

Abstract
The classic pathologic hallmarks of Alzheimer’s disease (AD) are amyloid plaques and neurofibrillary tangles (AD neuropathologic changes, or ADNC). However, brains from individuals clinically diagnosed with “AD-type” (amnestic) dementia usually harbor heterogeneous neuropathologies in addition to, or other than, ADNC. We hypothesized that some AD-type dementia associated genetic single nucleotide variants (SNVs) identified from large genomewide association studies (GWAS) were associated with non-ADNC neuropathologies. To test this hypothesis, we analyzed data from multiple studies with available genotype and neuropathologic phenotype information. Clinical AD/dementia risk alleles of interest were derived from the very large GWAS by Bellenguez et al. (2022) who reported 83 clinical AD/dementia-linked SNVs in addition to the APOE risk alleles. To query the pathologic phenotypes associated with variation of those SNVs, National Alzheimer’s disease Coordinating Center (NACC) neuropathologic data were linked to AD Sequencing Project (ADSP) and AD Genomics Consortium (ADGC) data. Separate data were obtained from the harmonized Religious Orders Study and the Rush Memory and Aging Project (ROSMAP). A total of 4811 European participants had at least ADNC neuropathology data and also genotype data available; data were meta-analyzed across cohorts. As expected, a subset of dementia-associated SNVs were associated with ADNC risk in Europeans—e.g., BIN1, PICALM, CR1, MME, and COX7C. Other gene variants linked to (clinical) AD dementia were associated with non-ADNC pathologies. For example, the associations of GRN and TMEM106B SNVs with limbic-predominant age-related TDP-43 neuropathologic changes (LATE-NC) were replicated. In addition, SNVs in TNIP1 and WNT3 previously reported as AD-related were instead associated with hippocampal sclerosis pathology. Some genotype/neuropathology association trends were not statistically significant at P < 0.05 after correcting for multiple testing, but were intriguing. For example, variants in SORL1 and TPCN1 showed trends for association with LATE-NC whereas Lewy body pathology trended toward association with USP6NL and BIN1 gene variants. A smaller cohort of non-European subjects (n = 273, approximately one-half of whom were African-Americans) provided the basis for additional exploratory analyses. Overall, these findings were consistent with the hypothesis that some genetic variants linked to AD dementia risk exert their affect by influencing non-ADNC neuropathologies.
Keywords: NACC, SNP, Pleiotropy, PLCG2, ABCA7, APH1B, TDP-43, Hippocampal sclerosis
1. Introduction
Approximately 80% of Alzheimer’s disease (AD) risk is heritable according to twin studies (Gatz et al., 2006), yet the biological mechanisms underlying that heritability remain poorly understood. A key goal of genetic analyses is to elucidate the individual genetic variants (“genotypes”) that are associated with altered risk for the disease phenotype. However, it is increasingly clear that the clinical syndrome of AD (amnestic dementia) is influenced by many genes and multiple different neuropathologies. Thus, to understand the biology underlying the heritability of AD requires a cataloging of different genetic variants associated with dementia risk, and then a systematic analytic approach must be executed to identify how those genetic variants are associated with disease-driving phenotypes.
A definition of terms is required to avoid phenotypic ambiguity. During life, persons with amnestic dementia are usually given the clinical diagnosis of “Probable AD” (McKhann et al., 2011), or AD-type dementia (Mehta and Schneider, 2021). At autopsy, or as predicted by biomarkers, the neuropathological hallmarks of AD are amyloid-β (Aβ) plaques and tau neurofibrillary tangles (NFTs) – AD neuropathologic changes (ADNC) (Knopman et al., 2018; Montine et al., 2012).
Although the gold standard for disease instantiation and severity is neuropathologic diagnoses, it is the clinical diagnoses (lacking biomarker data) that have been used for many large surveys of AD genetics. A recent genome-wide association study (GWAS), which analyzed genomes of almost 800,000 individuals, reported 83 dementia-related single-nucleotide variants (SNVs) in addition to the APOE risk alleles (Bellenguez et al., 2022). The use of clinical diagnoses in this study for disease phenotyping had advantages – this strategy enabled a large sample size and facilitated inclusion of a broad range of participants. However, there are limitations, and the potential for confusion, resulting from this study design. To address these challenges requires an understanding of the underlying causes of the clinical syndrome of AD dementia.
The classification of dementia-associated neuropathologic phenotypes has been refined extensively in the past several years, partly because neuropathologies other than ADNC commonly underlie the clinical disease of AD dementia. More specifically, previous studies revealed that the clinical diagnosis of AD dementia was imperfectly predictive of ADNC (Beach et al., 2012), and only ~20% of dementia cases are “pure” ADNC at autopsy (Karanth et al., 2021; Shim et al., 2013). In attributable risk analyses of a large community-based cohort that factored in all known pathologies, ADNC accounted for <40% of all identified AD-type dementia risk (Boyle et al., 2021; Nelson et al., 2019a).
What other neuropathologies can underlie AD-type dementia? An important dementia-associated brain disease of aging is limbic-predominant age-related TDP-43 encephalopathy (LATE). The pathological hallmark of LATE neuropathologic changes (LATE-NC) is TDP-43 proteinopathy (Neumann et al., 2006) that is mostly restricted to the medial temporal lobes. Approximately one-third of people aged 85 or older have LATE-NC (Nelson et al., 2022). People with LATE-NC are also at elevated risk for comorbid hippocampal sclerosis (HS), a diagnostic term that implies atrophy and cell loss in the hippocampal formation, and HS is associated with added cognitive impairment (Boyle et al., 2019; Gauthreaux et al., 2022; Nelson et al., 2010). Another major subtype of pathology associated with amnestic dementia is Lewy body disease (Boyle et al., 2019). Lewy body pathologies comprise a heterogeneous group of neurological disorders with the common element of misfolded α-synuclein protein (Attems et al., 2021).
The neurodegenerative disease phenotypes are even more complex because findings at brain autopsy are usually characterized by neuropathologic combinations as opposed to “pure” isolated neuropathologies (Brenowitz et al., 2017; Jack Jr et al., 2016; James et al., 2016; Jellinger et al., 2015; Kryscio et al., 2016; Nelson et al., 2007; Nelson et al., 2016; Neltner et al., 2016; Whitwell et al., 2007). Genetics may contribute to these phenomena because specific genetic variants often influence more than one subtype of neuropathology. As a conspicuous example, different neuropathologies have been linked to gene variants of the APOE dementia-associated ε4 risk allele (Cykowski et al., 2022; Dugan et al., 2021; Kukull et al., 1996; O’Meara et al., 1997; Tsuang et al., 2005).
For the reasons stated above, it is clear that a considerable proportion of amnestic dementia in the human population is associated with neuropathologies other than ADNC. Yet only a few studies have examined the influence of genetic risk factors on non-ADNC neuropathologies underlying AD-type dementia (Beecham et al., 2014; Farfel et al., 2016). In the present study, we hypothesized that some of the SNVs reported to be associated with risk for AD-type dementia by Bellenguez et al. (Bellenguez et al., 2022) are actually associated with other pathologies. We gathered extensive genotype and neuropathologic phenotype data from >4000 research participants to evaluate the associations between AD dementia-linked SNVs and the neuropathologies that each SNV was associated with.
2. Material and methods
2.1. Participants
The National Alzheimer’s Coordinating Center (NACC) phenotype data were derived from 37 different United States (U.S.) Alzheimer’s Disease Research Centers (ADRCs) with autopsy data scored using NACC Neuropathology (NP) v10–11 forms through the March 2022 data freeze (https://www.alz.washington.edu/). Autopsies were performed within each of the contributory ADRCs. The NACC NP data were linked to ADRC genetic data including whole genome sequencing (WGS) data produced under the Alzheimer’s Disease Sequencing Project (ADSP) and genotype data were provided by the Alzheimer’s Disease genetic Consortium (ADGC). These data sets were described in detail previously (Crane et al., 2017; Dugan et al., 2021; Naj et al., 2018; Naj et al., 2017). Duplicated participants were removed from the ADGC genotype data, and thus the study sample in the WGS data was independent of those in the ADGC genotype data (hereafter referred to as “ADSP WGS” and “ADGC”, respectively). Harmonized data from two cohorts (the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP)) are referred to as the ROSMAP study (Bennett et al., 2018). Again, duplicates were removed so that the study samples were non-overlapping. We performed principal component analysis (PCA) implemented in PLINK v1.90a (Chang et al., 2015; Purcell et al., 2007) with the “–pca” option using a linkage disequilibrium (LD) pruned subset of markers (pairwise r2 < 0.2) merged to 1000 Genomes Project Phase 3 (Genomes Project et al., 2010) data after removing symmetric SNVs and flipping SNVs discordant for DNA strands between the two datasets. Based on the first and second PC plot, we split participants into two ancestry groups: European ancestry and other ancestries (Supplementary Figs. 1–3, Supplementary Tables 1–3). For our primary analysis, we excluded participants who had at least one of 19 rare brain diseases diagnosed at autopsy (see Fig. 1 and (Katsumata et al., 2020)) from ADSP WGS and ADGC datasets in people with European ancestry. The excluded rare brain diseases included frontotemporal lobar degeneration (FTLD), chronic traumatic encephalopathy, multiple sclerosis, multiple system atrophy, amyotrophic lateral sclerosis, triplet repeat (e. g., Huntington’s and other) diseases, and prion diseases. These exclusions involved removing 98 participants from initial analyses. Data from all participants (including those with rare disease) were investigated as a sensitivity analysis. Similar exclusion criteria were not applied to ROSMAP due to lack of data availability and to ADSP WGS and ADGC in people with other ancestries due to small sample sizes.
Fig. 1.
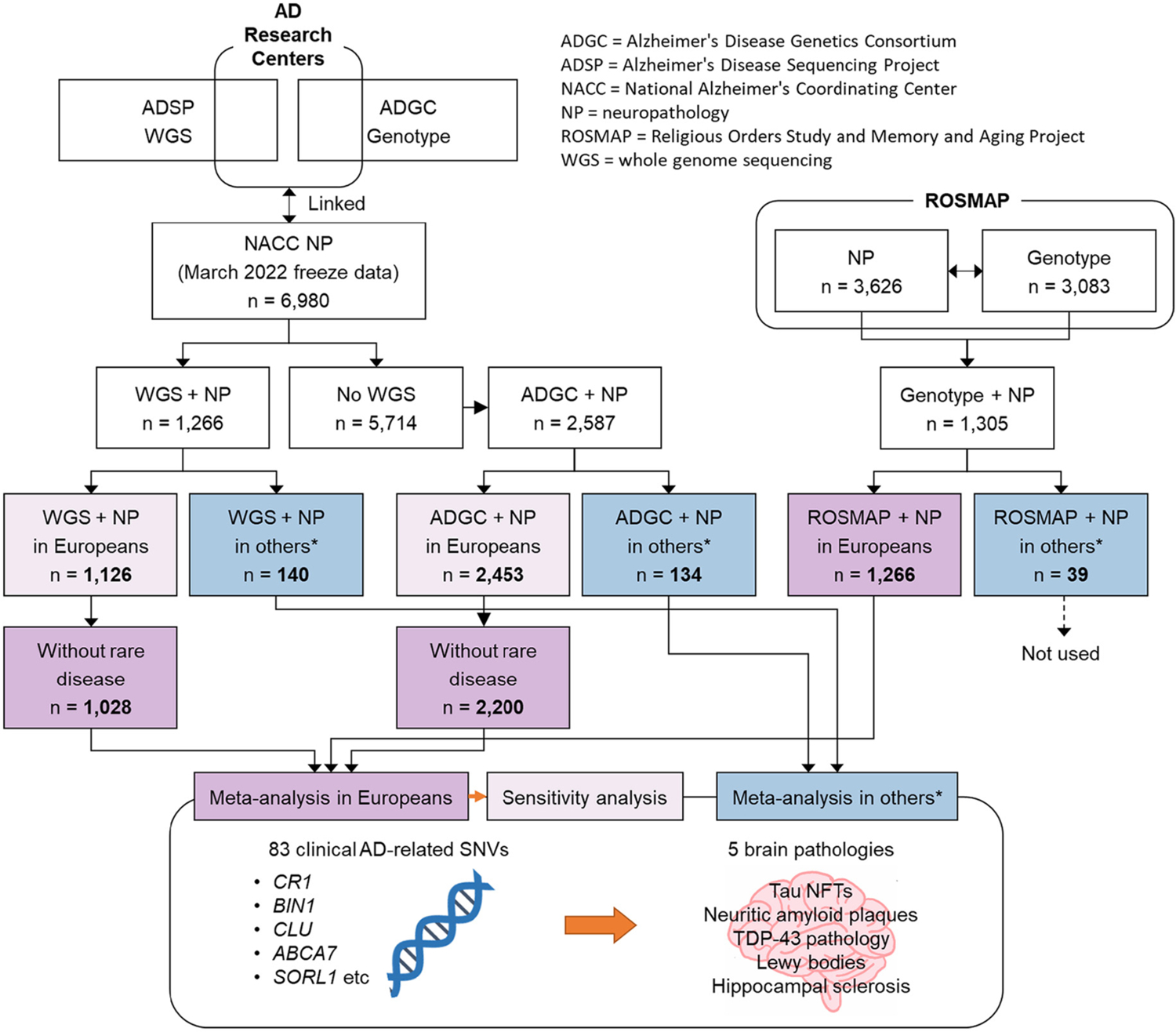
Work flow diagram of the present study. * “Others” includes other ancestries than Europeans (i.e., Africans, Admixed Americans, East Asians, and South Asians).
2.2. Neuropathology data
Data were included related to the following neuropathologic features: ADNC including neocortical neuritic plaques (Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) rating (Mirra, 1997)) and Braak NFT stages (Braak and Braak, 1991), Lewy body pathologies identified using α-synuclein immunohistochemistry (Attems et al., 2021), whereas LATE-NC was operationalized by TDP-43 pathology (Dugan et al., 2021; Nelson et al., 2019a; Neumann et al., 2006), which is often comorbid with (but separate from) HS (Dugan et al., 2021; Nelson et al., 2011). The neuropathological data were dichotomized: ADNC/NFT was represented by 0 = Braak NFT stage 0 to IV and 1 = Braak NFT stage V or VI (Braak and Braak, 1991); neocortical neuritic plaques with 0 = none to moderate and 1 = frequent (Mirra et al., 1991); TDP-43 pathology with 0 = none and 1 = present in any brain regions including amygdala, hippocampus, entorhinal/inferior temporal cortex, and neocortex; Lewy body pathology with 0 = none and 1 = present in any brain regions including brainstem, limbic, amygdala, olfactory bulb, and neocortex; and HS with 0 = none and 1 = present either unilaterally or bilaterally. Additional post hoc sensitivity analyses were performed, using different cut-points for diagnostic operationalizations, to evaluate the findings when other pathologic criteria were applied.
2.3. Genetic data
ADSP WGS data were provided from NIH/ADSP collaborators. We obtained variant calling data (n = 16,785) for SNVs and short insertions/deletions in VCF data file formats mapped to Genome Reference Consortium Human Build 38 (GRCh38). We also obtained ADGC and ROSMAP genotype data (PLINK format file sets). These genotype data were imputed using the TOPMed Imputation Server (https://imputation.biodatacatalyst.nhlbi.nih.gov/) (Taliun et al., 2019) based on GRCh38. We reran the PCA for each neuropathology outcome to derive orthogonal PCs which were used as covariates in the subsequent analyses. When SNV data were missing, we used the proxy which was in perfect LD (i.e., r2 = 1 and D′ = 1) identified using LDLink (ldlink.nci.nih.gov) (Machiela and Chanock, 2015) by querying the LDproxy Tool in the relevant population.
2.4. Statistical analysis
For each of the neuropathology outcomes, we performed association tests of clinical AD-related SNVs reported in the Bellenguez et al. (2022) study (Supplementary Table 4) under an additive mode of inheritance using logistic regression adjusted for age at death, sex, and the top three PCs computed in PLINK v1.90a. We then combined results across the three datasets (ADSP WGS, ADGC, and ROSMAP) for European ancestry in a fixed-effect meta-analysis with weighted Z-score-based p-values implemented in PLINK v1.90a with the “–meta-analysis weighted-z” option. Since the sample size in the other ancestries from the ROSMAP dataset was too small, we performed meta-analysis for other ancestries using the results from the two datasets, ADSP WGS and ADGC. We created clustered heatmaps for p-values with the pheatmap R package. We set statistical significance at false discovery rate (FDR) adjusted p-value (i.e., Q-value) < 0.05 using the Benjamini-Hochberg procedure in each of the neuropathology outcomes.
3. Results
The numbers of included participants are shown in Fig. 1. Since there were missing values, the samples sizes differed for each of the neuropathologies (Supplementary Table 1). The ROSMAP study included more females, fewer APOE ε4 carriers, fewer people with high ADNC, more people with TDP-43 pathology, and fewer people with Lewy body pathology and hippocampal sclerosis compared with the ADSP WGS and ADGC studies in both participants with European and other ancestries (Table 1 and Supplementary Table 3).
Table 1.
Characteristics in subjects of European ancestry.
| Characteristic | ADRC | ROSMAP (n = 1266) | |
|---|---|---|---|
| ADSP WGS (n = 1028) | ADGC (n = 2200) | ||
| Age at death, mean ± SD | 82.5 ± 10.5 | 82.2 ± 10.6 | 89.7 ± 6.5 |
| Years in education, mean ± SD | 16.5 ± 9.1 | 16.3 ± 8.2 | 16.3 ± 3.7 |
| Sex, n (%) | |||
| Male | 529 (51.5) | 1173 (53.3) | 412 (32.5) |
| Female | 499 (48.5) | 1027 (46.7) | 854 (67.5) |
| APOE, n (%) | |||
| −/− | 526 (51.2) | 1155 (52.5) | 952 (75.2) |
| −/ε4 | 438 (42.6) | 798 (36.3) | 293 (23.1) |
| ε4/ε4 | 64 (6.2) | 247 (11.2) | 21 (1.7) |
| Braak NFT stage, n (%) | |||
| 0 – IV | 374 (36.5) | 909 (41.5) | 916 (72.4) |
| V/VI | 652 (63.5) | 1282 (58.5) | 350 (27.6) |
| Neocortical neuritic plaques, n (%) | |||
| No – moderate | 466 (45.3) | 1072 (48.8) | 840 (66.4) |
| Frequent | 562 (54.7) | 1123 (51.2) | 426 (33.6) |
| TDP-43 in any brain regions, n (%) | |||
| No | 273 (75.0) | 520 (65.2) | 554 (47.1) |
| Yesa | 91 (25.0) | 277 (34.8) | 622 (52.9) |
| Lewy bodies in any brain regions, n | (%) | ||
| No | 661 (64.6) | 1388 (63.5) | 927 (75.7) |
| Yesb | 362 (35.4) | 797 (36.5) | 298 (24.3) |
| Hippocampal sclerosis, n (%) | |||
| No | 418 (87.4) | 962 (86.7) | 1133 (91.1) |
| Yesc | 60 (12.6) | 147 (13.3) | 111 (8.9) |
Abbreviations: ADGC = Alzheimer’s Disease Genetics Consortium; ADRC = Alzheimer’s Disease Research Center; ADSP = Alzheimer’s Disease Sequencing Project; NFT = neurofibrillary tangle; ROSMAP = Religious Orders Study (ROS) and Memory and Aging Project (MAP); SD = standard deviation; TDP-43 = TAR DNA binding protein 43 kDa; WGS = whole genome sequencing.
Observed in any regions including amygdala, hippocampus, entorhinal/inferior temporal cortex, and neocortex.
Observed in any brain regions including brainstem-predominant, limbic, neocortical, amygdala predominant, and olfactory bulb.
Observed in either unilateral or bilateral.
The associations of the non-APOE SNVs with the individual neuropathologies are displayed in Supplementary Tables 5–10 in people with primarily European ancestry and Supplementary Tables 11–16 in people with other ancestries. The meta-analyzed association results in people with European ancestry are shown in Table 2 (summarized results) and Supplementary Table 10 (full results). In the meta-analysis for European ancestry, rs6733839 in BIN1 was strongly associated with AD-related neuropathology (OR = 1.30 and P-value = 2.6 × 10−8 in Braak NFT stage and OR = 1.21 and P-value = 3.9 × 10−5 in neocortical neuritic plaques). The SNVs in MME and EED/PICALM were also associated with both Braak NFT stage and neocortical neuritic plaques. The SNV in TMEM106B was detected as a signal of TDP-43 pathology (OR = 0.78 and P-value = 1.0 × 10−4 in the A allele of rs13237518) and HS (OR = 0.64 and P-value = 9.3 × 10−7 in the A allele of rs13237518). The T allele of rs5848 in GRN was associated with HS (OR = 1.53 and P-value = 2.1 × 10−6). The G allele of rs74685827 in SORL1 and the C allele of rs6489896 in TPCN1 were rather genetic risk factors of TDP-43 pathology; however, these associations were not statistically significant after FDR adjustment (Q-values were 0.062 and 0.085, respectively). The SNVs in WNT3 and TNIP1 were significantly associated with HS but not associated with AD-related neuropathologies (Table 2). These SNVs were separated into TDP-43/HS related clusters including TMEM106B, GRN, TPCN1, SORL1, WNT3, TNIP1, ACE, and SCIMP and AD-related clusters including FERMT2, RBCK1, PTK2B, INPP5D, APH1B, EED/PICALM, SPI1, COX7C, MME, and CR1 (Fig. 2 and Supplementary Fig. 4–5).
Table 2.
Summary of association from meta-analysis in people with European ancestry.
| Gene | Variant | MAFa | AD-ORb | Meta-analysis | ||
|---|---|---|---|---|---|---|
| NP-OR | P-value | Q-valuec | ||||
| Braak NFT stage | ||||||
| BIN1 | rs6733839 | 0.38 | 1.17 | 1.30 | 2.6 × 10−8 | 1.5 × 10−6 |
| CR1 | rs679515 | 0.17 | 1.13 | 1.24 | 3.8 × 10−4 | 0.011 |
| MME | rs16824536 | 0.032 | 0.92 | 0.70 | 9.8 × 10−4 | 0.016 |
| COX7C | rs62374257 | 0.21 | 1.07 | 1.20 | 0.0011 | 0.016 |
| SPI1 | rs10437655 | 0.38 | 1.06 | 1.13 | 0.0064 | 0.071 |
| EED/PICALM | rs3851179 | 0.37 | 0.90 | 0.88 | 0.0089 | 0.075 |
| INPP5D | rs10933431 | 0.23 | 0.93 | 0.86 | 0.0094 | 0.075 |
| Neocortical neuritic plaques | ||||||
| BIN1 | rs6733839 | 0.38 | 1.17 | 1.21 | 3.9 × 10−5 | 0.0022 |
| EED/PICALM | rs3851179 | 0.37 | 0.90 | 0.85 | 0.0010 | 0.021 |
| MME | rs16824536 | 0.032 | 0.92 | 0.70 | 0.0011 | 0.021 |
| APH1B | rs117618017 | 0.14 | 1.11 | 1.23 | 0.0020 | 0.026 |
| RBCK1 | rs1358782 | 0.27 | 0.95 | 0.85 | 0.0023 | 0.026 |
| FERMT2 | rs17125924 | 0.080 | 1.10 | 1.25 | 0.0047 | 0.044 |
| PTK2B | rs73223431 | 0.35 | 1.07 | 1.13 | 0.0088 | 0.071 |
| TDP-43 in any brain regions | ||||||
| TMEM106B | rs13237518 | 0.40 | 0.96 | 0.78 | 1.0 × 10−4 | 0.0059 |
| SORL1 | rs74685827 | 0.015 | 1.19 | 1.85 | 0.0042 | 0.092 |
| GRN | rs5848 | 0.30 | 1.07 | 1.22 | 0.0049 | 0.092 |
| TPCN1 | rs6489896 | 0.094 | 1.08 | 1.42 | 0.0071 | 0.10 |
| Lewy bodies in any brain regions | ||||||
| BIN1 | rs6733839 | 0.38 | 1.17 | 1.16 | 0.0012 | 0.068 |
| USP6NL | rs7912495 | 0.45 | 1.06 | 1.14 | 0.0043 | 0.12 |
| Hippocampal sclerosis | ||||||
| TMEM106B | rs13237518 | 0.40 | 0.96 | 0.64 | 9.3 × 10−7 | 5.2 × 10−5 |
| GRN | rs5848 | 0.30 | 1.07 | 1.53 | 2.1 × 10−6 | 5.8 × 10−5 |
| WNT3 | rs199515 | 0.22 | 0.94 | 0.69 | 0.0014 | 0.025 |
| TNIP1 | rs871269 | 0.34 | 0.96 | 0.75 | 0.0018 | 0.025 |
| ACE | rs4277405 | 0.37 | 0.94 | 1.12 | 0.0082 | 0.089 |
| SCIMP | rs7225151 | 0.14 | 1.08 | 1.36 | 0.0095 | 0.089 |
Abbreviations: AD = Alzheimer’s disease; MAF = minor allele frequency; NFT = neurofibrillary tangle; NP = neuropathology; OR = odds ratio; TDP-43 = TAR DNA binding protein 43 kDa.
MAFs are calculated from 1000 genome phase 3 in Europeans (EUR) population.
AD-OR represents clinical AD related odds ratios reported in “New insights into the genetic etiology of Alzheimer’s disease and related dementias” (Bellenguez et al., 2022).
Q-value indicates the false discovery rate (FDR) adjusted p-value with the Benjamini-Hochberg procedure.
Fig. 2.
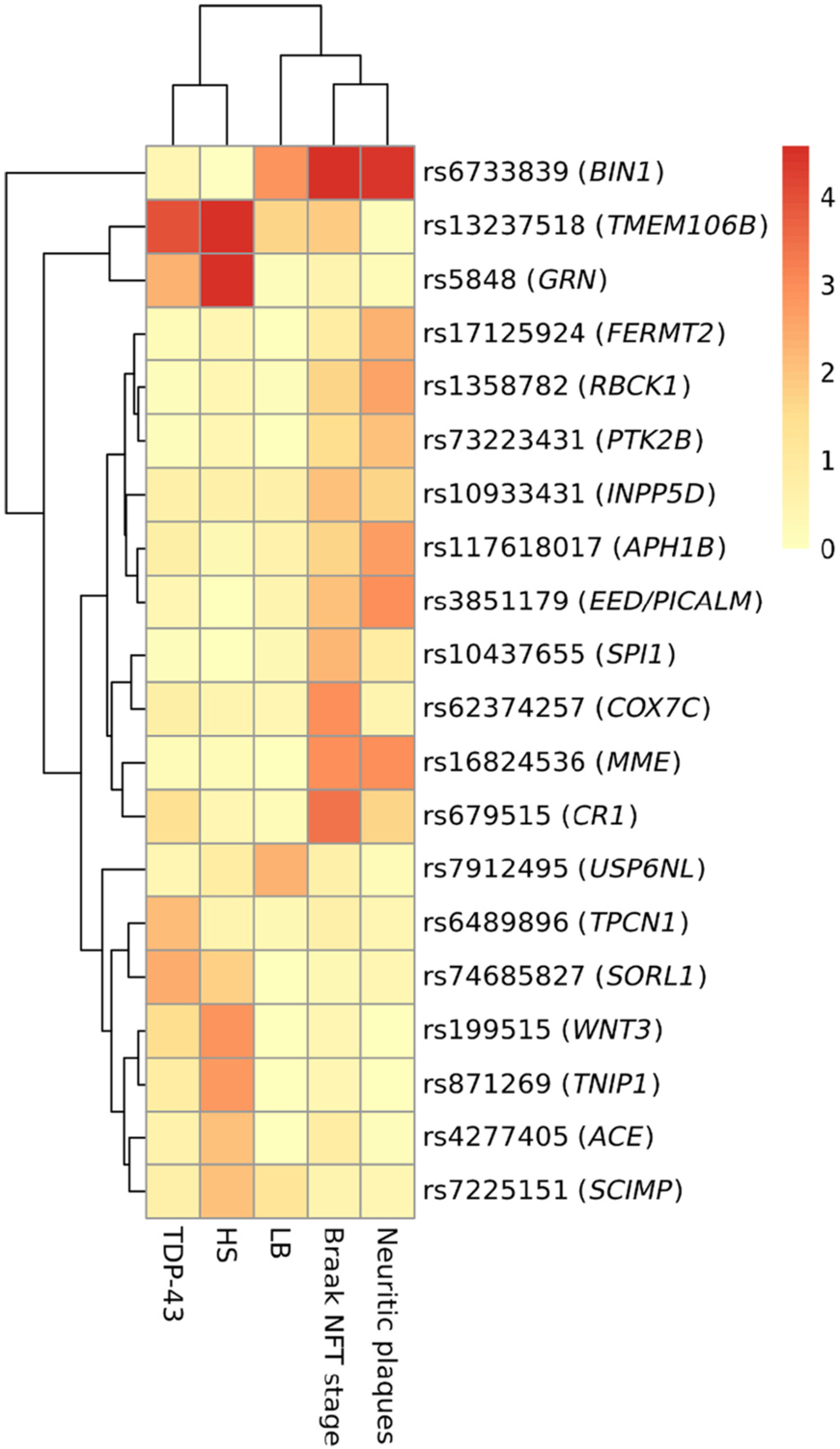
Heatmap for -log10 transformed p-values of single nucleotide variants with p < 0.01 among people with European ancestry. Abbreviations: TDP-43 = TAR DNA binding protein 43 kDa; HS = hippocampal sclerosis; LB = Lewy bodies; NFT = neurofibrillary tangle.
It is possible that some genetic variants underlie the tendency for ADNC and LATE-NC to co-exist in the same brains. In a post-hoc analysis, we examined the two SNVs (linked nominally to LATE-NC risk) in SORL1 and TPCN1 for comorbid phenotypes with LATE-NC and ADNC using ADSP WGS data. As shown in Table 3, the G allele of rs74685827 in SORL1 was associated with both (comorbid) widespread NFTs (Braak NFT stages V/VI) and TDP-43 pathology (P-value = 0.034) in this sample.
Table 3.
Participant counts comparing single nucleotide variants in SORL1 and TPCN1 and comorbid neuropathologies of limbic-predominant age-related TDP-43 encephalopathy (LATE) and Alzheimer’s disease (AD) in ADSP WGS participants with European ancestry.
| LATE + AD | Others | P-valuea | |
|---|---|---|---|
| rs74685827 in SORL1 | |||
| T/T | 71 | 283 | 0.034 |
| G/T | 5 | 5 |
P-value calculated with Fisher’s exact test.
There were different analytic strategies that may have been used. Therefore, we performed two relevant post hoc sensitivity analyses. The first sensitivity analysis used data from all participants with or without rare disease (i.e., included all subjects before applying exclusion criteria) in people with European ancestry. The ORs from the sensitivity analyses were similar with those in people who did not have any rare disease (Supplementary Table 17). In the second sensitivity analysis, we changed the operationalization (cut-points) for severities in neuropathologies: 0 = Braak NFT stage 0 to III and 1 = Braak NFT stage IV to VI; neocortical neuritic plaques with 0 = none or sparse and 1 = moderate or frequent; TDP-43 pathology with 0 = none or present in amygdala (i. e. LATE-NC stages 0/1) and 1 = present in hippocampus, entorhinal/inferior temporal cortex, or neocortex (LATE-NC stages 2/3); Lewy body pathology data with 0 = none or present in other regions and 1 = present in neocortex. The results of this sensitivity analysis are shown in Supplementary Table 18. Notably, the odds ratios were larger, and P-values smaller, for GRN (rs5848) and SORL1 (rs74685827) SNVs’ association with LATE-NC using the revised operationalization (i.e., LATE-NC stages 0/1 vs 2/3) of TDP-43 pathology.
An ad hoc grouping of non-European research subjects was composed of approximately one-half African-Americans in addition to other ethnoracial groups as defined by genetic PCA analyses (Supplemental Figs. 1–3). Among this heterogeneous subsample, the numbers of subjects were small, and no SNV was associated with any of the surveyed neuropathology phenotypes after FDR adjustment (Table 4 for summary results, Supplementary Tables 11–15 for individual neuropathologies, and full results are shown in Supplementary Table 16). Notably, rs12151021 in ABCA7 was the top SNV in ADNC (plaques and tangles) neuropathology for non-Europeans. The C allele of CLU was risk for HS rather than protective. ANK3, ABCA7, and BIN1 made an ADNC-related cluster, MME, RBCK1, and USP6NL were in a cluster for Braak NFT stage, and PLCG2, ANKH, and CLU were separated into the TDP-43/HS cluster (Fig. 3 and Supplementary Fig. 6).
Table 4.
Summary of association from meta-analysis in people with other ancestries.
| Gene | Variant | MAFa | AD-ORb | Meta-analysis | ||
|---|---|---|---|---|---|---|
| NP-OR | P-value | Q-valuec | ||||
| Braak NFT stage | ||||||
| ABCA7 | rs12151021 | 0.38 | 1.1 | 1.68 | 0.0072 | 0.42 |
| MME | rs16824536 | 0.12 | 0.92 | 0.54 | 0.021 | 0.53 |
| RBCK1 | rs1358782 | 0.14 | 0.95 | 0.58 | 0.029 | 0.53 |
| BIN1 | rs6733839 | 0.40 | 1.17 | 1.49 | 0.046 | 0.53 |
| USP6NL | rs7912495 | 0.37 | 1.06 | 1.48 | 0.049 | 0.53 |
| Neocortical neuritic plaques | ||||||
| ABCA7 | rs12151021 | 0.38 | 1.1 | 1.73 | 0.0046 | 0.18 |
| BIN1 | rs6733839 | 0.40 | 1.17 | 1.74 | 0.0063 | 0.18 |
| ANK3 | rs7068231 | 0.46 | 0.95 | 0.59 | 0.012 | 0.23 |
| TDP-43 in any brain regions | ||||||
| PLCG2 | rs12446759 | 0.49 | 0.95 | 0.35 | 0.0042 | 0.23 |
| Lewy bodies in any brain regions | ||||||
| MS4A4A | rs1582763 | 0.23 | 0.91 | 0.45 | 0.0078 | 0.45 |
| BIN1 | rs6733839 | 0.40 | 1.17 | 1.60 | 0.020 | 0.58 |
| Hippocampal sclerosis | ||||||
| ANKH | rs112403360 | 0.058 | 1.09 | 5.82 | 0.0048 | 0.27 |
| CLU | rs11787077 | 0.38 | 0.91 | 2.83 | 0.0096 | 0.27 |
Abbreviations: AD = Alzheimer’s disease; MAF = minor allele frequency; NFT = neurofibrillary tangle; NP = neuropathology; OR = odds ratio; TDP-43 = TAR DNA binding protein 43 kDa.
MAFs are calculated from 1000 genome phase 3 in other populations (AFR, African; AMR, Admixed American; EAS, East Asian; SAS, South Asian).
AD-OR represents clinical AD related odds ratios reported in “New insights into the genetic etiology of Alzheimer’s disease and related dementias” (Bellenguez et al., 2022).
Q-value indicates the false discovery rate (FDR) adjusted p-value with the Benjamini-Hochberg procedure.
Fig. 3.
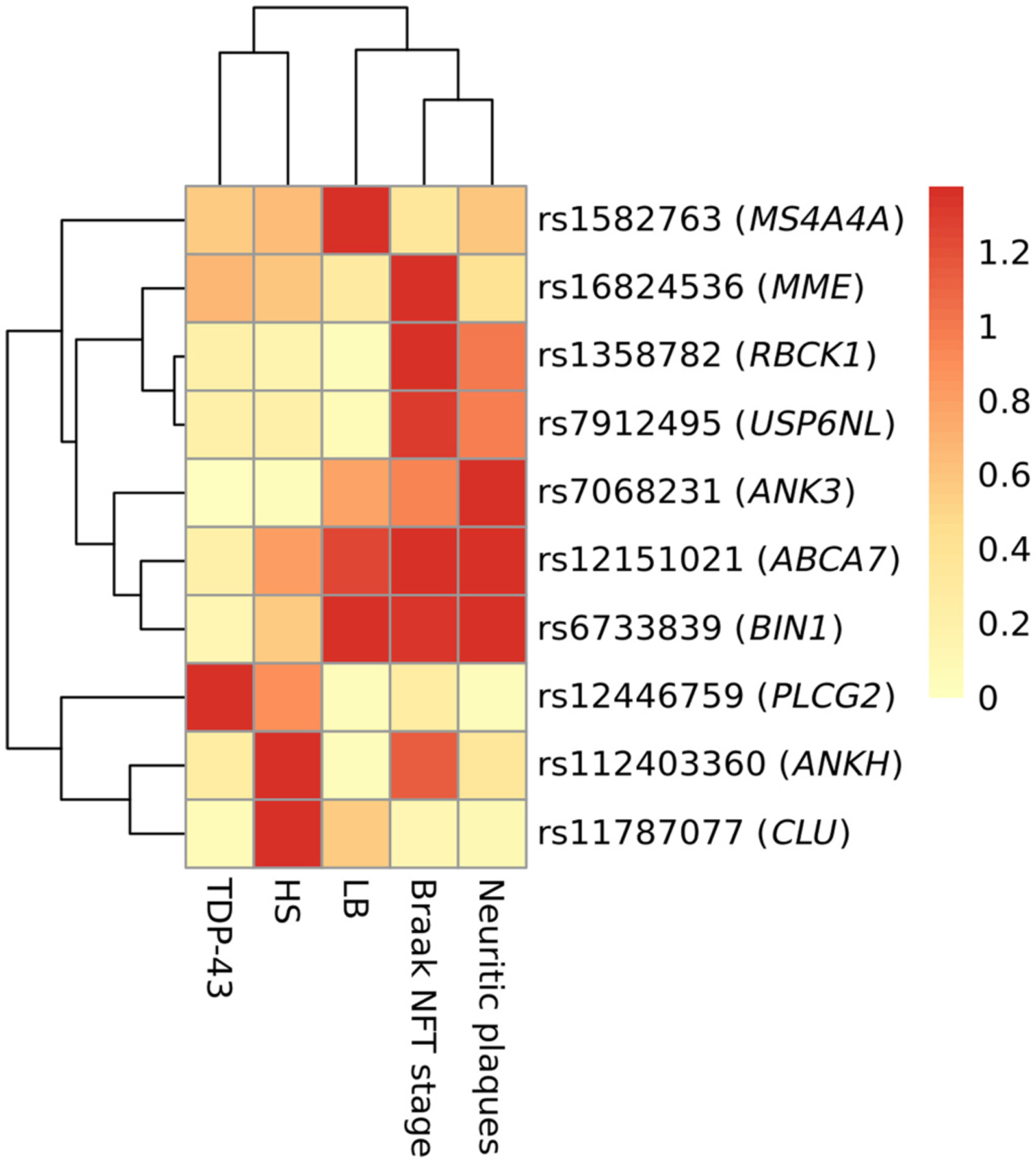
Heatmap for −log10 transformed p-values of single nucleotide variants with p < 0.05 among people with non-European ancestries. Abbreviations: TDP-43 = TAR DNA binding protein 43 kDa; HS = hippocampal sclerosis; LB = Lewy bodies; NFT = neurofibrillary tangle.
Finally, we confirmed the associations between APOE diplotype determined with rs429358 and rs7412 and each of the NPs (Supplementary Table 19). As we expected, the ε2/ε3 diplotype had protective effects on Braak NFT stage and neocortical neuritic plaques, but did not have on TDP-43, Lewy body, and HS pathologies. The ε4 allele was strongly associated with all the neuropathologies in people with European ancestry, and with ADNC in people with other ancestries.
4. Discussion
In the current study, genotype and pathologic endophenotype data were meta-analyzed together based on the AD-related SNVs identified and/or replicated in the large GWAS of (Bellenguez et al., 2022). More specifically, we combined neuropathologic and genotype data from three high-quality data sets: ADSP, ADGC, and ROSMAP. As expected, some of the dementia-linked SNVs were indeed associated with ADNC risk, e.g., SNVs in or near BIN1, PICALM, and CR1. Also replicated were the SNVs associated previously with LATE-NC risk in GRN and TMEM106B genes. In terms of novel findings, SNVs in or near TNIP1 and WNT3 that were previously reported as AD-related variants were instead associated with HS pathology. Overall, our findings underscore that non-ADNC neuropathologies can contribute substantially to AD-type amnestic dementia clinically.
There have been several prior GWA-based studies focusing on multiple AD dementia-related risk alleles and/or non-ADNC related neuropathologic endophenotypes (Beecham et al., 2014; Farfel et al., 2016; Yang et al., 2020). For example, Beecham et al. previously confirmed the expected associations between ADNC phenotypes (Braak NFT stages and CERAD neuritic amyloid plaques scores) for BIN1, PICALM, and CR1. That study also included some phenotypes related to cerebrovascular pathologies (e.g., cerebral amyloid angiopathy and “vascular brain injury”), that were not included in the current study. Beecham et al. also reported evidence of a novel gene variant in KCNMB2 (rs9637454) linked to HS risk (Beecham et al., 2014), that was not evaluated in the current study but was assessed in a similar data set as was analyzed in the current study (Dugan et al., 2021).
In a separate study of ROSMAP data, the associations were analyzed between various neuropathologies and 22 gene variants linked to AD dementia by GWAS (Farfel et al., 2016). The endophenotypes assessed also included cerebrovascular pathologies. Several suggestive findings were made in this study including an association between ZCWPW1 SNV and HS pathology, whereas a SNV in the CELF1 gene was associated with both micro-infarcts and Lewy body pathology. This study (Farfel et al., 2016) also emphasized that far larger sample sizes may be required for confident hypothesis-testing to link dementia-associated SNVs with neuropathologic features.
The current study built on prior published work by incorporating additional parameters in both the genotype and phenotype portions of the analytic equation, due to new information being made available in the past several years. In terms of SNVs linked to AD-type (amnestic) dementia risk, the paper by Bellenguez et al. (2022) identified dozens of novel putative risk alleles. It should also be emphasized that the prior AD/dementia-associated genetic variants were mostly replicated by this study (Bellenguez et al., 2022).
As to the neuropathologic phenotypes, a relatively recent development is the classification of LATE-NC (Nelson et al., 2019a), which is a common pathology linked to the amnestic dementia clinical syndrome. Here we focus on several of the novel putative risk alleles that were associated with LATE-NC or HS risk in the current study. Each of these provides opportunities for further research. The estimated effect sizes of these genetic variants’ linkages to neuropathologies were in the “same direction” (same risk-associated allele) and effect sizes ascertained were larger than those of clinical diagnosed AD reported (Bellenguez et al., 2022). We discuss these associations agnostic about the impacts of the genetic variants themselves, mindful of the fact that GWAS can identify proxy markers that are indicative of a co-inherited genetic characteristic (e.g., structural variants or repeat sequences), may affect splicing, or could modulate expression of another gene or genes (possibly far away on the chromosome) underlying the phenotype(s). With those caveats in mind, we were intrigued by findings of putative risk-associated genetic variation in or near the genes SORL1, TNIP/GPX3, and WNT3.
The SORL1 (Sortilin Related Receptor 1) gene is located on human Chromosome 11 and SORL1 is a well-known AD dementia-linked gene (Barthelson et al., 2020; Campion et al., 2019). This gene stands out because both highly-penetrant SORL1 rare genetic variants are associated with early-onset AD, and other more common SORL1 genetic variants are linked to late-onset “sporadic” AD dementia–including pathologically confirmed ADNC cases (Thonberg et al., 2017). The SORL1 protein is involved in amyloid-β clearance so has a highly credible functional link to ADNC (Willnow and Andersen, 2013). SORL1 mutations have also been linked to FTLD (Benussi et al., 2021). In the current study, we found a nominally statistically significant signal for association between SORL1 variation and LATE-NC, but no association was found between the SORL1 SNVs and ADNC. Note that despite our null result for the association between ADNC and SORL1 variants in this dataset, this is not an “either/or” scenario; rather, SORL1 variant(s) may drive a subtype of LATE-NC with comorbid ADNC. Consistent with that hypothesis, a post-hoc analysis indicated that the rs74685827 SORL1 risk allele was associated with the ADNC+LATE-NC phenotype.
Another genetic locus associated with a non-ADNC pathology was the SNV rs199515 in the WNT3 (Wnt Family Member 3) gene, which was associated with HS risk. HS is an important pathological phenotype indicating neuronal cell death in the hippocampus, often seen at autopsy in brains with comorbid LATE-NC (Amador-Ortiz et al., 2007), and also associated with substantial additional clinical impairment (Gauthreaux et al., 2022). There has been prior indications that genetic variation associated with LATE-NC and HS pathologies overlap incompletely (Dugan et al., 2021; Nelson et al., 2014; Nelson et al., 2015). The WNT3 protein serves as a ligand for members of the frizzled receptors family and functions in the canonical Wnt signaling pathway that results in activation of transcription factors relevant to neurodevelopment (Katoh, 2008; Min et al., 2022). Previously linked to FTLD and Parkinson’s disease risk (Ferrari et al., 2017; Li et al., 2022; Liu et al., 2011), WNT3 resides in an intriguing portion of human Chromosome 17, ~1 MB in the telomeric direction from the potentially pathogenetic MAPT locus, which is an additional ~1 MB distance telomeric from the GRN gene.
A third intriguing genetic association identified in the current study is the SNV rs871269 in the Chromosome 5 TNIP1 (TNFAIP3 Interacting Protein 1) gene, which also was linked to altered risk for HS. Mutations in the TNIP1 gene region were previously associated with altered risk for amyotrophic lateral sclerosis (ALS) (Benyamin et al., 2017; Restuadi et al., 2022). The ALS-linked SNV in TNIP1 (rs10463311) is located ~20 kb away from the AD/dementia-associated SNV evaluated in the current study. One prior study concluded that the TNIP1-region genetic risk allele rs10463311 may exert its effect on ALS risk by modulating expression of the nearby GPX3 (Glutathione Peroxidase 3) gene (Restuadi et al., 2022). Functionally, the TNIP1 protein is an inflammation modulatory protein that exerts its influence by regulating nuclear factor kappa-B activation (Shamilov and Aneskievich, 2018). The GPX3 protein, by contrast, is active in protecting cells from reactive oxygen species and has been connected functionally with superoxide dismutase (Chang et al., 2020).
There were a number of limitations in our study design. Potential confounding factors may bias ascertainment of neuropathologic endophenotypes and thereby distort our genetic association results. We ignored cognitive status in this study, and were only concerned with the correlations between SNV status and neurodegenerative pathologies. We also note that the present work is not a replication or validation study, because many of the cases included from ADGC and NACC data sets were also incorporated in the Bellenguez et al. (2022), albeit studying non-identical phenotypes and the numbers of overlap being <1% of the overall sample size of the latter study. We note however that the large majority of the Bellenguez sample were not neuropathologically characterized as far as we are aware.
There is no autopsy cohort with perfect (or even near-perfect) population representation or 100% autopsy rate. Moreover, sex, race, and socioeconomic factors are known to influence subject recruitment, and practices vary among neuropathologists (Haneuse et al., 2009). The analyses performed on non-Europeans was largely exploratory due to restricted sample sizes (<300 subjects combining cases and controls) and caution is required in over-interpreting such data (Ighodaro et al., 2017). However, the findings were suggestive: the SNV which with the strongest nominal association with dementia-related neuropathology was in the ABCA7 gene, which previously was linked to AD dementia risk, particularly in African-Americans (Hohman et al., 2016; Liao et al., 2014; Logue et al., 2011; Reitz et al., 2013). We also highlighted intriguing possible novel associations such as the finding of a trend for association between PLCG2 SNV variation and LATE-NC risk in non-Europeans.
In addition to recruitment biases, there are other potential confounders that pertain to this study. >30 different research centers contributed research subject data for this study. Neuropathologic practices (specific techniques, diagnostic cut-points applied, etc.) differ between research centers and add variation to the results (Besser et al., 2018). The nosology of LATE-NC is a somewhat contentious area (Josephs et al., 2019; Nelson et al., 2019b). “Border zones” between LATE-NC, FTLD, and ADNC are not universally agreed on; we are mindful of other perspectives and recognize that terminology and underlying scientific assumptions will probably continue to evolve.
From a statistical perspective, we applied multiple statistical tests with a sample size in the low thousands of European cases and controls. Thus, even with applying the sharpened endophenotype of the diagnostic “gold standard” (neuropathology), relatively few genotype/phenotype associations were robust enough to survive correction for multiple comparisons. Future studies may also be required to generate a better analytic framework for polygenic risk profiles for each pathological endophenotype. Nonetheless, there were multiple indications of a valid analytic workflow with suggestive results: (1) The strongest association signals were seen in the expected genetic loci that have been replicated consistently with the same specific neuropathologies in the past, e.g., BIN1 and EED/PICALM SNVs were associated with ADNC risk, whereas TMEM106B and GRN SNVs were associated with risk for LATENC; (2) In the subset of SNVs that trended to association with nominal P < 0.01, the genetic variation was consistently associated with dementia-related pathologies in the same direction (same risk allele), but with larger effect sizes (odds ratios) in comparison to the prior large AD/dementia (mostly clinical data) GWAS; and, (3) The pattern of “hit” SNVs related to ADNC, Lewy body pathology, and LATE-NC/HS conformed generally to the known distribution of attributable risk for AD-type dementia in community-based cohorts (Boyle et al., 2019).
In conclusion, some gene variants that were previously identified as contributing to clinical dementia risk appear to exert their influence via non-ADNC neuropathologies. Among the new findings of our study were novel associations between specific AD-linked genes and the LATE-NC/ HS phenotype(s). Although further replication of these results is required, our findings were consistent with the hypothesis that non-ADNC neuropathologies can contribute substantially to amnestic dementia, and to its heritability.
Supplementary Material
Acknowledgements
We are extremely grateful to the research volunteers, clinicians, and staff who made this study possible.
Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24-AG041689-01); NACC, U01 AG016976; NIA LOAD, U24 AG026395, R01AG041797; Banner Sun Health Research Institute P30 AG019610; Boston University, P30 AG013846, U01 AG10483, R01 CA129769, R01 MH080295, R01 AG017173, R01 AG025259, R01AG33193; Columbia University, P50 AG008702, R37 AG015473; Duke University, P30 AG028377, AG05128; Emory University, AG025688; Group Health Research Institute, UO1 AG006781, UO1 HG004610, UO1 HG006375; Indiana University, P30 AG10133; Johns Hopkins University, P50 AG005146, R01 AG020688; Massachusetts General Hospital, P50 AG005134; Mayo Clinic, P50 AG016574; Mount Sinai School of Medicine, P50 AG005138, P01 AG002219; New York University, P30 AG08051, UL1 RR029893, 5R01AG012101, 5R01AG022374, 5R01AG013616, 1RC2AG036502, 1R01AG035137; Northwestern University, P30 AG013854; Oregon Health & Science University, P30 AG008017, R01 AG026916; Rush University, P30 AG010161, R01 AG019085, R01 AG15819, R01 AG17917, R01 AG30146; TGen, R01 NS059873; University of Alabama at Birmingham, P50 AG016582; University of Arizona, R01 AG031581; University of California, Davis, P30 AG010129; University of California, Irvine, P50 AG016573; University of California, Los Angeles, P50 AG016570; University of California, San Diego, P50 AG005131; University of California, San Francisco, P50 AG023501, P01 AG019724; University of Kentucky, P30 AG028383, AG05144; University of Michigan, P50 AG008671; University of Pennsylvania, P30 AG010124; University of Pittsburgh, P50 AG005133, AG030653, AG041718, AG07562, AG02365; University of Southern California, P50 AG005142; University of Texas Southwestern, P30 AG012300; University of Miami, R01 AG027944, AG010491, AG027944, AG021547, AG019757; University of Washington, P50 AG005136; University of Wisconsin, P50 AG033514; Vanderbilt University, R01 AG019085; and Washington University, P50 AG005681, P01 AG03991.
The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA-funded ADRCs: P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30 AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Neil Kowall, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI Frank LaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PI Eric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI Allan Levey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30 AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD).
The Alzheimer’s Disease Sequencing Project (ADSP) is comprised of two Alzheimer’s Disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer’s Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non-governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Schellenberg, Farrer, Pericak-Vance, Mayeux, and Haines); U01AG049505 to Dr. Seshadri; U01AG049506 to Dr. Boerwinkle; U01AG049507 to Dr. Wijsman; and U01AG049508 to Dr. Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate, U01AG052410 to Dr. Pericak-Vance and U01 AG052409 to Drs. Seshadri and Fornage.
Sequencing for the Follow Up Study (FUS) is supported through U01AG057659 (to Drs. PericakVance, Mayeux, and Vardarajan) and U01AG062943 (to Drs. Pericak-Vance and Mayeux). Data generation and harmonization in the Follow-up Phase is supported by U54AG052427 (to Drs. Schellenberg and Wang). The FUS Phase analysis of sequence data is supported through U01AG058589 (to Drs. Destefano, Boerwinkle, De Jager, Fornage, Seshadri, and Wijsman), U01AG058654 (to Drs. Haines, Bush, Farrer, Martin, and Pericak-Vance), U01AG058635 (to Dr. Goate), RF1AG058066 (to Drs. Haines, Pericak-Vance, and Scott), RF1AG057519 (to Drs. Farrer and Jun), R01AG048927 (to Dr. Farrer), and RF1AG054074 (to Drs. Pericak-Vance and Beecham).
The ADGC cohorts include: Adult Changes in Thought (ACT) (UO1 AG006781, UO1 HG004610, UO1 HG006375, U01 HG008657), the Alzheimer’s Disease Centers (ADC) (P30 AG019610, P30 AG013846, P50 AG008702, P50 AG025688, P50 AG047266, P30 AG010133, P50 AG005146, P50 AG005134, P50 AG016574, P50 AG005138, P30 AG008051, P30 AG013854, P30 AG008017, P30 AG010161, P50 AG047366, P30 AG010129, P50 AG016573, P50 AG016570, P50 AG005131, P50 AG023501, P30 AG035982, P30 AG028383, P30 AG010124, P50 AG005133, P50 AG005142, P30 AG012300, P50 AG005136, P50 AG033514, P50 AG005681, and P50 AG047270), the Chicago Health and Aging Project (CHAP) (R01 AG11101, RC4 AG039085, K23 AG030944), Indianapolis Ibadan (R01 AG009956, P30 AG010133), the Memory and Aging Project (MAP) (R01 AG17917), Mayo Clinic (MAYO) (R01 AG032990, U01 AG046139, R01 NS080820, RF1 AG051504, P50 AG016574), Mayo Parkinson’s Disease controls (NS039764, NS071674, 5RC2HG005605), University of Miami (R01 AG027944, R01 AG028786, R01 AG019085, IIRG09133827, A2011048), the Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study (MIRAGE) (R01 AG09029, R01 AG025259), the National Cell Repository for Alzheimer’s Disease (NCRAD) (U24 AG21886), the National Institute on Aging Late Onset Alzheimer’s Disease Family Study (NIA- LOAD) (R01 AG041797), the Religious Orders Study (ROS) (P30 AG10161, R01 AG15819), the Texas Alzheimer’s Research and Care Consortium (TARCC) (funded by the Darrell K Royal Texas Alzheimer’s Initiative), Vanderbilt University/Case Western Reserve University (VAN/CWRU) (R01 AG019757, R01 AG021547, R01 AG027944, R01 AG028786, P01 NS026630, and Alzheimer’s Association), the Washington Heights-Inwood Columbia Aging Project (WHICAP) (RF1 AG054023), the University of Washington Families (VA Research Merit Grant, NIA: P50AG005136, R01AG041797, NINDS: R01NS069719), the Columbia University HispanicEstudio Familiar de Influencia Genetica de Alzheimer (EFIGA) (RF1 AG015473), the University of Toronto (UT) (funded by Wellcome Trust, Medical Research Council, Canadian Institutes of Health Research), and Genetic Differences (GD) (R01 AG007584). The CHARGE cohorts are supported in part by National Heart, Lung, and Blood Institute (NHLBI) infrastructure grant HL105756 (Psaty), RC2HL102419 (Boerwinkle) and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193.
The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS-Family study, and the Prospective Dementia Registry-Austria (ASPS/PRODEM-Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Erasmus Rucphen Family Study (ERF), the Framingham Heart Study (FHS), and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF) grant number P20545-P05 and P13180 and the Medical University of Graz. The ASPS-Fam is funded by the Austrian Science Fund (FWF) project I904),the EU Joint Programme - Neurode-generative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science) and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM-Austria is supported by the Austrian Research Promotion agency (FFG) (Project No. 827462) and by the Austrian National Bank (Anniversary Fund, project 15435. ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN2682011 00012C). Neurocognitive data in ARIC is collected by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA and NIDCD), and with previous brain MRI examinations funded by R01-HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01-HC-25195 and HHSN268201500001I. This study was also supported by additional grants from the NIA (R01s AG054076, AG049607 and AG033040 and NINDS (R01 NS017950). The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4- 2007-201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002- 01254). High-throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the municipality of Rotterdam. Genetic data sets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project 050-060-810. All studies are grateful to their participants, faculty and staff. The content of these manuscripts is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Health and Human Services.
The FUS cohorts include: the Alzheimer’s Disease Centers (ADC) (P30 AG019610, P30 AG013846, P50 AG008702, P50 AG025688, P50 AG047266, P30 AG010133, P50 AG005146, P50 AG005134, P50 AG016574, P50 AG005138, P30 AG008051, P30 AG013854, P30 AG008017, P30 AG010161, P50 AG047366, P30 AG010129, P50 AG016573, P50 AG016570, P50 AG005131, P50 AG023501, P30 AG035982, P30 AG028383, P30 AG010124, P50 AG005133, P50 AG005142, P30 AG012300, P50 AG005136, P50 AG033514, P50 AG005681, and P50 AG047270), Alzheimer’s Disease Neuroimaging Initiative (ADNI) (U19AG024904), Amish Protective Variant Study (RF1AG058066), Cache County Study (R01AG11380, R01AG031272, R01AG21136, RF1AG054052), Case Western Reserve University Brain Bank (CWRUBB) (P50AG008012), Case Western Reserve University Rapid Decline (CWRURD) (RF1AG058267, NU38CK000480), CubanAmerican Alzheimer’s Disease Initiative (CuAADI) (3U01AG052410), Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) (5R37AG015473, RF1AG015473, R56AG051876), Genetic and Environmental Risk Factors for Alzheimer’s disease Among African Americans Study (GenerAAtions) (2R01AG09029, R01AG025259, 2R01AG048927), Gwangju Alzheimer’s and Related Dementias Study (GARD) (U01AG062602), Hussman Institute for Human Genomics Brain Bank (HIHGBB) (R01AG027944, Alzheimer’s Association “Identification of Rare Variants in Alzheimer Disease”), Ibadan Study of Aging (IBADAN) (5R01AG009956), Mexican Health and Aging Study (MHAS) (R01AG018016), Multi-Institutional Research in Alzheimer’s Genetic Epidemiology (MIRAGE) (2R01AG09029, R01AG025259, 2R01AG048927), Northern Manhattan Study (NOMAS) (R01NS29993), Peru Alzheimer’s Disease Initiative (PeADI) (RF1AG054074), Puerto Rican 1066 (PR1066) (Wellcome Trust (GR066133/GR080002), European Research Council (340755)), Puerto Rican Alzheimer Disease Initiative (PRADI) (RF1AG054074), Reasons for Geographic and Racial Differences in Stroke (REGARDS) (U01NS041588), Research in African American Alzheimer Disease Initiative (REAAADI) (U01AG052410), Rush Alzheimer’s Disease Center (ROSMAP) (P30AG10161, P30AG72975, R01AG15819, R01AG17919, U01AG46152, U01AG61356), University of Miami Brain Endowment Bank (MBB), and University of Miami/Case Western/North Carolina A&T African American (UM/CASE/NCAT) (U01AG052410, R01AG028786).
The four LSACs are: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659), and the Washington University Genome Institute (U54HG003079).
Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Cell Repository for Alzheimer’s Disease (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA funded Alzheimer’s Disease Centers (ADCs), and the National Alzheimer’s Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA This research was supported in part by the Intramural Research Program of the National Institutes of health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations.
Funding
This work was supported by grants the National Institute on Aging R56AG057191, R01AG057187, R21AG061551, R01AG054060, and the UK-ADC P30AG072946.
The National Institutes of Health, National Institute on Aging (NIH-NIA) supported this work through the following grants: ADGC, U01 AG032984, RC2 AG036528; Samples from the National Cell Repository for Alzheimer’s Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study.
Abbreviations:
- ADGC
Alzheimer’s disease Genomics Consortium
- ADNC
Alzheimer’s disease neuropathologic change
- ADRC
Alzheimer’s Disease Research Center
- ADSP
Alzheimer’s Disease Sequencing Project
- ALS
amyotrophic lateral sclerosis
- FDR
false discovery rate
- FTLD
frontotemporal lobar degeneration
- HS
hippocampal sclerosis
- LATE-NC
limbic-predominant age-related TDP-43 encephalopathy neuropathologic change
- LD
linkage disequilibrium
- NACC
National Alzheimer’s Coordinating Center
- NFT
neurofibrillary tangle
- OR
odds ratio
- PCA
principal component analysis
- SNV
single nucleotide (genetic) variant
- ROSMAP
Rush University Religious Orders Study and the Memory and Aging Project
- WGS
whole genome sequencing
Footnotes
CRediT authorship contribution statement
Yuriko Katsumata: Conceptualization, Data curation, Formal analysis, Funding acquisition, Project administration, Resources, Writing – original draft, Writing – review & editing. Lincoln M. Shade: Data curation, Formal analysis, Writing – review & editing. Timothy J. Hohman: Data curation, Writing – review & editing. Julie A. Schneider: Data curation, Writing – review & editing. David A. Bennett: Data curation, Project administration, Writing – review & editing. Jose M. Farfel: Data curation, Writing – review & editing. Walter A. Kukull: Data curation, Project administration, Resources, Writing – review & editing. David W. Fardo: Conceptualization, Data curation, Formal analysis, Funding acquisition, Project administration, Resources, Writing – original draft, Writing – review & editing. Peter T. Nelson: Conceptualization, Data curation, Formal analysis, Funding acquisition, Project administration, Resources, Writing – original draft, Writing – review & editing.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105880.
Data availability
Data will be made available on request.
References
- Amador-Ortiz C, et al. , 2007. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol 61, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attems J, et al. , 2021. Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-Centre study. Acta Neuropathol. 141, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelson K, et al. , 2020. Sorting out the role of the Sortilin-related receptor 1 in Alzheimer’s disease. J. Alzheimers Dis. Rep 4, 123–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, et al. , 2012. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer disease centers, 2005–2010. J. Neuropathol. Exp. Neurol 71, 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beecham GW, et al. , 2014. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 10, e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellenguez C, et al. , 2022. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet 54, 412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, et al. , 2018. Religious orders study and rush memory and aging project. J. Alzheimers Dis 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benussi L, et al. , 2021. Investigating the Endo-lysosomal system in major neurocognitive disorders due to Alzheimer’s disease, frontotemporal lobar degeneration and Lewy body disease: evidence for SORL1 as a cross-disease gene. Int. J. Mol. Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyamin B, et al. , 2017. Cross-ethnic meta-analysis identifies association of the GPX3TNIP1 locus with amyotrophic lateral sclerosis. Nat. Commun 8, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser LM, et al. , 2018. The revised National Alzheimer’s coordinating Center’s neuropathology form-available data and new analyses. J. Neuropathol. Exp. Neurol 77, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, et al. , 2019. Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann. Neurol 85, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, et al. , 2021. To what degree is late life cognitive decline driven by age-related neuropathologies? Brain 144 (7), 2166–2175. 10.1093/brain/awab092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Alzheimer’s disease affects limbic nuclei of the thalamus. Acta Neuropathol. 81, 261–268. [DOI] [PubMed] [Google Scholar]
- Brenowitz WD, et al. , 2017. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimers Dement. 13, 654–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion D, et al. , 2019. SORL1 genetic variants and Alzheimer disease risk: a literature review and meta-analysis of sequencing data. Acta Neuropathol. 138, 173–186. [DOI] [PubMed] [Google Scholar]
- Chang C, et al. , 2020. Extracellular glutathione peroxidase GPx3 and its role in Cancer. Cancers (Basel). 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, et al. , 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane PK, et al. , 2017. Alzheimer’s disease sequencing project discovery and replication criteria for cases and controls: data from a community-based prospective cohort study with autopsy follow-up. Alzheimers Dement. 13, 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cykowski MD, et al. , 2022. Patterns of amygdala region pathology in LATE-NC: subtypes that differ with regard to TDP-43 histopathology, genetic risk factors, and comorbid pathologies. Acta Neuropathol. 143 (5), 531–545. 10.1007/s00401-022-02416-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan AJ, et al. , 2021. Analysis of genes (TMEM106B, GRN, ABCC9, KCNMB2, and APOE) implicated in risk for LATE-NC and hippocampal sclerosis provides pathogenetic insights: a retrospective genetic association study. Acta Neuropathol. Commun 9, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel JM, et al. , 2016. Relation of genomic variants for Alzheimer disease dementia to common neuropathologies. Neurology. 87, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, et al. , 2017. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 88, 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, et al. , 2006. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. [DOI] [PubMed] [Google Scholar]
- Gauthreaux KM, et al. , 2022. Limbic-predominant age-related TDP-43 encephalopathy: medical and pathologic factors associated with comorbid hippocampal sclerosis. Neurology. 98, e1422–e1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project, Consortium, et al. , 2010. A map of human genome variation from population-scale sequencing. Nature. 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneuse S, et al. , 2009. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology. 32, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohman TJ, et al. , 2016. Global and local ancestry in African-Americans: implications for Alzheimer’s disease risk. Alzheimers Dement. 12, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ighodaro ET, et al. , 2017. Challenges and considerations related to studying dementia in blacks/African Americans. J. Alzheimers Dis 60, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., et al. , 2016. Suspected non-Alzheimer disease pathophysiology–concept and controversy. Nat. Rev. Neurol 12, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James BD, et al. , 2016. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain. 139, 2983–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, et al. , 2015. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 129, 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, et al. , 2019. LATE to the PART-y. Brain. 142, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanth SD, et al. , 2021. Four common late-life cognitive trajectories patterns associate with replicable underlying Neuropathologies. J. Alzheimers Dis 82, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M, 2008. WNT signaling in stem cell biology and regenerative medicine. Curr. Drug Targets 9, 565–570. [DOI] [PubMed] [Google Scholar]
- Katsumata Y, et al. , 2020. Distinct clinicopathologic clusters of persons with TDP-43 proteinopathy. Acta Neuropathol. 140, 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, et al. , 2018. The National Institute on Aging and the Alzheimer’s Association research framework for Alzheimer’s disease: perspectives from the research roundtable. Alzheimers Dement. 14, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryscio RJ, et al. , 2016. Self-reported memory complaints: a comparison of demented and unimpaired outcomes. J. Prev. Alzheimers Dis 3, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukull WA, et al. , 1996. Apolipoprotein E in Alzheimer’s disease risk and case detection: a case-control study. J. Clin. Epidemiol 49, 1143–1148. [DOI] [PubMed] [Google Scholar]
- Li C, et al. , 2022. Shared genetic links between frontotemporal dementia and psychiatric disorders. BMC Med. 20, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YC, et al. , 2014. ABCA7 gene and the risk of Alzheimer’s disease in Han Chinese in Taiwan. Neurobiol. Aging 35, 2423 e7–2423 e13. [DOI] [PubMed] [Google Scholar]
- Liu X, et al. , 2011. Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med. Genet 12, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue MW, et al. , 2011. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch. Neurol 68, 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, Chanock SJ, 2015. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 31, 3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, et al. , 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RI, Schneider JA, 2021. What is ‘Alzheimer’s disease’? The neuropathological heterogeneity of clinically defined Alzheimer’s dementia. Curr. Opin. Neurol 34, 237–245. [DOI] [PubMed] [Google Scholar]
- Min JK, et al. , 2022. Cross-talk between Wnt signaling and Src tyrosine kinase. Biomedicines. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, 1997. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol. Aging 18, S91–S94. [DOI] [PubMed] [Google Scholar]
- Mirra SS, et al. , 1991. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 41, 479–486. [DOI] [PubMed] [Google Scholar]
- Montine TJ, et al. , 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, et al. , 2017. Genomic variants, genes, and pathways of Alzheimer’s disease: an overview. Am. J. Med. Genet. B Neuropsychiatr. Genet 174, 5–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, et al. , 2018. Quality control and integration of genotypes from two calling pipelines for whole genome sequence data in the Alzheimer’s disease sequencing project. Genomics. 111 (4), 808–818. 10.1016/j.ygeno.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2007. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J. Neuropathol. Exp. Neurol 66, 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2010. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 20, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2011. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 134, 1506–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2014. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol. 127, 825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2015. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J. Neuropathol. Exp. Neurol 74, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2016. “new old pathologies”: AD, PART, and cerebral age-related TDP-43 with sclerosis (CARTS). J. Neuropathol. Exp. Neurol. 75, 482–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2019a. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2019b. Reply: LATE to the PART-y. Brain. 142, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 2022. Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol. 144 (1), 27–77. 10.1007/s00401-022-02444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neltner JH, et al. , 2016. Brain pathologies in extreme old age. Neurobiol. Aging 37, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, et al. , 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314, 130–133. [DOI] [PubMed] [Google Scholar]
- O’Meara ES, et al. , 1997. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am. J. Epidemiol 146, 373–384. [DOI] [PubMed] [Google Scholar]
- Purcell S, et al. , 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, et al. , 2013. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA. 309, 1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restuadi R, et al. , 2022. Functional characterisation of the amyotrophic lateral sclerosis risk locus GPX3/TNIP1. Genome Med. 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamilov R, Aneskievich BJ, 2018. TNIP1 in autoimmune diseases: regulation of toll-like receptor signaling. J Immunol Res 2018, 3491269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim YS, et al. , 2013. Clinicopathologic study of Alzheimer’s disease: Alzheimer mimics. J. Alzheimers Dis 35, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliun D, et al. , 2019. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv, 563866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thonberg H, et al. , 2017. Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol. Commun 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang DW, et al. , 2005. Genetic association between the APOE*4 allele and Lewy bodies in Alzheimer disease. Neurology. 64, 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, et al. , 2007. Rates of cerebral atrophy differ in different degenerative pathologies. Brain. 130, 1148–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willnow TE, Andersen OM, 2013. Sorting receptor SORLA–a trafficking path to avoid Alzheimer disease. J. Cell Sci 126, 2751–2760. [DOI] [PubMed] [Google Scholar]
- Yang HS, et al. , 2020. Genetics of gene expression in the aging human brain reveal TDP-43 Proteinopathy pathophysiology. Neuron. 107, 496–508 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.