

Abstract
Gastric cancer (GC) is a common malignancies with unfavourable prognosis. As one of the most common RNA modifications in nature, alternative polyadenylation (APA) plays a critical role in the progression of carcinomas. CPSF1 is a critical APA-related factor and is involved in many cancers. Nevertheless, the roles and underlying mechanisms of CPSF1 remain unclear in GC. In this work, we identified that CPSF1 is significantly upregulated in GC and that high CPSF1 expression indicates an unfavourable prognosis in GC patients. Moreover, CPSF1 expression levels were closely associated with tumour size, TNM stage and lymph node metastasis. CPSF1 depletion dramatically weakened GC cell proliferation and metastasis. We then performed RNA sequencing and found numerous downstream genes involved the regulation of CPSF1 with remarkable changes in 3’UTR length, among which NSDHL was positively regulated by CPSF1 and promoted GC progression. In addition, rescue assays demonstrated that NSDHL mediated the carcinogenic effect of CPSF1, and this process potentially involved APA. Therefore, this study showed that CPSF1 promotes GC progression, at least in part, by enhancing NSDHL and offered new insights into therapeutic targets for GC.
Keywords: CPSF1, NSDHL, APA, gastric cancer, progression
Introduction
Gastric cancer (GC) is a common malignancies with unfavourable prognosis. One million new cases were diagnosed and 784000 deaths occurred worldwide in 2018 [1]. The incidence of GC has fallen slightly, but more gastric cancer cases may occur in the future due to ageing and the expansion of the world population [2]. At present, numerous therapeutic methods, including chemotherapy, radiotherapy, surgery, immunotherapy, and targeted therapy, are available for GC; nevertheless, the prognosis of patients remains poor due to the high heterogeneity and relapse rate [3]. Consequently, it is urgent to explore the nosogenesis and identify efficient therapeutic targets for GC.
Recently, increasing evidence has shown that RNA modification participates in many biological processes, especially the initiation and development of malignant tumours [4]. As one of the most common RNA modifications in nature, APA regulates the generation of several mRNA isoforms containing the 3’ untranslated region (3’UTR) of different lengths by cleaving pre-mRNA and adding a poly(A) tail at different sites, which subsequently affects mRNA translation, stability, nuclear export, and cellular localization [5,6]. Interestingly, the selection of the sites is regulated cooperatively by several APA-related molecules, such as CPSF, CSTF, CFI, and CFII complexes [7]. In recent years, numerous studies have verified that APA-related factors influence the progression of carcinomas through APA mechanisms [8-12]. CPSF1 (also known as CPSF160), the largest component of the CPSF complex, is one of the key members of APA-related factors. Wang et al. [13] reported that CPSF1 promotes the progression of breast cancer and the occurrence of APA events involved in proliferation, tumorigenesis, chemosensitivity and metastasis. Chen et al. [14] reported that CPSF1 overexpression was closely associated with unfavourable clinical outcomes in hepatocellular carcinoma (HCC) patients by changing APA and alternative splicing events. Van Etten et al. [15] found that the silencing of CPSF1 led to a splice switch that inhibited the expression of androgen receptor variants and blocked androgen-independent growth of castration-resistant prostate cancer cells. However, as far as we know, the underlying mechanisms of CPSF1 remain unclear in GC and need to be further investigated. Consequently, we performed the study to explore CPSF1-mediated mechanistic models in GC.
In this research, we systematically explored the biological function of CPSF1 in GC. As expected, we found that CPSF1 exhibited tumour-promoting activities. Compared to normal gastric tissues or cells, CPSF1 was upregulated in GC. High CPSF1 expression showed a worse prognosis. Moreover, CPSF1 expression levels were closely associated with tumour size, lymph node metastasis and TNM stage. Depletion of CPSF1 suppressed GC cell growth and metastasis, promoted GC cell apoptosis and induced G1/G0 phase arrest. RNA sequencing data demonstrated that CPSF1 knockdown resulted in remarkable changes in the expression and 3’UTR length of numerous downstream genes, among which the NSDHL gene caught our attention. We observed that CPSF1 silencing caused 3’UTR lengthening of NSDHL and inhibited NSDHL expression. In addition, forced NSDHL expression reversed the phenotypes induced by CPSF1 knockdown. Together, our study demonstrated that CPSF1 exhibited tumorigenic activities mediated by NSDHL in GC and that targeting CPSF1 potentially represents a new treatment method for GC.
Materials and methods
Clinical samples
In our research, human paraffin-embedded GC tissues and matched nontumor tissues were collected from the First Affiliated Hospital of Anhui Medical University (Hefei, Anhui, China). None of the patients received chemotherapy before operation, and all patients were followed up for more than five years. Moreover, all patients signed informed consent. The present research was approved by the Institutional Review Boards of Anhui Medical University.
Immunohistochemical (IHC) assay
Tissue samples from gastric cancer patients or mice were paraffin embedded and sectioned. Haematoxylin-eosin (HE) staining and IHC assays were implemented in accordance with the manufacturer’s instructions. The antibodies against CPSF1 (1:200, ab81552, Abcam) or Ki-67 (1:200, MA5-14520, Thermo Scientific) were used. The stained pictures were obtained using an Olympus microscope (Olympus, Japan). For CPSF1 analysis, tissues with greater than 10% positively stained cells were defined as CPSF1 positive and conversely CPSF1 negative.
Cell culture
All human gastric cancer cells (AGS, MGC-803, SGC7901, MKN-28, HGC-27) and normal gastric cells (GES1) were obtained from the Shanghai Cell Bank (Shanghai, China), and cultured in RPMI-1640 or DMEM with 10% foetal bovine serum (Gibco) at 37°C in a 5% CO2 humidified atmosphere as recommended.
Western blotting
Western blotting assays were implemented as described in our previous study [16]. Antibodies against CPSF1 (1:2000 dilution, ab81552, Abcam), NSDHL (1:1000, 15111-1-AP, Proteintech) or GAPDH (1:10000, 10494-1-AP, Proteintech) were used.
RT-qPCR
Total RNA was extracted with a TRIzol Plus RNA Purification Kit (CAT #12183555, Invitrogen) and reverse transcribed into cDNA using PrimeScript RT Master Mix (Perfect Real Time) (CAT #RR036A, TAKARA) in accordance with the manufacturer’s protocol. Then, RT-qPCR was performed with TB Green Premix Ex Taq II (Tli RNaseH Plus) (CAT #RR820A, TAKARA) and a StepOnePlus Real-Time PCR System (ABI). Finally, we analysed the data using the 2-ΔΔCT method. GAPDH was used as an internal control. The primer sequences are as follows: CPSF1 (F: CGCAGCTCTACGTGTACCG, R: GGACATGACGTTGCCAAAGAA); NSDHL (F: CAAGTCGCACGGACTCATTTG, R: ACTGTGCATCTCTTGGCCTG) and GAPDH (F: AGAAGGCTGGGGCTCATTTG, R: AGGGGCCATCCACAGTCTTC).
Cell function assays
In our study, Cell viability was detected using the CCK8 assay. First, cells (2×103 cells per well) were inoculated into 96-well plates. After 24 h, CCK8 detection solution was added to each well for a 2-hour incubation. Then, the absorbance at 450 nm wavelength was measured by a microplate reader. CCK8 assays were performed every 24 h for five consecutive days.
For the colony formation assay, cells (600 cells per well) were plated into 6-well plates and cultured for 2 weeks. The medium was replaced every three days. After 2 weeks, the cells were fixed, stained, and photographed. Finally, the colonies were counted.
For the wound healing assay, AGS or MGC-803 cells were added into 12-well plates, and a scratch wound was created when the cell confluence reached approximately 90%. Then, the wounds were captured at 0, 24 and 48 h. Finally, the migration ratio was analysed by measuring the width difference of the wound area.
For transwell assays, 5-10×104 AGS or MGC-803 cells were inoculated into the upper chambers without serum. For the invasion assay, the upper chambers contained Matrigel. For the migration assay, upper chambers without Matrigel were used. The inserts were rinsed and stained after 24-48 hours. Images were obtained using an Olympus IX-70 microscope.
Flow cytometry assays
The cell cycle and apoptosis were detected by flow cytometry assays according to the manufacturer’s protocols. For apoptosis assays, the harvested cells were double-stained with propidium iodide (PI) in accordance with the protocol from an Annexin V-APC/PI apoptosis kit (22837, AAT Bioquest). Then, the cells were analysed by a flow cytometer (BD FACSCanto, BD Biosciences). For the cell cycle assay, the harvested cells were stained with PI using a cell cycle analysis kit (C1052, Beyotime), and the percentage of cells in each phase was analysed using a flow cytometer.
RNA-sequencing
To investigate the downstream genes related to the function of CPSF1 in GC, we amplified MGC-803-shCPSF1 and MGC-803-shCtrl cells and performed RNA sequencing with the Genefund Biotechnology Company (Shanghai, China). PolyASite V2.0, GENCODE V35 and Ensembl V101 were used to build the 3’UTR database. Proximal Poly(A) Usage (PPAU) was used for APA difference analysis, and |ΔPPAU|>20 was adopted as APA difference criteria.
Xenograft assay
The in vivo experiments were approved by the Animal Ethics Committee of Anhui Medical University. We randomly divided male nude mice (five weeks old) into two groups (MGC-803-shCtrl, MGC-803-shCPSF1 or MGC-803-shNSDHL), and six mice were used in each group. Briefly, 500×104 MGC-803 cells were subcutaneously injected into the flanks or tail veins of nude mice. For the tumorigenicity assay, the volumes of the tumours were continuously measured every three days after two weeks. For the tumour metastasis assay, five weeks after tail vein injection, we performed a HE staining assay or quantified the metastasis sites using an in vivo imaging system (IVIS Lumina III, PerkinElmer).
Statistical analyses
In the study, the results are presented as the means ± standard deviation. Student’s t tests were used to analyse the differences between groups. The correlation between CPSF1 expression and clinicopathological parameters was analysed using Pearson’s chi-square test. Kaplan-Meier plots were used to analyse the overall survival (OS) rates in GC patients, and the differences were compared using log-rank tests. SPSS 16.0 and GraphPad Prism 8.0.1.244 software were used for statistical analysis. P<0.05 was considered a statistically significant difference.
Results
CPSF1 is significantly upregulated in GC, and high CPSF1 expression indicates an unfavourable prognosis in GC patients
We first analysed the expression of 22 major APA-related factors in GC using The Cancer Genome Atlas (TCGA) database and found that CPSF1 mRNA was dramatically upregulated in GC tissues compared with normal gastric tissues (Figure 1A). To further explore the level of CPSF1 mRNA in other cancers, we performed a pan-cancer analysis, and the results indicated that CPSF1 was upregulated in most tumour types, including gastric cancer (Figure 1B). Therefore, we hypothesized that CPSF1 may be an oncogene. Using TCGA databases, we found that CPSF1 expression levels in GC tissues (n=375) were significantly higher compared with gastric normal tissues (n=32) (P=7.288e-10), and we obtained the same results in 27 matched GC and surrounding nontumor tissues (P=4.014e-09) (Figure 1C, 1D). Interestingly, we further confirmed these findings by analysing Gene Expression Omnibus (GEO) datasets (GSE66229, GSE33335) (Figure 1E, 1F).
Figure 1.

High CPSF1 expression indicated an unfavourable prognosis in GC. A. Expression of 22 APA-related factors in GC and nontumor tissues was analysed using TCGA data through online website (https://www.aclbi.com/static/index.html#/batch). B. Pan-cancer analysis of CPSF1 was performed through online website (http://ualcan.path.uab.edu/analysis.html). C. Expression of CPSF1 mRNA in TCGA GC (n=375) and normal samples (n=32). D. Analysis of CPSF1 mRNA in 27 matched TCGA GC and adjacent nontumor tissues. E. CPSF1 expression in GC (n=300) and normal tissues (n=100) in the GSE66229 dataset. F. CPSF1 expression in 25 matched GC and surrounding normal tissues in the GSE33335 dataset. G. Expression of CPSF1 in 60 matched GC and surrounding nontumor tissues were detected by IHC. Scale bar, 50 μm. H. Kaplan-Meier analysis assessing OS according to the CPSF1 expression levels in GC patients. I. Kaplan-Meier OS curve of GC patients from online website (http://kmplot.com/analysis/). **P<0.01.
To verify the above bioinformatics results, we collected 60 matched GC and adjacent nontumor tissues. As shown in Figure 1G, the IHC assay showed that CPSF1 protein levels were obviously increased in GC tissues compared with adjacent nontumor tissues. Then, we analysed the clinical implications of CPSF1. As described in Table 1, we observed that CPSF1 expression was associated with TNM stage (P=0.03), lymph node metastasis (P=0.002) and tumour size (P=0.023) in GC. In addition, we analysed the correlation between CPSF1 expression levels and survival in 60 GC patients. The results demonstrated that high CPSF1 expression indicates an unfavourable prognosis (Figure 1H). Interestingly, we observed the same results using bioinformatics analysis (P=6.2e-11) (http://kmplot.com/analysis/) (Figure 1I). Consequently, the findings showed that CPSF1 is significantly upregulated in GC and that high CPSF1 expression indicates an unfavourable prognosis in GC patients.
Table 1.
Correlation between CPSF1 expression and clinicopathologic parameters in GC patients
| Parameter | CPSF1 expression (N) | X2 | p-value | ||
|---|---|---|---|---|---|
|
| |||||
| N | Positive | Negative | |||
| Age (years) | |||||
| ≥60 | 32 | 25 | 7 | 0.357 | 0.550 |
| <60 | 28 | 20 | 8 | ||
| Gender | |||||
| Male | 33 | 24 | 9 | 0.202 | 0.653 |
| Female | 27 | 21 | 6 | ||
| Tumor size | |||||
| ≥5 cm | 35 | 30 | 5 | 5.143 | 0.023* |
| <5 cm | 25 | 15 | 10 | ||
| Differentiation | |||||
| Well, moderately | 36 | 28 | 8 | 0.370 | 0.543 |
| Poorly, undifferentiated | 24 | 17 | 7 | ||
| Lymph node metastasis | |||||
| Yes | 40 | 35 | 5 | 10.000 | 0.002** |
| No | 20 | 10 | 10 | ||
| TNM stage | |||||
| I+II | 22 | 13 | 9 | 4.689 | 0.030* |
| III+IV | 38 | 32 | 6 | ||
P<0.05;
P<0.01.
CPSF1 promotes cell proliferation and inhibits apoptosis in GC
We then investigated CPSF1 protein levels in normal gastric cells (GES1) and human GC cell lines (AGS, MGC-803, SGC7901, MKN-28, HGC-27). The results showed that CPSF1 expression levels were higher in GC cell lines compared with normal gastric cell lines, especially in AGS and MGC-803 cells (Figure 2A). Consequently, we selected these two cell lines for subsequent research. To clarify the function of CPSF1 in GC, we depleted CPSF1 with shRNAs and conducted loss-of-function assays. The degree of CPSF1 knockdown were verified using qRT-PCR and western blotting (Figure 2B, 2C). Cell proliferation and CCK8 assays indicated that the depletion of CPSF1 dramatically weakened gastric cancer cell proliferation and viability (Figure 2D, 2E), and colony formation assays indicated that CPSF1 knockdown dramatically decreased the numbers of colonies (Figure 2F). Conversely, the protein expression level of CPSF1 in HGC-27 cells was significantly increased after CPSF1-overexpression plasmid-mediated overexpression of CPSF1. Cell proliferation, CCK8 and colony formation assays indicated that overexpression of CPSF1 enhanced cell proliferation, cell viability and the number of colonies formed (Figure S1A-D).
Figure 2.

CPSF1 stimulated human GC cell proliferation. (A) Western blotting detecting the protein levels of CPSF1 in GC cells (AGS, MGC-803, SGC7901, MKN-28, HGC-27) and normal gastric cells (GES1). (B) RT-qPCR examining the mRNA levels of CPSF1. AGS and MGC-803 cells were stably transfected with shCPSF1-1, shCPSF1-2 or shCtrl. (C) Western blotting examining the protein levels of CPSF1. (D) Proliferation curves of AGS and MGC-803 cells. (E) CCK8 assays and (F) colony formation assays in AGS and MGC-803 cells. (G) The images of xenograft tumours that were harvested from MGC-803 cells stably transfected with shCtrl or shCPSF1. (H) The tumour volumes and weights were measured and analyzed. (I) IHC analysis of Ki-67 expression in xenograft tumours. All experiments were repeated at least three times. *P<0.05 and **P<0.01.
To further assess whether CPSF1 affects GC growth in vivo, we injected MGC-803 cells (shCPSF1 or shCtrl group) into the flanks of male nude mice. The results demonstrated that the tumours in the shCPSF1 group were dramatically smaller than those in the shCtrl group (Figure 2G). Additionally, the depletion of CPSF1 resulted in a significant reduction in tumour volume and weight (Figure 2H). Moreover, the population of Ki-67-positive cells derived from those tumours formed in vivo was dramatically reduced in the shCPSF1 group compared with the shCtrl group (Figure 2I).
Next, we explored the impacts of CPSF1 on the cell apoptosis and cycle. As shown in Figure 3A, 3B, the percentage of apoptotic GC cells with CPSF1 knockdown was dramatically higher compared with the control cells. In addition, cell cycle assays demonstrated that CPSF1 participated in regulating cell cycle. Compared to the shCtrl group, the G1 phase percentage of GC cells with CPSF1 knockdown was obviously increased and the S phase percentage of GC cells with CPSF1 knockdown was dramatically decreased. Additionally, the population of MGC-803 cells in the G2/M phase after shRNA-mediated knockdown of CPSF1 was also obviously decreased (Figure 3C, 3D).
Figure 3.

CPSF1 regulated the GC cell apoptosis and cycle. A, B. Flow cytometry assays assessing the impacts of CPSF1 on GC cell apoptosis. C, D. Cell cycle assays assessing the impacts of CPSF1 on GC cell cycle. All experiments were repeated at least three times. *P<0.05 and **P<0.01.
Consequently, these findings demonstrated that CPSF1 enhances GC cell growth in vitro and in vivo, regulates the cell cycle and inhibits apoptosis.
Knockdown of CPSF1 suppresses GC cell metastasis
Considering that CPSF1 is related to TNM stages in GC patients, wound healing and transwell assays were implemented to investigate the impacts of CPSF1 on regulating GC cell migration and invasion. CPSF1 knockdown resulted in reduced wound healing (Figure 4A). In addition, Transwell assays demonstrated that the depletion of CPSF1 dramatically inhibited GC cell migration and invasion (Figure 4B). Conversely, overexpression of CPSF1 significantly facilitated GC cells migration and invasion (Figure S1E). Therefore, the findings revealed that CPSF1 enhances cell migration and invasion in GC.
Figure 4.

CPSF1 promoted GC cell metastasis. A. Wound healing assays of GC cells. B. Transwell assays of GC cells. C. In vivo biofluorescence imaging assays assessing the impacts of CPSF1 on GC cell metastasis in vivo. Representative images were shown. D. Representative pictures of haematoxylin-eosin staining of lungs and incidence of lung metastasis from mice injected with MGC-803 cells stably transfected with shCPSF1 or shCtrl. Red arrow indicates the lung metastasis. All experiments were repeated at least three times. *P<0.05 and **P<0.01.
Then, we injected MGC-803 cells (shCtrl, shCPSF1) via the tail vein to assess whether CPSF1 modulates lung metastasis in vivo. In vivo biofluorescence imaging showed that the total fluorescence intensity in the CPSF1 knockdown group was obviously lower compared with the shCtrl group (Figure 4C). Additionally, HE staining assays indicated significantly fewer mice with lung metastases in the shCPSF1 group (1/6) compared with the control group (6/6) (Figure 4D). Hence, CPSF1 may play a critical role in enhancing cell metastasis in GC.
CPSF1 modulates 3’UTR alteration in GC cells
CPSF1, as a critical APA-related factor, contributes to the regulation of pre-mRNA processing. Given the function of CPSF1 in APA and its oncogene roles in GC described above, we sought to determine whether CPSF1 contributes to GC progression by influencing APA and altering the 3’UTR length of downstream genes. To verify this hypothesis, we performed RNA sequencing in the CPSF1 knockdown and control groups to examine all changes in gene expression and 3’UTR length. RNA sequencing results demonstrated that there were 1764 upregulated and 718 downregulated genes after CPSF1 knockdown (filter conditions: |log2FC|>1 and q value <0.05) (Figure 5A, 5B). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that the potential downstream targets of CPSF1 were mainly enriched in cytokine-cytokine receptor interactions, cell adhesion molecules and amino acid metabolism (Figure 5C). Gene Ontology (GO) analysis also identified multiple related biological processes, such as organic acid and amino acid transport, regulation of lipid biosynthetic process and cell-cell adhesion (Figure 5D). In addition, we performed KEGG analysis on down- and upregulated transcripts (Figure S2). The results further indicated that downstream targets were mainly enriched in cytokine-cytokine receptor interactions and biometabolic processes.
Figure 5.

NSDHL was regulated by CPSF1 in GC cells. (A) Hierarchical clustering analysis of RNA sequencing of the MGC-803-shCPSF1 and the MGC-803-shCtrl group. (B) Volcano plot showed the differentially expressed genes (DEGs) in MGC-803-shCPSF1 and MGC-803-shCtrl cells. (C) KEGG analysis and (D) GO analysis for all changed genes after CPSF1 knockdown. (E) Volcano plot showed the genes with significant alteration in the length of 3’UTR. (F) Candidate genes with both 3’UTR lengthening and downregulated expression after CPSF1 knockdown. (G) Western blotting detecting the protein levels of NSDHL. All experiments were repeated at least three times.
Then, we performed APA quantification and difference analysis on RNA sequencing data using the QAPA tool [17]. The results showed that CPSF1 knockdown resulted in widespread 3’UTR alterations in GC cells, including 220 lengthened and 158 shortened genes (Figure 5E). A study found that 3’UTR shortening of oncogenes may promote cancer progression by escaping the inhibitory effect of miRNAs and producing more protein [18]. Therefore, to determine whether CPSF1 plays a carcinogenic role by affecting the shortening of the 3’UTR of downstream oncogenes, we focused on the genes with both 3’UTR lengthening and downregulated expression after CPSF1 knockdown. As described in Figure 5F, 12 genes matched the above conditions. Among them, the NSDHL gene captured our attention. As shown in Figure 5G, NSDHL protein levels were significantly downregulated after CPSF1 knockdown.
NSDHL promotes cell proliferation and metastasis in GC
Given that the function of NSDHL in GC has not been reported, we constructed stable cell lines with shRNAs targeting NSDHL or control shRNA. As shown in Figure 6A, similar to CPSF1, NSDHL protein levels were higher in GC cells compared with normal gastric cells, especially in AGS and MGC-803 cells. Consequently, we selected these two cell lines for the biological function research of NSDHL. RT-qPCR and western blotting were implemented to examined the degree of NSDHL knockdown (Figure 6B, 6C). NSDHL depletion resulted in an obvious decrease in cell number, viability, and clonogenicity (Figure 6D-F). To further clarify the impact of NSDHL on the proliferation ability of GC cells in vivo, we injected MGC-803 cells (shNSDHL or shCtrl group) into the flanks of male nude mice. As described in Figure 6G-I, NSDHL silencing caused a obvious decrease in tumour volume and weight. Moreover, the percentage of Ki-67-positive cells derived from those tumours formed in vivo was dramatically reduced in the shNSDHL group.
Figure 6.

NSDHL promoted human GC cell proliferation. (A) Western blotting detecting the protein levels of NSDHL in GC cells (AGS, MGC-803, SGC7901, MKN-28, HGC-27) and normal gastric cells (GES1). (B) RT-qPCR examining mRNA levels of NSDHL. AGS and MGC-803 cells were stably transfected with shNSDHL-1, shNSDHL-2 or shCtrl. (C) Western blotting examining the protein levels of NSDHL. (D) Proliferation curves of AGS and MGC-803 cells. (E) CCK8 assays and (F) colony formation assays in AGS and MGC-803 cells. (G, H) The volumes and weights of xenograft tumours that were harvested from MGC-803 cells stably transfected with shCtrl or shNSDHL were measured and analyzed. (I) IHC analysis of Ki-67 expression in xenograft tumours. All experiments were repeated at least three times. **P<0.01.
Additionally, we explored the function of NSDHL in the GC cell metastasis in vitro and vivo. Transwell assays demonstrated that NSDHL knockdown dramatically weakened GC cell metastasis in vitro (Figure 7A, 7B). HE staining assays indicated that fewer mice in the shNSDHL group (2 out of 6) showed obvious lung metastasis than that in the shCtrl group (6 out of 6) (Figure 7C). Consequently, these results showed that NSDHL also promotes GC cell proliferation and metastasis in vitro and in vivo.
Figure 7.
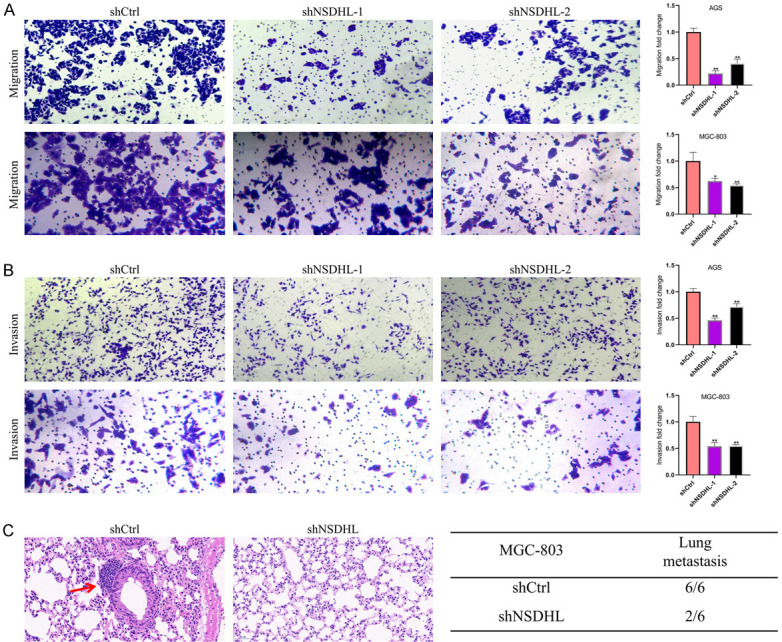
NSDHL stimulated GC cell metastasis. (A) Migration and (B) invasion assays in GC cells. (C) Representative images of haematoxylin-eosin staining of lungs and incidence of lung metastasis from mice injected with MGC-803 cells stably transfected with shNSDHL or shCtrl. Red arrow indicates the lung metastasis. All experiments were repeated at least three times. **P<0.01.
As described above, CPSF1 serves as an oncogene in GC and may regulate NSDHL expression by altering the 3’UTR length of NSDHL. To investigate whether CPSF1 promotes gastric cancer progression through NSDHL, we performed rescue experiments. As shown in Figure 8A, CPSF1 knockdown decreased NSDHL protein levels, and these decreases were restored by cotransfection with shCPSF1 and NSDHL-overexpressing plasmids (OE-NSDHL). Consistent with the above results, knockdown of CPSF1 decreased the proliferation, viability, clonogenicity, migration and invasion of GC cells. However, NSDHL gene overexpression abrogated the decreases induced by silencing CPSF1 (Figure 8B-F). Therefore, these findings showed that CPSF1 enhances GC cell proliferation and metastasis by regulating NSDHL (Figure 9).
Figure 8.
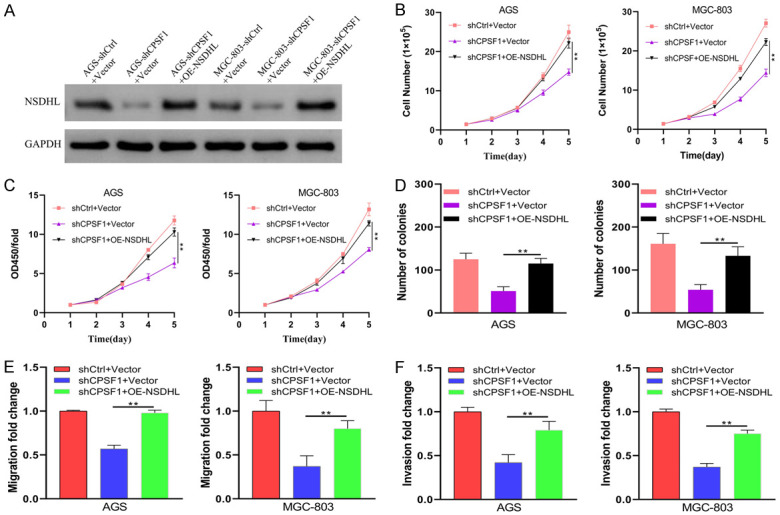
Forced NSDHL expression abrogated the phenotypes induced by CPSF1 silencing. AGS and MGC-803 cells were transfected with shCtrl plus Vector, shCPSF1 plus Vector or shCPSF1 plus OE-NSDHL. (A) Western blotting assessing the protein levels of NSDHL. (B) Proliferation curves of AGS and MGC-803 cells. (C) CCK8 assays and (D) colony formation assays in AGS and MGC-803 cells. (E) Transwell migration and (F) invasion assays were performed to assess cell metastasis. All experiments were repeated at least three times. **P<0.01.
Figure 9.
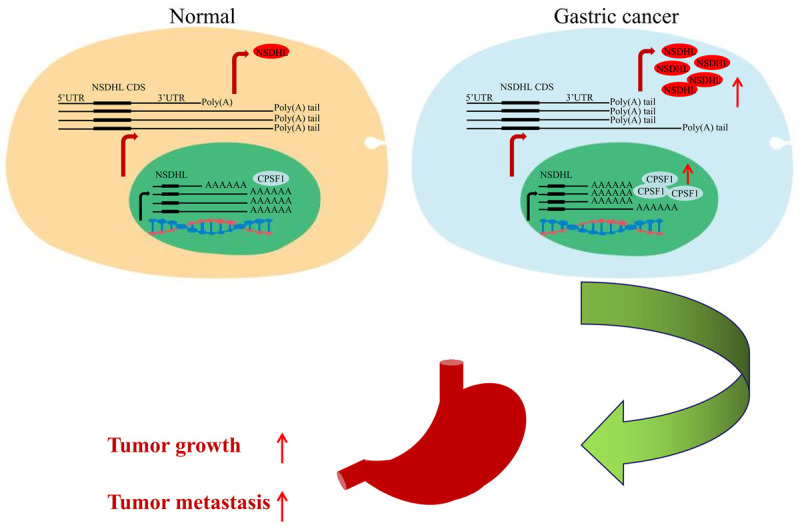
Schematic diagram depicting the mechanism of CPSF1 in enhancing GC progression.
Discussion
With increasing research on the aetiology and mechanisms of carcinoma, a growing body of studies have demonstrated that APA mechanisms mediated by APA factors are closely connected with a variety of carcinomas. Tan et al. [9] found that CPSF6 promoted hepatocellular carcinoma progression by upregulating NQO1 expression through APA. Xing et al. [10] confirmed that NUDT21 inhibited cervical cancer progression by regulating APA of genes involved in fatty acid metabolism and the Wnt and NF-κB signalling pathways. Wang et al. [12] found that CPSF4 enhanced the progression of hepatocellular carcinoma by hindering the formation of circRNAs with a polyadenylation signal sequence, reducing the accumulation of miRNA and disrupting miRNA-mediated gene silencing. As one of the 22 major factors that regulate APA progression, CPSF1 is involved in multiple carcinomas and other diseases [13-15,19-25], but its function in GC remains unknown to date.
In our research, we systematically assessed the function of CPSF1 in GC. We first assessed the expression of CPSF1 by analysing the data from TCGA and GEO databases and further confirmed the overexpression of CPSF1 in GC tissues/cells. GC patients with higher CPSF1 expression showed worse OS rates and clinicopathological parameters. Through functional assays, we demonstrated that depletion of CPSF1 not only significantly inhibited cell growth and metastatic ability but also enhanced apoptosis in GC. As reported previously, Sakai et al. [21] reported that CPSF1 was upregulated in head and neck cell cancer tissues, and aberrant expression of CPSF1 promoted tumour progression. The results are consistent with the findings achieved in this research. Moreover, CPSF1 was overexpressed in ovarian cancer tissues, and CPSF1 silencing decreased cell viability and clonogenic capacity by enhancing cell apoptosis and G0/G1 phase arrest [19]. Intriguingly, Wang et al. [13] achieved similar results and found that CPSF1 silencing suppressed cell proliferation, enhanced apoptosis, and caused cell cycle redistribution in breast cancer. These data are consistent with our findings obtained in this study. Thus, CPSF1 is upregulated in the many cancers, including gastric cancer. However, the molecular mechanisms underlying the upregulation of CPSF1 remain unclear. Recently, Davis et al. [26] illustrated that eIF4E drives the protein expression of CPSF1 and physically interacts with CPSF3, CPSF1, and uncleaved target RNA to stimulate 3’-end cleavage of selected RNAs. eIF4E, as a eukaryotic translation initiation factor and oncogene, plays an important role in the export and translation of specific transcripts [27]. We hypothesized that eIF4E might contribute to the upregulation of CPSF1 in gastric cancer by promoting its translation. Nevertheless, further experiments are needed to verify the specific mechanism. Additionally, some studies have demonstrated that CPSF1 acts as an oncogene by inducing the occurrence of abnormal APA events in downstream genes. For example, Chen et al. [14] found that CPSF1 regulated the widespread 3’UTR changes through an APA mechanism and enhanced cell growth in HCC. Therefore, to determine whether CPSF1 serves as a tumour driver by influencing APA events of downstream genes, we performed high-throughput RNA sequencing.
Given the role of CPSF1 in APA regulation and GC, we hypothesized that CPSF1 exerts carcinogenic effects in GC, at least in part, by affecting the 3’UTR of downstream genes. We next implemented APA quantification and difference analysis on RNA sequencing data, and NSDHL was chosen for further study. RNA sequencing results showed that CPSF1 knockdown caused the downregulation of NSDHL expression and the generation of NSDHL mRNA isoforms with long 3’UTRs. We further determined that NSDHL was positively regulated by CPSF1 using a western blotting assay. In addition, NSDHL depletion dramatically inhibited GC cell proliferation and metastasis. As described above, CPSF1 knockdown decreased the GC cell growth, migration and invasion, whereas rescue assays demonstrated that forced NSDHL expression abrogated the phenotypes induced by CPSF1 silencing. Consequently, NSDHL mediated the carcinogenic role of CPSF1 in GC. At present, there are no reports on the mechanism of NSDHL in GC. As an essential enzyme in the cholesterol synthesis pathway, NSDHL is closely associated with tumour growth [28]. It has been reported that the inhibition of NSDHL decreases the expression of EGFR to suppress EGFR-dependent signalling and EGFR-driven carcinomas [29,30]. Xiao et al. [31] reported that NSDHL was overexpressed in GC tissues and could act as a diagnostic biomarker for early GC. These results supported our findings in this study. In addition, Yoon et al. [32] reported that NSDHL promoted breast cancer cell growth and metastasis. Interestingly, Chen et al. [33] obtained similar results and found that NSDHL accelerated triple-negative breast cancer metastasis by activating the TGFβ signalling pathway and cholesterol biosynthesis. These results were consistent with those of the present study. Therefore, we herein reported that CPSF1 may positively regulate NSDHL through APA to promote GC progression. Nevertheless, further research is needed to confirm the mechanisms involved in APA.
Conclusion
Collectively, we revealed that CPSF1 and NSDHL serve as oncogenes to promote gastric cancer cell proliferation and metastasis. NSDHL was positively regulated by CPSF1 and mediated the carcinogenic effect of CPSF1, which may be involved in APA. However, the detailed APA mechanism remains to be further confirmed. Overall, these findings may offer new insights into therapeutic targets for gastric cancer.
Acknowledgements
The authors thank all anonymous reviewers for their valuable comments and suggestions. The present study was supported by the Natural Science Research Projects at Higher Institutions in Anhui Province (grant no. KJ2021ZD0025).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635–648. doi: 10.1016/S0140-6736(20)31288-5. [DOI] [PubMed] [Google Scholar]
- 3.Joshi SS, Badgwell BD. Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71:264–279. doi: 10.3322/caac.21657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gruber AJ, Zavolan M. Alternative cleavage and polyadenylation in health and disease. Nat Rev Genet. 2019;20:599–614. doi: 10.1038/s41576-019-0145-z. [DOI] [PubMed] [Google Scholar]
- 5.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 6.Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011;43:853–866. doi: 10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18:18–30. doi: 10.1038/nrm.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan S, Li H, Zhang W, Shao Y, Liu Y, Guan H, Wu J, Kang Y, Zhao J, Yu Q, Gu Y, Ding K, Zhang M, Qian W, Zhu Y, Cai H, Chen C, Lobie PE, Zhao X, Sun J, Zhu T. NUDT21 negatively regulates PSMB2 and CXXC5 by alternative polyadenylation and contributes to hepatocellular carcinoma suppression. Oncogene. 2018;37:4887–4900. doi: 10.1038/s41388-018-0280-6. [DOI] [PubMed] [Google Scholar]
- 9.Tan S, Zhang M, Shi X, Ding K, Zhao Q, Guo Q, Wang H, Wu Z, Kang Y, Zhu T, Sun J, Zhao X. CPSF6 links alternative polyadenylation to metabolism adaption in hepatocellular carcinoma progression. J Exp Clin Cancer Res. 2021;40:85. doi: 10.1186/s13046-021-01884-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xing Y, Chen L, Gu H, Yang C, Zhao J, Chen Z, Xiong M, Kazobinka G, Liu Y, Hou T. Downregulation of NUDT21 contributes to cervical cancer progression through alternative polyadenylation. Oncogene. 2021;40:2051–2064. doi: 10.1038/s41388-021-01693-w. [DOI] [PubMed] [Google Scholar]
- 11.Xiong M, Chen L, Zhou L, Ding Y, Kazobinka G, Chen Z, Hou T. NUDT21 inhibits bladder cancer progression through ANXA2 and LIMK2 by alternative polyadenylation. Theranostics. 2019;9:7156–7167. doi: 10.7150/thno.36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Dong J, Li X, Cheng Z, Zhu Q. CPSF4 regulates circRNA formation and microRNA mediated gene silencing in hepatocellular carcinoma. Oncogene. 2021;40:4338–4351. doi: 10.1038/s41388-021-01867-6. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Lang G, Xue M, Yang L, Chen L, Yao L, Li X, Wang P, Hu X, Shao Z. Dissecting the heterogeneity of the alternative polyadenylation profiles in triple-negative breast cancers. Theranostics. 2020;10:10531–10547. doi: 10.7150/thno.40944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, Zhu Z, Yang X, Liu L, He Y, Yang M, Guan X, Wang X, Yun J. Cleavage and polyadenylation specific factor 1 promotes tumor progression via alternative polyadenylation and splicing in hepatocellular carcinoma. Front Cell Dev Biol. 2021;9:616835. doi: 10.3389/fcell.2021.616835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Etten JL, Nyquist M, Li Y, Yang R, Ho Y, Johnson R, Ondigi O, Voytas DF, Henzler C, Dehm SM. Targeting a single alternative polyadenylation site coordinately blocks expression of androgen receptor mRNA splice variants in prostate cancer. Cancer Res. 2017;77:5228–5235. doi: 10.1158/0008-5472.CAN-17-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Zheng Q, Pan S, Chen W, Huang J, Cao Y, Tu Y, Li Z, Yu C, Jie Z. The RNA-binding protein NELFE promotes gastric cancer growth and metastasis through E2F2. Front Oncol. 2021;11:677111. doi: 10.3389/fonc.2021.677111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ha KCH, Blencowe BJ, Morris Q. QAPA: a new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol. 2018;19:45. doi: 10.1186/s13059-018-1414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Liu Y, Liu D, Yang L. Targeting cleavage and polyadenylation specific factor 1 via shRNA inhibits cell proliferation in human ovarian cancer. J Biosci. 2017;42:417–425. doi: 10.1007/s12038-017-9701-x. [DOI] [PubMed] [Google Scholar]
- 20.Kiehl S, Zimmermann T, Savai R, Pullamsetti SS, Seeger W, Bartkuhn M, Dammann RH. Epigenetic silencing of downstream genes mediated by tandem orientation in lung cancer. Sci Rep. 2017;7:3814–3896. doi: 10.1038/s41598-017-04248-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakai A, Ando M, Fukusumi T, Ren S, Liu C, Qualliotine J, Haft S, Sadat S, Saito Y, Guo TW, Xu G, Sasik R, Fisch KM, Gutkind JS, Fertig EJ, Molinolo AA, Califano JA. Aberrant expression of CPSF1 promotes head and neck squamous cell carcinoma via regulating alternative splicing. PLoS One. 2020;15:e233380. doi: 10.1371/journal.pone.0233380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du Y, Hultquist JF, Zhou Q, Olson A, Tseng Y, Zhang T, Hong M, Tang K, Chen L, Meng X, McGregor MJ, Dai L, Gong D, Martin-Sancho L, Chanda S, Li X, Bensenger S, Krogan NJ, Sun R. mRNA display with library of even-distribution reveals cellular interactors of influenza virus NS1. Nat Commun. 2020;11:2449. doi: 10.1038/s41467-020-16140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouyang J, Sun W, Xiao X, Li S, Jia X, Zhou L, Wang P, Zhang Q. CPSF1 mutations are associated with early-onset high myopia and involved in retinal ganglion cell axon projection. Hum Mol Genet. 2019;28:1959–1970. doi: 10.1093/hmg/ddz029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Zhang X, Zou Y, Han F. CPSF1 mediates retinal vascular dysfunction in diabetes mellitus via the MAPK/ERK pathway. Arch Physiol Biochem. 2022;128:708–715. doi: 10.1080/13813455.2020.1722704. [DOI] [PubMed] [Google Scholar]
- 25.Evsyukova I, Bradrick SS, Gregory SG, Garcia-Blanco MA. Cleavage and polyadenylation specificity factor 1 (CPSF1) regulates alternative splicing of interleukin 7 receptor (IL7R) exon 6. RNA. 2013;19:103–115. doi: 10.1261/rna.035410.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis MR, Delaleau M, Borden KLB. Nuclear eIF4E stimulates 3’-end cleavage of target RNAs. Cell Rep. 2019;27:1397–1408. doi: 10.1016/j.celrep.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carroll M, Borden KL. The oncogene eIF4E: using biochemical insights to target cancer. J Interferon Cytokine Res. 2013;33:227–238. doi: 10.1089/jir.2012.0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabitova L, Restifo D, Gorin A, Manocha K, Handorf E, Yang D, Cai KQ, Klein-Szanto AJ, Cunningham D, Kratz LE, Herman GE, Golemis EA, Astsaturov I. Endogenous sterol metabolites regulate growth of EGFR/KRAS-dependent tumors via LXR. Cell Rep. 2015;12:1927–1938. doi: 10.1016/j.celrep.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D, Cho S, Lee K, Cheon S, Yoon H, Lee J, Kim D, Shin K, Koh C, Koo JS, Choi Y, Lee HH, Oh Y, Jeong Y, Chung S, Baek M, Jung K, Lim HJ, Kim HS, Park SJ, Lee J, Lee SJ, Lee B. Crystal structures of human NSDHL and development of its novel inhibitor with the potential to suppress EGFR activity. Cell Mol Life Sci. 2021;78:207–225. doi: 10.1007/s00018-020-03490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sukhanova A, Gorin A, Serebriiskii IG, Gabitova L, Zheng H, Restifo D, Egleston BL, Cunningham D, Bagnyukova T, Liu H, Nikonova A, Adams GP, Zhou Y, Yang D, Mehra R, Burtness B, Cai KQ, Klein-Szanto A, Kratz LE, Kelley RI, Weiner LM, Herman GE, Golemis EA, Astsaturov I. Targeting C4-demethylating genes in the cholesterol pathway sensitizes cancer cells to EGF receptor inhibitors via increased EGF receptor degradation. Cancer Discov. 2013;3:96–111. doi: 10.1158/2159-8290.CD-12-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Y, Xie J, Liu L, Huang W, Han Q, Qin J, Liu S, Jiang Z. NAD(P)-dependent steroid dehydrogenase-like protein and neutral cholesterol ester hydrolase 1 serve as novel markers for early detection of gastric cancer identified using quantitative proteomics. J Clin Lab Anal. 2021;35:e23652. doi: 10.1002/jcla.23652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon S, Kim HS, Kim RN, Jung S, Hong BS, Kang EJ, Lee H, Moon H, Noh D, Han W. NAD(P)-dependent steroid dehydrogenase-like is involved in breast cancer cell growth and metastasis. BMC Cancer. 2020;20:375. doi: 10.1186/s12885-020-06840-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen M, Zhao Y, Yang X, Zhao Y, Liu Q, Liu Y, Hou Y, Sun H, Jin W. NSDHL promotes triple-negative breast cancer metastasis through the TGFβ signaling pathway and cholesterol biosynthesis. Breast Cancer Res Treat. 2021;187:349–362. doi: 10.1007/s10549-021-06213-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.