Usami S, Nakazeki F, Ide Y, Koyama S, Kimura M, Sowa N, Ohno S, Aoki H, Hasegawa K, Sakamoto K, Minatoya K, Kimura T, Ono K. Genetic ablation of microrna-33 attenuates inflammation and abdominal aortic aneurysm formation via several anti-inflammatory pathways. Arterioscler Thromb Vasc Biol. 2017;37:2161–2170 [DOI] [PubMed] [Google Scholar]
- 39.Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C, Fullerton MD, Cecchini K, Rayner KJ, Steinberg GR, Zamore PD, Fisher EA, Loke P, Moore KJ. Microrna-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125:4334–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, Gulati M, Homer RJ, Russell T, van Rooijen N, Elias JA, Hogaboam CM, Herzog EL. Tgf-beta driven lung fibrosis is macrophage dependent and blocked by serum amyloid p. Int J Biochem Cell Biol. 2011;43:154–162 [DOI] [PubMed] [Google Scholar]
- 41.Gratchev A, Guillot P, Hakiy N, Politz O, Orfanos CE, Schledzewski K, Goerdt S. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein betaig-h3. Scand J Immunol. 2001;53:386–392 [DOI] [PubMed] [Google Scholar]
- 42.Bourlier V, Zakaroff-Girard A, Miranville A, De Barros S, Maumus M, Sengenes C, Galitzky J, Lafontan M, Karpe F, Frayn KN, Bouloumie A. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation. 2008;117:806–815 [DOI] [PubMed] [Google Scholar]
- 43.Hourigan ST, Solly EL, Nankivell VA, Ridiandries A, Weimann BM, Henriquez R, Tepper ER, Zhang JQJ, Tsatralis T, Clayton ZE, Vanags LZ, Robertson S, Nicholls SJ, Ng MKC, Bursill CA, Tan JTM. The regulation of mirnas by reconstituted high-density lipoproteins in diabetes-impaired angiogenesis. Sci Rep. 2018;8:13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen MA, Wyatt H, Susser L, Geoffrion M, Rasheed A, Duchez AC, Cottee ML, Afolayan E, Farah E, Kahiel Z, Cote M, Gadde S, Rayner KJ. Delivery of micrornas by chitosan nanoparticles to functionally alter macrophage cholesterol efflux in vitro and in vivo. ACS Nano. 2019;13:6491–6505 [DOI] [PubMed] [Google Scholar]
- 45.Yung BC, Li J, Zhang M, Cheng X, Li H, Yung EM, Kang C, Cosby LE, Liu Y, Teng L, Lee RJ. Lipid nanoparticles composed of quaternary amine-tertiary amine cationic lipid combination (qtsome) for therapeutic delivery of antimir-21 for lung cancer. Mol Pharm. 2016;13:653–662 [DOI] [PubMed] [Google Scholar]
- 46.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of mir-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.