

Abstract
Geographic atrophy (GA) secondary to age-related macular degeneration (AMD) is a retinal neurodegenerative disorder. Human genetic data support the complement system as a key component of pathogenesis in AMD, which has been further supported by pre-clinical and recent clinical studies. However, the involvement of the different complement pathways (classical, lectin, alternative), and thus the optimal complement inhibition target, has yet to be fully defined. There is evidence that C1q, the initiating molecule of the classical pathway, is a key driver of complement activity in AMD. C1q is expressed locally by infiltrating phagocytic cells and C1q-activating ligands are present at disease onset and continue to accumulate with disease progression. The accumulation of C1q on photoreceptor synapses with age and disease is consistent with its role in synapse elimination and neurodegeneration that has been observed in other neurodegenerative disorders. Furthermore, genetic deletion of C1q, local pharmacologic inhibition within the eye, or genetic deletion of downstream C4 prevents photoreceptor cell damage in mouse models. Hence, targeting the classical pathway in GA could provide a more specific therapeutic approach with potential for favorable efficacy and safety.
Keywords: Age-related macular degeneration, Complement, geographic atrophy, C1q, Neurodegeneration
Background
Geographic atrophy (GA) secondary to age-related macular degeneration (AMD) is a chronic progressive neurodegenerative disorder of the retina involving the loss of photoreceptor cells (neurons) and supportive retinal pigmented epithelial cells (RPE) [1]. GA affects approximately 5 million people world-wide and is a common cause of blindness in the developed world [2]. The complement system has been implicated in GA by human genetics, retinal pathology, genomic and proteomic analyses of human tissues, and by work in animal models [3–11]. However, these studies do not address the identity of the pathway for complement activation in disease—classical, lectin, or alternative. While the alternative pathway has been implicated in numerous studies, its specific role is unclear since components of the alternative pathway are integral to the amplification of all three pathways. Further, targeting alternative pathway components has not been clinically successful [12]. Inhibition of C3 or C5 as core components of all 3 pathways has proven more promising in clinical studies, but targeted inhibition within a single pathway may provide greater safety and efficacy. In this regard, C1q, the initiating molecule of the classical complement pathway, may play an important role in GA pathogenesis. C1q and its activating substrates are present in nearly all layers of the outer retina and accumulate throughout disease progression. Further, C1q plays a unique role in synapse elimination and neurodegeneration in multiple disorders of the central nervous system [13–19] and may likewise contribute to the loss of photoreceptor cells in GA. Finally, genetic deletion of C1q or downstream C4, as well as pharmacological inhibition of C1q are protective in animal models of photoreceptor cell loss [6, 11]. Thus, C1q and the classical pathway are well positioned to be important drivers of complement activation and pathology in GA.
Overview of the complement system
The complement system is comprised of a core enzymatic cascade that has three major routes of activation—the classical, lectin, and alternative pathways (Fig. 1; [20]). Each pathway is defined by distinct initiating molecules that interact with different target substrates for activation, but all produce 3 main outcomes: target opsonization, release of anaphylatoxins, and target membrane damage (Fig. 1). Opsonization refers to coating the target surface with complement activation products, or opsonins (e.g., C1q, C4b, C3b, iC3b), that are recognized by phagocytic immune cells. Anaphylatoxins are soluble activation products (C4a, C3a, C5a) that increase vascular permeability, regulate local cell activity, and recruit immune cells. The membrane attack complex is comprised of complement components (C5b, C6-C9) that form a pore structure resulting in membrane dysfunction or cell lysis. Due to a high degree of pathway regulation, upstream activation can lead to varying levels of immune cell recruitment and opsonization without resulting in downstream lysis.
Fig. 1.

Key Components of the Complement System: C1q is the initiating molecule of the classical pathway. Binding to any of a number of substrates triggers its activation, leading to an enzymatic cascade with cleavage of the core solution-phase components C4, C2, C3, and C5 to generate activated fragments. The lectin pathway is initiated by MBL or related ficolin or collectin family members [22]. They activate the same cascade (C4, C3, and C5) as C1q by binding to a different but overlapping set of substrates, including carbohydrate and acetylated structures [21]. The alternative pathway is initiated by spontaneous solution-phase hydrolysis of C3 to generate C3b-H2O, which then becomes associated with the activated protease Bb to cleave other C3 molecules, generating C3b [23]. If close enough to a surface, C3b can covalently attach to initiate the surface amplification loop described above. It is important to note the distinction between the alternative complement pathway and the complement amplification loop. The alternative pathway is defined by its initiating mechanism—the spontaneous activation of C3 via non-enzymatic hydrolysis. This event leads to production of C3b, which then engages factor B and the amplification loop. However, since all three complement pathways lead to production of C3b and engage the amplification loop, this loop functions as a general “complement amplification loop” or “C3b amplification loop.” Hence, unless specifically affecting the spontaneous hydrolysis rate of C3, genetic polymorphisms or mutations that affect amplification impact the activity of all three pathways (e.g., FH, FI, FB, FD, C3, and FHR1/3). Also of note, the classical and lectin pathways will remain active as long as their substrates are present
C1q is the initiating molecule of the classical pathway, while mannose-binding lectin and the highly related molecules collectin-10 and -11 and ficolin-1, -2, and -3 (together referred to as “MBL” in this review) are the initiating molecules of the lectin pathway [21, 22]. They function as pattern recognition molecules that bind to a broad spectrum of substrates found on pathogens, damaged tissue, and tissue debris (Fig. 1). They are designed to focus robust complement activation on a select set of targets. In contrast, the alternative pathway is stochastic, with spontaneous solution-phase hydrolysis of complement component C3 that leads to random tagging of nearby surfaces with C3b for pathway activation [23]. The alternative pathway functions to detect pathogens or substrates that may not otherwise be recognized by more selective immune receptors. While diffuse activation of the alternative pathway is ongoing throughout the body, classical and lectin pathway activation is initiated by specific substrates and ceases when the substrates are cleared. However, if activating substrates are persistent (produced faster than can be removed or resisting removal) ongoing complement activation can occur, resulting in significant tissue damage. In GA, numerous C1q-activating substrates are present at the earliest stages of disease and continue to accumulate with disease progression as components of drusen and drusenoid deposits, as well as normal and stress-related components of cell membranes. As shown in Fig. 1, these substrates include pentraxins such as C-reactive protein (CRP) and serum amyloid protein (SAP), phospholipids such as phosphatidyl serine and lysophospholipids, hydroxyapatite, immunoglobulins, and amyloid material (discussed further below; [22, 24–29]). Hence, ongoing classical complement activation is a likely component of the chronic inflammatory response in GA.
It is important to note that all 3 complement pathways lead to the cleavage of C3 to form C3b as a central component of the complement system. C3b can engage the protease factor B to cleave additional C3 and generate more C3b. This central amplification loop functions to amplify the amount of C3b bound to the substrate surface and is an integral part of all 3 complement pathways, thereby enhancing their impact [22, 30]. Since the amplification loop involves factors B and D, it is often confused with the alternative complement pathway, which is initiated by spontaneous, solution-phase hydrolysis of C3 (C3-H2O) and depends upon factors B and D for amplification. However, the amplification loop is a component of all three pathways, and genetic mutations that affect the amplification loop (including C3, complement factor H-related (FHR) 1/3, and factors B, D, I, and H) affect the activity of all three pathways. Greater detail of these pathways is provided in the legend to Fig. 1 and in the text below.
Human genetics implicate the complement system in GA
Human genetics strongly implicate the involvement of the complement system in GA. Variants in at least 6 complement-related genes are associated with disease, including CFH, CFHR1/3, C2/CFB, C3, CFI, C9 [3, 4, 32–35]. Together, variants in complement-related genes account for more than half of AMD genetic risk [3, 36], and the altered protein products of these genes have been directly linked to regulation of complement activity (Table 1). Other genetic loci are strongly and independently linked to GA, but can work in conjunction with complement variants to augment GA progression. The ARM2/HTRA1 locus, in particular, has been associated with increased neovascular AMD [37–40].
Table 1.
Genes associated with GA/AMD
| Gene | Protein | Activity | Impact of mutation/polymorphism on protein product function | Mutation impact on complement activity | GA risk |
|---|---|---|---|---|---|
| CFH | Factor H (FH) | Cofactor for FI-mediated cleavage of C3b; Competes C1q binding; Competes FHR1 | Decrease surface binding of FH decreases FI cleavage; allows C1q binding | Increase | Increase OR = 2.7–7.4 |
| CFHR1/3 | FHR1 and / or FHR3 | Stabilizers C3b; enhances C1q binding; Competes FH | Gentic deletion allows more FH binding, greater FI cleavage of C3b, less C1q binding | Decrease | Decrease OR = 0.35 |
| CFI | Factor I (FI) | Cleaves C3b to inhibit cascade activity and to produce Ic3b for opsonization/phagocytosis | Reduced FI levels / activity | Increase | Increase OR up to 22 |
| C2 | C2 | Drives classical / lectin activation of C3 | Uncharacterized | Increase | Decrease OR = 0.47–0.49 |
| CFB | Factor B (FB) | Drives alternative / AP amplification via activation of C3 | Decrease activity | Decrease | Decrease OR = 0.41–0.54 |
| C3 | C3 | Activation leads to C3b as opsonin and plateform for AP amplification or C5 cleavage | Resistance to inactivation by FI cleavage | Increase | Increase OR 2.8 |
| C9 | C9 | Component of membrane attack complex(MAC) | Increased MAC polymerization | Increase | Increase OR 2.2 |
Reference for Table 1: CFH: 23, 37, 38, 41–44; CFHR1/3: 32, 39, 45–48; FI: 35, 49, 50, 54; C2/CFB: 51, 52, 53, 55-57; C3: 35; C9: 58
The most compelling aspect of the genetic evidence is that variants known to increase complement activity are associated with increased risk of AMD, while those that decrease complement activity provide protection. For example, a common variant of CFH (Y402H) that generally leads to enhanced complement activity is associated with a 3- to sevenfold increased risk of AMD [41, 42], and a common genetic deletion of FHR1 that decreases complement activity reduces risk of AMD by 60% [32, 43, 44]. The impact of genetic polymorphisms in AMD is further discussed in Table 1.
While human genetics point to increased complement activity in GA, they do not specifically address how complement is being activated in disease. The alternative pathway has been historically implicated in AMD pathobiology because many of the affected genes impact the stability of C3b, the initiating component of the amplification loop. However, as discussed above, C3b and its amplification loop are central components of all 3 complement pathways, with C3b functioning as a key opsonin and a critical element of both C5 convertases (i.e., C3b working in conjunction with C4b2a or C3bBb for C5 convertase activity; Fig. 1).
Complement in GA pathology
GA affects the outer most layers of the retina—the photoreceptor cells, RPE cells, and vascular choroid layer—and involves the presence or abnormal accumulation of complement-activating substrates within these layers ([4, 5, 36]; Fig. 2). The unique oxidative and metabolic environment of the outer retina leads to damage of light-collecting membranes, the photoreceptor cell outer segments (POS), that undergo a process of distal shedding and proximal renewal [59]. Shed POS exhibit phosphatidyl serine on their outer membrane, facilitating their recognition by RPE for uptake and removal. However, in AMD, damaged membranes and other lipid-rich cellular components accumulate as drusen and drusenoid deposits below and above the RPE [36, 60, 61]. These deposits contain a number of complement-activating substrates that are specifically recognized by C1q, including CRP, SAP, phosphatidyl serine, hydroxyapatite, lysophospholipids, immunoglobulins, and amyloid material [8, 22, 24–29, 62, 63]. A compromise in the blood-retinal barrier due to RPE dysfunction allows leakage of serum proteins, including C1q and other complement components, into the retina from the underlying choriocapillaris [6]. C1q, either alone or in conjunction with CRP and other pentraxins, binds to phosphatidyl serine exposed on POS and on damaged RPE membranes [6], as well as to the numerous substrates present in drusen. Local complement activation leads to recruitment of macrophages and microglia, which produce additional C1q and other complement components. POS normally lack expression of membrane-bound complement regulatory proteins (such as CD46, CD55; [6]), while expression of these proteins is down-regulated on the surface of damaged RPE, making membranes within both retinal layers exposed to complement attack [64]. With ineffective clearance and continued accumulation of drusen and drusenoid deposits, C1q binding and classical complement activation will continue, along with engagement of the downstream amplification loop. Therefore, the unique structure and function of the outer retina—with highly metabolic, membrane-rich cells that are exposed to light in an oxygen-rich environment—makes it particularly vulnerable to dysregulation of the complement system.
Fig. 2.

Expression and Deposition of C1q in Outer Layers of the Retina in GA: C1q is deposited in multiple locations within the outer retina, including on photoreceptor cell synapses within the OPL, on photoreceptor cell outer segments below the ONL, and on drusen within the RPE and BM. C1q is expressed locally within these layers by infiltrating microglial cells and / or macrophages
Animal models of GA
To study the role of complement in RPE and photoreceptor cell damage, numerous animal models have been developed through direct damage to these cell types, including exposure to blue light to damage RPE, white light to induce photoreceptor cell damage, NaIO3 administration to selectively kill RPE, focal laser damage to disrupt the blood/retinal barrier, and immunization against photo-oxidative metabolites to cause direct immune attack [6, 65, 66]. These studies consistently show increased retinal expression of complement components within the classical, amplification loop, and lytic pathways (RNA and protein; [6, 10, 67, 68]). Results suggest an important role for both the classical and alternative/amplification pathways, with dominance by the alternative/amplification pathway, in many models [6, 68–70]. Again, it is impossible to distinguish between the alternative pathway and the central amplification loop in these studies because components identified in this work, such as C3 and factors B, D, H, and I, are common to both.
While these results are often cited as support for the alternative pathway as a key driver of GA, it is important to note that many studies focus on acute induction of disease and tissue damage over a period of days to weeks rather than chronic disease in which ongoing accumulation of specific complement substrates may induce persistent complement activation. It is possible that amplification loop plays an outsized role in disease initiation by enhancing a complement signal generated through early accumulation of damaged cells or specific complement substrates. Further, the activation or regulation of the classical pathway in mice is distinct from that in humans and other species, with the classical cascade generally being highly restrained in mice [7, 71–74]. Consistent with this idea, exposure of POS to human serum in vitro leads to C3 deposition on their surface that is driven in large extent by the classical pathway, while C3 deposition upon exposure to mouse serum is entirely dependent on the alternative pathway [6].
Clinical trials of complement inhibitors
The “alternative pathway hypothesis” was rigorously tested in AMD in 2 large phase 3 studies of lampalizumab, a potent factor D (FD) inhibitor dosed via intravitreal administration [12]. FD inhibition would block alternative pathway initiation as well as the amplification loop (Fig. 1). Patients received intravitreal lampalizumab (10 mg/eye/month) or sham injections for 48 weeks, tracking GA lesion size and visual function. Unfortunately, both phase 3 studies were negative, even in a subset of patients that carried genetic variants of complement factor I associated with AMD risk [12]. A separate and smaller study done with another inhibitor of the alternative pathway was also negative (CLG-561, anti-properdin Fab; NCT02515942). There are many potential explanations for these results, including a need to capture patients before advanced disease ensues, for systemic rather than local inhibition, for different/more potent drug molecules that better penetrate the retina, or the need to inhibit additional or different complement pathways upstream of the amplification loop. Likewise, targeting downstream C5—either with intravitreal (LFG316, NCT01527500; [75]) or systemic (eculizumab; NCT00935883; [76]) administration—or by targeting C5 in combination with the alternative pathway (LFG316 + CLG561; NCT02515942) were also not successful in early phase clinical studies, potentially suggesting that the alternative or common lytic pathways are not key drivers of pathology in AMD; however, these latter studies were either small (n < 40/arm; LFG316/CLG561) or of limited duration (6 months; eculizumab).
Inhibition of C3, the central component of all three complement pathways, has been evaluated in one phase 2 [78] and two phase 3 studies with pegcetacoplan, an anti-C3 peptide conjugated to 40 kD polyethylene glycol (PEG). At 24 months, compared to sham, monthly and every-other-month treatment with intravitreal pegcetacoplan significantly reduced GA lesion growth by 22% (p = 0.0001) and 18% (p = 0.0002), respectively in the OAKS studyand by 19%% (p = 0.00004) and 16% (p = 0.003), respectively, in the DERBY study (company press release).
In GATHER1, a trial investigating the efficacy of C5 inhibition with a different compound (avacincaptad pegol; NCT02686658), there was a statistically significant 27.4% (p = 0.0072) reduction in GA lesion development at 12 months [77]. The second trial, GATHER2 showed a 14.3% (p = 0.0064) reduction in mean rate of growth at 12 months (company press release).
Treatment with either drug, however, resulted in an increase in development of exudative AMD/neovascularization in the treated eye (4–6% with C3 inhibition vs. 2% sham, and 10% with C5 inhibition vs. 3% sham). A potential explanation may be that, although C3 and C5 inhibition prevent neovascularization in several acute models used for GA, it can promote neovascularization in other models of retinal damage [79] or can prevent tissue repair [80–84]. Also, C3-mediated phagocytosis has been shown to be protective of RPE in a murine model of retinal damage [85]. Alternatively, the PEG carrier molecule common to both drugs may itself induce complement activation and vascular growth, as shown with direct subretinal injection [86]. However, in this model, PEG injections were administered in the subretinal space and induced direct tissue damage, unlike the scenario in clinical studies which employed intravitreal PEGylated drugs. Overall, the results of these clinical trials are encouraging, indicating that intravitreal administration of inhibitors of C3 or C5 can be effective, each providing close to 30% reduction in lesion growth at 1 year. Nevertheless, since these approaches fully inhibit the downstream components of all three complement pathways, they do not address the questions posed by human genetics: how C3 is being activated in AMD and how best to modulate it for therapeutic benefit.
Potential role of the classical complement pathway in GA
Multiple lines of evidence suggest that the classical pathway may be a key initiator of complement activation in GA. First, C1q is present in all layers of the outer retina in GA along with numerous C1q-activating ligands that accumulate throughout disease initiation and progression. Second, C1q and the classical pathway are implicated by retinal pathology in GA and in animal models of GA. Third, in RPE culture models, C1q triggers C5b-9 deposition on basement membrane components and contributes to induction of the inflammasome and cell necrosis. Fourth, inhibition of C1q and downstream C4 is protective in animal models of photoreceptor cell loss. Fifth, C1q accumulates on photoreceptor cell synapses in mice with age and light-induced injury, consistent with the unique role it plays in synapse elimination and neurodegeneration in other disorders. Each of these points is discussed in greater detail below.
C1q and C1q-activating ligands are present in the retina with GA
C1q is highly expressed in infiltrating macrophages within all layers of the outer retina (OPL, POS, RPE, and choroid layers) in GA (Fig. 2; [6, 11, 87]). In addition, disruption of the RPE cells and their ability to maintain a tight blood-retina barrier allows increased influx of C1q from the circulation along with other blood proteins [6]. Activating ligands for C1q in GA include surface bound CRP, SAP, membrane-exposed phosphatidyl serine, hydroxyapatite, lysophospholipids, immunoglobulins, and amyloid material [8, 22, 24–29, 62, 63]. Many of these ligands are associated with RPE and Bruch’s membrane or are components of drusen and drusenoid deposits (Figs. 1, 2, 3; [88–90]). C1q activation on CRP is increased by FHR1, a soluble complement regulatory protein that has been genetically linked to AMD. FHR1 enhances C1q activation and stabilizes downstream C3b (Table 1; [51]). Decreased expression of FHR1 reduces complement activation potential and is protective against AMD. Further, C1q activation may be enhanced by autoantibodies associated with disease [91–93]. In mice, generation of antibodies against oxidation products unique to the outer retina leads to classical complement activation, with downstream C3 deposition in Bruch’s membrane, macrophage infiltration and RPE damage [50, 66]. In addition, the presence of these oxidation products with C1q in drusen activates the NLPR3 inflammasome in cultured RPE, contributing to RPE necrosis [94]. Interestingly, genetic deletion of C1q prevented inflammasome activation in a mouse model of photoreceptor cell damage [67].
Fig. 3.
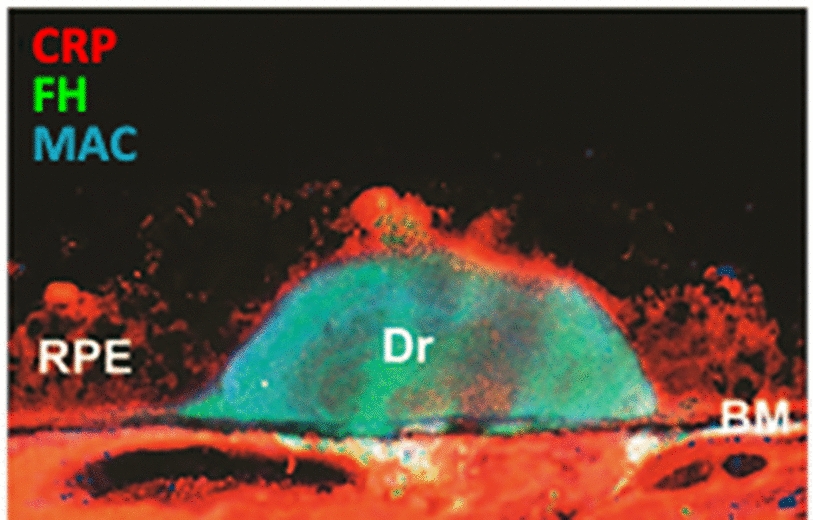
CRP Associates with drusen and RPE in GA: Confocal image of the retina of a patient with AMD showing drusen (Dr) stained for CRP (red) and factor H (green) [88]
C1q/classical pathway implicated in GA by human and animal pathology
POS exhibit the C1q-activating ligand phosphatidyl serine on their surface as part of the daily light cycle [95]. This ligand is normally recognized by soluble proteins that bridge POS, as they are shed from photoreceptors, to integrin receptors on RPE for phagocytosis. However, aberrant exposure to C1q, either from serum proteins across the blood-retinal barrier or through local production, may help drive complement activation in GA lesion development or expansion. Following retinal damage in mice, POS rapidly become coated by C1q and downstream complement activation products C4b and C3b (Fig. 4; [6]). The POS are then phagocytosed by infiltrating macrophages, and photoreceptor cells are lost. Genetic deletion of C4 as part of the classical/lectin pathway and inhibition of FD within the alternative/amplification pathway provided full protection against photoreceptor cell loss [52–58]. When isolated POS are exposed to human serum in vitro, they activate C4 deposition via the classical pathway and C3 deposition by both the classical and alternative pathways to facilitate phagocytosis by macrophages. There is no apparent involvement of the lectin pathway in this in vitro system [78]. In tissue from patients with GA, C4 and C3 are deposited on POS of both rods and cones in areas of healthy retina just outside the expanding edge of lesions, supporting the idea that C1q recognition of POS is contributing to GA lesion development or expansion (Fig. 4; [6]).
Fig. 4.

Complement Deposition on POS in Mouse Models of GA and in GA Patient Tissue: Panel A: Image of mouse retinal Sect. 5 h after RPE damage caused by intravenous injection of NaIO3. Immunofluorescence shows C1q deposition on POS, which occurs in conjunction with C3 prior to decrease in retinal function by day 7; Panels B and C: Images of retinal sections from patients with GA. Immunofluorescence shows C4 and C3 deposition on photoreceptor cell outer segments (POS) in a healthy region of retina just outside the edge of an expanding lesion, consistent with complement activation preceding loss of photoreceptor neurons. C4 deposition directly implicates the classical and/or lectin pathways since it occurs upstream of C3 and the alternative or amplification pathways [6]
C1q triggers C5b-9 deposition and contributes to induction of the inflammasome in cultures of RPE
In vitro culture of RPE cells results in deposition of apolipoprotein E (ApoE)-rich drusen-like material that binds and activates C1q, leading to complement activation and deposition of C5b-9 [96]. Complement activation was dependent on the classical, but not the alternative pathway, as determined by serum depletion of either C1q or factor B (Fig. 5). In a separate study, in vitro treatment of lipoparticles (containing ApoE) with hepatic lipase led to production of lysophospholipids that bound C1q and induced complement activation. Again, this activity was dependent on the classical, but not the alternative pathway, and C1q activation was facilitated by CRP [28]. Lysophospholipids are present at high levels in drusen, while genetic variants of ApoE and lipase C, hepatic type (LIPC) gene are both linked to GA. Polymorphisms that result in reduced levels of hepatic lipase are associated with GA protection. In a separate in vitro model, C1q worked in conjunction with other drusen components to induce the inflammasome in RPE cells, leading to RPE necrosis [94].
Fig. 5.
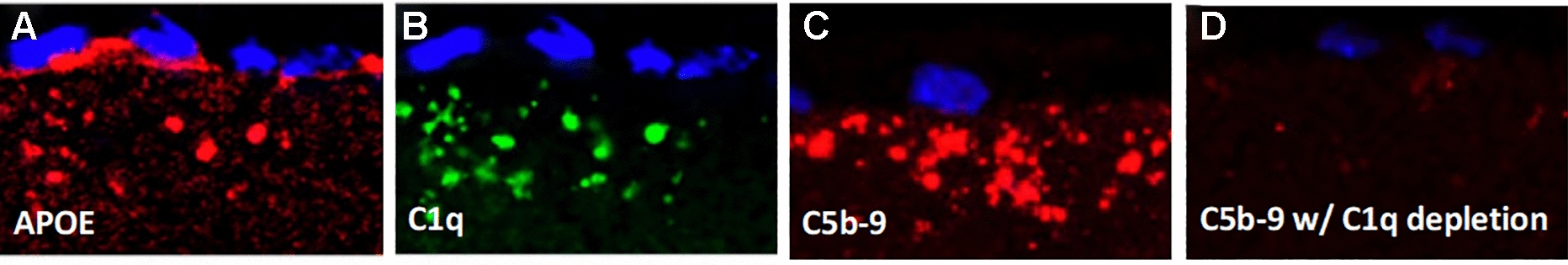
ApoE-rich Deposits That Form Below Cultured RPE Cells Activate the Classical Complement Pathway:Primary human RPE cells grown on porous supports produce globular extracellular deposits. Cultures were exposed to normal or complement-depleted human serum and then processed for immunohistochemistry with various antibody markers. A. Anti-ApoE with no serum exposure; B. Anti-C1q after exposure to normal human serum; C. Anti-C5b-9 after exposure to normal human serum; D. Anti-C5b-9 after expsoure to C1q-depleted human serum. Depletion of factor B had no impact on C5b-9 staining, implicating C1q-dependent/classical cascade activation [96]
C1q/classical cascade inhibition is protective in animal models of disease
In a model of photoreceptor cell damage involving prolonged exposure to white light, macrophages infiltrate the outer retina where they express high levels of C1q. Genetic deletion or pharmacological inhibition of C1q provided protection against photoreceptor cell loss and preserved receptor cell function, even when inhibition was provided after damage had occurred (Fig. 6; [11] also see Fig. 7 for expression of C1q in a similar model). Interestingly, while local (intravitreal) administration of an anti-C1q antibody was protective, systemic administration was not—presumably due to local production of C1q in the retina and to lower levels of antibody penetration into the eye with systemic dosing. Further support is provided by 2 additional animal models, where genetic deletion of C4 was protective against photoreceptor cell loss following RPE damage induced by NaIO3 or by direct laser damage [6].
Fig. 6.
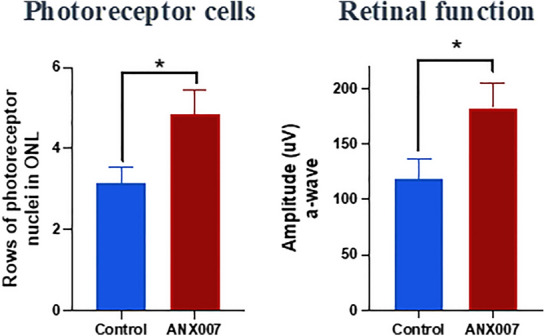
C1q Inhibition Protects Photoreceptor Cells and Retinal Function: Anti-C1q was given via intravitreal administration 7 days after initiation of photo-oxidative damage. Measures were taken one week after dosing. Amplitude measures for a wave (left graph) and b wave (not shown) were taken with flash intensity of 1.9 log cd.s/m2 [11]
Fig. 7.

C1q Binds Photoreceptor Cell Synapses With Age or Retinal: Left images: Mice were aged from 3 to 24 months. Retinal sections were stained with anti-synaptophysin and anti-C1q (antibody 4.8, Abcam); Annexon, data on file. Right images: Balb/c mice were exposed to white light (28-29 k lux) for 4 h, and after an additional 20 h, retinal sections were prepared and stained with anti-C1q as above. Note C1q staining of both synapses (arrows) and infiltrating cells (asterisk); adjacent retinal section stained with bassoon (presynaptic marker: red) and IBA-1 (infiltrating macrophages or microglia: magenta). Note C1q overlap with synapses of the photoreceptor cells (arrows) in the OPL but not synapses within the IPL (arrow heads) [108]. Abbreviations, see figure.
C1q’s unique role in synapse elimination and neurodegeneration
Photoreceptor cell stress in GA caused by decline in RPE function and other factors may lead to a process of synapse elimination, neuroinflammation and neuronal loss that is common in multiple neurodegenerative disorders. In development, C1q recognizes the surface of weaker supernumerary synapses, activating the classical complement pathway and recruiting microglial cells to prune the opsonized synapses away from neurons [13, 97]. The role of C1q in sculpting neuronal circuitry was first shown in the visual pathway examining synaptic connections in the lateral geniculate and has more recently been shown to occur during development with synapses in the outer retina [98]. After development, C1q accumulates on synapses with normal aging, increasing up to 300-fold in aged mice relative to younger animals [15]. Age-related accumulation may reflect changes on the synapse surface that resemble those in development and appears to place synapses in the adult at risk for aberrant removal. In GA, with declining RPE function and neuronal stress, C1q on synapses may activate the classical pathway, and as in other neurodegenerative disorders, result in compromised synapse function and microglial cell recruitment [99]. Microglial cells then prune complement-coated synapses away from neurons, depriving neurons of trophic support, and cause neuroinflammation that adds to neuronal damage and loss. C1q inhibition has been shown to be protective in multiple animal models of neurodegeneration by blocking complement-mediated synapse elimination in the central nervous system (CNS), including models of Alzheimer’s disease [16, 19, 100], spinal muscular atrophy [101], glaucoma [14, 18], photoreceptor cell damage [11], frontotemporal dementia [17], traumatic brain injury [102, 103], and Huntington’s disease [104]. The fact that C1q is involved in synapse pruning in the outer retina during development [98] and that genetic deletion or pharmacological inhibition of C1q is protective against synapse loss and neuronal damage in several models of glaucoma demonstrates that this mechanism is relevant to the retina [14, 18]. Consistent with a potential role in GA, we have found that C1q accumulates in the OPL on photoreceptor cell synapses in mice with both age and acute light damage. C1q is also strongly expressed by phagocytic cells that infiltrate the OPL following light damage (Fig. 7) and in other animal models of photoreceptor cell loss [6, 11]. Thus, declining RPE function and photoreceptor cells stress may augment the decline of retinal function and integrity through a previously-described process of complement-mediated neurodegeneration [45–49].
Advantages of targeting C1q in GA
The initial success of clinical trials with C3 and C5 inhibitors in GA extends insights from human genetics, indicating a role for complement in GA progression beyond disease initiation. Further, it suggests that efficacy can be achieved in the outer retina through intravitreal administration of a complement inhibitor. However, C3 and C5 inhibitors block the central and terminal aspects of all three complement pathways. Animal studies have shown that these approaches can negatively impact retinal health and facilitate neovascularization following retinal damage. While upstream targeting of the alternative pathway has not been successful to date, targeting upstream components of the classical pathway, in particular C1q, may provide a therapeutic advantage. In chronic diseases such as GA where classical cascade substrates accumulate and persist, there will be continuous opportunity for C1q binding and classical complement activation, with C4b, C3b, and C5b-9 production (Fig. 1). Following alternative pathway inhibition, C3b levels would be reduced [105], but there would still be activation of C3 and C5 through the C4b2a convertase, particularly in a chronic disease setting (Fig. 8A). Remarkably, even with C3 or C5 inhibition, chronic or excessive accumulation of C4b can drive C5b-9 activation directly (Fig. 8B; [31]). In contrast, preventing the focused binding of C1q to its substrates would block all downstream classical cascade components and their amplification to reduce overall complement activation, while leaving the clearance and tissue repair functions of the lectin and alternative complement pathways in place. Given supporting evidence that C1q is produced locally in the retina, that activating substrates for C1q are present in all layers of the outer retina in GA, and that C1q plays a unique role in synapse elimination and neuronal damage in multiple neurodegenerative disorders, the classical pathway is well positioned for therapeutic intervention in GA. In a recent phase 1 clinical study, intravitreal administration of an inhibitory antibody against C1q was able to fully reduce free C1q levels for at least one month, as measured in aqueous humor [106]. These findings were supported by preclinical studies in nonhuman primates, which additionally showed penetration of the drug into the outer layers of the retina with elimination of free C1q in the retina, optic nerve head and choroid [107]. Hence, the rationale for targeting the classical pathway as a mediator of tissue damage in GA is currently being tested in phase 2/3 studies.
Fig. 8.

Activation of C5 in the Presence of C3, C5, or Alternative Pathway Inhibitors
Summary
There are strong human genetic data demonstrating a role for complement in AMD pathobiology that is further supported by in vitro and in vivo model studies, and, importantly, by clinical data. These studies indicate that the complement system is a key component of pathology, but they do not address how complement is being activated in AMD—via classical, lectin, or alternative pathways. Clinical studies focused on the alternative pathway have been unsuccessful to date, while studies targeting downstream activity of all complement pathways (C3 and C5) have shown promise. C1q is well positioned to be a key driver of complement activity in AMD, with local expression by infiltrating phagocytic cells and numerous C1q–activating ligands that are present at disease initiation and accumulate with AMD progression. Genetic deletion of C1q, local pharmacologic inhibition within the eye, or genetic deletion of downstream C4 provides protection in several mouse models of photoreceptor cell damage. C1q’s presence on photoreceptor synapses with age and damage is consistent with its role in synapse elimination and neurodegeneration in many other CNS disorders. While blocking downstream components of all three pathways has provided promise in clinical studies, targeting the classical pathway in GA would provide a more selective approach with the potential for increased efficacy and safety.
Acknowledgements
Not applicable.
Abbreviations
- AMD
Age-related macular degeneration
- ApoE
Apolipoprotein E
- C3-H2O
Hydrolysis of C3
- CNS
Centra nervous system
- CRP
C-reactive protein
- FD
Factor D
- GA
Geographic atrophy
- LIPC
Lipase C, hepatic type
- MBL
Mannose-binding lectin and the highly related molecules collectin-10 and -11 and ficolin-1, -2, and -3
- OPL
Outer plexiform layer
- PEG
Polyethylene glycol
- POS
Photoreceptor cell outer segments
- RPE
Retinal pigmented epithelial cells
- SAP
Serum amyloid protein
Author contributions
TY, DF and EML made substantial contributions to the conception of the work. TY provided the first draft, but all authors substantively revised it. All authors have read and approved the final manuscript.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Availability of data and materials
Not applicable. The paper is a review paper.
Declarations
Ethics approval and consent to participate
Not applicable. This is a review paper.
Consent for publication
The authors consent for publication.
Competing interests
EL has received consultant honoraria from Annexon Biosciences, Roche, Novartis, Apellis, Allegro, Alexion, Retrotope, Galimedix, IVERIC Bio, NGM Biopharmaceuticals, Janssen, Thea Laboratoires. Research Funding: Roche, Novartis, Apellis, and LumiThera. TY and DF are employees of Annexon Biosciences.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fleckenstein M, Mitchell P, Freund KB, Sadda S, Holz FG, Brittain C, et al. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology. 2018 doi: 10.1016/j.ophtha.2017.08.038. [DOI] [PubMed] [Google Scholar]
- 2.Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng CY, Wong TY. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014 doi: 10.1016/S2214-109X(13)70145-1. [DOI] [PubMed] [Google Scholar]
- 3.Fritsche LG, Igl W, Bailey JNC, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016 doi: 10.1038/ng.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan PL, Bowes Rickman C, Katsanis N. AMD and the alternative complement pathway: genetics and functional implications. Hum Genomics. 2016 doi: 10.1186/s40246-016-0079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyer DS, Schmidt-Erfurth U, Van Lookeren CM, Henry EC, Brittain C. The pathophysiology of geographic atrophy secondary to age-related macular degeneration and the complement pathway as a therapeutic target. Retina. 2017 doi: 10.1097/IAE.0000000000001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katschke KJ, Jr, Xi H, Cox C, Truong T, Malato Y, Lee WP, et al. Classical and alternative complement activation on photoreceptor outer segments drives monocyte-dependent retinal atrophy. Sci Rep. 2018 doi: 10.1038/s41598-018-25557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennis A, Gorgels TGMF, ten Brink JB, van der Spek PJ, Bossers K, Heine VM, Bergen AA. Comparison of mouse and human retinal pigment epithelium gene expression profiles: potential implications for age-related macular degeneration. PLoS ONE. 2015 doi: 10.1371/journal.pone.0141597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hageman GS, Mullins RF. Molecular composition of drusen as related to substructural phenotype. Mol Vis. 1995;5:28. [PubMed] [Google Scholar]
- 9.Mullins RF, Faidley EA, Daggett HT, Jomary C, Lotery AJ, Stone EM. Localization of complement 1 inhibitor (C1INH/SERPING1) in human eyes with age-related macular degeneration. Exp Eye Res. 2009 doi: 10.1016/j.exer.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schäfer N, Grosche A, Schmitt SI, Braunger BM, Pauly D. Complement components showed a time-dependent local expression pattern in constant and acute white light-induced photoreceptor damage. Front Mol Neurosci. 2017 doi: 10.3389/fnmol.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiao H, Rutar M, Fernando N, Yednock T, Sankaranarayanan S, Aggio-Bruce R, Provis J, Natoli R. Subretinal macrophages produce classical complement activator C1q leading to the progression of focal retinal degeneration. Mol Neurodegener. 2018 doi: 10.1186/s13024-018-0278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holz FG, Sadda SR, Busbee B, Chew EY, Mitchell P, Tufail A, et al. Chroma and spectri study investigators. efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: chroma and spectri phase 3 randomized clinical trials. JAMA Ophthalmol. 2018 doi: 10.1001/jamaophthalmol.2018.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007 doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 14.Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. 2011 doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013 doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in alzheimer mouse models. Science. 2016 doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016 doi: 10.1016/j.cell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams PA, Tribble JR, Pepper KW, Cross SD, Morgan BP, Morgan JE, John SWM, Howell GR. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol Neurodegener. 2016 doi: 10.1186/s13024-016-0091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, et al. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by c1q antibodies. Neuron. 2018 doi: 10.1016/j.neuron.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov. 2019 doi: 10.1038/s41573-019-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, Skjoedt MO. A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. 2016 doi: 10.1111/imr.12468. [DOI] [PubMed] [Google Scholar]
- 22.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I—molecular mechanisms of activation and regulation. Front Immunol. 2015 doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toomey CB, Johnson LV, Bowes RC. Complement factor H in AMD: bridging genetic associations and pathobiology. Prog Retin Eye Res. 2018 doi: 10.1016/j.preteyeres.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGrath FDG, Brouwer MC, Arlaud GJ, Daha MR, Hack CE, Roos A. Evidence that complement protein C1q interacts with C-reactive protein through its globular head region. J Immunol. 2006 doi: 10.4049/jimmunol.176.5.2950. [DOI] [PubMed] [Google Scholar]
- 25.Tacnet-Delorme P, Chevallier S, Arlaud GJ. β-Amyloid fibrils activate the C1 complex of complement under physiological conditions: evidence for a binding site for a β on the C1q globular regions. J Immunol. 2001 doi: 10.4049/jimmunol.167.11.6374. [DOI] [PubMed] [Google Scholar]
- 26.Martin M, Leffler J, Blom AM. Annexin A2 and A5 serve as new ligands for C1q on apoptotic cells. J Bio Chem. 2012 doi: 10.1074/jbc.M112.341339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson RB, Reffatto V, Bundy JG, Kortvely E, Flinn JM, Lanzirotti A, et al. Identification of hydroxyapatite spherules provides new insight into subretinal pigment epithelial deposit formation in the aging eye. Proc Natl Acad Sci USA. 2015 doi: 10.1073/pnas.1413347112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma W, Paik DC, Barile GR. Bioactive lysophospholipids generated by hepatic lipase degradation of lipoproteins lead to complement activation via the classical pathway. Invest Ophthalmol Vis Sci. 2014 doi: 10.1167/iovs.14-14352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014 doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009 doi: 10.1016/S0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 31.Mannes M, Dopler A, Zolk O, Lang SJ, Halbgebauer R, Höchsmann B, et al. Complement inhibition at the level of C3 or C5: mechanistic reasons for ongoing terminal pathway activity. Blood. 2021 doi: 10.1182/blood.2020005959. [DOI] [PubMed] [Google Scholar]
- 32.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006 doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 33.Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006 doi: 10.1016/j.survophthal.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD) Hum Mol Genet. 2010 doi: 10.1093/hmg/ddq399. [DOI] [PubMed] [Google Scholar]
- 35.Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013 doi: 10.1038/ng.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014 doi: 10.1146/annurev-genom-090413-025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitz-Valckenberg S, Fleckenstein M, Zouache MA, et al. Progression of age-related macular degeneration among individuals homozygous for risk alleles on chromosome 1 (CFH-CFHR5) or chromosome 10 (ARMS2/HTRA1) or both. JAMA Ophthalmol. 2022;140(3):252–260. doi: 10.1001/jamaophthalmol.2021.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sobrin L, Yang JY. Not all genes are created equal in age-related macular degeneration. JAMA Ophthalmol. 2022;140(3):260–261. doi: 10.1001/jamaophthalmol.2021.6069. [DOI] [PubMed] [Google Scholar]
- 39.Thee EF, Colijin JM, Cougnard-Gregorie A, et al. The Phenotypic course of age-related macular degeneration for ARMS2/HTRA1. the EYE-RISK consortium. Ophthalmology. 2022;129:752–764. doi: 10.1016/j.ophtha.2022.02.026. [DOI] [PubMed] [Google Scholar]
- 40.Williams BL, Seager NA, Gardiner JD, et al. Chromosome 10q26-driven age-related macular degeneration is associated with reduced levels of HTRA1 in human retinal pigment epithelium. Proc Natl Acad Sci USA. 2021;118(30):e2103617118. doi: 10.1073/pnas.2103617118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005 doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005 doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 43.Cantsilieris S, Nelson BJ, Huddleston J, Baker C, Harshman L, Penewit K, et al. Recurrent structural variation, clustered sites of selection, and disease risk for the complement factor H (CFH) gene family. Proc Natl Acad Sci USA. 2018 doi: 10.1073/pnas.1717600115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cserhalmi M, Papp A, Brandus B, Uzonyi B, Józsi M. Regulation of regulators: role of the complement factor H-related proteins. Semin Immunol. 2019 doi: 10.1016/j.smim.2019.101341. [DOI] [PubMed] [Google Scholar]
- 45.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005 doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005 doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 47.Nayak A, Pednekar L, Reid KBM, Kishore U. Complement and non-complement activating functions of C1q: a prototypical innate immune molecule. Innate Immun. 2012 doi: 10.1177/1753425910396252. [DOI] [PubMed] [Google Scholar]
- 48.Kishore U, Sim RB. Factor H as a regulator of the classical pathway activation. Immunobiology. 2012 doi: 10.1016/j.imbio.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 49.Gershov D, Kim S, Brot N, Elkon KB. C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med. 2000 doi: 10.1084/jem.192.9.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alic L, Papac-Milicevic N, Czamara D, Rudnick RB, Ozsvar-Kozma M, Hartmann A, et al. A genome-wide association study identifies key modulators of complement factor H binding to malondialdehyde-epitopes. Proc Natl Acad Sci U S A. 2020 doi: 10.1073/pnas.1913970117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Csincsi ÁI, Szabó Z, Bánlaki Z, Uzonyi B, Cserhalmi M, Kárpáti É, et al. FHR-1 binds to C-reactive protein and enhances rather than inhibits complement activation. J Immunol. 2017 doi: 10.4049/jimmunol.1600483. [DOI] [PubMed] [Google Scholar]
- 52.Kárpáti É, Papp A, Schneider AE, Hajnal D, Cserhalmi M, Csincsi ÁI, Uzonyi B, Józsi M. Interaction of the factor H family proteins FHR-1 and FHR-5 with DNA and dead cells: implications for the regulation of complement activation and opsonization. Front Immunol. 2020 doi: 10.3389/fimmu.2020.01297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Java A, Baciu P, Widjajahakim R, Sung YJ, Yang J, Kavanagh D, Atkinson J, Seddon J. Functional analysis of rare genetic variants in complement Factor I (CFI) using a serum-based assay in advanced age-related macular degeneration. Trans Vis Sci Technol. 2020 doi: 10.1167/tvst.9.9.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van de Ven JPH, Nilsson SC, Tan PL, Buitendijk GHS, Ristau T, Mohlin FC, et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat Genet. 2013 doi: 10.1038/ng.2640. [DOI] [PubMed] [Google Scholar]
- 55.Thakkinstian A, McEvoy M, Chakravarthy U, Chakrabarti S, McKay GJ, Ryu E, et al. The association between complement component 2/complement factor B polymorphisms and age-related macular degeneration: a HuGE review and meta-analysis. Am J Epidemiol. 2012 doi: 10.1093/aje/kws031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, et al. AMD genetics clinical study group variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006 doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montes T, Tortajada A, Morgan BP, de Rodríguez Córdoba S, Harris CL. Functional basis of protection against age-related macular degeneration conferred by a common polymorphism in complement factor B. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0812584106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McMahon O, Hallam TM, Patel S, Harris CL, Menny A, Zekek WM, et al. The rare C9 P167S risk variant for age-related macular degeneration increases polymerization of the terminal component of the complement cascade. Hum Mol Genet. 2021 doi: 10.1093/hmg/ddab086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pujol-Lereis LM, Schäfer N, Kuhn LB, Rohrer B, Pauly D. Interrelation between oxidative stress and complement activation in models of age-related macular degeneration. Adv Exp Med Biol. 2016 doi: 10.1007/978-3-319-17121-0_13. [DOI] [PubMed] [Google Scholar]
- 60.Curcio CA, Johnson M, Rudolf M, Huang JD. The oil spill in ageing Bruch membrane. Br J Ophthalmol. 2011 doi: 10.1136/bjophthalmol-2011-300344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spaide RF, Ooto S, Curcio CA. Subretinal drusenoid deposits AKA pseudodrusen. Surv Ophthalmol. 2018 doi: 10.1016/j.survophthal.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 62.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000 doi: 10.1096/fasebj.14.7.835. [DOI] [PubMed] [Google Scholar]
- 63.Païdassi H, Tacnet-Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, Arlaud GJ, Frachet P. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. 2008 doi: 10.4049/jimmunol.180.4.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ebrahimi KB, Fijalkowski N, Cano M, Handa JT. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J Pathol. 2013 doi: 10.1002/path.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Organisciak DT, Vaughan DK. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010 doi: 10.1016/j.preteyeres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008 doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Natoli R, Jiao H, Barnett NL, Fernando N, Valter K, Provis JM, Rutar M. A model of progressive photo-oxidative degeneration and inflammation in the pigmented C57BL/6J mouse retina. Exp Eye Res. 2016 doi: 10.1016/j.exer.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 68.Rohrer B, Guo Y, Kunchithapautham K, Gilkeson GS. Eliminating complement factor D: reduces photoreceptor susceptibility to light-induced damage. Invest Opthalmol Vis Sci. 2007 doi: 10.1167/iovs.07-0282. [DOI] [PubMed] [Google Scholar]
- 69.Rohrer B, Coughlin B, Kunchithapautham K, Long Q, Tomlinson S, Takahashi K, Holers VM. The alternative pathway is required, but not alone sufficient, for retinal pathology in mouse laser-induced choroidal neovascularization. Mol Immunol. 2011 doi: 10.1016/j.molimm.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Woodell A, Jones BW, Williamson T, Schnabolk G, Tomlinson S, Atkinson C, Rohrer B. A targeted inhibitor of the alternative complement pathway accelerates recovery from smoke-induced ocular injury. Invest Opthalmol Vis Sci. 2016 doi: 10.1167/iovs.15-18471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ratelade J, Verkman AS. Inhibitor(s) of the classical complement pathway in mouse serum limit the utility of mice as experimental models of neuromyelitis optica. Mol Immunol. 2014 doi: 10.1016/j.molimm.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McGonigal R, Cunningham ME, Yao D, Barrie JA, Sankaranarayanan S, Fewou SN, et al. C1q-targeted inhibition of the classical complement pathway prevents injury in a novel mouse model of acute motor axonal neuropathy. Acta Neuropathol Commun. 2016 doi: 10.1186/s40478-016-0291-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka S, Suzuki T, Nishioka K. Assay of classical and alternative pathway activities of murine complement using antibody-sensitized rabbit erythrocytes. J Immunol Methods. 1986 doi: 10.1016/0022-1759(86)90448-5. [DOI] [PubMed] [Google Scholar]
- 74.Galvan MD, Luchetti S, Burgos AM, Nguyen HX, Hooshmand MJ, Hamers FPT, Anderson AJ. Deficiency in complement C1q improves histological and functional locomotor outcome after spinal cord injury. J Neurosci. 2008 doi: 10.1523/JNEUROSCI.2823-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kassa E, Ciulla TA, Hussain RM, Dugel PU. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin Biol Ther. 2019 doi: 10.1080/14712598.2019.1575358. [DOI] [PubMed] [Google Scholar]
- 76.Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, Gregori G, Penha FM, Moshfeghi AA, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLE study. Ophthalmology. 2014 doi: 10.1016/j.ophtha.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jaffe GJ, Westby K, Csaky KG, Monés J, Pearlman JA, Patel SS, et al. C5 inhibitor avacincaptad pegol for geographic atrophy due to age-related macular degeneration: a randomized pivotal phase 2/3 trial. Ophthalmology. 2021 doi: 10.1016/j.ophtha.2020.08.027. [DOI] [PubMed] [Google Scholar]
- 78.Liao DS, Grossi FV, El Mehdi D, Gerber MR, Brown DM, Heier JS, et al. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: a randomized phase 2 trial. Ophthalmology. 2020 doi: 10.1016/j.ophtha.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 79.Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010 doi: 10.1182/blood-2010-01-261503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rohrer B. Anaphylatoxin signaling in retinal pigment and choroidal endothelial cells: characteristics and relevance to age-related macular degeneration. Adv Exp Med Biol. 2018 doi: 10.1007/978-3-319-75402-4_6. [DOI] [PubMed] [Google Scholar]
- 81.Parsons N, Annamalai B, Obert E, Schnabolk G, Tomlinson S, Rohrer B. Inhibition of the alternative complement pathway accelerates repair processes in the murine model of choroidal neovascularization. Mol Immunol. 2019 doi: 10.1016/j.molimm.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haynes T, Luz-Madrigal A, Reis ES, Echeverri Ruiz NP, Grajales-Esquivel E, Tzekou A, et al. Complement anaphylatoxin C3a is a potent inducer of embryonic chick retina regeneration. Nat Commun. 2013 doi: 10.1038/ncomms3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Natarajan N, Abbas Y, Bryant DM, Gonzalez-Rosa JM, Sharpe M, Uygur A, et al. Complement receptor C5aR1 plays an evolutionarily conserved role in successful cardiac regeneration. Circulation. 2018 doi: 10.1161/CIRCULATIONAHA.117.030801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu M, Zou W, Peachey NS, McIntyre TM, Liu J. A novel role of complement in retinal degeneration. Invest Opthalmol Vis Sci. 2012 doi: 10.1167/iovs.12-10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Silverman SM, Ma W, Wang X, Zhao L, Wong WT. C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J Exp Med. 2019 doi: 10.1084/jem.20190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lyzogubov VV, Tytarenko RG, Liu J, Bora NS, Bora PS. Polyethylene glycol (PEG) induced mouse model of choroidal neovascularization. J Biol Chem. 2011 doi: 10.1074/jbc.M110.204701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fletcher EL. Contribution of microglia and monocytes to the development and progression of age related macular degeneration. Ophthalmic Physiol Opt. 2020 doi: 10.1111/opo.12671. [DOI] [PubMed] [Google Scholar]
- 88.Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, et al. Y402H polymorphism of complement factor h affects binding affinity to C-reactive protein. J Immunol. 2007 doi: 10.4049/jimmunol.178.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bhutto IA, Baba T, Merges C, Juriasinghani V, McLeod DS, Lutty GA. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br J Ophthalmol. 2011 doi: 10.1136/bjo.2010.199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sivaprasad S, Bird A, Nitiahpapand R, Nicholson L, Hykin P, Chatziralli I. Moorfields UCL AMD consortium. perspectives on reticular pseudodrusen in age-related macular degeneration. Surv Ophthalmol. 2016 doi: 10.1016/j.survophthal.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 91.Morohoshi K, Patel N, Ohbayashi M, Chong V, Grossniklaus HE, Bird AC, Ono SJ. Serum autoantibody biomarkers for age-related macular degeneration and possible regulators of neovascularization. Exp Mol Pathol. 2012 doi: 10.1016/j.yexmp.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 92.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011 doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, Saloman RG. Carboxyethylpyrrole Protein Adducts and Autoantibodies, Biomarkers for Age-related Macular Degeneration. J Biol Chem. 2003 doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- 94.Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012 doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ruggiero L, Connor MP, Chen J, Langen R, Finnemann SC. Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5-/- or Mfge8-/- mouse retina. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1121101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Johnson LV, Forest DL, Banna CD, Radeke CM, Maloney MA, Hu J, Spencer CN, Walker AM, Tsie MS, Bok D, Radeke MJ, Anderson DH. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci USA. 2011;108(45):18277–18282. doi: 10.1073/pnas.1109703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012 doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burger CA, Jiang D, Li F, Samuel MA. C1q regulates horizontal cell neurite confinement in the outer retina. Front Neural Circuits. 2020 doi: 10.3389/fncir.2020.583391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012 doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 100.Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of alzheimer’s disease. J Neurosci. 2004 doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vukojicic A, Delestrée N, Fletcher EV, Pagiazitis JG, Sankaranarayanan S, Yednock TA, Barres BA, Mentis GZ. The classical complement pathway mediates microglia-dependent remodeling of spinal motor circuits during development and in SMA. Cell Rep. 2019 doi: 10.1016/j.celrep.2019.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krukowski K, Chou A, Xi Feng, Tiret B, Paladini MS, Riparip LK, et al. Traumatic brain injury in aged mice induces chronic microglia activation, synapse loss, and complement-dependent memory deficits. Int J Mol Sci. 2018 doi: 10.3390/ijms19123753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Holden SS, Grandi FC, Aboubakr O, Higashikubo B, Cho FS, Chang AH, et al. Complement factor C1q mediates sleep spindle loss and epileptic spikes after mild brain injury. Science. 2021 doi: 10.1126/science.abj2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stevens B. Dynamic microglia states and functions in health and disease. 2019. Presentation at Keystone Symposia on neural environment in disease: glial responses and neuroinflammation; Keystone, CO.
- 105.Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004 doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun Y, Wirta D, Mathur V, Sankaranarayanan S, Yednock T, Keswani S, Goldberg JL. Safety, tolerability and target engagement of intravitreal (IVT) ANX007 injections in primary open-angle glaucoma (POAG). 2020. AAO presentation.
- 107.Grover A, Sankaranarayanan S, Mathur V, Suri P, Andrews-Zwilling Y, Mease K, et al. Pharmacokinetics and target engagement of intravitreal administration of ANX007, an anti-C1q antibody fragment, in nonhuman primates. IOVS. 2021;62:8. doi: 10.1167/iovs.64.2.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tassoni A, Vereen J, Andrews-Zwilling Y, et al. C1q mediates microglial pruning of photoreceptor synapses in a light damage model of photoreceptor degeneration. IOVS. 2022;63(7):4608–F0400. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable. The paper is a review paper.