

Abstract
The complexity of polymer–protein interactions makes rational design of the best polymer architecture for any given biointerface extremely challenging, and the high throughput synthesis and screening of polymers has emerged as an attractive alternative. A porphyrin-catalysed photoinduced electron/energy transfer–reversible addition-fragmentation chain-transfer (PET-RAFT) polymerisation was adapted to enable high throughput synthesis of complex polymer architectures in dimethyl sulfoxide (DMSO) on low-volume well plates in the presence of air. The polymerisation system shows remarkable oxygen tolerance, and excellent control of functional 3- and 4-arm star polymers. We then apply this method to investigate the effect of polymer structure on protein binding, in this case to the lectin concanavalin A (ConA). Such an approach could be applied to screen the structure–activity relationships for any number of polymer–protein interactions.
Keywords: click chemistry, glycopolymers, oxygen tolerance, PET-RAFT, photomediated radical polymerization
Synthetic polymers are widely used to interface with cells and proteins using rationally designed approaches. However, the multitude of unknowns at this biointerface makes this design process very challenging. Often, the combinatorial probing of structure–activity relationships is the best approach and has enabled many researchers to identify biomaterials that better support cell growth and differentiation than rationally designed substrates.[1–5] In each of these applications, the materials are prepared by uncontrolled polymerization techniques, as precise control of molecular weight and polydispersity is less important than the bulk compositional characteristics. At the protein level, however, precise control of polymer size, shape, and binding motif presentation becomes very important. Many studies have shown that the multivalent presentation of ligands to soluble proteins and cell receptors is highly dependent on ligand density, spacing, and orientation.[6–12]
There has therefore been growing interest in methods that will enable the high throughput synthesis and screening of well-defined polymers. Early works showed the possibility of using automatic synthesisers to conduct conventional controlled radical polymerisations (CRPs) in inert atmospheres,[13–17] but the recent development of various oxygen tolerant CRP mechanisms including singlet oxygen trapping,[18–23] enzyme degassing,[24–29] among others,[14,30–33] have now been applied to the synthesis of polymer libraries in low volumes and open to the atmosphere.[34–39] These techniques have been used to prepare polymers with complex architectures such as arm first stars,[36,37] and block copolymers,[39] and have been applied to the investigation of polymerization-induced self-assembly (PISA) systems.[37,38] However, the synthesis of well-defined star polymers or bioactive functional polymers via such a combinatorial route has not been shown, nor have they been applied to the investigation of biological structure–activity relationships.
Most of the oxygen tolerant combinatorial CRP systems to date have been conducted in water, where solubility concerns limit the polymers and architectures that can be accessed. We recently reported a new photoinduced RAFT (PET-RAFT) polymerization technique using zinc tetraphenylporphyrin (ZnTPP) as a catalyst, which can enable oxygen tolerant polymerisations in organic solvents such as DMSO.[21–23] When we applied this system to flow polymerisations, the ZnTPP was able to perform two functions in the polymerisation when activated by visible light (at 560 nm); 1) converting triplet oxygen to singlet oxygen which is irreversibly trapped by the DMSO solvent, and 2) activating the RAFT agent to provide a source of initiation for the CRP. Given the system’s apparent tolerance to oxygen in an excellent solvent for multifunctional polymer synthesis, we hypothesised that this method could be applied to prepare combinatorial libraries of functional linear and star polymers. In this work we therefore investigated the kinetics and oxygen tolerance of the ZnTPP PET-RAFT technique for the polymerisation of a range of acrylamides in well plates at low volumes and monomer concentrations to high conversion (Scheme 1). We chose to test the hypothesis that such a method would allow screening of bioactivity by screening the effect of polymer structure on binding to a model lectin ConA. The binding of glycopolymers to lectins is well-studied,[40–47] and data on the affinity of mannose star polymers to ConA exist to validate our findings.[46,47] To this end we prepared libraries of mannose functionalised linear, 3-arm and 4-arm star glycopolymers by post-polymerisation modification of clickable polymer scaffolds,[48] and screened them for their binding affinity to ConA.
Scheme 1.
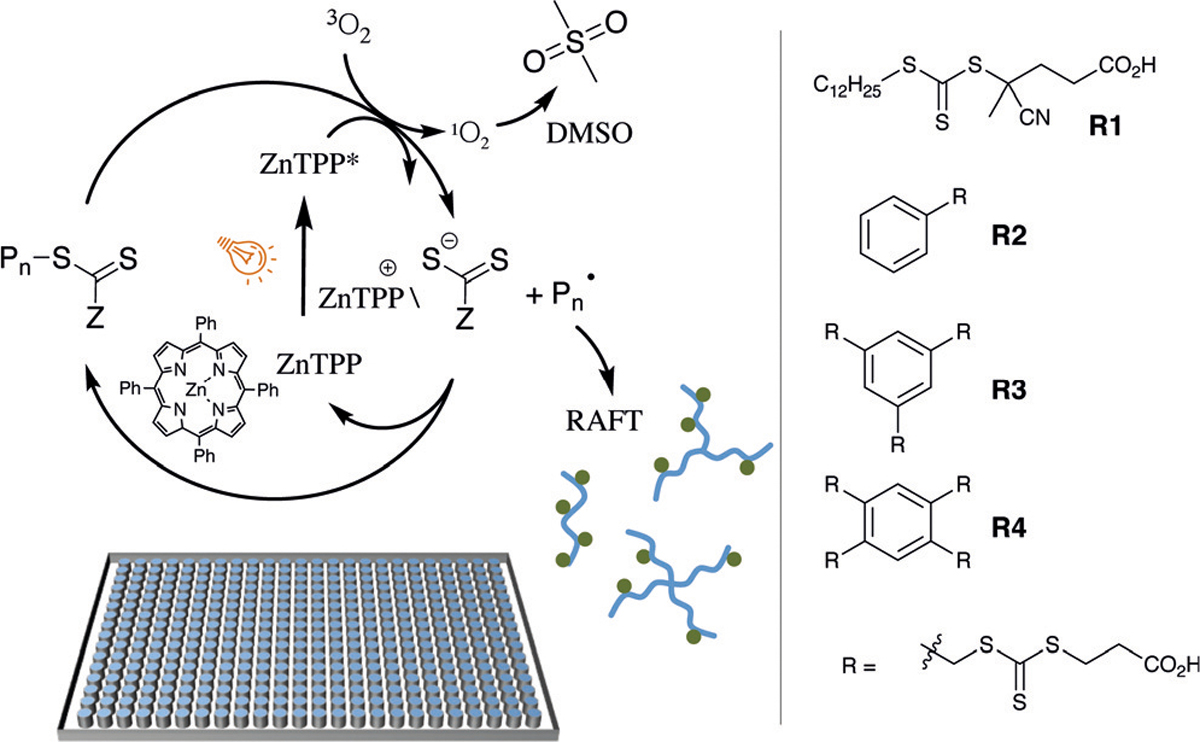
Representation showing the ZnTPP polymerisation mechanism and RAFT agents used in the library design.
We began by investigating the kinetics of a set of 1m N,N-dimethylacrylamide (DMA) polymerisations using ZnTPP in 96- and 364-well plates, at volumes of 300 μL and 40 μL respectively. Following previous studies, we held the ratio of ZnTPP to RAFT at 0.01–0.02, and irradiated the well plates at room temperature under 560 nm LED light (9.7 mWcm−2). Figure 1a shows the kinetics of a set of polymerisations with a target degree of polymerisation (DP) of 200 using a typical trithiocarbonate RAFT agent R1. At these concentrations of ZnTPP, no significant difference in polymerization rate was observed between the 40 μL and 300 μL polymerisations. A much longer inhibition time was observed in the 40 μL format, which was attributed to the lower area of light irradiation (relative to the sample volume) in these wells. MALDI-TOF analysis of crude polymer from a typical 40 μL polymerisation after 5 h of irradiation shows only the expected polymer peaks (Supporting Information, Figure S1) and all polymerisations, regardless of the format, resulted in products with low dispersity and the expected molecular weight as measured by GPC (Figure 1b). These results indicate that no detrimental side reactions occur during the inhibition period at the start of the polymerisations. Increasing the light intensity by addition of LED lamps at 590 nm led to faster kinetics but also the development of a small high molecular weight shoulder in the GPC traces, not present in the products of the slower polymerisations (Supporting Information, Figures S2,S3), indicating that some chain-chain coupling can occur if the light intensity is greater than 9.7 mWcm−2.
Figure 1.
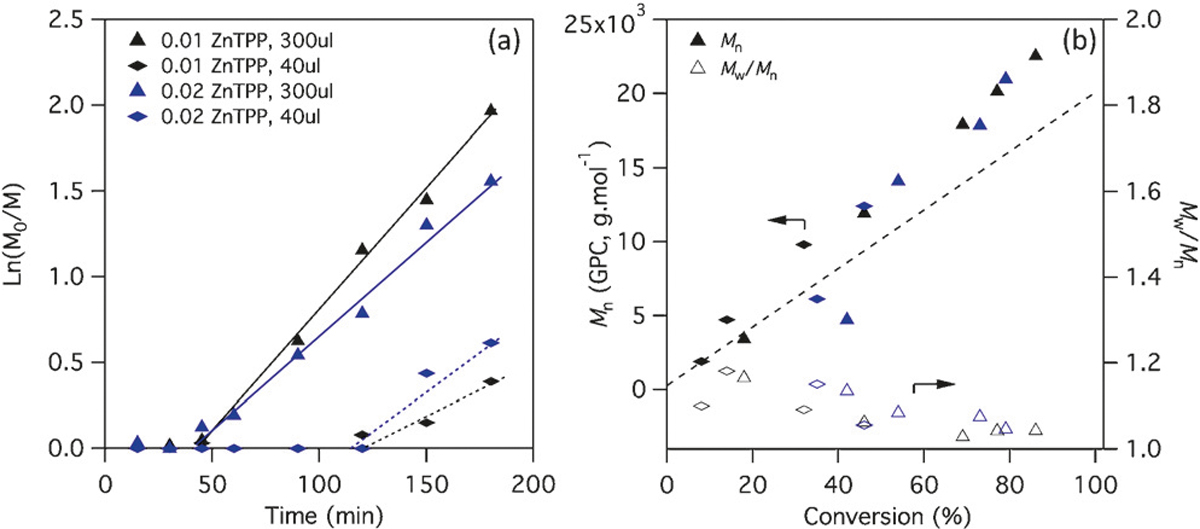
a) Pseudo first-order kinetics and b) molecular weight evolution of DMA polymerisations using RAFT agent R1 and either 0.01 or 0.02 equiv ZnTPP relative to RAFT, in well plates with 300 μL and 40 μL volumes. Target DP = 200, [DMA] = 1m.
We had assumed that the long inhibition period evident at the start of these low-volume polymerisations was due to a slower consumption of oxygen by the ZnTPP catalyst, but this was found not to be the case. In fact, the polymerisation kinetics were not noticeably affected by the presence of oxygen either at the start of the reaction, or when it is introduced during the polymerisation. Figure 2a shows the kinetics of a 1m DMA polymerisation (target DP = 200), conducted in 96-well plates either sealed in the presence of oxygen or after aliquoting the freeze–pump–thaw degassed polymerisation mixture to the plate in a glove box and Figure 2b the kinetics of the same polymerisations performed in a 1 mL cuvette (Figure 2b; Supporting Information, Table S1). Conversion was followed by NMR in the well-plate polymerisations, and by NIR in the cuvette polymerisations. In both formats these polymerisations show identical inhibition periods and rates of polymerisation with and without oxygen. Furthermore, no new inhibition period is observed after the bubbling air through the reaction mixture at various points during the polymerisation (as indicated by the arrows in Figure 2b). This can be attributed to the high efficiency with which ZnTPP converts triplet oxygen to singlet oxygen (Φ = 0.96), and the speed of singlet oxygen trapping by DMSO.[22] The slight differences in rates of polymerisations between the cuvette and the well-plate polymerisations is likely to be due to differences in effective light intensity due to the changed surface-area/volume ratio, angle and distance from the lamp that is unavoidable when changing the polymerisation format. When we substituted the RAFT agent for a different trithiocarbonate (R2, Figure 2b), we observed a change in the inhibition period, suggesting that inhibition is related to the activation of the RAFT agent rather than to do with the consumption of oxygen. We confirmed this by performing a chain extension experiment of a macroRAFT agent (pDMA15, Table S1 entry 1) with DMA (Figure 2b; Supporting Information, Table S1, entry 2), which resulted in no inhibition period, even after addition of air throughout the course of the polymerisation. The control of these polymerisations is very high, as evidenced by the narrow dispersity of the polymers (which remained below 1.15 even after chain extension), and complete conversion of the macroRAFT agent (Figure 2c; Supporting Information, Table S1). When left overnight in 96-well plates, polymerisations of a range of other acrylamides also proceeded to near full conversion with similarly excellent control (Figure 2d; Supporting Information, Table S2).
Figure 2.
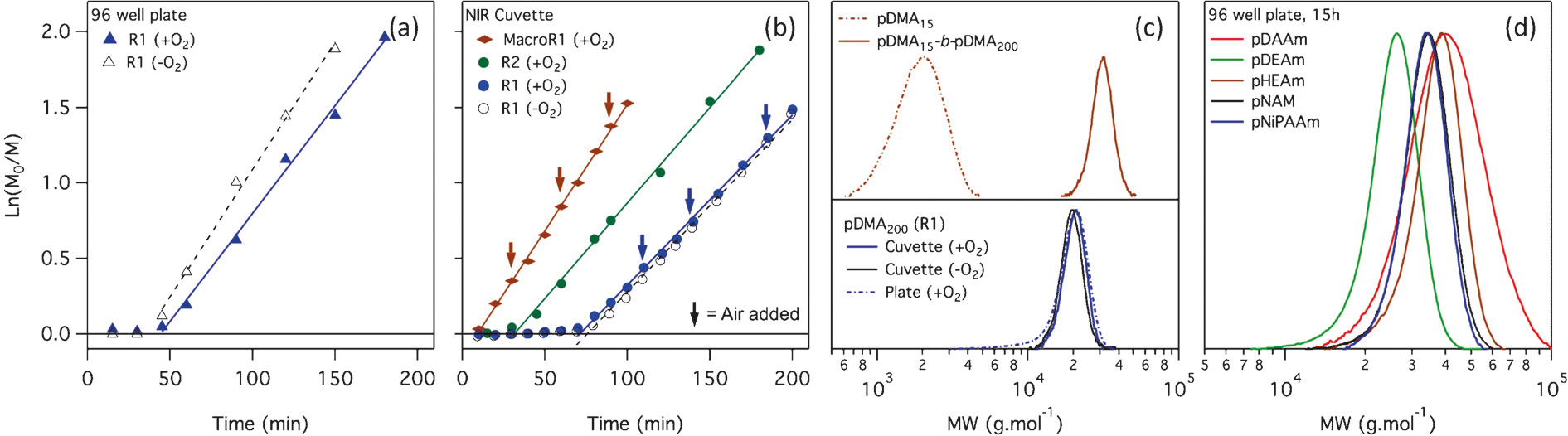
a),b) Pseudo first-order kinetics of polymerisations with [DMA]/[RAFT]/[ZnTPP] = 200:1:0.01 at 1m DMA in a a) 96-well plate and b) an NIR cuvette, with and without removal of oxygen prior to polymerisation. MacroR1 = DP 15 DMA macroRAFT agent prepared from R1 and purified prior to chain extension. Arrows indicate the bubbling of air through the cuvette with a pipette after measurement. c),d) GPC molecular weight distributions from the chain extension experiment and final kinetic samples (c) as well as from five other acrylamides at full conversion (d).
To test the hypothesis that the ZnTPP method could provide a simple method for the synthesis and screening of a library of complex architectures, we prepared libraries of pDMA based polymers from linear (R2), 3-arm (R3), and 4-arm (R4) RAFT agents at DPs of 20–960- in 96-well plates. We wanted to introduce bioactive functional groups, in this case mannose, using a versatile strategy that would allow any desired functionality to be incorporated into the polymer structure. For this reason, we decided to post-functionalise the polymers rather than polymerise the desired functionality into the polymer directly. Taking advantage of the organic solvent, we polymerised copolymer libraries of 90% (mol) DMA/10% (mol) NHS acrylate to incorporate a functional handle randomly into the backbone. Acetic acid (1 equiv/NHS) was mixed in with the monomer stock solution to prevent hydrolysis of the activated ester during polymerisation. Analysis by 1H NMR showed the activated ester was completely stable for at least 72 h under these conditions (Supporting Information, Figure S4). At a monomer concentration of 0.5m, overnight polymerisation was required to reach full conversion (>90%) with target DPs of 20–640. Despite the presence of the NHS acrylate and the long reaction times, the linear, 3-arm and 4-arm RAFT agents gave polymers with very low dispersities (Figure 3a; Supporting Information, Table S3). Minimal chain–chain coupling was observed, suggesting that not only is the ZnTPP methodology highly oxygen tolerant, but appears also to facilitate better control than is typically achieved with bulk thermally activated RAFT polymerisations. Similar results were obtained when the polymerisation was conducted in a 40 μL format on a 384-well plate (Supporting Information, Figure S9, Tables S7,S8). When the monomer concentration was increased to 1m on 96-well plates (Figure 3b; Supporting Information, Table S5), a shorter polymerization time of 5 h was required to reach high conversion, and similarly well-controlled linear, 3-arm and 4-arm stars could be obtained with either 10% or 20% (mol) NHS acrylate relative to DMA.
Figure 3.
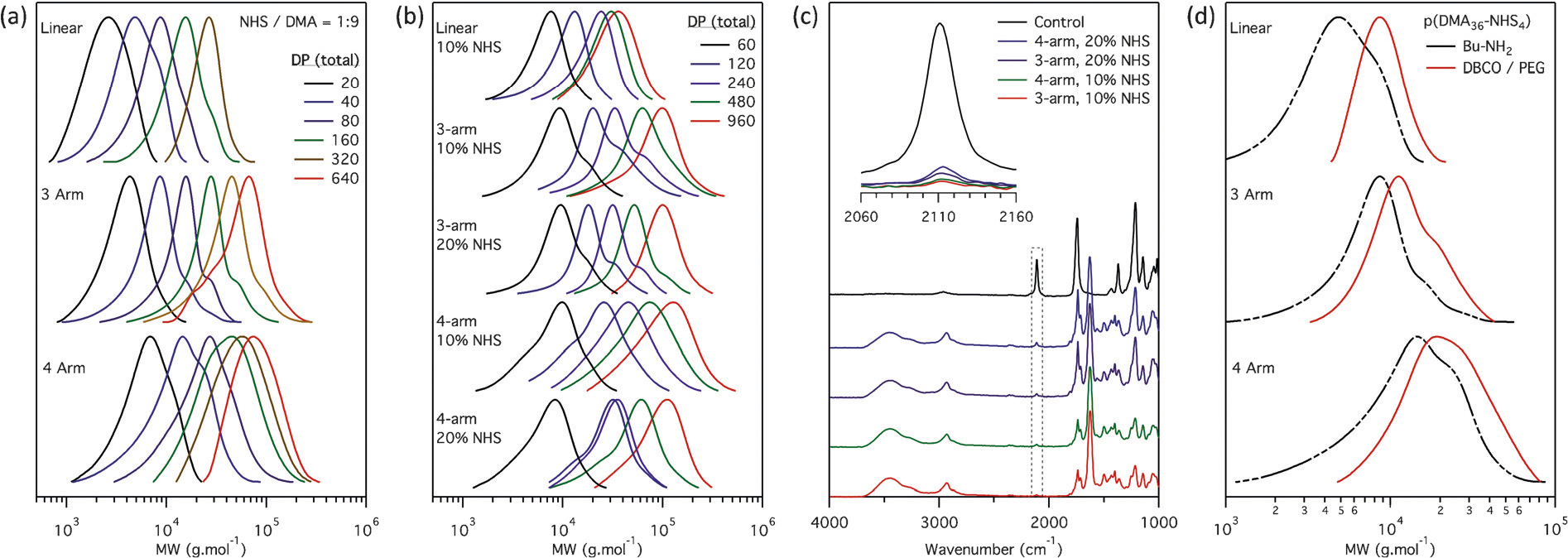
a),b) GPC molecular weight distributions of DMA/NHS polymer library polymerised at a monomer concentration of a) 0.5m, [DMA]/[NHS] = 9:1 (Supporting Information, Table S3) and b) 1m, [DMA]/[NHS] = 9:1 and 8:2 (Supporting Information, Table S5). c) FTIR on crude polymers from 0.5m polymerisation (Supporting Information, Table S6) after functionalization with DBCO-NH2 and Man(OAc)-N3 showing almost full functionalization. d) GPC molecular weight distributions of DP40 polymers before and after functionalization with DBCO-NH2 and PEG7-N3 (Supporting Information, Table S4). All polymers containing NHS were reacted with butyl amine before running the GPCs.
After polymerisation, the scaffolds were functionalised with a strained alkyne (dibenzocyclooctyne–amine, DBCO-NH2, 1 equiv/NHS) in the presence of dimethylaminopyridine (DMAP, 1 equiv/NHS), which should allow further functionalisation with any desired azide via the strain-promoted azide–alkyne cycloaddition (SPAAC) reaction.[49,50] To confirm the efficiency of this reaction, we monitored the reaction of DBCO-NH2 to the NHS acrylate monomer by 1H NMR under similar conditions as used on the polymer. Coupling proceeded to completion within 8 h, as can be seen by the downfield shift in both the vinyl and NHS protons (Supporting Information, Figure S5). As it was more difficult to monitor the reaction on the activated ester polymers by NMR, we instead used HPLC. Complete functionalisation of a range of linear, 3-arm, and 4-arm activated ester polymers (DP60–960, 10% and 20% NHS relative to DMA; Supporting Information, Table S6) with DBCO was observed within 6 h by the disappearance of the DBCO peak in the HPLC trace (Supporting Information, Figure S7). These clickable scaffolds were then functionalised with mannose by adding one equivalent of acetylated azido-mannose per strained alkyne to the crude mixture. Again, a model reaction performed using DBCO and azido-mannose showed completion of the azide–cyclooctyne click reaction within 8 h by the upfield shift in the sugar protons by NMR spectroscopy (Supporting Information, Figure S6). FTIR was used to confirm completion of the reaction on the DBCO functionalised polymer scaffolds by the near total disappearance of the azide signal (Figure 3c). After purification of a representative polymer (a linear DP 960 polymer, Supporting Information Table S6, entry 5) by precipitation in diethyl ether, the expected amount of mannose and DBCO relative to DMA could be observed by NMR (Supporting Information, Figure S8), confirming the success of this post-polymerisation modification approach. The successful functionalization of the scaffolds could also be observed by reacting DBCO functionalised polymers with PEG7-N3, which resulted in a clear upward shift in apparent molecular weight by GPC (Figure 3d; Supporting Information, Table S4).
After functionalisation, the acetyl protecting groups on the mannose were removed using sodium methoxide, and after neutralisation with acetic acid the polymers were diluted in PBS for measurement of their binding affinity to ConA. While the conversion was close to 100%, to ensure no free mannose from unincorporated NHS acrylate (or pipetting error) remained in the sample, the polymers were purified prior to use. We screened the functionalised polymers for their ability to bind ConA using a modified version of an enzyme linked lectin assay reported for wheat germ lectin.[44,45] In this assay the polymers were incubated at varying concentrations with a fixed concentration of ConA–horseradish peroxidase (ConA-HRP) conjugate, and then transferred onto a well plate pre-coated with a copolymer of acrylic acid and mannose acrylamide. Free ConA-HRP, which was not bound to the functional polymer in the incubation step, was allowed to adhere to the well, and was then quantified by development of the HRP after washing away any unbound material. Figure 4 shows the inhibition of ConA resulting from polymer–ConA binding against polymer concentration for the linear, 3-arm star, and 4-arm star series. At low polymer concentration, low binding to ConA was observed, and stronger binding was observed as the concentration increased. IC50 values, defined as the polymer concentration at which 50% of the lectin was bound were extracted from the sigmoidal fits and are shown in Figure 4d, normalised to mannose content (rather than polymer concentration). The 3-arm star polymers showed the strongest binding affinity at any given size (DP), and linear polymers performed similar or better than the 4-arm polymers for any given DP. Relative to polymer molarity binding affinity increased with polymer size (Figure 4a–c), but relative to total mannose concentration the smaller polymers actually bound the lectin slightly better (Figure 4d). While none of the binding affinities measured were particularly high, relative to polymer concentration they are within the very broad range of IC 50 values for other lectin binding polymers in the literature.[44,46,47] Our aim in this work was not primarily to find a strongly binding polymer but rather to investigate whether the method could provide a simple way to screen for fine effects of changes to the polymer size and structure on binding efficiency. The success of the approach is evident from the differences between the polymers observed in these data.
Figure 4.
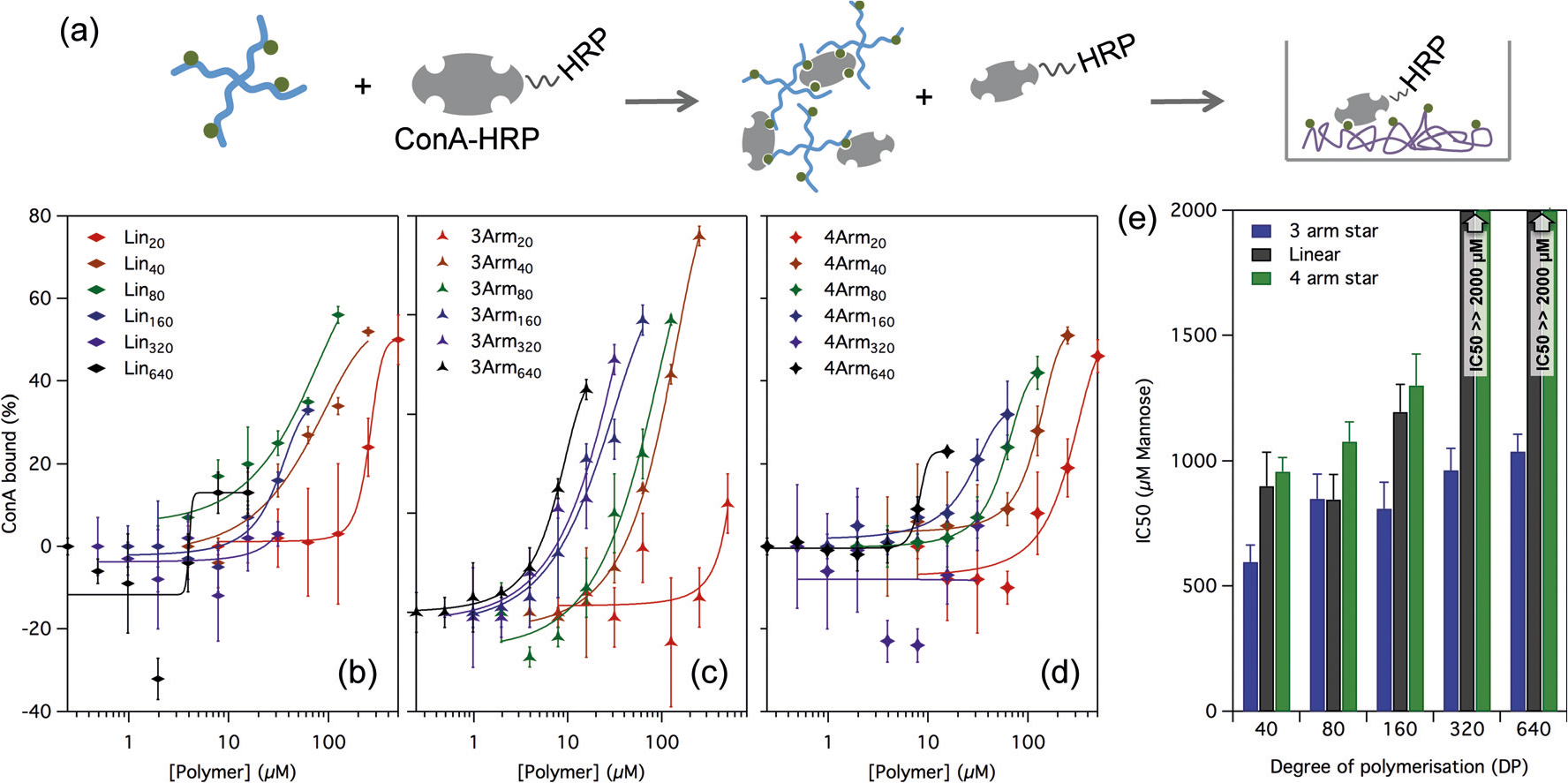
(a) Enzyme-linked lectin binding assay. Polymer is incubated with the lectin-HRP conjugate at varying concentrations. Free unbound Con-A-HRP adheres to a mannose-coated well plate, and the HRP developed to determine the fraction of Con-A bound to the polymer. b)–d) Binding curves for linear (b), 3 arm star (c), and 4 arm star (d) polymer series to ConA, showing the percentage of unbound lectin vs. polymer concentration. e) IC50 values extracted from the sigmoidal fit, normalised to mannose content showing the effect of size and structure on binding affinity. Error bars correspond to ±1 standard deviation from the mean in triplicate measurements. The IC50 for the linear and 4-arm polymers at DP 320 and 640 could not be determined as minimal inhibition was observed at the concentration range tested, but were found to be much greater than 2000 μm mannose.
In conclusion, we have demonstrated the very high oxygen tolerance of ZnTPP mediated PET-RAFT polymerisations. This reaction allows for the high throughput synthesis of polymer libraries with complex architectures in organic solvents such as DMSO and in low volumes. We have applied this approach to make clickable polymer libraries with linear, 3-arm, and 4-arm stars, which we functionalised with mannose. The library allows the relationships between polymer structure and protein binding, in this case to the lectin ConA. We anticipate that such an approach could be applied to screen the structure–activity relationships for any number of polymer-protein interactions
Experimental Section
Full synthetic details of all of the materials used in this work are given in the Supporting Information. The RAFT agent 4-cyano-4-[(dodecylsulfanylthiocarbonyl) sulfanyl] pentanoic acid (CDTPA, R1) was purchased from Boron Scientific and used as supplied. S-benzyl S’-propionic acid trithiocarbonate (BSPA, R2), was synthesised according to literature procedures and isolated as a yellow solid. With the exception of NHS acrylate, all monomers were purchased from Sigma Aldrich and deinhibited prior to use by passing them over MEHQ inhibitor removal resin. 1,3,4,6-tetra-O-acetyl-2-azido-2-deoxy-α-d-mannopyranose (Man(OAc)-N3) was purchased from Carbosynth (UK). Zinc tetraphenyl porphyrin and all other reagents, including the ConA–horseradish peroxidase conjugate, were purchased from Sigma Aldrich and used as supplied.
General procedure for RAFT polymerisations in well plates: Stock solutions of RAFT agent (0.2m), ZnTPP (0.004m), DMA and NHS acrylate (2m) were prepared in DMSO and mixed to the desired volume in either clear 96-well flat-bottom tissue culture plates (Corning), or low volume 384-well clear flat-bottom black plates (Corning), using micropipettes. Acetic acid (1 equiv/NHS) was included in the NHS acrylate stock solution to prevent hydrolysis from any water present in the DMSO. 96-well plates were filled with 150 or 300 μL of reaction mixture, and 384-well plates with 40 μL. Plates were then covered with PCR tape to prevent evaporation of the monomer, and exposed to 560 nm LED light from an Oriel LSS-7120 solar simulator (9.7 mWcm−2). All kinetics were performed at a monomer concentration of 1m, and at a RAFT/M ratio of 1:200. Kinetic samples were taken after removing the plate from the light source and piercing the top of the tape with a needle, using a separate well for each timepoint. For the degassed experiment, the reaction mixture was first mixed, degassed by five freeze–pump–thaw cycles, and transferred to the well plates in a glovebox. Libraries were prepared by polymerisation in well plates for 18 h under the same conditions with 0.01 equiv ZnTPP/RAFT agent, and were found by 1H NMR to proceed to close to full conversion (>95% in most cases).
Post-polymerisation modification and purification of polymer scaffolds: After polymerisation, the polymer solution (100 μL) was added to a mixture of DBCO-NH2 (1 equiv/NHS) and dimethylaminopyridine (1 equiv/NHS) in DMSO (20 μL) and left for at least 8 h. Part of this mixture (20 μL) was then added to either PEG7-N3 (1 equiv/DBCO) or Man(OAc)-N3 (1 equiv/DBCO) in DMSO (10 μL) and left overnight. The mannose functional polymers were then deprotected by addition of sodium methoxide in methanol (4 equiv NaOMe/mannose, 5 μL total volume), left for 1 h and neutralised by addition of acetic acid in water (4 equiv/mannose, 5 μL total volume). The polymers were then diluted in PBS such that the concentration of mannose was 2 mm (total volume 415 μL), and purified by passing through a spin column of sephadex G15. In this procedure a 2 mL spin column was loaded with sephadex (0.5 mL in PBS), and the eluent removed by spinning the column at 6000 rpm for 5 s. The polymer was then added to the top of the column and it was spun again for 8 s. No change in volume was observed.
Preparation of mannose coated plates for binding assay: Mannose acrylamide (200 mg, 60 equiv), acrylic acid (130 mg, 140 equiv), and azobisisobutyronitrile (AIBN, 1.8 mg, 1 equiv) were dissolved in methanol (2 mL), degassed by bubbling with N2, and polymerised overnight at 60°C to full conversion. The polymer was purified by dialysis against water (MWCO = 3.5 kDa), filtered (PES, 0.45 μm) and freeze dried. The polymer was redissolved in PBS (pH 7.4) at 1 mg mL−1 and incubated in standard 96-well tissue-culture plates (Corning, 50 μL/well, overnight). The solution was then removed and the plates were blocked with BSA (5 mg mL−1, 150 μL/well, 30 min) and washed (3 × PBST 0.01% w/v, 3 × PBS) before use in the binding assay.
Enzyme linked lectin binding assay: The polymer samples were diluted in PBS to the desired concentration and incubated with a ConA–horseradish peroxidase conjugate (ConA-HRP, 0.2 μg mL−1, PBS) in 96-well tissue-culture plates (180 μL/well, plates pre-blocked with BSA and washed as above) for 1 h. The mixture was then transferred to mannose-coated well plates in triplicate (50 μL/well) and incubated for another 1 h for the free ConA-HRP, not bound to the polymer in the first step, to bind to the surface of the plate. The mannose-coated plates were then washed (3 × PBST 0.01% w/v, 3 × PBS), and the amount of ConA-HRP bound to the plate was quantified by developing the HRP with 3,3’,5,5’-tetramethylbenzidine (TMB, 0.5 mm) and hydrogen peroxide (0.01% v/v) in citrate buffer (100 μL, 50 mm, pH 5.5) for 10–20 min. After development the reaction was stopped by addition of H2SO4 (1m, 10 μL), and the absorbance was read at 450 nm. The average binding of polymer to ConA was determined at each concentration relative to a control sample of ConA-HRP not incubated with any free polymer, and this data was used to construct IC50 curves using a sigmoidal fit.
Supplementary Material
Acknowledgements
A.J.G. was supported by a Busch Biomedical Seed Grant and an American Cancer Society—Institutional Research Grant Early Investigator Pilot Award. R.C. is grateful to UNSW for funding through the Vice Chancellors Research fellowship, and to the Australian Research Council (ARC) for funding through the Discovery Early Career Research Award (DE170100315). C.B. acknowledges the ARC for his Future Fellowship (FT12010096).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and ORCID(s) from the author(s) for this article can be found under: https://doi.org/10.1002/anie.201711044.
Contributor Information
Adam J. Gormley, Department of Biomedical Engineering, Rutgers, NJ (USA)
Jonathan Yeow, Australian Centre for Nanomedicine, UNSW, Sydney (Australia); Centre for Advanced Macromolecular Design, School of Chemical Engineering, UNSW, Sydney (Australia).
Gervase Ng, Australian Centre for Nanomedicine, UNSW, Sydney (Australia); Centre for Advanced Macromolecular Design, School of Chemical Engineering, UNSW, Sydney (Australia).
Órla Conway, Australian Centre for Nanomedicine, UNSW, Sydney (Australia); Centre for Advanced Macromolecular Design, School of Chemistry, UNSW, Sydney (Australia).
Cyrille Boyer, Australian Centre for Nanomedicine, UNSW, Sydney (Australia); Centre for Advanced Macromolecular Design, School of Chemical Engineering, UNSW, Sydney (Australia).
Robert Chapman, Australian Centre for Nanomedicine, UNSW, Sydney (Australia); Centre for Advanced Macromolecular Design, School of Chemistry, UNSW, Sydney (Australia).
References
- [1].Lynn GM, Laga R, Darrah PA, Ishizuka AS, Balaci AJ, Dulcey AE, Pechar M, Pola R, Gerner MY, Yamamoto A, et al. , Nat. Biotechnol. 2015, 33, 1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mei Y, Saha K, Bogatyrev SR, Yang J, Hook AL, Kalcioglu ZI, Cho S-W, Mitalipova M, Pyzocha N, Rojas F, et al. , Nat. Mater. 2010, 9, 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Green JJ, Langer R, Anderson DG, Acc. Chem. Res. 2008, 41, 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Goldberg M, Mahon K, Anderson D, Adv. Drug Delivery Rev. 2008, 60, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Simon CG, Lin-Gibson S, Adv. Mater. 2011, 23, 369–387. [DOI] [PubMed] [Google Scholar]
- [6].Cairo CW, Gestwicki JE, Kanai M, Kiessling LL, J. Am. Chem. Soc. 2002, 124, 1615–1619. [DOI] [PubMed] [Google Scholar]
- [7].Kane RS, Langmuir 2010, 26, 8636–8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Webber MJ, Tongers J, Newcomb CJ, Marquardt K-T, Bauersachs J, Losordo DW, Stupp SI, Proc. Natl. Acad. Sci. USA 2011, 108, 13438–13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Moon JJ, Hahn MS, Kim I, Nsiah BA, West JL, Tissue Eng. Part A 2009, 15, 579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pashuck ET, Duchet BJR, Hansel CS, Maynard SA, Chow LW, Stevens MM, ACS Nano 2016, 10, 11096–11104. [DOI] [PubMed] [Google Scholar]
- [11].Kanai M, Mortell KH, Kiessling LL, J. Am. Chem. Soc. 1997, 119, 9931–9932. [Google Scholar]
- [12].Conway A, Vazin T, Spelke DP, Rode NA, Healy KE, Kane RS, Schaffer DV, Nat. Nanotechnol. 2013, 8, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guerrero-Sanchez C, Moad G, Haven JJ, Guerrero-Sanchez C, Keddie DJ, Moad G, Thang SH, Schubert US, Polym. Chem. 2014, 5, 5236–5246. [Google Scholar]
- [14].Pan X, Lathwal S, Mack S, Yan J, Das SR, Matyjaszewski K, Angew. Chem. Int. Ed. 2017, 56, 2740–2743; Angew. Chem. 2017, 129, 2784–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guerrero-Sanchez C, Keddie DJ, Saubern S, Chiefari J, ACS Comb. Sci. 2012, 14, 389–394. [DOI] [PubMed] [Google Scholar]
- [16].Guerrero-Sanchez C, OQBrien L, Brackley C, Keddie DJ, Saubern S, Chiefari J, Polym. Chem. 2013, 4, 1857. [Google Scholar]
- [17].Haven JJ, Guerrero-Sanchez C, Keddie DJ, Moad G, Macromol. Rapid Commun. 2014, 35, 492–497. [DOI] [PubMed] [Google Scholar]
- [18].Shanmugam S, Xu J, Boyer C, J. Am. Chem. Soc. 2015, 137, 9174–9185. [DOI] [PubMed] [Google Scholar]
- [19].Xu J, Jung K, Boyer C, Macromolecules 2014, 47, 4217–4229. [Google Scholar]
- [20].Xu J, Jung K, Atme A, Shanmugam S, Boyer C, J. Am. Chem. Soc. 2014, 136, 5508–5519. [DOI] [PubMed] [Google Scholar]
- [21].Shanmugam S, Xu J, Boyer C, Macromolecules 2017, 50, 1832–1846. [Google Scholar]
- [22].Corrigan N, Rosli D, Warren J, Jones J, Xu J, Boyer C, Macromolecules 2016, 49, 6779–6789. [Google Scholar]
- [23].Yeow J, Shanmugam S, Corrigan N, Kuchel RP, Xu J, Boyer C, Macromolecules 2016, 49, 7277–7285. [Google Scholar]
- [24].Gormley AJ, Chapman R, Stevens MM, Nano Lett. 2014, 14, 6368–6373. [DOI] [PubMed] [Google Scholar]
- [25].Zhang B, Wang X, Zhu A, Ma K, Lv Y, Wang X, An Z, Macromolecules 2015, 48, 7792–7802. [Google Scholar]
- [26].Chapman R, Gormley AJ, Herpoldt K, Stevens MM, Macromolecules 2014, 47, 8541–8547. [Google Scholar]
- [27].Lv Y, Liu Z, Zhu A, An Z, J. Polym. Sci. Part A 2017, 55, 164–174. [Google Scholar]
- [28].Liu Z, Lv Y, An Z, Angew. Chem. Int. Ed. 2017, 56, 13852–13856; Angew. Chem. 2017, 129, 14040–14044. [DOI] [PubMed] [Google Scholar]
- [29].Enciso AE, Fu L, Russell AJ, Matyjaszewski K, Angew. Chem. Int. Ed. 2017, DOI: 10.1002/anie.201711105; [DOI] [PubMed] [Google Scholar]
- [30].Matyjaszewski K, Coca S, Gaynor SG, Wei M, Wood-worth BE, Macromolecules 1998, 31, 5967–5969. [Google Scholar]
- [31].Gody G, Barbey R, Danial M, Perrier S, Polym. Chem. 2015, 6, 1502–1511. [Google Scholar]
- [32].Reyhani A, McKenzie TG, Ranji-Burachaloo H, Fu Q, Qiao GG, Chem. Eur. J. 2017, 23, 7221–7226. [DOI] [PubMed] [Google Scholar]
- [33].Fu Q, Xie K, McKenzie TG, Qiao GG, Polym. Chem. 2017, 8, 1519–1526. [Google Scholar]
- [34].Wu H, Yang L, Tao L, Polym. Chem. 2017, 8, 5679–5687. [Google Scholar]
- [35].Siegwart DJ, Leiendecker M, Langer R, Anderson DG, Macromolecules 2012, 45, 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cosson S, Danial M, Saint-Amans JR, Cooper-white JJ, Macromol. Rapid Commun. 2017, 38, 1600780. [DOI] [PubMed] [Google Scholar]
- [37].Yeow J, Chapman R, Xu J, Boyer C, Polym. Chem. 2017, 8, 5012–5022. [Google Scholar]
- [38].Tan J, Liu D, Bai Y, Huang C, Li X, He J, Xu Q, Zhang L, Macromolecules 2017, ASAP. [Google Scholar]
- [39].Chapman R, Gormley AJ, Stenzel MH, Stevens MM, Angew. Chem. Int. Ed. 2016, 55, 4500–4503; Angew. Chem. 2016, 128, 4576–4579. [DOI] [PubMed] [Google Scholar]
- [40].Abdouni Y, Yilmaz G, Becer CR, Macromol. Rapid Commun. 2017, 38, 1700212. [DOI] [PubMed] [Google Scholar]
- [41].Becer CR, Macromol. Rapid Commun. 2012, 33, 742–752. [DOI] [PubMed] [Google Scholar]
- [42].Zhang Q, Collins J, Anastasaki A, Wallis R, Mitchell DA, Becer CR, Haddleton DM, Angew. Chem. Int. Ed. 2013, 52, 4435–4439; Angew. Chem. 2013, 125, 4531–4535. [DOI] [PubMed] [Google Scholar]
- [43].Li X, Chen G, Polym. Chem. 2015, 6, 1417–1430. [Google Scholar]
- [44].Bojarová P, Chytil P, Mikulova B, Bumba L, Konefał R, Pelantová H, Krejzová J, Slámová K, Petrásková L, Kotrchová L, et al. , Polym. Chem. 2017, 8, 2647–2658. [Google Scholar]
- [45].Fiore M, Berthet N, Marra A, Gillon E, Dumy P, Dondoni A, Imberty A, Renaudet O, Org. Biomol. Chem. 2013, 11, 7113. [DOI] [PubMed] [Google Scholar]
- [46].Chen Y, Chen G, Stenzel MH, Macromolecules 2010, 43, 8109–8114. [Google Scholar]
- [47].Chen Y, Lord MS, Piloni A, Stenzel MH, Macromolecules 2015, 48, 346–357. [Google Scholar]
- [48].Slavin S, Burns J, Haddleton DM, Becer CR, Eur. Polym. J. 2011, 47, 435–446. [Google Scholar]
- [49].Agard NJ, Prescher JA, Bertozzi CR, J. Am. Chem. Soc. 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- [50].Iha RK, Wooley KL, Nystrom AM, Burke DJ, Kade MJ, Hawker CJ, Chem. Rev. 2009, 109, 5620–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.